

Abstract
Mitochondria participate in various metabolic pathways, and their dysregulation results in multiple disorders, including aging-related diseases. However, the metabolic changes and mechanisms of mitochondrial disorders are not fully understood. Here, we found that induced pluripotent stem cells (iPSCs) from a patient with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) showed attenuated proliferation and survival when glycolysis was inhibited. These deficits were rescued by taurine administration. Metabolomic analyses showed that the ratio of the reduced (GSH) to oxidized glutathione (GSSG) was decreased; whereas the levels of cysteine, a substrate of GSH, and oxidative stress markers were upregulated in MELAS iPSCs. Taurine normalized these changes, suggesting that MELAS iPSCs were affected by the oxidative stress and taurine reduced its influence. We also analyzed the retinal pigment epithelium (RPE) differentiated from MELAS iPSCs by using a three-dimensional culture system and found that it showed epithelial mesenchymal transition (EMT), which was suppressed by taurine. Therefore, mitochondrial dysfunction caused metabolic changes, accumulation of oxidative stress that depleted GSH, and EMT in the RPE that could be involved in retinal pathogenesis. Because all these phenomena were sensitive to taurine treatment, we conclude that administration of taurine may be a potential new therapeutic approach for mitochondria-related retinal diseases.
Keywords: Epithelial mesenchymal transition, Induced pluripotent stem cells, Metabolomics, Mitochondria, Retinal pigment epithelium, Taurine
Abbreviations: 2DG, 2-deoxy-d-glucose; AMD, age-related macular degeneration; ANOVA, analysis of variance; CE-TOFMS, capillary electrophoresis-time-of-flight mass spectrometry; DAP, 2,2-dichloroacetophenone; DAPI, 4′,6-diamidino-2-phenyindole; DCA, sodium dichloroacetate; ECAR, extracellular acidification rate; EMT, epithelial mesenchymal transition; FBS, fetal bovine serum; GSH, reduced glutathione; GSSG, oxidized glutathione; iPSCs, induced pluripotent stem cells; MEF, mouse embryonic fibroblast; MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes; MMC, mitomycin C; mtDNA, mitochondrial DNA; ND6, NADH: ubiquinone oxidoreductase core subunit 6; OCR, oxygen consumption rate; OXPHOS, oxidative phosphorylation; PBS, phosphate-buffered saline; qRT-PCR, quantitative reverse transcription-polymerase chain reaction; PEP, phosphoenolpyruvic acid; PPP, pentose phosphate pathway; RPE, retinal pigment epithelium; TCA, tricarboxylic acid; WT, wild-type
Graphical abstract

Highlights
-
•
iPS cell lines were derived from a MELAS patient with the mtDNA A3243G mutation.
-
•
Decreased proliferation and survival of MELAS iPSCs were rescued by taurine.
-
•
Reduction in GSH/GSSG ratio in MELAS iPSCs was suppressed by taurine.
-
•
EMT in MELAS iPSC-derived retinal pigment epithelium was suppressed by taurine.
-
•
Oxidative stress markers in MELAS iPSCs and RPE were suppressed by taurine.
1. Introduction
Mitochondria play a key role in metabolism, and their impairments cause various diseases, ranging from specific mitochondrial disorders [1] to aging-related diseases, such as age-related macular degeneration (AMD) [2,3]. Mitochondrial defects affect various metabolic pathways, including oxidative phosphorylation (OXPHOS) [1]; however, the associated changes in various metabolite levels and the exact mechanisms of mitochondrial disorders are not fully understood.
Mitochondrial DNA (mtDNA) encodes 13 proteins related to the OXPHOS system, two ribosomal RNAs, and 22 transfer RNAs, which are used for mitochondrial RNA translation [4]. Although every mitochondrion has several different mtDNA sequences, a phenomenon called heteroplasmy, accumulation of mutant mtDNA can cause mitochondrial diseases, such as mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) [1]. The mutation of A to G at position 3243 of the mtDNA (A3243G) producing mutant tRNALeu(UUR) is the most prevalent cause of MELAS and accounts for 80% of all cases [5]. The mitochondrial dysfunction caused by this mutation is explained by the decreased translation of mitochondrial RNA transcripts, especially that of Leu-rich proteins, such as NADH: ubiquinone oxidoreductase (complex I in the respiratory chain) core subunit 6 (ND6) [6].
Although a recent clinical trial showed that oral supplementation of taurine, a sulfur-containing amino acid, reduced the recurrence of stroke-like episodes in MELAS patients [7], whether and how that treatment affected metabolism and cellular phenotypes of mitochondrial disorders have not been clarified.
MELAS patients show retinal pigment epithelium (RPE) atrophy [[8], [9], [10], [11], [12], [13]]. Because the RPE forms a barrier between the neural retina, including photoreceptors, and their feeding connective tissue, choroid, as well as has critical roles in photoreceptor survival [14], MELAS patients exhibit progressive photoreceptor death that causes a vision loss.
Moreover, mitochondrial dysfunction is also deeply involved in AMD [2,3], which is caused by the RPE dysregulation through inflammatory signaling [15,16] and resulting epithelial-mesenchymal transition (EMT) [17,18]. However, the relationship between mitochondrial functions and EMT in the RPE is still obscure.
To investigate the mechanism of these mitochondrial diseases, the cytoplasmic hybrid (cybrid) fusion has been used to generate cellular mtDNA disease models [19]. More recently, several lines of induced pluripotent stem cells (iPSCs) derived from patients with mitochondrial diseases have been generated [[20], [21], [22], [23], [24], [25]]. However, the heteroplasmy levels, and the ratio of pathological to wild-type (WT) mitochondria in those cells complicated the interpretation of in vitro phenotypes, so further studies in the cells with high levels of mutant mtDNA were warranted.
Here, we generated MELAS patient-derived iPSC lines harboring the mtDNA A3243G mutation and expressing high levels of the mutant mtDNA. We documented metabolic changes in MELAS iPSCs, demonstrated the phenotype of the RPE differentiated from these iPSCs [[25], [26], [27], [28], [29]], and analyzed the protective effects of taurine. The undifferentiated pluripotent stem cells (PSCs) possess functional OXPHOS machinery; however, it is decoupled from glycolysis, which is the major energy source in PSCs, similarly to the situation in cancer cells [[30], [31], [32]]. This reduces the electron transport chain coupling, production of reactive oxygen species, and oxidative stress, which minimizes potential genome damages [31,[33], [34], [35]]. We reasoned that the analysis of metabolites in iPSCs that are highly homogeneous and stable, carrying a high fraction of mutant mtDNA will help to understand the mechanisms of mitochondrial disorders, including MELAS, under the condition of decoupled glycolysis. In contrast, differentiated cells employing OXPHOS as the main energy generation source [30,31] are exposed to oxidative stress even under physiological conditions. The phenotypic analysis of the RPE affected by a mitochondrial impairment would be important to explore new therapeutic approaches. Based on this reasoning, in the current study, we have investigated the pathogenic mechanisms of MELAS at the molecular and biochemical levels by using patient-derived undifferentiated iPSCs and the iPSC-derived RPE to advance our understanding of the pathogenesis of mitochondrial disorders and the therapeutic effects of taurine.
2. Materials and methods
2.1. Generation of MELAS patient-derived iPSC lines
The skin fibroblasts derived from a MELAS patient were used to generate iPSCs as described previously [36]. Briefly, 5 × 105 fibroblast cells were seeded on 6-well plates and infected with a Sendai virus vector carrying OCT3/4, SOX2, KLF4, and c-MYC at the multiplicity of infection of three. After 7 days in culture, the infected cells were harvested by trypsin and plated on a 60-mm dish with mitomycin C (MMC)-treated mouse embryonic fibroblast (MEF) feeder cells. After culturing for 18–25 days, the colonies were picked and cultured in human iPSC medium containing DMEM/F12 (Sigma, St. Louis, MO, USA), 20% Knockout serum replacement (KSR; Invitrogen, CA, USA), 5 ng/μl basic fibroblast growth factor (Wako, Japan), 0.1 mM non-essential amino acids (Sigma), 2 mM l-glutamine (Life Technologies, USA), 0.1 mM 2-mercaptoethanol (Sigma), and 0.5% penicillin-streptomycin (Nacalai Tesque, Japan). To remove the Sendai virus, the iPSCs were cultured at 38 °C for 3 days at passage 1 or 2 and then maintained on MMC-MEF with human iPSC medium.
