

Abstract
Hepatitis B (HB) is a viral infectious disease that seriously endangers human health, and since there are no radical drugs to counter this, effective and safe therapies urgently need to be developed. HB virus (HBV) mainly infects hepatocytes (HCs), while the drugs are easily phagocytosed by Kupffer cells (KCs). In this study, the glutathione concentration difference between HCs and KCs was examined and utilized in an ideal drug-release strategy. Here, galactosylated chitosan-oligosaccharide-SS-octadecylamine (Gal-CSSO) was prepared to accurately deliver 10-23 DNAzyme DrzBC (blocking HBeAg expression) or DrzBS (blocking HBsAg expression) in targeted HB therapy. In vitro Gal-CSSO systems exhibited low cytotoxicity, endosomal escape, and glutathione responsiveness. The HBeAg and HBsAg secretion of HepG2.2.15 was significantly decreased by Gal-CSSO systems, and the maximum inhibition rates were 1.82-fold and 2.38-fold greater than those of commercial Lipofectamine 2000 (Lipo2000) systems. In vivo Gal-CSSO systems exhibited HC targeting and HC microenvironmental responsiveness without noticeable hepatotoxicity or systemic toxicity. The HBeAg and HBsAg titers of the HBV-infected mice were evidently decreased by Gal-CSSO systems, and the inhibition rates were 1.52-fold and 1.22-fold greater than those of Lipo2000 systems. This study presents a kind of glycolipid-like polymer micelles that promise efficient and safe gene therapy of HB.
Keywords: glycolipid-like polymer micelles, hepatocyte target, hepatocyte microenvironmental response, hepatitis B, gene therapy
Graphical abstract
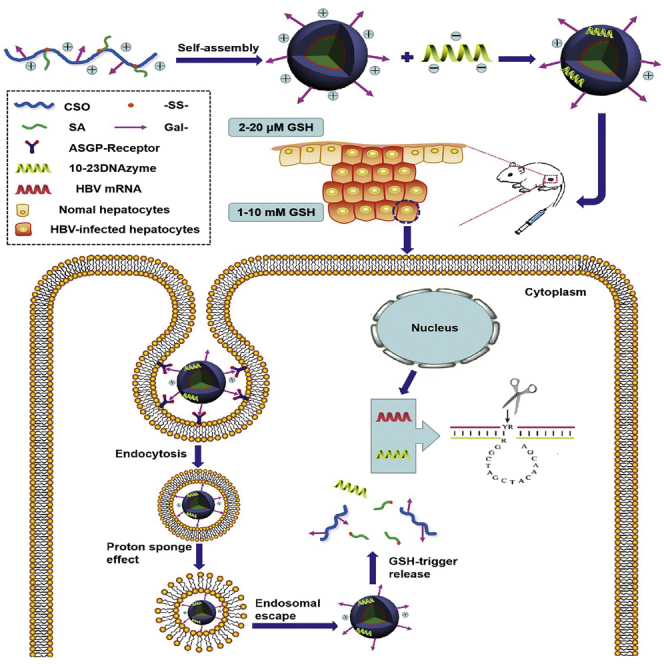
Wu, Hu, and colleagues developed a kind of glycolipid-like polymer micelles to accurately deliver DNAzyme in targeted hepatitis B therapy to block viral gene expression. This drug delivery system exhibited hepatocyte targeting and microenvironmentally responsive cargo release without hepatotoxicity or systemic toxicity, suggesting a potential gene therapy for hepatitis B.
Introduction
Hepatitis B (HB) is a viral infectious disease that seriously endangers human health. Every year, nearly one million people die of liver failure, cirrhosis, and liver cancer caused by HB.1 Today, the drugs for HB mainly include nucleoside analogs and interferon, which have many problems including a long course of treatment, high dosage, low efficacy, drug resistance, and relapse after drug withdrawal.2,3 Therefore, genetic drugs have recently been formulated to target the life cycle of the hepatitis B virus (HBV) directly.4, 5, 6 Among them, deoxyribozyme 10-23 DNAzyme has attracted much attention from researchers because of its unique advantages in the treatment of viral infectious diseases.7,8 10-23 DNAzyme is the 23rd clone from round 10 during the course of 10 rounds of in vitro screening.9 Here, we designed two kinds of 10-23 DNAzymes specific to HBV e-gene, ORF A1816UG (DrzBC), and s-gene, ORF A157UG (DrzBS). DrzBC and DrzBS can specifically block the expression of HBeAg (the component of the HBV core antigen and an indicator of HBV replication) and HBsAg (the HBV coat protein antigen and an indicator of HBV infection), respectively. The molecular structure and functional mechanism of DrzBC and DrzBS are shown in the Supplemental information (Figure S1).
However, the practical applications of DNAzyme are still restricted by the difficulty of delivery due to the high molecular weight,10 the possibility of degradation by various enzymes in vivo,11 and the lack of focused and selective infection targeting. Even if 10-23 DNAzyme is delivered into the blood with certain vectors, such as the common commercially available Lipofectamine 2000 (Lipo2000), it is easily phagocytosed by the reticuloendothelial system and only partially delivered into the liver. However, there are many non-parenchymal cells (mainly Kupffer cells [KCs]) in the liver, and the drugs delivered into the blood are inclined to be phagocytosed by KCs, limiting the efficacy of the drugs targeted to hepatocytes (HCs). Considering that the main host of HBV is HCs,12 the nonspecific uptake of 10-23 DNAzyme in non-target cells may reduce the gene therapy effect and lead to side effects such as hepatotoxicity. Therefore, for efficient and safe gene therapy of HB, it is particularly important to design a functional vector vehicle for delivering and releasing 10-23 DNAzyme into HCs.
The asialoglycoprotein receptors (ASGPRs) have been validated as potential targets for selective drugs delivered to the liver because of the specific expression and high capacity for receptor-mediated endocytosis. Since ASGPRs can specifically recognize β-d-galactose or N-acetylgalactosamine residues of desialylated glycoproteins, the polymers with galactosyl residues on their surfaces can enhance the drug accumulation in the liver tissue through active targeting.13,14 Moreover, as ASGPRs are expressed primarily on the sinusoidal surfaces of HCs,15 the polymers modified by galactosyl residues have the potential to achieve HC targeting.
For releasing drugs into HCs selectively, the microenvironment of the liver is a crucial factor. According to Rodrigues and Percival16 and Jones et al.,17 liver tissue is the main site for glutathione (GSH) synthesis. The concentration of GSH in the liver is ∼1–10 mM, while in the extracellular fluid and systemic circulation it is ∼2–20 μM. However, the GSH concentration in HCs, particularly when they are infected by HBV, has not been explained or addressed yet. In this study, we tried to extract HCs and KCs from the liver and determine the GSH concentration. In the liver of HBV-infected mice, an obvious GSH concentration difference was observed between HCs and KCs, and the GSH concentration in HCs was >1 mM. Therefore, a GSH-responsive vector may be conducive to drug delivery and may be able to target infections accurately.