2.2. Alkaline staining and immunostaining
Staining was performed as previously described [37]. Briefly, alkaline phosphatase staining was performed by using a Leukocyte Alkaline Phosphatase kit (Sigma). For immunostaining, cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min at 4 °C. The cells were then washed three times with the blocking buffer (PBS containing 2% fetal bovine serum) and incubated overnight at 4 °C with primary antibodies (anti-NANOG, at 1/500, CST, MA, USA; anti-OCT3/4, at 1/500, Santa Cruz Biotechnology, TX, USA; anti-TRA-1-60, at 1/500, Millipore, MA, USA; anti-Ki67, at 1/500, Abcam, UK; anti-cleaved caspase 3, at 1/500, CST) diluted in the blocking buffer. The RPE were plated on Matrigel-coated 4-well or 8-well glass chamber slides (Matsunami, Japan). For staining, the cells were fixed with 4% paraformaldehyde for 15 min at room temperature. After the fixation, the cells were treated with a 0.05% Tween20 solution in PBS (PBS-T), and a 0.3% Triton-X solution in PBS twice for membrane permeabilization. Then, the cells were treated with a blocking solution containing 5% bovine serum albumin in PBS for 1 h at room temperature and incubated at 4 °C overnight with the primary antibodies (anti-OTX2, at 1/500, Sigma; anti-ZO-1, at 1/250, Invitrogen; anti-α-SMA, at 1/1000, Sigma; anti-NRF2, at 1/500, Abcam) in PBS containing 1% goat serum. Then, samples were washed with PBS-T and incubated for 1 h at room temperature with the secondary anti-mouse or anti-rabbit IgG antibody conjugated with Alexa 488/594 at 1:500 (Life Technologies, CA, USA) and 4′,6-diamidino-2-phenyindole (DAPI at 1:500; Nacalai Tesque) dissolved in PBS containing 1% goat serum. After washing with PBS-T, the samples were mounted in the mountant (Permafluor, Thermo Scientific), and fluorescent images were acquired with a fluorescence (BZ-X800, KEYENCE, Japan) or a confocal (LSM710, Carl Zeiss, Germany) microscope.
2.3. Human iPSC maintenance and RPE differentiation culture
MELAS patient-derived (A24#1–3) and control human iPSCs (454E2, RIKEN BRC, Japan) [38] were cultured in a feeder-free condition as described previously [27]. Cells were maintained in Nutristem (Riprocell, Japan) on Matrigel (human embryonic stem cell-qualified Matrigel, BD, NJ, USA) coated 6-well plates. Cells were dissociated with a 0.5 mM ethylenediaminetetraacetic acid (Nacalai Tesque) solution in Dulbecco's phosphate-buffered saline (Nacalai Tesque) for passaging or differentiation. For retinal organoid differentiation, dissociated cells were re-aggregated in V-bottom cell-repellent 96 well plates (Greiner Bio-One, Germany) at 9000 cells per well in 100 μl of the Glasgow's Minimal Essential Medium (#11710-035, Gibco) containing 10% knockout serum replacement (Gibco), 0.1 mM non-essential amino acids (Wako), 1 mM pyruvate (Sigma, USA), 100 U/ml penicillin-100 μg/ml streptomycin cocktail (Nacalai Tesque), 0.05 mM 2-mercaptoethanol (Wako) supplemented with 10 μM Y-27632 (Nacalai Tesque) and 3 μM IWR-1-endo (Millipore). On day 2 after differentiation, 100 μl of the differentiation medium supplemented with 10 μM Y-27632, 3 μM IWR-1-endo, and 2% Matrigel (growth factor reduced Matrigel, BD) was added to each well. On day 6, half of the total medium volume in each well was replaced by fresh differentiation medium supplemented with 3 μM IWR-1-endo, 1.5 nM BMP4, and 2% Matrigel. On day 12, cell aggregates that had grown into embryoid bodies were transferred to petri dishes (Asnol Sterilization Plate, AS ONE, Japan), and the medium was changed to the differentiation medium containing 10% FBS (Gibco) and 100 nM Smoothened agonist (SAG, Enzo Life Science, NY, USA). On day 18, the medium was changed to the maintenance medium (DMEM/F12 + GlutaMax (#10565-018, Gibco) containing 1% N2 supplement with transferrin (Holo, Wako), 0.5 μM retinoic acid (Nacalai Tesque) and 100 U/ml penicillin-100 μg/ml streptomycin cocktail. Differentiated retinal cells were cultured in the RPE medium containing 1% FBS (Gibco), 1% N1 supplement (Sigma), 2 mM l-glutamine (Sigma), 0.1 mM NEAA (Wako) in MEM-alpha (Sigma), and the medium was changed every 3 or 4 days.
2.4. Human iPSC survival assay
The iPSCs were dissociated and plated on Matrigel (human embryonic stem cell-qualified Matrigel, BD, USA) coated 96-well plates at 20,000 cells per well in 100 μl of the Nutristem medium containing 10 μM Y-27632 (Nacalai Tesque). On day 1, an additional 100 μl of the Nutristem medium with the drug(s) was added to each well. On day 2, 100 μl of the Nutristem medium with 4 μg/ml calcein-AM (Dojindo, Japan) was added to each well, and the plate was incubated for 30 min at 37 °C. The cells stained with calcein were imaged by a fluorescence microscope (BZ-X800, KEYENCE, Japan). After thresholding the images, the stained cell areas were calculated by using Image J (https://imagej.nih.gov/ij/index.html, NIH, USA). 2-Deoxy-d-glucose (2DG, Sigma), JX 06 (Tocris, USA), sodium dichloroacetate (DCA, Sigma), 2, 2-dichloroacetophenone (DAP, Sigma), and taurine (Sigma) were added to the culture medium when needed.
2.5. Metabolome analysis
The iPSCs were plated on 100 mm culture dishes (Falcon, USA) for 3 days to 80% confluence. Taurine (1 mM) was added to the medium of the taurine-treated group 4 h before sampling. Metabolites were extracted from the cells with a solvent containing methanol, distilled water, and chloroform (1:0.5:1 v/v/v). The mixture was centrifuged at 12,000×g for 15 min at 4 °C, and the upper aqueous layer was centrifugally filtered through a 5-kDa cutoff filter (Human Metabolome Technologies, Tsuruoka, Japan) to remove proteins. The filtrate was lyophilized and dissolved in 50 μl of the Milli-Q water with reference compounds (3-aminopyrrolidine and trimesate at 300 μM each) prior to the capillary electrophoresis-time-of-flight mass spectrometry (CE-TOFMS) analysis. Metabolites were analyzed by using an Agilent CE-TOFMS system equipped with an Agilent G7100A CE instrument and an Agilent 6530 Q-TOF LC/MS system (Agilent Technologies, Waldbronn, Germany). The details were described in our previous reports [[39], [40], [41]].
2.6. Electron microscopy
The iPSCs were plated on Matrigel-coated chamber slides. For the observation, the cells were fixed with 4% paraformaldehyde for 15 min at room temperature and with 2.5% glutaraldehyde overnight at 4 °C. After washing with 0.1 M Phosphate buffer for 5 min, the samples were fixed with 1% osmium tetroxide for 90 min at 4 °C. Thereafter, the samples were dehydrated in 50%/70%/80%/90%/100% ethanol solutions (for 5 min in each solution) twice at 4 °C. The samples were then treated with 100% acetone for 5 min, with QY-1 (99% butyl glycidyl ether) for 5 min twice, with Epon (Epon Plastic Solution; in Weight: 27.0 MNA, 51.3 EPOK-812, 21.9 DDSA, and 1.1 DMP-30) and QY-1 in a 1:1 ratio mixture for 1 h, and with 100% Epon overnight. The samples were then sectioned with a diamond knife at a thickness of 70 nm. The sections were stained with uranyl acetate for 10 min, washed with dH2O for 1 min three times, and incubated in lead citrate for 10 min. After the wash with dH2O for 1 min three times, the sections were dried for 1 h. The cells in the sections were observed by using electron microscopy (model 1200 EXII; JEOL Ltd., Tokyo, Japan) in the Electron Microscopy Laboratory of the Keio University School of Medicine.
2.7. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Total RNA was prepared from cultured cells that were lysed in TRIzol (Thermo Fisher Scientific, MA, USA). RNA was extracted with chloroform, precipitated with isopropyl alcohol, washed with 75% ethanol, and air-dried briefly. The RNA pellet was re-suspended in RNase-free water and then reverse-transcribed with a SuperScript IV VILO Master Mix (Thermo Fisher Scientific). The resulting cDNA was amplified with gene-specific primers listed in Supplementary Table 1 and a SYBR Green Master Mix (Thermo Fisher Scientific). Quantitative gene expression analysis was performed by using a StepOnePlus real-time PCR instrument (Applied Biosystems, Thermo Fisher Scientific). Changes in gene expression levels are reported relative to the actin-β (ACTB) gene expression level.