Usually, the GSH-responsive vectors have disulfide linkages in a hydrophilic shell, in a hydrophobic core, or in the cross-linker.18,19 In extracellular fluid and systemic circulation, the vectors with disulfide linkages are stable; however, after cellular uptake, the disulfide linkages are degraded, responding to the intracellular high GSH level. Owing to the high variance of GSH level between tumor cells and normal cells, a variety of vectors with disulfide bonds were developed to deliver and release anti-tumor drugs.20, 21, 22 Few studies have assessed the GSH concentration difference in the different liver cells to develop HC-targeting, microenvironmentally responsive vectors.
In this study, we designed a glycolipid-like polymer named galactosylated chitosan oligosaccharide-SS-octadecylamine (Gal-CSSO) that could self-assemble into micelles in an aqueous medium. By electrostatic interaction, Gal-CSSO micelles could couple with DrzBC and DrzBS to form Gal-CSSO/DrzBC and Gal-CSSO/DrzBS systems, respectively. Gal-CSSO gene delivery systems could specifically target HCs owing to the modification of galactosyl residues, achieve endosomal escape owing to the chitosan oligosaccharide (CSO), and realize HC microenvironmentally responsive fast drug release owing to the structure of –SS–. The physicochemical characteristics of Gal-CSSO, Gal-CSSO/DrzBC, and Gal-CSSO/DrzBS were investigated. The anti-HBV efficacy and safety of Gal-CSSO/DrzBC and Gal-CSSO/DrzBS systems were evaluated both in vitro and in vivo.
The current study presents a kind of HC-targeting and microenvironmentally responsive glycolipid-like polymer micelles that promise efficient and safe gene therapy of HB.
Results
Synthesis and characterization of Gal-CSSO
Gal-CSSO was prepared by a two-step procedure, as shown in Figure 1A. First, 3,3¢-dithiodipropionic acid (DTPA) was used as the coupling agent, and one side of the carboxyl groups was modified with octadecylamine (ODA) at a molar ratio of 1:1. Thereafter, the remaining carboxyl group was reacted with the free amino groups of the chitosan by amine-reactive coupling to produce CSSO. For realizing HC targeting, lactobionic acid (LA) was coupled with the amino groups of CSSO via an amidation reaction.
Figure 1.

Synthesis of Gal-CSSO
(A) Synthetic scheme of Gal-CSSO. (B) 1H NMR spectra of Gal-CSSO.
The 1H NMR spectra of CSSO, LA, and Gal-CSSO are shown in Figure 1B. In the Gal-CSSO spectra, the proton signals at 3.45–3.95 ppm are attributed to the proton peaks of the sugar ring of LA; the proton signals at 5.02 ppm represent the proton peaks of LA labeled as 1 and 2; the proton signal at 5.10 ppm represents the proton peak of LA labeled as 3; and the proton signal at 5.30 ppm represents the proton peak of LA labeled as 4. The results confirm the successful grafting of the galactosyl of LA onto CSSO.
As shown in Table S1, the substitution degree of amino groups (SD%) of the CSO chain on Gal-CSSO was measured as 11.91% ± 0.21%. The synthesized Gal-CSSO could self-assemble easily into nano-sized micelles with an average size of 138.33 ± 3.68 nm and a zeta potential of 16.93 ± 0.42 mV. The variations in the I1/I3 ratio against the logarithmic concentration (log C) of Gal-CSSO are shown in Figure S2A, and the inflection point corresponding to the critical micelle concentration (CMC) value of Gal-CSSO was calculated as 81.06 ± 1.32 μg⋅mL−1.
Optimal mass ratio selection
The coupling ability of Gal-CSSO with 10-23 DNAzyme was assessed with a denaturing 7 M urea polyacrylamide gel electrophoresis assay. Considering the similarity in physical properties between DrzBC and DrzBS, we chose to only examine DrzBC in this experiment.
As shown in Figure 2A, the electrophoretic bands in lanes 3–6 were slightly different in intensity compared with those of naked DrzBC (lane 1). However, because the compaction of DrzBC was influenced by any retained DrzBC content in the sample well, we mainly investigated differences in the sample wells and not the electrophoretic bands per se. Differences were seen among lanes 3–6 in the sample wells in which the ratio of Gal-CSSO/DrzBC (w/w) was varied. When the ratio was 10:1 (lane 3), a relatively small amount of DrzBC was retained in the sample well, whereas when the ratio was 20:1 (lane 4), the retention of DrzBC in the sample well was more obvious, suggesting that polymers were able to compact DrzBC better when the mass ratio was 20:1 or greater. When the ratio was increased to 60:1 (lane 5), the electrophoretic band was slightly brighter, but the retention in the loading well was also enhanced compared with that of the naked control and lower ratios, indicating that the composite force of Gal-CSSO and DrzBC increased. When the ratio was increased to 100:1 (lane 6), the electrophoretic band was not diluted; however, the retention in the sample well also was not significantly greater (brighter) than that observed for the 60:1 sample. From these results, we concluded that when the ratio was 60:1 DrzBC was close to completely blocked in the sample well.
Figure 2.

Optimal mass ratio determination
(A) Gel retardation analysis of Gal-CSSO/DrzBC prepared at different mass ratios. Lane 1, naked DrzBC; lane 2, Lipo/DrzBC; lanes 3–6, Gal-CSSO/DrzBC prepared at mass ratios of 10, 20, 60, and 100, respectively. (B) In vivo cellular uptake of Gal-CSSO/DrzBC by hepatocytes. The P6 gate is the BV421-positive cells in the Cy5-positive cells. x axis, fluorescence intensity; y axis, side scatter area (SSC-A).
Regarding lane 2 (Lipo2000/DrzBC), it is noteworthy that neither the sample well nor the electrophoretic band exhibited DrzBC staining at any significant level. The reason may be the powerful ability of Lipo2000 to compact DrzBC, leading to the difficulty in staining DrzBC by ethidium bromide (EB). However, if the compaction ability is too powerful, it would make it difficult to release DrzBC.
To further screen the optimal mass ratio of Gal-CSSO/DrzBC, the uptake of complexes by HCs at mass ratios of 20:1, 60:1, and 100:1 was investigated. As Figure 2B shows, the value in the P6 gate, which shows the BV421-positive cells in the Cy5-positive cells and represents the percentage of HCs in the Cy5-positive cells, was highest in the group of Gal-CSSO/DrzBC at a mass ratio of 60:1, indicating the most effective uptake. Therefore, 60:1 was determined to be the optimal ratio, and the complexes were prepared at this mass ratio in our following studies. Because of the limited detection time during the experiment, the number of cells collected was different but sufficient to ensure the accuracy of the results.
Preparation and characterization of Gal-CSSO/10-23DNAzyme
Gal-CSSO/10-23 DNAzyme was prepared by mixing Gal-CSSO with DrzBC or DrzBS at the mass ratio of 60:1. The characteristics of Gal-CSSO/DrzBC and Gal-CSSO/DrzBS are exhibited in Table 1. The zeta potentials of Gal-CSSO/10-23 DNAzyme were all positive, and the average diameters were 240–260 nm with low polydispersity indexes (PDIs) that were larger than those of Gal-CSSO, resulting from 10-23 DNAzyme adherence on the surface of polymers via electrostatic interaction. There was no obvious difference between Gal-CSSO/DrzBC and Gal-CSSO/DrzBS with respect to zeta potential or average diameter because of the similarity in physical properties between DrzBC and DrzBS.
Table 1.