2.8. Statistical analysis
Metabolomic data were shown in mean ± standard error, and other data were shown in mean ± standard deviation. Statistical analysis was performed by using the Welch's t-test or one-way analysis of variance (ANOVA) followed by the post hoc Dunnett's or Tukey's test for multiple comparisons. Differences were considered statistically significant if P < 0.05.
3. Results
3.1. Characteristics of iPSCs derived from a MELAS patient
Three iPSC lines derived from a MELAS patient (A24#1–3) that had the A3243G mutation in the MTTL1 mtDNA encoding tRNALeu(UUR) (Fig. 1A) expressed pluripotent markers such as OCT4, NANOG, TRA1-60, and alkaline phosphatase (Fig. 1B). Using the primers specific for the WT or A3243G point mutated mtDNA sequence (Fig. 1A), we confirmed the levels of the WT and mutant mtDNAs in each iPSC line by using real-time PCR (Fig. 1C). Mutant mtDNA was hardly found in the control iPSCs (454E2), however, it was present in considerable quantities in the MELAS iPSCs (A24#1–3) (Fig. 1C).
Fig. 1.

Characteristics of iPSCs derived from a MELAS patient. (A) The sequence at the MELAS mutation site of the tRNA gene in mtDNA. The mutated MTTL1 gene that encodes for tRNALeu (UUR) contained a substitution of A to G at mtDNA position 3243 (A3243G). Forward WT and Mut primers, as well as the reverse primer for qPCR analysis, are shown. (B) Staining for the pluripotency markers (OCT4, NANOG, TRA1-60, green; alkaline phosphatase activity, red) in MELAS (A24#1–3) iPSC lines. (C) Copy numbers of WT and Mut mtDNA in the control (454E2) and MELAS (A24#1–3) iPSCs were quantified by qPCR and normalized to nuclear DNA (FBXO15) levels (4 replicates). *P < 0.05: comparison of WT mtDNA levels between control (454E2) and MELAS (A24#1–3) iPSCs; #P < 0.05: comparison of Mut mtDNA levels between control (454E2) and MELAS (A24#1–3) iPSCs. (D) Changes in the number of the control (454E2) and MELAS (A24#1–3) iPSCs during 3 days of maintenance in culture (n = 3; *P < 0.05). (E) Lack of changes in the mitochondrial area in electron microscopy images (n = 30–42). WT, wild-type; Mut, mutant. Scale bar, 100 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The increase in cell number after dissociation was significantly attenuated in the three MELAS iPSC lines compared with that in the control iPSC line (Fig. 1D). However, the sizes of mitochondria measured in electron microscopy images were not different between control and MELAS iPSCs (Fig. 1E).
3.2. Respiratory deficit in MELAS iPSCs
Because mitochondria have a critical role in OXPHOS that contributes to ATP production by utilizing oxygen, the oxygen consumption rate (OCR) in each iPSC line was measured by using a flux analyzer (Fig. 2A–C). At baseline, the OCR was significantly reduced in all three MELAS iPSCs (A24#1–3) (Fig. 2B), indicating that mitochondrial respiration was suppressed in MELAS iPSCs. The application of 1 μM oligomycin, an ATP synthase inhibitor that suppresses OXPHOS, decreased the OCR in both control and MELAS iPSCs, suggesting the MELAS iPSCs retained OCR, which depends on ATP synthase activity. The application of 10 μM BAM15, a mitochondrial protonophore uncoupler that induces mitochondrial membrane depolarization and maximizes OCR, demonstrated that maximal OCR was also reduced in all three MELAS iPSCs compared with that in control iPSCs (Fig. 2C). In the same experiment, the extracellular acidification rate (ECAR) that describes the formation of lactate generated by glycolysis (Fig. 2D–F) was also reduced both at baseline (Fig. 2E) and after oligomycin administration (Fig. 2F), which indicated that respiratory systems outside the mitochondria were also impaired and unable to compensate for the OXPHOS deficiency in all MELAS iPSC lines (A24#1–3).
Fig. 2.

Metabolic deficits in MELAS iPSCs. (A–C) The oxygen consumption rate (OCR) and (D–F) extracellular acidification rate (ECAR) in the control (454E2) and MELAS (A24#1–3) iPSCs measured by an extracellular flux analyzer. Oligomycin (an ATP synthase inhibitor, 1 μM), BAM15 (a mitochondrial protonophore uncoupler, 10 μM), and R/A (rotenone, a mitochondrial electron transport chain complex I inhibitor, 2 μM; antimycin A, a mitochondrial electron transport chain complex III inhibitor, 2 μM) were sequentially added to the wells. OCRs in the control (454E2) and MELAS (A24#1–3) iPSCs were compared at baseline (B, at 1 min 23 s) and the maximal oxygen consumption condition after adding BAM15 (C, at 53 min 20 s). ECARs in the control (454E2) and MELAS (A24#1–3) iPSCs were compared at baseline (E, at 1 min 23 s) and the maximal extracellular acidification condition after adding oligomycin (F, at 27 min 21 s). (A–F) n = 3–4. (G–I) mRNA levels of the mtDNA gene encoding NADH dehydrogenase 6 (ND6, G), pyruvate dehydrogenase E1 subunit alpha 1 (PDHA1, H), and lactate dehydrogenase-B (LDHB, I) (n = 3). (J) Schematic of the energy metabolic pathway changes in MELAS iPSCs. (K–M) Examples of staining of live cells by calcein (green, K), proliferating cells by anti-Ki67 (red, arrowheads, L), and dying cells by anti-cleaved caspase 3 (green, arrowheads, M) with counterstaining of the nuclei by DAPI (blue in L and M) with or without 2.5 mM 2-deoxy-d-glucose (2DG) and 1 mM taurine (Tau) in the control (454E2) and MELAS (A24#2) iPSCs. 2DG reduced the number of MELAS iPSC by decreasing proliferation and increasing the number of dying cells, whereas taurine counteracted both these processes (n = 3). *P < 0.05. Scale bar, 100 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
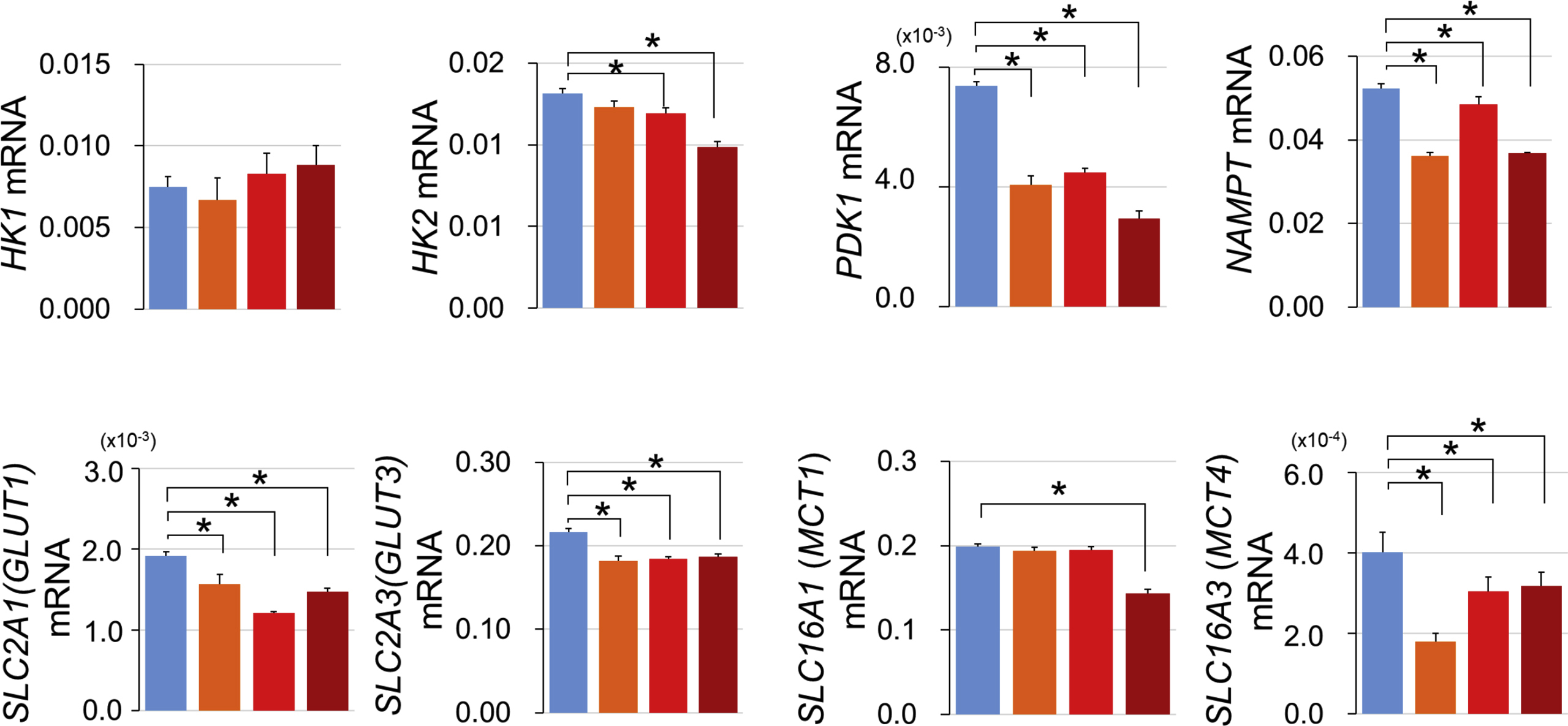
At baseline, mRNA levels of the mitochondrial gene ND6, (Fig. 2G), pyruvate dehydrogenase E1 subunit alpha 1 (PDHA1, Fig. 2H), and lactate dehydrogenase (LDHB, Fig. 2I) were downregulated in the MELAS iPSC lines. Other genes, encoding respiratory system-related proteins, such as hexokinase-2 (HK2), pyruvate dehydrogenase kinase 1 (PDK1), nicotinamide phosphoribosyltransferase (NAMPT), glucose transporters 1 and 3 (SLC2A1, SLC2A3), and monocarboxylate transporter 4 (SLC16A3) were also expressed at a lower level in MELAS iPSC lines (Supplementary material, Fig. S1). The roles of the abovementioned molecules are schematically shown in Fig. 2J.