Characterization of Gal-CSSO gene delivery systems
| Micelles | Diameter (nm) | PDI | Zeta potential (mV) | EE% | DL% |
|---|---|---|---|---|---|
| Gal-CSSO/DrzBC | 246.73 ± 6.73 | 0.22 ± 0.06 | 13.93 ± 1.05 | 94.85 ± 0.31 | 1.555 ± 0.005 |
| Gal-CSSO/DrzBS | 253.00 ± 8.65 | 0.24 ± 0.03 | 13.55 ± 0.87 | 95.52 ± 0.34 | 1.566 ± 0.006 |
PDI, EE, and DL values represent the polydispersity index, drug encapsulation efficiency, and drug loading, respectively. Data represent the mean ± SD (n = 3).
As shown in Table 1, the encapsulation efficiencies (EEs%) of Gal-CSSO/Cy5-DrzBC and Gal-CSSO/Cy5-DrzBS complexes were 94.85% ± 0.31% and 95.52% ± 0.34%, respectively, demonstrating the excellent encapsulation ability of Gal-CSSO. The drug loading percentages (DLs%) of the complexes were 1.555% ± 0.005% and 1.566% ± 0.006%, respectively, because of the preparation mass ratio of 60:1, leading to the low proportion of 10-23 DNAzyme in the Gal-CSSO gene system.
Transmission electron microscopy (TEM) images (Figure 3A) showed the spherical and irregular morphology of Gal-CSSO polymer micelles that may be related to the negative dyeing process of TEM observation. The surface of Gal-CSSO/DrzBC and Gal-CSSO/DrzBS looked relatively rough, with particle size significantly increased from that of Gal-CSSO (Figure S2B). This is probably due to the multiple composition of the complexes and the solid state of the complexes after negative dyeing. In addition, the average diameters of complexes in deionized (DI) water were larger than those shown in TEM images because of the severe shrinkage of the complexes after vacuum extraction.
Figure 3.

Characterization of complexes in vitro
(A) Transmission electron microscopy (TEM) images of Gal-CSSO/DrzBC and Gal-CSSO/DrzBS. (B) DNase I protection assay: −/+ represent the complexes untreated/treated with DNase I. Complexes were prepared at a mass ratio of 60:1. (C and D) DrzBC (C) and DrzBS (D) release behavior in Gal-CSSO gene delivery systems (n = 3).
The protective effects of CSSO and Gal-CSSO on DrzBC were studied by incubation with DNase I. Figure 3B shows that naked DrzBC incubated with DNase I did not exhibit an obvious band, indicating that DrzBC was completely degraded by DNase I. CSSO/DrzBC and Gal-CSSO/DrzBC incubated with DNase I exhibited obvious bands, very similar to DrzBC without DNase I incubation. This indicated that DrzBC was not degraded; CSSO and Gal-CSSO could effectively protect DrzBC, significantly reducing nuclease degradation and thereby ensuring the effectiveness of gene drugs.
GSH-responsive release behavior
With the structure of reducible disulfide bonds, –SS–, the glycolipid-like nanocarrier Gal-CSSO is redox-responsive to high GSH concentration. We had isolated HCs and KCs from the livers of HBV-infected mice, and the GSH concentration of HCs was determined as 1.04 mM, while the GSH concentration of KCs was merely 0.03 mM. Therefore, it may be concluded that the glutathione-responsive release behavior of the Gal-CSSO gene delivery system might have been triggered by the HCs’ microenvironment. As shown in Figures 3C and 3D, the 10-23 DNAzyme release from the Gal-CSSO gene delivery system accelerated with the increase in GSH concentration. Compared with the release rate in the release medium containing 0 mM GSH, the 10-23 DNAzyme release rate from the Gal-CSSO gene delivery system slightly increased in the 0.1 mM GSH medium; however, the release rate significantly increased in the release medium containing 1.0 mM and 10 mM GSH. The results confirmed the sensitive and sufficient 10-23 DNAzyme release by the Gal-CSSO system while targeting HCs with >1.0 mM GSH.
In vitro cytotoxicity
The methylthiazole tetrazolium (MTT) assay was conducted to evaluate the cytotoxicity of Gal-CSSO and Gal-CSSO/10-23 DNAzyme against HepG2.2.15 cells. As shown in Figure S3, when the concentration of Gal-CSSO was 200 μg⋅mL−1, which is higher than the in vitro administration dosage, HepG2.2.15 cell viability was >80%. This implied low cytotoxicity and safe applicability potential of Gal-CSSO. Further, the cytotoxicity of the Gal-CSSO/10-23 DNAzyme complexes is exhibited in Figures 4A and 4B. When the 10-23 DNAzyme concentration in the complexes was 2 μg⋅mL−1, which is much higher than the administration dosage for anti-HBV, the HepG2.2.15 cell viability was >80%, suggesting the low cytotoxicity of Gal-CSSO/10-23 DNAzyme complexes.
Figure 4.

In vitro cytotoxicity and intracellular trafficking
(A and B) Cytotoxicity of Gal-CSSO/DrzBC (A) and Gal-CSSO/DrzBS (B) against HepG2.2.15 cells after treatment for 48 h (n = 3). (C) Intracellular localization of FITC-Gal-CSSO/Cy5-DrzBC in HepG2.2.15 cells after 1, 4, and 12 h. Lysosomes were labeled with LysoTracker (blue), Gal-CSSO was labeled with FITC (green), DrzBC was labeled with Cy5 (red), Merge 1 is the merged image of the red and blue channels, and Merge 2 is the merged image of the red and green channels.
Intracellular trafficking
To ensure high efficiency of 10-23 DNAzyme delivery, escape from the endosomes is an important criterion.23,24 To investigate whether Gal-CSSO/10-23 DNAzyme could escape from the endosomes to achieve efficient 10-23 DNAzyme delivery after cellular uptake, the intracellular distribution of Gal-CSSO/DrzBC in HepG2.2.15 cells was observed after 1, 4, and 12 h with confocal laser scanning microscopy (CLSM). Fluorescein isothiocyanate (FITC)-labeled Gal-CSSO and Cy5-labeled DrzBC were used to prepare complexes by vortex mixing, and the lysosomes were stained with LysoTracker Blue. If the Cy5-labeled DrzBC (red) and LysoTracker-labeled lysosome (blue) coincided in the merge graph, a blue-purple fluorescence was generated. If the Cy5-labeled DrzBC (red) and FITC-labeled Gal-CSSO (green) coincided in the merge graph, a yellow-green fluorescence was generated.
As shown in merged image 1 in Figure 4C, after 1 h some red fluorescence spots of Cy5-DrzBC were distinguishable from the blue fluorescence spots of lysosomes, suggesting that some DrzBC escaped from lysosomes. In merged image 2, some red fluorescence spots of Cy5-DrzBC were distinguishable from the green fluorescence spots of FITC-Gal-CSSO, suggesting that some DrzBC was released from the Gal-CSSO gene delivery system. In merged image 1 after 4 h and 12 h, the number of free red fluorescence spots did not show obvious difference; however, in merged image 2 there was still a certain amount of red-green fluorescence overlap at 4 h, indicating that Cy5-DrzBC release from FITC-Gal-CSSO was limited. In contrast, in merged image 2 at 12 h there was little red-green fluorescence overlap, indicating that a large amount of DrzBC escaped from the lysosomes and was released into the cytoplasm through the Gal-CSSO gene delivery system.
In vitro anti-HBV efficacy
As shown in Table 2, the HBeAg secretion inhibition rates (IRs) of the free DrzBC solution were between 3% and 5%, indicating that DrzBC could barely demonstrate its efficacy without an effective delivery carrier. The HBeAg secretion IRs of Lipo2000/DrzBC reached a peak value of 47.29% ± 1.86% after 48 h; however, they decreased to 23.98% ± 0.81% after 72 h, indicating a high cytotoxicity of Lipo2000. In comparison with the Lipo2000/DrzBC IRs, the HBeAg secretion IRs of Gal-CSSO/DrzBC were significantly improved to 75.39% ± 1.81% after 48 h (p < 0.001) and further increased to 85.89% ± 2.57% after 72 h (p < 0.0001).
Table 2.
HBeAg secretion inhibition rates of different DrzBC systems
| Time (h) | DrzBC (%) | Lipo2000/DrzBC (%) | CSSO/DrzBC (%) | Gal-CSSO/DrzBC (%) |
|---|---|---|---|---|
| 24 | 3.84 ± 0.22 | 35.12 ± 0.77 | 68.30 ± 2.79 | 69.97 ± 2.15 |
| 48 | 4.21 ± 0.25 | 47.29 ± 1.86 | 73.86 ± 1.77 | 75.39 ± 1.81 |
| 72 | 4.89 ± 0.19 | 23.98 ± 0.81 | 83.83 ± 2.34 | 85.89 ± 2.57 |
Data represent the mean ± SD (n = 3).
Moreover, the maximum IR of Gal-CSSO/DrzBC was 1.82-fold greater than that of Lipo2000/DrzBC.
Similarly, as shown in Table 3, because of the lack of effective delivery carriers, the HBsAg secretion IRs of free DrzBS solution were merely between 2% and 5%. The HBsAg secretion IRs of Lipo2000/DrzBS reached a peak of 33.58% ± 0.72% after 48 h but decreased to 22.76% ± 0.67% after 72 h. The HBsAg secretion IRs of Gal-CSSO/DrzBS reached 69.34% ± 1.95% after 48 h and continued to rise to 79.86% ± 1.87% after 72 h. The IRs of Gal-CSSO/DrzBS were significantly higher (48 h, p < 0.001; 72 h, p < 0.0001) and the maximum IR of Gal-CSSO/DrzBS was 2.38-fold greater than that of Lipo2000/DrzBS.
Table 3.
HBsAg secretion inhibition rates of different DrzBS systems
| Time (h) | DrzBS (%) | Lipo2000/DrzBS (%) | CSSO/DrzBS (%) | Gal-CSSO/DrzBS (%) |
|---|---|---|---|---|
| 24 | 2.25 ± 0.29 | 19.35 ± 0.31 | 55.84 ± 1.03 | 56.98 ± 1.08 |
| 48 | 4.56 ± 0.41 | 33.58 ± 0.72 | 67.80 ± 2.51 | 69.34 ± 1.95 |
| 72 | 3.95 ± 0.15 | 22.76 ± 0.67 | 76.79 ± 2.18 | 79.86 ± 1.87 |
Data represent the mean ± SD (n = 3).
In vivo distribution
To verify whether Gal-CSSO could be an effective liver-targeting carrier to deliver 10-23 DNAzyme, the in vivo distributions of CSSO/Cy5-DrzBC and Gal-CSSO/Cy5-DrzBC were investigated in BALB/c mice. As shown in Figure 5A, the fluorescence signals of Cy5-DrzBC in the Gal-CSSO/Cy5-DrzBC group were primarily observed in the liver, while in the Cy5-DrzBC and CSSO/Cy5-DrzBC groups only weak fluorescence signals were observed in the liver. Further, the fluorescence enrichment of Gal-CSSO/Cy5-DrzBC in the liver was verified by observing the dissected tissue and eliminating the interference of fluorescence penetration in vivo. The results showed that the accumulation of Gal-CSSO/Cy5-DrzBC in the liver was enhanced with the modification of LA, confirming the active targeting ability of Gal-CSSO.
Figure 5.

In vivo distribution and cellular uptake selectivity
(A) Distribution in BALB/c mice of Gal-CSSO/Cy5-DrzBC after intravenous injection for 6, 12, and 24 h. The color bar indicates the average fluorescence intensity. (B) Quantitative analysis of HCs and KCs that absorbed Cy5-DrzBC in different systems. HCs and KCs were isolated from livers of mice after intravenous injection for 12 h and analyzed by flow cytometry.
In vivo cellular uptake selectivity
To confirm whether the cellular uptake of Gal-CSSO/Cy5-DrzBC could be attributed to endocytosis by HCs or phagocytosis by KCs, the in vivo cellular uptake by HCs and KCs was investigated via immunofluorescence assay. In the Gal-CSSO/Cy5-DrzBC group (Figure S4A), a considerable number of Cy5-DrzBC red points were colocalized with HCs (green points) to generate a yellow-green fluorescence. In contrast, only a few Cy5-DrzBC red points overlapped with HCs in the CSSO/Cy5-DrzBC group. As shown in Figure S4B, neither CSSO/Cy5-DrzBC nor Gal-CSSO/Cy5-DrzBC showed any obvious co-localization of Cy5-DrzBC with KCs. These results indicated that CSSO/Cy5-DrzBC and Gal-CSSO/Cy5-DrzBC were mainly absorbed by HCs rather than KCs; Gal-CSSO/Cy5-DrzBC, in particular, has active HC-targeting ability.
To further quantitatively analyze the in vivo HC-targeting of Gal-CSSO/DrzBC, DrzBC was labeled with Cy5, and the percentages of HCs and KCs exhibiting uptake of Cy5-DrzBC were measured via flow cytometry. Based on the results shown in Figure 5B, the number of HCs that absorbed Gal-CSSO/DrzBC was 7.78-fold greater than the number of KCs, indicating the Gal-receptor-mediated active targeted uptake.25 Owing to the lack of Gal receptors on the cellular surface, the primary uptake mechanism of KCs was phagocytosis.26 The results further confirmed the HC-targeting ability of the in vivo Gal-CSSO gene delivery system.
In vivo toxicity
The body weights of mice were measured every day after administration of the corresponding complexes on day 0 (Figures 6A and 6B). The results showed a gradual increase in the body weights of the mice treated with complexes, similar to the trend observed in the saline group mice, demonstrating that none of the complexes had any effect on the body weight of the mice.
Figure 6.

In vivo toxicity and anti-HBV efficacy
(A and B) Weight changes in mice treated with (A) DrzBC and (B) DrzBS delivery systems. (C and D) Representative H&E staining of mouse organs after treatment with different (C) DrzBC and (D) DrzBS delivery systems.(E and F) HBeAg (E) and HBsAg (F) secretion of HBV-infected mice after being administered the corresponding formulations on day 7. (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001). All data are expressed as the mean ± SD (n = 6).
Organs, including heart, liver, spleen, lung, and kidney, were collected for hematoxylin and eosin (H&E) analysis to evaluate the in vivo toxicities (Figures 6C and 6D). The similarity in H&E staining demonstrated the systemic safety of Gal-CSSO gene delivery systems.