3.3. Cytoprotective effects of taurine in MELAS iPSCs
To identify potential cytoprotective drugs that could be effective in mitochondrial disorders, we established a drug screening system (Fig. 2K). The number of live cells was evaluated by the staining with calcein followed by binarization of the images and measurement of the stained area using ImageJ program. First, to activate the mitochondria in MELAS iPSCs, PDK inhibitors such as JX06, DCA, and DAP were added to the culture medium. We found that these drugs were rather toxic to MELAS iPSCs, while some increased the number of control iPSCs (Supplementary material, Fig. S2). Then, we analyzed the effect of taurine; for this experiment, we added 2DG at a concentration of 2.5 mM, which decreased the number of live MELAS iPSCs but not of the control iPSCs (Fig. 2K). This system, using calcein staining under 2DG administration showed that the decrease in live MELAS iPSCs by 2DG was significantly attenuated by taurine administration (Fig. 2K).
Then, we analyzed whether the 2DG-induced decrease in the number of live MELAS iPSCs and its rescue by taurine were due to changes in cell proliferation and/or cell death. Immunostaining for Ki67 indicated that taurine augmented the number of proliferating cells, which was attenuated by 2DG, in all three MELAS iPSC lines (Fig. 2L). The staining with an antibody against cleaved caspase 3 showed that 2DG-induced increase in apoptosis in MELAS A24#2 and A24#3 iPSCs was attenuated by taurine administration (Fig. 2M). The degrees of cell proliferation and cell death were not changed by 2DG alone or in combination with taurine in control iPSCs (454E2). Thus, taurine counteracted the decrease in cell proliferation and the increase in cell death selectively in MELAS iPSCs.
3.4. Taurine increases the GSH/GSSG ratio in MELAS iPSCs
To understand the mechanisms of the protective effects of taurine, we performed the metabolome analysis of control (454E2) and MELAS (A24#2) iPSCs grown in the presence or absence of taurine. We found that a substantial decrease in the reduced (GSH) and the increase in oxidized (GSSG) glutathione, as well as the resulting decrease in the GSH/GSSG ratio in MELAS iPSCs, were all rescued by taurine (Fig. 3A). The levels of cysteine were increased in MELAS iPSCs, and mRNA expression of the genes encoding glutamate-cysteine ligase catalytic subunit (GCLC) and glutathione synthetase (GSS) were upregulated in MELAS iPSCs (Supplementary material, Fig. S3), suggesting that GSH synthesis was increased. Whereas, GSSG was increased in MELAS iPSCs (Fig. 3A). Taken together, it indicated that GSH was extensively synthesized, however, its utilization may have been substantially enhanced and overwhelmed the production in MELAS iPSCs, which decreased the GSH/GSSG ratio. This was most likely because MELAS iPSCs underwent higher oxidative stress as evidenced by the upregulated expression of the genes encoding antioxidant enzymes (Fig. 3B), heme oxygenase 1 (HMOX1, HO-1, Fig. 3C), catalase (CAT, Fig. 3D), and glutathione reductase (GSR, Fig. 3E). However, the GSH/GSSG ratio was increased while cysteine was decreased by taurine treatment, suggesting that taurine reduced the level of oxidative stress and GSH consumption. Consistently, taurine reduced not only mRNA levels of HMOX1 (Fig. 3C), CAT (Fig. 3D), and GSR (Fig. 3E) but also those of glutathione peroxidase 1 (GPX1, Fig. 3F) and superoxide dismutase 2 (SOD2, Fig. 3G) in either MELAS or control iPSCs, or both WT and mutant lines. The level of hypotaurine, an intermediate product of taurine that has a reducing capacity, was not increased (Fig. 3A), and the genes that encode the enzymes producing hypotaurine, CDO1, CSAD, and ADO were all downregulated in taurine-treated MELAS iPSCs (Supplementary material, Fig. S3), supporting the idea that the antioxidant effect of hypotaurine was not required after the taurine treatment.
Fig. 3.

Restoration of the decreased GSH/GSSG ratio by taurine in MELAS iPSCs. (A) Quantities of methylated products measured by the metabolome assay. The GSH/GSSG ratio was reduced in MELAS iPSCs, however, it was restored by the treatment with 1 mM taurine. Data from control iPSCs (454E2), MELAS iPSCs (A24#2), and MELAS iPSCs (A24#2) treated with taurine are illustrated by blue, red, and green bars, respectively (n = 6). (B) Schematic of the redox pathways in MELAS iPSCs in the absence and presence of taurine. (C–G) mRNA levels of the antioxidant enzymes heme oxygenase 1 (HMOX, C), catalase (CAT, D), glutathione reductase (GSR, E), glutathione peroxidase 1 (GPX1, F), and superoxide dismutase 2 (SOD2, G) (n = 3). *P < 0.05 for comparisons between control and MELAS iPSCs; #P < 0.05 for comparisons of the values obtained in the presence and absence of taurine treatment in MELAS iPSCs (ANOVA followed by the post hoc Tukey's test). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The activity of the pentose phosphate pathway (PPP) that generates NADPH and increases GSH levels was upregulated in MELAS iPSCs (Supplementary material, Fig. S4), suggesting that the PPP was induced to increase GSH generation under oxidative stress conditions. However, this increase in the PPP activity was not sufficient most likely due to the abnormally high level of oxidative stress that consumed GSH. Accumulation of phosphoenolpyruvic acid (PEP) was observed in MELAS iPSCs, which was suppressed by taurine treatment (Supplementary material, Fig. S4). Despite mRNA levels of genes encoding pyruvate kinase M1 and M2 (PKM1 and PKM2) were increased, the inactivation of PKM1 and PKM2 by oxidative stress [42] may have suppressed the conversion of PEP to pyruvate, whereas the reduction in oxidative stress by taurine allowed the conversion (Supplementary material, Fig. S4).
The levels of tricarboxylic acid (TCA) cycle metabolites were not changed (Supplementary material, Fig. S4), although the OCR was reduced in MELAS iPSCs. Activation of the PPP could induce nucleotide production through the nitrogen-shift pathway [43]. Expression of the gene encoding phosphoribosyl pyrophosphate amidotransferase (PPAT), which is a key enzyme in nucleotide biosynthetic pathways, was increased (Supplementary material, Fig. S4), and indeed, the nucleic acid levels were preserved or even increased in MELAS iPSCs, with the latter effect being sensitive to taurine (Supplementary material, Fig. S5). Amino acids were also increased in MELAS iPSCs, and this elevation was reduced by taurine treatment (Supplementary material, Fig. S5). Considering that the mRNA expression of the autophagy-related molecule, LC3B [44] was increased, which was downregulated by taurine treatment, in a MELAS iPSC line (A24#2) (Supplementary material, Fig. S6), autophagy could have been affected in MELAS iPSCs. Expression levels of the genes encoding amino acid transporters SLC7A5, SLC7A8, SLC43A1, SL43A2, SLC7A11, and SLC3A2 were changed in MELAS iPSCs in the absence or presence of taurine, suggesting that changes in amino acid levels may have been at least in part related to the changes in uptake of amino acids by MELAS iPSCs (Supplementary material, Fig. S6). Metabolites of amino acids in the urea cycle were increased in MELAS iPSCs (Supplementary material, Fig. S7), indicating that amino acids could be utilized as an energy source in conditions of OXPHOS dysregulation. In contrast, amino acid levels were decreased by taurine, suggesting that this treatment may have normalized energy demand.
3.5. Characteristics of the MELAS iPSC-derived RPE
Because MELAS patients have been reported to exhibit RPE degeneration [[8], [9], [10], [11], [12], [13]], the RPE samples differentiated from the retinal organoid cultures of control (454E2) and MELAS (A24#1–3) iPSCs (Fig. 4A) were analyzed. After 60 days in culture, clusters of pigmented cells found in the organoids were picked and plated on Matrigel-coated dishes, followed by several passages for purification (Fig. 4B). Although both control and MELAS iPSC-derived RPE had pigmentation and expressed the RPE marker OTX2 and the tight junction marker ZO-1 (Fig. 4B), regular hexagons bounded by ZO-1 staining generally observed in the RPE were less frequently observed in the MELAS iPSC-derived RPE than in the control iPSC-derived RPE (Fig. 4B and C). Moreover, the size of the RPE cells derived from MELAS iPSCs were larger than those of control (Fig. 4D). Under the condition, the levels of the mutant mtDNA in the MELAS iPSC-derived RPE were confirmed to be maintained (Fig. 4E).
Fig. 4.

Structural irregularity of the MELAS iPSC-derived RPE. (A) Procedures of the three-dimensional retinal organoid and RPE differentiation culture. (B) The pigmented cells derived from the control (454E2) and MELAS (A24#2) iPSCs were purified through several passages to generate the RPE. The RPE cells showed the hexagonal cell shape and expressed tight junction (ZO-1, green) and RPE (OTX2, red) markers. (C) Cell shapes of the RPE derived from the control (454E2) and MELAS (A24#2) iPSCs were quantified by calculating their circularity. MELAS iPSC-derived RPE had irregular shape (n = 6, Over 1000 cells were analyzed and averaged in each group. *P < 0.05). (D) Size of the cell body of the RPE derived from the control (454E2) and MELAS (A24#2) iPSCs were quantified by the areas surrounded by ZO-1. n = 1000, *P < 0.05. (E) Copy numbers of WT and Mut mtDNAs in the RPE derived from the control (454E2) and MELAS (A24#1–3) iPSCs determined by qPCR (4 replicates). *P < 0.05 for comparisons of WT mtDNA levels between control and MELAS iPSCs; #P < 0.05 for comparison of Mut mtDNA levels between control (454E2) and MELAS (A24#1–3) RPE. (ANOVA followed by the post hoc Tukey's test). WT, wild-type; Mut, mutant. Scale bar, 100 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.6. Respiratory deficit and EMT of the MELAS iPSC-derived RPE
The OCR was reduced in MELAS iPSC-derived RPE (Fig. 5A–C) both at baseline (Fig. 5B) and in the presence of the mitochondrial protonophore uncoupler, BAM15 (Fig. 5C). The ECAR was also reduced in MELAS iPSC-derived RPE (Fig. 5D–F) both at baseline (Fig. 5E) and after OXPHOS suppression by oligomycin (Fig. 5F), indicating that as in the MELAS iPSCs, OXPHOS impairment was not compensated by glycolysis in the differentiated RPE. The mRNA expression level of the mtDNA encoding gene ND6 was downregulated in MELAS iPSC-derived RPE (Fig. 5G), whereas catalase mRNA was significantly upregulated (Fig. 5H), which collectively suggested a higher level of oxidative stress in MELAS iPSC-derived RPE.
Fig. 5.

Respiratory deficit in the MELAS iPSC-derived RPE. (A–C) The oxygen consumption rate (OCR) and the (D–F) extracellular acidification rate (ECAR) in the control (454E2) and MELAS (A24#1–3) iPSC-derived RPE samples measured by an extracellular flux analyzer. Oligomycin (1 μM), BAM15 (10 μM), and R/A (rotenone, 2 μM; antimycin A, 2 μM) were sequentially added to the well. OCRs in the control (454E2) and MELAS (A24#1–3) iPSC-derived RPE were compared at baseline (B, at 1 min 23 s) and the maximal oxygen consumption condition after adding BAM15 (C, at 53 min 20 s). ECARs in the control (454E2) and MELAS (A24#1–3) iPSC-derived RPE were compared at baseline (E, at 1 min 23 s) and at the maximal extracellular acidification condition after the addition of oligomycin (F, at 27 min 21 s). n = 3–4. (G–N) mRNA levels of the mtDNA gene encoding NADH dehydrogenase 6 (ND6, G), the antioxidant enzyme catalase (CAT, H), as well as genes encoding epithelial-mesenchymal transition markers ZEB1 (I), SNAI1 (J), TWIST1 (K), ACTA (L), CDH2 (M), and CDH3 (N) in the control (454E2) and MELAS (A24#1–3) iPSC-derived RPE. n = 4. *P < 0.05.
The RPE is a single-layered epithelium that contributes to the barrier between photoreceptors and the choroid. However, when EMT occurs, retinal diseases such as age-related macular degeneration are promoted [17]. We found that mRNA levels of the EMT markers zinc finger E-box-binding homeobox 1 (ZEB1, Fig. 5I), snail family zinc finger 1 (SNAI1, Fig. 5J), twist-related protein 1 (TWIST1, Fig. 5K), and actin alpha 2 (ACTA2, Fig. 5L), were increased in the MELAS iPSC-derived RPE compared with those in the control (454E2) iPSC-derived RPE. Moreover, mRNA level of cadherin 2 (CDH2, Fig. 5M) was increased and cadherin 3 (CDH3, Fig. 5N) was decreased, consistent with the previous report that showed changes in cadherin expression levels during the EMT of the RPE in animal models [18]. The enlargement of the cell body (Fig. 4D) was consistent with the propensity of EMT, and resulting migration out of the MELAS iPSC-derived RPE cells from epithelial tissue, given that the RPE cell body enlarges when the adjacent cells are lost [45,46].
3.7. Suppressive effects of taurine on EMT in the MELAS iPSC-derived RPE
The effects of taurine on EMT-related changes in the iPSC-derived RPE were then investigated. The increases in the expression levels of ZEB1 (Fig. 6A), SNAI1 (Snail, Fig. 6B), TWIST1 (Fig. 6C), and ACTA2 (αSMA, Fig. 6D) in the MELAS iPSC-derived RPE, were all reverted by taurine treatment (Fig. 6A–D).
Fig. 6.

Protective effect of taurine on the epithelial-mesenchymal transition processes in MELAS iPSC-derived RPE. (A–F) mRNA levels of the genes encoding epithelial-mesenchymal transition markers ZEB1 (A), SNAI1 (B), TWIST1 (C), ACTA (D), TGFB1 (E), and TGFB2 (F) in the control (454E2) and MELAS (A24#2) iPSC-derived RPE in the absence or presence of the treatment with taurine (n = 3). (G–J) Staining of the cells for ACTA (αSMA) (G, green; H), and NRF2 (I, green, counterstaining with DAPI in magenta; J) in MELAS (A24#2) iPSC-derived RPE in the absence or presence of the treatment with taurine (n = 6). *P < 0.05. Scale bar, 100 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
EMT is generally induced by TGFβ [47], and the upregulation of TGFB1 (Fig. 6E) and TGFB2 (Fig. 6 F) mRNA levels in the MELAS iPSC-derived RPE was suppressed also by taurine administration. Along with these EMT-related changes, the number of ACTA2 (αSMA)-positive cells in the MELAS iPSC-derived RPE was also suppressed by taurine (Fig. 6G and H). Also, we observed that the nuclei of the MELAS iPSC-derived RPE exhibited higher accumulation of the antioxidant transcription factor NRF2 that translocates into the nucleus under oxidative stress [48], and this effect was attenuated by taurine treatment (Fig. 6I and J), suggesting that oxidative stress in the MELAS iPSC-derived RPE was reduced by taurine.