In vivo evaluation of anti-HBV efficacy
As shown in Figures 6E and 6F, compared with the control group, all of the gene delivery systems mediated by Gal-CSSO, CSSO, and Lipo2000 significantly inhibited HBV antigen secretion. Moreover, the order of IRs was as follows: Gal-CSSO/DrzBC (53.8%) > Lipo2000/DrzBC (35.4%) > CSSO/DrzBC (17.3%) and Gal-CSSO/DrzBS (89.2%) > Lipo2000/DrzBS (73.4%) > CSSO/DrzBS (57.3%). Compared with Lipo2000 systems, the IRs of HBeAg and HBsAg secretion increased 1.52-fold and 1.22-fold, respectively, in the case of the Gal-CSSO system, suggesting the anti-HBV efficacy of the Gal-CSSO system. It was noteworthy that the IRs of HBeAg and HBsAg secretion due to the Gal-CSSO system were 3.11-fold and 1.56-fold greater, respectively, relative to the CSSO systems. As shown in Tables 2 and 3, for the in vitro anti-HBV efficacy, there was no obvious difference between CSSO and Gal-CSSO systems, while LA modification significantly improved the in vivo anti-HBV efficacy.
Discussion
In this study, we designed 10-23 DNAzymes, DrzBC and DrzBS, to inhibit the expression of HBeAg and HBsAg, respectively, which depend on exogenous delivery. To realize efficient gene therapy of HB, we synthesized the polymer Gal-CSSO, which could self-aggregate in an aqueous medium to form micelles. At an optimized mass ratio of 60:1, Gal-CSSO could conveniently couple with DrzBC and DrzBS via electrostatic interactions to form Gal-CSSO/DrzBC and Gal-CSSO/DrzBS systems, respectively, in the nano-size range of 240–260 nm. Gal-CSSO/DrzBC and Gal-CSSO/DrzBS exhibited low cytotoxicity and systemic toxicity, thereby efficiently protecting DrzBC and DrzBS from enzymatic degradation.
Gal-CSSO/DrzBC and Gal-CSSO/DrzBS exhibited an anti-HBV effect in the following three ways: First, because of the galactosyl residues specifically recognized by ASGPRs, Gal-CSSO achieved active liver targeting and HC targeting. Second, after target uptake, Gal-CSSO exhibited effective endosomal escape into the cytoplasm. Finally, owing to the reducible disulfide bonds –SS–, the structure of Gal-CSSO/DrzBC and Gal-CSSO/DrzBS responded to the high-GSH level microenvironment of HCs with rapid drug release. Therefore, compared with the traditional commercial carrier Lipo2000, both in vitro and in vivo, Gal-CSSO/DrzBC and Gal-CSSO/DrzBS showed a significant increase in HBeAg and HBsAg secretion inhibition ability, respectively.
However, as shown in Tables 2 and 3, it seemed that the in vitro anti-HBV activity (the IR values) of the nontargeted system (CSSO) was similar to that of the targeted system (Gal-CSSO). According to our previous research,27 CSSO also exhibited efficient cellular uptake by HepG2.2.15. Therefore, although Gal-CSSO could accelerate cellular uptake, the uptake was not increased significantly. As a result, there was no obvious difference in in vitro anti-HBV activity between the CSSO and Gal-CSSO systems. However, in vivo the Gal-CSSO system had an advantage in anti-HBV activity over CSSO due to selective targeting of the HBV-infected hepatocytes.
Considering the characteristics of HBV infection and the redox microenvironment of HCs, Gal-CSSO—as a HC-targeting and microenvironmentally responsive glycolipid-like polymer—has promising application potential. As there are no radical drugs to counter HB, the Gal-CSSO gene delivery system provides an effective and safe gene therapy alternative for treatment of HB.
Materials and methods
Materials
CSO with a low molecular weight (MW = 18.9 kDa) was obtained by enzymatic degradation of 95% deacetylated chitosan (MW = 450 kDa, Halobios Products Factory, Yuhuan, China). ODA was purchased from Fluka (Milwaukee, WI, USA). Reagents were purchased from the companies listed as follows: DTPA (Tokyo Chemical Industry, Tokyo, Japan); LA, N-hydroxysuccinimide (NHS), and pyrene (Aladdin Reagent, Shanghai, China); FITC, MTT, Tetramethylethylenediamine (TEMED) and 2,4,6-trinitrobenzene sulfonic acid (TNBS; Sigma, St. Louis, MO, USA); 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC; Shanghai Medpep, Shanghai, China); Lipo2000 (Invitrogen, Carlsbad, CA, USA); fetal bovine serum (Sijiqing Biology Engineering Materials, Zhejiang, China); DrzBC and DrzBS (Sangon Biotech, Shanghai, China); anti-ASGPR1 (BD Biosciences, Franklin Lakes, NJ, USA); and Brilliant Violet 421 Donkey anti-rabbit IgG (minimal x-reactivity) antibody, PE/Cy7 anti-mouse F4/80, and the Multi-Analyte Flow Assay Kit (BioLegend, San Diego, CA, USA).
Synthesis of Gal-CSSO
Gal-CSSO was produced by the two-step process shown in Figure 2A.
In step 1, CSSO was synthesized as described in a previous study.28 In brief, ODA was coupled with DTPA via amide bonding between the amino groups on ODA and the carboxyl groups on DTPA. The reaction was performed at 60°C for 24 h in a nitrogen atmosphere and subsequently filtered to remove by-products. Thereafter, the mixture was activated by dropwise addition of EDC/NHS to the chitosan solution for 30 min. The mixture was stirred for 8 h at 60°C and then dialyzed against DI water for 2 days. The solution was then centrifuged, and subsequently the supernatant was lyophilized. The lyophilized product was washed with hot ethanol to remove any unreacted reagent. Finally, the product was re-dispersed in DI water and lyophilized to obtain CSSO.
In step 2, LA was attached to the amino groups of CSSO via an amidation reaction. LA (220 mg) was dissolved in DI water (10 mL), and after the pH was adjusted to 5.0, EDC (220 mg) and NHS (110 mg) were added. After the LA solution was fully mixed and activated for 30 min, the pH value was adjusted to 8.0 with TEMED. The obtained LA solution was added dropwise into the CSSO solution (250 mg CSSO dissolved in 30 mL DI water) by stirring for 72 h, followed by dialyzing against DI water for 3 days and, finally, lyophilizing.
LA, CSSO, and Gal-CSSO were dissolved in Deuterium oxide (D2O), and the chemical structures were characterized by 1H NMR with an AVANCE DMX 500 NMR spectrometer (Bruker, Rheinstetten, Germany).
Preparation of complexes
Gal-CSSO/10-23 DNAzyme complexes were prepared by mixing Gal-CSSO with DrzBC or DrzBS at mass ratios of 20:1, 60:1, and 100:1. First, Gal-CSSO was dissolved in DI water; thereafter, either DrzBC or DrzBS solution was added to the Gal-CSSO solution, followed by vortex mixing and incubating for 30 min.
Selection of the optimal mass ratio for preparation of the complexes
The 10-23 DNAzyme condensation ability of Gal-CSSO was evaluated with denaturing 7 M urea polyacrylamide gel electrophoresis. Considering the similar molecular weights and similar physical and chemical properties of DrzBC and DrzBS, DrzBC was used as a prototype. Gal-CSSO/DrzBC complexes were prepared at ratios of 20:1, 60:1, and 100:1 (Gal-CSSO:DrzBC, w/w) and brought to 10 μL prior to loading. Electrophoresis was then conducted at 100 V for 40 min in a TBE buffer solution (1 M Tris, 0.9 M boric acid, and 0.01 M EDTA). DrzBC retardation was observed by staining with EB under a UV lamp. As a control, pristine DrzBC and Lipo2000/DrzBC were also observed by denaturing 7 M urea polyacrylamide gel electrophoresis.
To screen the optimal mass ratio of Gal-CSSO/DrzBC, the fluorochrome Cy5 was used to label DrzBC; Gal-CSSO:Cy5-DrzBC at different mass ratios of 20:1, 60:1, and 100:1 was prepared and injected intravenously into the BALB/c mice. The livers were isolated 12 h after injection, and HC suspensions were prepared according to the method described in a previous study.29 In brief, six male BALB/c mice (average weight 22.1 ± 1.4 g) were perfused with Hanks’ balanced salt solution without Ca2+ and Mg2+ under anesthetized conditions and then perfused with Hanks’ balanced salt solution containing collagenase (type IV, 0.05%), Ca2+, and Mg2+. Subsequently, the livers were isolated for HC and KC extraction. HCs were labeled with anti-ASGPR1 and -BV421, and the HC uptake of Gal-CSSO/Cy5-DrzBC was determined by multicolor flow cytometry, according to the percentage of HCs in the Cy5-positive cells.
Characterization of Gal-CSSO and Gal-CSSO/10-23 DNAzyme
The degree of amino acid substitution (SD%) of Gal-CSSO polymers was determined by the TNBS method.30 The CMC of Gal-CSSO micelles was determined via fluorescence spectroscopy (F-2500, Hitachi, Japan) using pyrene as a probe. The particle sizes and zeta potentials of Gal-CSSO and Gal-CSSO/10-23 DNAzyme were measured through dynamic light scattering (DLS, Zetasizer 3000HS, Malvern Instruments, Malvern, UK). The morphology of the micelles was observed via TEM (Stereoscan, Leica, UK).
DNase I protection ability
Gal-CSSO/DrzBC prepared at a mass ratio of 60:1 was incubated for 30 min at 20 ± 5°C, followed by the addition of DNase I. After 30 min of incubation, the samples were further treated with EDTA for 10 min to terminate the activity of DNase I, followed by incubation in heparin for 30 min to replace DrzBC in Gal-CSSO/DrzBC. As a control, pristine DrzBC and CSSO/DrzBC were also treated with DNase I under the same conditions.
EE% and DL% determination
10-23 DNAzymes were labeled with Cy5 to prepare Gal-CSSO/Cy5-DrzBC and Gal-CSSO/Cy5-DrzBS complexes. After ultra-centrifugation, unbound Cy5-DrzBC and Cy5-DrzBS were determined with a fluorescence spectrophotometer (F-2500 fluorescence spectrophotometer, Hitachi, Japan). The excitation wavelength (λex) was 646 nm, the emission wavelength (λem) was 662 nm for DrzBC and 667 nm for DrzBS, and the slit size was 5 nm. The EE (%) and DL (%) were calculated by the following equations:
| (1) |
| (2) |
where Wa is the amount of total 10-23 DNAzyme added to the system, Ws is the analyzed amount of unbound 10-23 DNAzyme in the supernatant after ultra-centrifugation, and WL is the weight of Gal-CSSO added to the system.
GSH concentration determination
The HCs and KCs in the livers of HBV-infected mice were isolated according to the method described above. Subsequently, the GSH concentrations in HCs and KCs were determined with a reduced glutathione assay kit (Jiancheng Bioengineering Institute, Nanjing, China), according to the instruction manual.
GSH-responsive release behavior investigation
Cy5-DrzBC and Cy5-DrzBS standard solutions with concentration gradients of 0, 0.5, 1.0, 2.5, and 5.0 μg⋅mL−1 were prepared. Fluorescence intensity of the standard solution was determined via fluorescence spectrophotometry (λex = 649 nm, λem = 662 nm for Cy5-DrzBC, λem = 667 nm for Cy5-DrzBS). By linear regression of the DNA concentration to fluorescence intensity, a standard curve was constructed. Gal-CSSO/Cy5-DrzBC or Gal-CSSO/Cy5-DrzBS complexes were centrifugated for 10 min at 13,000 rpm; the supernatant was discarded, and thereafter Tris-HCl acid buffer (1 mL) solutions, each containing 0, 0.1, 1.0, or 10 mM GSH (pH 7.2), were added to disperse the precipitate. Subsequently, the solutions were gently shaken at 37°C under horizontal shaking (60 rpm) in a shaking incubator. At predetermined time points, the samples were centrifuged at 13,000 rpm for 10 min, and 0.6 mL of supernatant was removed to measure the fluorescence intensity. The cumulative release of DrzBC or DrzBS was calculated according to the standard curve.
Cell culture
HepG2.2.15 was obtained from the Cell Resource Center (Chinese Academy of Medical Sciences, Beijing, China) and cultured in RPMI-1640 supplemented with 10% fetal bovine serum, 100 U⋅mL−1 penicillin, 100 U⋅mL−1 streptomycin, and 380 μg⋅mL−1 geneticin G418 in a humidified atmosphere containing 5% CO2 at 37°C.
In vitro cytotoxicity
Cytotoxicity of Gal-CSSO, DrzBC, DrzBS, Gal-CSSO/DrzBC, and Gal-CSSO/DrzBS was determined with an MTT assay. HepG2.2.15 cells were seeded at a density of 4 × 103 cells/well in 96-well culture plates and cultured for 24 h under 5% CO2 at 37°C. The polymer solution with Gal-CSSO concentrations of 10, 50, 70, 100, and 200 μg⋅mL−1 and Gal-CSSO/DrzBC or Gal-CSSO/DrzBS solution with DrzBC or DrzBS concentrations of 0.1, 0.5, 1.0, 2.0, 3.0, 4.0, and 5.0 μg⋅mL−1 were added to the 96-well culture plates. A blank culture medium was treated as a control. After 48 h, 20 mL of MTT solution (5.0 mg⋅mL−1) was added and the solution was incubated for another 4 h. Thereafter, the medium was replaced by 200 mL of DMSO to dissolve the purple formazan crystals. The sample absorbance of each well was measured at 570 nm by a microplate reader (Model 680, Bio-Rad, Berkeley, CA, USA). Cell viability was calculated with reference to the cells incubated in the culture medium alone. Dose-effect curves were plotted from the data corresponding to the triplicate assays.
Intracellular localization
To evaluate the intracellular localization in HepG2.2.15 cells, DrzBC was labeled with Cy5 and Gal-CSSO was labeled with FITC, according to the methods described previously.31 HepG2.2.15 cells were seeded at a density of 2 × 103 cells/well in 24-well culture plates. After incubation for 12 h, cells were treated with the prepared Gal-CSSO/DrzBC for 24 h. To label the lysosomes, the cells were incubated in prewarmed media (37°C) containing LysoTracker Blue (LysoTracker Blue DND-26, Invitrogen, Carlsbad, CA, USA). The corresponding fluorescent images were obtained by CLSM (Ix81-FV1000, Olympus, Tokyo, Japan).