4. Discussion
We demonstrated that MELAS patient-derived iPSCs had a deficient mitochondrial respiratory function, which was not compensated by glycolysis. Diminished proliferation and survival of the MELAS iPSCs were rescued by the administration of taurine that normalized the decreased GSH/GSSG ratio and increased oxidative stress markers in MELAS iPSCs. In the RPE derived from MELAS iPSCs, hexagonal structure, as well as respiratory function was disturbed. The EMT markers were increased in the RPE derived from MELAS iPSCs, and all these changes were reverted by taurine administration. Moreover, the accumulation of nuclear NRF2 in MELAS iPSCs was also suppressed by taurine.
4.1. Energy metabolism in MELAS iPSCs
In the current study, the basal and maximal OCRs were reduced in MELAS iPSCs, which was consistent with the results of the previous reports [22]. This could be related to impaired iPSC growth after the dissociation. We focused on the mitochondrial respiration by suppressing glycolysis with 2DG and found that cell proliferation and survival were both impaired in MELAS iPSCs, whereas the same dose of 2DG did not affect control iPSCs. Because both glycolysis and mitochondrial respiratory system were impaired in MELAS iPSCs, the minimum reduction of glycolytic ability may be critical for survival. Importantly, taurine treatment rescued the cells, whereas PDH activation by PDK inhibitors undertaken to increase the levels of TCA cycle substrates did not. Instead, the overload with TCA cycle substrates may have been cytotoxic in the MELAS iPSCs, supporting the idea that MELAS pathogenesis may involve excessive stress on the abnormally working mitochondrial respiratory system. Because the system remained functional in MELAS iPSCs, albeit at a lower level as shown by the OCR data, inappropriate reactions could have increased oxidative stress. Future studies to realize the details of this process would be of interest.
4.2. Oxidative stress in MELAS iPSCs and its reduction by taurine
Remarkably, MELAS iPSCs retained low levels of GSH and had high levels of GSSG, which resulted in the lower GSH/GSSG ratio. The levels of cysteine, a substrate of GSH, were increased and oxidative stress markers were elevated, suggesting that GSH was produced but consumed to adapt to the oxidative stress generated in MELAS iPSCs. The reduction in the ECAR, lactate, pyruvate, and the increase in the upstream metabolite PEP in MELAS iPSCs most likely have occurred because the oxidative stress inactivated PKM that converts PEP to pyruvate [42]. Such inactivation could be one of the compensatory alterations to reduce the TCA cycle reactions and OXPHOS to attempt to decrease oxidative stress. Upregulated PPP may have increased GSH levels in response, however, it was insufficient to prevent MELAS-related oxidative stress. The upregulated PPP may have coupled with glutamine metabolism to increase the nitrogen shift that produces nucleotides [43]. In general, an increase in nucleotides could induce cell proliferation, which is observed in cancer cells [43], however, in MELAS iPSCs, proliferation was not increased but rather impaired. Therefore, an increase in nucleotides could be a necessary, but insufficient condition for the augmented proliferation, with the increased oxidative stress due to the mitochondrial dysregulation having a negative effect on the proliferation. It has been reported that increased oxidative stress and reduction of GSH cause cell cycle arrest, interfering with cell proliferation [49]. Thus, enhanced oxidative stress, as well as GSH system dysregulation, might have resulted in the impaired proliferation of MELAS iPSCs despite their increased nucleotide biosynthesis. Collectively, the normal mitochondrial condition is required for regulating oxidative stress even when mitochondrial OXPHOS machinery is uncoupled from glycolysis in PSCs [[30], [31], [32]].
One of the cytoprotective effects of taurine in our experiments was the increase in the GSH/GSSG ratio in the absence of enhanced levels of cysteine or of the GSH synthesis enzyme expression. Considering that oxidative stress markers were decreased, taurine may have decreased oxidative stress levels through the pathway(s) different from those generating GSH.
Taurine is involved in a variety of biological processes and is found at high concentrations in specific tissues, including the retina, brain, and muscles, [50]. Taurine scavenges many reactive oxygen and nitrogen species [51], and the antioxidant potential may be enhanced by drinking taurine-containing water [52]. Taurine has been reported to activate NRF2 and induce its downstream antioxidant response element responsive genes, including HMOX1 (HO-1), in several cell lines [[53], [54], [55], [56]], but this was not observed in the current study. Additionally, taurine is taken up by mitochondria, where it modifies uridines with the formation of 5-taurinomethyluridine and 5-taurinomethyl-2-thiouridine of the mitochondrial tRNAs, which potentiates efficient codon binding [57], however, this mechanism of taurine action will require detailed future studies.
4.3. Propensity for EMT in mitochondrial disorders
The protective effect of taurine was also observed in the RPE derived from MELAS iPSCs, which showed the propensity to undergo EMT. As has been shown in human RPE samples attached to the choroidal neovascularization extracted by surgery, the EMT of the RPE is involved in AMD pathogenesis [17]. TGFβ, an inflammatory cytokine that induces EMT in the RPE [17], was upregulated in the MELAS iPSC-derived RPE, consistent with a previous report that showed augmented TGFB expression caused by the dysregulation of miRNAs in MELAS cybrids [58]. Mitochondrial dysfunction was reported to change intracellular Ca2+ signaling and nuclear gene expression in an RPE cell line [59].
Mitochondria have been recognized as key players in several cancer-related phenomena, including invasiveness and metastases [60], which are initiated by the EMT [61]. The interaction between oxidative stress and the EMT is well known [47]. Moreover, oxidative stress and inflammation interact to accelerate each other [15], and we found that TGFB was induced in the MELAS iPSC-derived RPE. Alternatively, EMT induction may be achieved by the changes in glutamine metabolism afforded by the knockdown of the mitochondrial pyruvate carrier [62]. Several pathways could be involved in EMT induction by mitochondrial disorders. Taurine reduced oxidative stress and normalized glutamine metabolism in MELAS iPSCs, suggesting that it likely acted on several pathways to suppress the EMT.
4.4. MELAS iPSCs as a drug screening system
We constructed a drug screening system that utilized mitochondrial dysfunction in MELAS iPSCs by suppressing glycolysis with 2DG administration, in which the effect was evaluated easily by measuring the area of live cells in the images. This system could enable the identification of the drugs effective for mitochondrial diseases in addition to taurine. Previous reports have described several potential drugs that might be effective for treating MELAS and mitochondrial dysfunction in other in vitro systems, e.g., I-BET 525762A, a pan-BET inhibitor [63,64], riboflavin and coenzyme Q10 [65], as well as β-lapachone, a substrate of NAD(P)H quinone oxidoreductase 1 [66]. It would be of interest to test these candidates in our drug screening system in future studies.
4.5. Merits of using taurine compared with gene therapies
Mutant mtDNA elimination was performed by using mitochondria-targeted ZFN, TALEN, and CRISPR/Cas9 [[67], [68], [69], [70], [71], [72], [73]], and the specific base editing by cytidine deaminase can change the cellular function, including OCR [74]. These gene-editing technologies could be applied to human iPSC-derived disease models [24,[75], [76], [77]]. However, these procedures may not be necessarily useful for the disease that affects multiple tissues and organs, and/or a wide area of a single tissue, in contrast to the effects of the systemic administration of drugs.
In summary, we have established MELAS patient-derived iPSC lines and analyzed the phenotypes in these cells as well as in the RPE differentiated from them. Treatment with taurine promoted the proliferation and survival of MELAS iPSCs and partly rescued the pathological EMT status of the MELAS iPSC-derived RPE. The current study will help future clinical trials of taurine in MELAS and other diseases accompanied by mitochondrial disorders.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank the members of the Laboratory of Retinal Cell Biology for their kind assistance. We also thank Dr. Shinsuke Shibata (Department of Physiology, Keio University School of Medicine/Division of Microscopic Anatomy Graduate School of Medical and Dental Sciences Niigata University) for supporting our electron microscopic experiments, Drs. Takako Hishiki and Makoto Suematsu (Department of Biochemistry and Integrative Medical Biology, Keio University School of Medicine) for the metabolome analysis.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.101921.
Funding
The study was partially supported by a grant from JSR corporation to KH and YO, the Mochida Memorial Foundation for Medical and Pharmaceutical Research to KH, Keio University Internal Research Funding for the Advancement of Next Generation Research Projects to YO, Keio University Global Research Institute (KGRI) Program for the Advancement of Research in Core Projects and a Grants-in-Aid for Scientific Research from Japan Society of the Promotion of Science to KH (19K09977) and YO (20K09833, 18K09422).
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Respiratory enzymes and substrate transporters in MELAS iPSCs. mRNA levels of hexokinase 1 (HK1), hexokinase 2 (HK2), 3-phosphoinositide-dependent protein kinase 1 (PDK1), nicotinamide phosphoribosyltransferase (NAMPT), glucose transporters SLC2A1 and SLC2A3, and lactate transporters SLC16A1 and SLC16A3 in the control (454E2) and MELAS (A24#1–3) iPSCs (n = 3). *P < 0.05.