In vitro evaluation of anti-HBV efficacy
HepG2.2.15 cells were seeded in a 24-well plate at a density of 3 × 104 cells/well and incubated at 37°C in a humid atmosphere with 5% CO2. After 24 h of incubation, the cells were supplemented with Opti-MEM I, followed by washing with serum-free culture medium. Thereafter, DrzBC, DrzBS, Lipo2000/DrzBC, Lipo2000/DrzBS, Gal-CSSO/DrzBC, and Gal-CSSO/DrzBS were added with the same final DrzBC or DrzBS concentration of 1.0 μmol⋅L−1. After incubation for a certain period of time, the cell supernatant was collected and the cells were continually cultured with fresh medium for different periods of time. The titers of HBeAg and HBsAg in the cell supernatant were determined with an Architect I4000 chemiluminescence immunoassay analyzer (Abbott Laboratories, Chicago, IL, USA).
Animals
Male BALB/c (6–8 weeks) mice were purchased from the Shanghai Silaike Laboratory Animal Limited Liability Company. rAAV8-1.3HBV (1 × 1011 Viral genome, vg) was purchased from PackGene Biotech and used to obtain HBV-infected animal models. Details of the construction and evaluation of the animal models are presented in the Supplemental information (Figure S5). All animal procedures were performed in accordance with the national regulations and were approved by the Animal Experiments Ethical Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University (reference no. 2020-821).
In vivo distribution
All of the studies were conducted according to the guidelines issued by the Ethical Committee of Zhejiang University. Fluorochrome Cy5 was used to label DrzBC, and the complexes encapsulating Cy5-DrzBC were prepared as described in section “Preparation and characterization of Gal-CSSO/10-23DNAzyme”. Cy5-DrzBC, CSSO/Cy5-DrzBC, or Gal-CSSO/Cy5-DrzBC was intravenously injected to investigate the bio-distribution. At the predetermined time after injection, the mice were observed with the in vivo Maestro Imaging System (CRi, Woburn, MA, USA).
In vivo cellular uptake selectivity
The complexes CSSO/Cy5-DrzBC and Gal-CSSO/Cy5-DrzBC were prepared by the same procedure described above. Cy5-DrzBC, CSSO/Cy5-DrzBC, or Gal-CSSO/Cy5-DrzBC was intravenously injected into mice. After 12 h, the HCs and KCs were isolated by the method described above. To evaluate the in vivo target selectivity, HCs and KCs were stained with specific antibodies and analyzed by flow cytometry (BD Fortessa) analysis. In addition, an immunofluorescence assay was used to investigate the in vivo cellular uptake of the complexes.
In vivo toxicity evaluation
Male BALB/c mice were randomly divided into nine groups (n = 6 each). The categorization of the groups was as follows: group 1, saline; group 2, DrzBC; group 3, DrzBS; group 4, Lipo2000/DrzBC; group 5, Lipo2000/DrzBS; group 6, CSSO/DrzBC; group 7, CSSO/DrzBS; group 8, Gal-CSSO/DrzBC; and group 9, Gal-CSSO/DrzBS. Except for group 1 (administered 200 μL saline), the mice in the other groups were administered 200 μL of the corresponding formulations containing 10 μg DrzBC or DrzBS by intravenous injection on day 0. The body weights of the mice were measured every day. To evaluate the in vivo toxicity, the heart, liver, spleen, lung, and kidney were collected on day 7 for H&E analysis.
In vivo evaluation of anti-HBV efficacy
As described for the in vivo toxicity evaluation, rAAV8-1.3HBV-infected male BALB/c mice were randomly divided into nine groups (n = 6 each) and administered the corresponding formulations by intravenous injection on day 0. On day 7, the peripheral blood of mice was sampled and the serum was segregated to detect the titers of HBeAg and HBsAg with an Architect I4000 chemiluminescence immunoassay analyzer.
Statistical analysis
All data are reported as mean ± SD. Differences between groups were tested with the two-tailed Student’s t test (Prism 8.0). Differences with p <0.05 were considered statistically significant.
Acknowledgments
The research was supported by the Joint Fund of Zhejiang Provincial Natural Science Foundation (grant nos. LQY20H300002, LQY19H300001, and LYY20H300002); the National Nature Science Foundation of China (grant no. 81973402); the Zhejiang Provincial Natural Science Foundation (grant no. LY19H310007); the Medical and Health Science and Technology Project of Zhejiang Province (grant no. 2018KY402); and the Hospital Pharmacy Special Research Project of Zhejiang Pharmaceutical Association (grant no. 2018ZYY02).
Author contributions
F.H. and J.W. directed the study. J.M., X.Y., and X.S. performed most of the experiments and data analysis. J.M., X.Y., and Q.L. wrote the manuscript. H.Y., T.M., and Y.H. contributed to the interpretation of results and provided critical insights into the significance of the work. Z.G. constructed the HBV-infected animal models and managed the harvesting of mouse samples.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2021.02.013.
Contributor Information
Jiaying Wu, Email: jiayingwu@zju.edu.cn.
Fuqiang Hu, Email: hufq@zju.edu.cn.
Supplemental information
References
- 1.European Association for the Study of the Liver EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017;67:370–398. doi: 10.1016/j.jhep.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 2.Terrault N.A., Bzowej N.H., Chang K.M., Hwang J.P., Jonas M.M., Murad M.H., American Association for the Study of Liver Diseases AASLD guidelines for treatment of chronic hepatitis B. Hepatology. 2016;63:261–283. doi: 10.1002/hep.28156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Terrault N.A., Lok A.S.F., McMahon B.J., Chang K.M., Hwang J.P., Jonas M.M., Brown R.S., Jr., Bzowej N.H., Wong J.B. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology. 2018;67:1560–1599. doi: 10.1002/hep.29800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gane E.J. Future anti-HBV strategies. Liver Int. 2017;37(Suppl 1):40–44. doi: 10.1111/liv.13304. [DOI] [PubMed] [Google Scholar]
- 5.Testoni B., Durantel D., Zoulim F. Novel targets for hepatitis B virus therapy. Liver Int. 2017;37(Suppl 1):33–39. doi: 10.1111/liv.13307. [DOI] [PubMed] [Google Scholar]
- 6.Soriano V. Hepatitis B Gene Therapy Coming to Age. AIDS Rev. 2018;20:125–127. [PubMed] [Google Scholar]
- 7.Fokina A.A., Stetsenko D.A., François J.C. DNA enzymes as potential therapeutics: towards clinical application of 10-23 DNAzymes. Expert Opin. Biol. Ther. 2015;15:689–711. doi: 10.1517/14712598.2015.1025048. [DOI] [PubMed] [Google Scholar]
- 8.Zhou W., Ding J., Liu J. Theranostic DNAzymes. Theranostics. 2017;7:1010–1025. doi: 10.7150/thno.17736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santoro S.W., Joyce G.F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. USA. 1997;94:4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin Y., Lu Y., Li X. Biological characteristics of exosomes and genetically engineered exosomes for the targeted delivery of therapeutic agents. J. Drug Target. 2020;28:129–141. doi: 10.1080/1061186X.2019.1641508. [DOI] [PubMed] [Google Scholar]
- 11.Shi B., Zheng M., Tao W., Chung R., Jin D., Ghaffari D., Farokhzad O.C. Challenges in DNA delivery and recent advances in multifunctional polymeric DNA delivery systems. Biomacromolecules. 2017;18:2231–2246. doi: 10.1021/acs.biomac.7b00803. [DOI] [PubMed] [Google Scholar]
- 12.Gudima S., He Y., Meier A., Chang J., Chen R., Jarnik M., Nicolas E., Bruss V., Taylor J. Assembly of hepatitis delta virus: particle characterization, including the ability to infect primary human hepatocytes. J. Virol. 2007;81:3608–3617. doi: 10.1128/JVI.02277-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H.X., Xiong M.H., Wang Y.C., Zhu J., Wang J. N-acetylgalactosamine functionalized mixed micellar nanoparticles for targeted delivery of siRNA to liver. J. Control. Release. 2013;166:106–114. doi: 10.1016/j.jconrel.2012.12.017. [DOI] [PubMed] [Google Scholar]
- 14.Patil S., Ujalambkar V., Rathore A., Rojatkar S., Pokharkar V. Galangin loaded galactosylated pluronic F68 polymeric micelles for liver targeting. Biomed. Pharmacother. 2019;112:108691. doi: 10.1016/j.biopha.2019.108691. [DOI] [PubMed] [Google Scholar]
- 15.Guo H., Zhang D., Li C., Jia L., Liu G., Hao L., Zheng D., Shen J., Li T., Guo Y., Zhang Q. Self-assembled nanoparticles based on galactosylated O-carboxymethyl chitosan-graft-stearic acid conjugates for delivery of doxorubicin. Int. J. Pharm. 2013;458:31–38. doi: 10.1016/j.ijpharm.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 16.Rodrigues C., Percival S.S. Immunomodulatory Effects of Glutathione, Garlic Derivatives, and Hydrogen Sulfide. Nutrients. 2019;11:295. doi: 10.3390/nu11020295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones D.P., Mody V.C., Jr., Carlson J.L., Lynn M.J., Sternberg P., Jr. Redox analysis of human plasma allows separation of pro-oxidant events of aging from decline in antioxidant defenses. Free Radic. Biol. Med. 2002;33:1290–1300. doi: 10.1016/s0891-5849(02)01040-7. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y., Luo Q., Gao L., Gao C., Du H., Zha G., Li X., Shen Z., Zhu W. A facile strategy to prepare redox-responsive amphiphilic PEGylated prodrug with high drug loading content and low critical micelle concentration. Biomater. Sci. 2014;2:1367–1376. doi: 10.1039/c4bm00065j. [DOI] [PubMed] [Google Scholar]
- 19.Li Y.Q., Sun W., Liu X.Y., Chen L.Q., Huang W., Lu Z.L., He L. Synthesis of glutathione (GSH)-responsive amphiphilic duplexes and their application in gene delivery. ChemPlusChem. 2019;84:1060–1069. doi: 10.1002/cplu.201900295. [DOI] [PubMed] [Google Scholar]
- 20.Wen L., Hu Y., Meng T., Tan Y., Zhao M., Dai S., Yuan H., Hu F. Redox-responsive polymer inhibits macrophages uptake for effective intracellular gene delivery and enhanced cancer therapy. Colloids Surf. B Biointerfaces. 2019;175:392–402. doi: 10.1016/j.colsurfb.2018.12.016. [DOI] [PubMed] [Google Scholar]
- 21.Luo Y., Yin X., Yin X., Chen A., Zhao L., Zhang G., Liao W., Huang X., Li J., Zhang C.Y. Dual pH/Redox-Responsive Mixed Polymeric Micelles for Anticancer Drug Delivery and Controlled Release. Pharmaceutics. 2019;11:176. doi: 10.3390/pharmaceutics11040176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo X., Cheng Y., Zhao X., Luo Y., Chen J., Yuan W.E. Advances in redox-responsive drug delivery systems of tumor microenvironment. J. Nanobiotechnology. 2018;16:74. doi: 10.1186/s12951-018-0398-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morachis J.M., Mahmoud E.A., Sankaranarayanan J., Almutairi A. Triggered rapid degradation of nanoparticles for gene delivery. J. Drug Deliv. 2012;2012:291219. doi: 10.1155/2012/291219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du L., Zhou J., Meng L., Wang X., Wang C., Huang Y., Zheng S., Deng L., Cao H., Liang Z. The pH-Triggered Triblock Nanocarrier Enabled Highly Efficient siRNA Delivery for Cancer Therapy. Theranostics. 2017;7:3432–3445. doi: 10.7150/thno.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmeister K.M., Falet H. Platelet clearance by the hepatic Ashwell-Morrell receptor: mechanisms and biological significance. Thromb. Res. 2016;141(Suppl 2):S68–S72. doi: 10.1016/S0049-3848(16)30370-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller C.M., Tanowitz M., Donner A.J., Prakash T.P., Swayze E.E., Harris E.N., Seth P.P. Receptor-mediated uptake of phosphorothioate antisense oligonucleotides in different cell types of the liver. Nucleic Acid Ther. 2018;28:119–127. doi: 10.1089/nat.2017.0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miao J., Yang X.Q., Gao Z., Li Q., Meng T.T., Wu J.Y., Yuan H., Hu F.Q. Redox-responsive chitosan oligosaccharide-SS-Octadecylamine polymeric carrier for efficient anti-Hepatitis B Virus gene therapy. Carbohydr. Polym. 2019;212:215–221. doi: 10.1016/j.carbpol.2019.02.047. [DOI] [PubMed] [Google Scholar]
- 28.Hu Y.W., Du Y.Z., Liu N., Liu X., Meng T.T., Cheng B.L., He J.B., You J., Yuan H., Hu F.Q. Selective redox-responsive drug release in tumor cells mediated by chitosan based glycolipid-like nanocarrier. J. Control. Release. 2015;206:91–100. doi: 10.1016/j.jconrel.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 29.Aparicio-Vergara M., Tencerova M., Morgantini C., Barreby E., Aouadi M. Isolation of Kupffer cells and hepatocytes from a single mouse liver. Methods Mol. Biol. 2017;1639:161–171. doi: 10.1007/978-1-4939-7163-3_16. [DOI] [PubMed] [Google Scholar]
- 30.Hu F.Q., Zhang Y.Y., You J., Yuan H., Du Y.Z. pH triggered doxorubicin delivery of PEGylated glycolipid conjugate micelles for tumor targeting therapy. Mol. Pharm. 2012;9:2469–2478. doi: 10.1021/mp300002v. [DOI] [PubMed] [Google Scholar]
- 31.Yan J., Du Y.Z., Chen F.Y., You J., Yuan H., Hu F.Q. Effect of proteins with different isoelectric points on the gene transfection efficiency mediated by stearic acid grafted chitosan oligosaccharide micelles. Mol. Pharm. 2013;10:2568–2577. doi: 10.1021/mp300732d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.