{kind=link}
Drug screening system and effects of PDK inhibitors that activate pyruvate dehydrogenase (A, B) A drug screening system based on the effects of 2-deoxy-D-glucose (2DG). (C–E) The relative areas occupied by live cells stained by calcein and treated with 3-phosphoinositide-dependent protein kinase 1 (PDK1) inhibitors JX06 (1, 15, 30 mM, C), DAP (10, 30, 60 μM, D), or DCA (10, 50, 100 μM, E) were compared with those of the DMSO-treated cells in the control (454E) and MELAS (A24#1-3) iPSCs.
{kind=link}
Enzymes for GSH generation in MELAS iPSCs. mRNA levels of glutamate-cysteine ligase catalytic subunit (GCLC), glutathione synthetase (GSS), cysteine dioxygenase type 1 (CDO1), cysteine sulfinic acid decarboxylase (CSAD), and 2-aminoethanthiol dioxygenase (ADO) in the control (454E2) and MELAS (A24#1–3) iPSCs (n = 3). Enzymatic cascades for GSH generation are schematically shown. *P < 0.05 for comparisons between the control and MELAS iPSCs; #P < 0.05 for comparisons of data obtained in the presence and absence of the treatment with taurine in MELAS iPSCs (ANOVA followed by the post hoc Tukey’s test in both cases).
{kind=link}
Glucose metabolism in MELAS iPSCs. Results of the metabolome assay measuring the levels of the components of glycolysis, pentose phosphate pathway (PPP), and TCA cycle are shown (N = 6). The nitrogen shift pathway that contributes to nucleotide production by activating PPP and glutamine metabolism is schematically shown. mRNA levels of pyruvate kinases M1 and M2 (PKM1 and PKM2), phosphoribosyl pyrophosphate amidotransferase (PPAT), as well as of glutaminases 1 and 2 (GLS1 and GLS2) in the control and MELAS iPSCs are illustrated (n = 3). *P < 0.05 for comparisons between control and MELAS iPSCs; #P < 0.05 for comparisons of data obtained in the presence and absence of the treatment with taurine in MELAS iPSCs (ANOVA followed by the post hoc Tukey’s test in both cases).
{kind=link}
Metabolism of nucleotides and amino acids in MELAS iPSCs. Results of the metabolome assay measuring the levels of nucleic acids and amino acids are shown (N = 6). *P < 0.05 for comparisons between control and MELAS iPSCs; #P < 0.05 for comparisons of data obtained in the presence and absence of the treatment with taurine in MELAS iPSCs (ANOVA followed by the post hoc Tukey’s test in both cases).
{kind=link}
Expression levels of an autophagy marker and amino acid transporters. mRNA levels of the genes encoding the autophagy marker LC3A, LC3B, the amino acid transporters SLC7A5, SLC7A8, SLC43A1, SL43A2, SLC7A11, and SLC3A2, were measured in each iPSC line in the presence or absence of the treatment with taurine (n = 3). The role of each molecule is schematically shown. *P < 0.05 for comparisons between the control and MELAS iPSCs; #P < 0.05 for comparisons of data obtained in the presence and absence of the treatment with taurine in iPSCs (ANOVA followed by the post hoc Tukey’s test in both cases).
{kind=link}
Urea cycle products in MELAS iPSCs. Results of the metabolome assay, measuring the levels of the urea cycle products are shown (N = 6). *P < 0.05 for comparisons between the control and MELAS iPSCs; #P < 0.05 for comparisons of data obtained in the presence and absence of the treatment with taurine in MELAS iPSCs (ANOVA followed by the post hoc Tukey’s test in both cases).
{kind=link}
References
- 1.El-Hattab A.W. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metabol. 2015;116(1–2):4–12. doi: 10.1016/j.ymgme.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Ferrington D.A. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp. Eye Res. 2016;145:269–277. doi: 10.1016/j.exer.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feher J. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging. 2006;27(7):983–993. doi: 10.1016/j.neurobiolaging.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 4.Wallace D.C., Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013;5(11):a021220. doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goto Y., Nonaka I., Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348(6302):651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- 6.Schaffer S.W. Role of taurine in the pathologies of MELAS and MERRF. Amino Acids. 2014;46(1):47–56. doi: 10.1007/s00726-012-1414-8. [DOI] [PubMed] [Google Scholar]
- 7.Ohsawa Y. Taurine supplementation for prevention of stroke-like episodes in MELAS: a multicentre, open-label, 52-week phase III trial. J. Neurol. Neurosurg. Psychiatry. 2019;90(5):529–536. doi: 10.1136/jnnp-2018-317964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu C.C., Traboulsi E.I., Parikh S. Ophthalmological findings in 74 patients with mitochondrial disease. Ophthalmic Genet. 2017;38(1):67–69. doi: 10.3109/13816810.2015.1130153. [DOI] [PubMed] [Google Scholar]
- 9.Phillips P.H., Newman N.J. Mitochondrial diseases in pediatric ophthalmology. J aapos. 1997;1(2):115–122. doi: 10.1016/s1091-8531(97)90010-0. [DOI] [PubMed] [Google Scholar]
- 10.Sue C.M. Pigmentary retinopathy associated with the mitochondrial DNA 3243 point mutation. Neurology. 1997;49(4):1013–1017. doi: 10.1212/wnl.49.4.1013. [DOI] [PubMed] [Google Scholar]
- 11.de Laat P. Mitochondrial retinal dystrophy associated with the m.3243A>G mutation. Ophthalmology. 2013;120(12):2684–2696. doi: 10.1016/j.ophtha.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 12.Smith P.R. Pigmentary retinal dystrophy and the syndrome of maternally inherited diabetes and deafness caused by the mitochondrial DNA 3243 tRNA(Leu) A to G mutation. Ophthalmology. 1999;106(6):1101–1108. doi: 10.1016/S0161-6420(99)90244-0. [DOI] [PubMed] [Google Scholar]
- 13.Daruich A., Matet A., Borruat F.X. Macular dystrophy associated with the mitochondrial DNA A3243G mutation: pericentral pigment deposits or atrophy? Report of two cases and review of the literature. BMC Ophthalmol. 2014;14:77. doi: 10.1186/1471-2415-14-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strauss O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005;85(3):845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 15.Ozawa Y. Oxidative stress in the light-exposed retina and its implication in age-related macular degeneration. Redox Biol. 2020:101779. doi: 10.1016/j.redox.2020.101779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagai N. Renin-angiotensin system impairs macrophage lipid metabolism to promote age-related macular degeneration in mouse models. Commun. Biol. 2020;3(1):767. doi: 10.1038/s42003-020-01483-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirasawa M. Transcriptional factors associated with epithelial-mesenchymal transition in choroidal neovascularization. Mol. Vis. 2011;17:1222–1230. [PMC free article] [PubMed] [Google Scholar]
- 18.Tamiya S., Liu L., Kaplan H.J. Epithelial-mesenchymal transition and proliferation of retinal pigment epithelial cells initiated upon loss of cell-cell contact. Invest. Ophthalmol. Vis. Sci. 2010;51(5):2755–2763. doi: 10.1167/iovs.09-4725. [DOI] [PubMed] [Google Scholar]
- 19.King M.P., Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246(4929):500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 20.Kodaira M. Impaired respiratory function in MELAS-induced pluripotent stem cells with high heteroplasmy levels. FEBS Open Bio. 2015;5:219–225. doi: 10.1016/j.fob.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin D.S. Oxidative insults and mitochondrial DNA mutation promote enhanced autophagy and mitophagy compromising cell viability in pluripotent cell model of mitochondrial disease. Cells. 2019;8(1) doi: 10.3390/cells8010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma H. Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature. 2015;524(7564):234–238. doi: 10.1038/nature14546. [DOI] [PubMed] [Google Scholar]
- 23.Pek N.M.Q. Mitochondrial 3243A > G mutation confers pro-atherogenic and pro-inflammatory properties in MELAS iPS derived endothelial cells. Cell Death Dis. 2019;10(11):802. doi: 10.1038/s41419-019-2036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klein Gunnewiek T.M. m.3243A > G-induced mitochondrial dysfunction impairs human neuronal development and reduces neuronal network activity and synchronicity. Cell Rep. 2020;31(3):107538. doi: 10.1016/j.celrep.2020.107538. [DOI] [PubMed] [Google Scholar]
- 25.Chichagova V. Human iPSC disease modelling reveals functional and structural defects in retinal pigment epithelial cells harbouring the m.3243A > G mitochondrial DNA mutation. Sci. Rep. 2017;7(1):12320. doi: 10.1038/s41598-017-12396-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osakada F. Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat. Biotechnol. 2008;26(2):215–224. doi: 10.1038/nbt1384. [DOI] [PubMed] [Google Scholar]
- 27.Homma K., Usui S., Kaneda M. Knock-in strategy at 3'-end of Crx gene by CRISPR/Cas9 system shows the gene expression profiles during human photoreceptor differentiation. Gene Cell. 2017;22(3):250–264. doi: 10.1111/gtc.12472. [DOI] [PubMed] [Google Scholar]
- 28.Osakada F. Stepwise differentiation of pluripotent stem cells into retinal cells. Nat. Protoc. 2009;4(6):811–824. doi: 10.1038/nprot.2009.51. [DOI] [PubMed] [Google Scholar]
- 29.Kuwahara A. Generation of a ciliary margin-like stem cell niche from self-organizing human retinal tissue. Nat. Commun. 2015;6:6286. doi: 10.1038/ncomms7286. [DOI] [PubMed] [Google Scholar]
- 30.Shyh-Chang N. Human pluripotent stem cells decouple respiration from energy production. EMBO J. 2011;30(24):4851–4852. doi: 10.1038/emboj.2011.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell. 2012;11(5):589–595. doi: 10.1016/j.stem.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsogtbaatar E. Energy metabolism regulates stem cell pluripotency. Front. Cell. Dev. Biol. 2020;8:87. doi: 10.3389/fcell.2020.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crespo F.L. Mitochondrial reactive oxygen species mediate cardiomyocyte formation from embryonic stem cells in high glucose. Stem Cell. 2010;28(7):1132–1142. doi: 10.1002/stem.441. [DOI] [PubMed] [Google Scholar]
- 34.Saretzki G. Downregulation of multiple stress defense mechanisms during differentiation of human embryonic stem cells. Stem Cell. 2008;26(2):455–464. doi: 10.1634/stemcells.2007-0628. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011;30(24):4860–4873. doi: 10.1038/emboj.2011.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamasaki M. Pathogenic mutation of ALK2 inhibits induced pluripotent stem cell reprogramming and maintenance: mechanisms of reprogramming and strategy for drug identification. Stem Cell. 2012;30(11):2437–2449. doi: 10.1002/stem.1221. [DOI] [PubMed] [Google Scholar]
- 37.Soga M. HPGCD outperforms HPBCD as a potential treatment for Niemann-Pick disease type C during disease modeling with iPS cells. Stem Cell. 2015;33(4):1075–1088. doi: 10.1002/stem.1917. [DOI] [PubMed] [Google Scholar]
- 38.Okita K. A more efficient method to generate integration-free human iPS cells. Nat. Methods. 2011;8(5):409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 39.Shiota M. Gold-nanofève surface-enhanced Raman spectroscopy visualizes hypotaurine as a robust anti-oxidant consumed in cancer survival. Nat. Commun. 2018;9(1):1561. doi: 10.1038/s41467-018-03899-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tohyama S. Glutamine oxidation is indispensable for survival of human pluripotent stem cells. Cell Metabol. 2016;23(4):663–674. doi: 10.1016/j.cmet.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto T. Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway. Nat. Commun. 2014;5:3480. doi: 10.1038/ncomms4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhardwaj V., He J. Reactive oxygen species, metabolic plasticity, and drug resistance in cancer. Int. J. Mol. Sci. 2020;21(10) doi: 10.3390/ijms21103412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kodama M. A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nat. Commun. 2020;11(1):1320. doi: 10.1038/s41467-020-15136-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klionsky D.J. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao J. Evidence for the activation of pyroptotic and apoptotic pathways in RPE cells associated with NLRP3 inflammasome in the rodent eye. J. Neuroinflammation. 2018;15(1):15. doi: 10.1186/s12974-018-1062-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grierson I. Development, repair and regeneration of the retinal pigment epithelium. Eye. 1994;8(Pt 2):255–262. doi: 10.1038/eye.1994.54. [DOI] [PubMed] [Google Scholar]
- 47.Giannoni E., Parri M., Chiarugi P. EMT and oxidative stress: a bidirectional interplay affecting tumor malignancy. Antioxidants Redox Signal. 2012;16(11):1248–1263. doi: 10.1089/ars.2011.4280. [DOI] [PubMed] [Google Scholar]
- 48.Okamoto T. Dietary spirulina supplementation protects visual function from photostress by suppressing retinal neurodegeneration in mice. Transl. Vis. Sci. Technol. 2019;8(6):20. doi: 10.1167/tvst.8.6.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Diaz-Vivancos P. Glutathione--linking cell proliferation to oxidative stress. Free Radic. Biol. Med. 2015;89:1154–1164. doi: 10.1016/j.freeradbiomed.2015.09.023. [DOI] [PubMed] [Google Scholar]
- 50.Seidel U., Huebbe P., Rimbach G. Taurine: a regulator of cellular redox homeostasis and skeletal muscle function. Mol. Nutr. Food Res. 2019;63(16) doi: 10.1002/mnfr.201800569. [DOI] [PubMed] [Google Scholar]
- 51.Oliveira M.W. Scavenging and antioxidant potential of physiological taurine concentrations against different reactive oxygen/nitrogen species. Pharmacol. Rep. 2010;62(1):185–193. doi: 10.1016/s1734-1140(10)70256-5. [DOI] [PubMed] [Google Scholar]
- 52.Nandhini A.T., Balakrishnan S.D., Anuradha C.V. Response of liver antioxidant system to taurine in rats fed high fructose diet. Indian J. Exp. Biol. 2002;40(9):1016–1019. [PubMed] [Google Scholar]
- 53.Cheong S.H., Lee D.S. Taurine chloramine prevents neuronal HT22 cell damage through nrf2-related heme oxygenase-1. Adv. Exp. Med. Biol. 2017;975 Pt 1:145–157. doi: 10.1007/978-94-024-1079-2_13. [DOI] [PubMed] [Google Scholar]
- 54.Sun Q. Nrf2 signaling pathway mediates the antioxidative effects of taurine against corticosterone-induced cell death in HUMAN SK-N-SH cells. Neurochem. Res. 2018;43(2):276–286. doi: 10.1007/s11064-017-2419-1. [DOI] [PubMed] [Google Scholar]
- 55.Park S.Y. Hepatoprotective effects of xylose-taurine reduced against hydrogen peroxide-induced oxidative stress in cultured hepatocytes. Adv. Exp. Med. Biol. 2017;975 Pt 1:621–631. doi: 10.1007/978-94-024-1079-2_48. [DOI] [PubMed] [Google Scholar]
- 56.Yang W. Taurine protects mouse spermatocytes from ionizing radiation-induced damage through activation of Nrf2/HO-1 signaling. Cell. Physiol. Biochem. 2017;44(4):1629–1639. doi: 10.1159/000485762. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki T. Taurine as a constituent of mitochondrial tRNAs: new insights into the functions of taurine and human mitochondrial diseases. EMBO J. 2002;21(23):6581–6589. doi: 10.1093/emboj/cdf656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meseguer S. The MELAS mutation m.3243A>G promotes reactivation of fetal cardiac genes and an epithelial-mesenchymal transition-like program via dysregulation of miRNAs. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2018;1864(9 Pt B):3022–3037. doi: 10.1016/j.bbadis.2018.06.014. [DOI] [PubMed] [Google Scholar]
- 59.Miceli M.V., Jazwinski S.M. Nuclear gene expression changes due to mitochondrial dysfunction in ARPE-19 cells: implications for age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2005;46(5):1765–1773. doi: 10.1167/iovs.04-1327. [DOI] [PubMed] [Google Scholar]
- 60.Guerra F. Mitochondrial dysfunction: a novel potential driver of epithelial-to-mesenchymal transition in cancer. Front. Oncol. 2017;7:295. doi: 10.3389/fonc.2017.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaffer C.L., Weinberg R.A. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 62.Takaoka Y. Mitochondrial pyruvate carrier 1 expression controls cancer epithelial-mesenchymal transition and radioresistance. Canc. Sci. 2019;110(4):1331–1339. doi: 10.1111/cas.13980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nicodeme E. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barrow J.J. Bromodomain inhibitors correct bioenergetic deficiency caused by mitochondrial disease complex I mutations. Mol. Cell. 2016;64(1):163–175. doi: 10.1016/j.molcel.2016.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garrido-Maraver J. Screening of effective pharmacological treatments for MELAS syndrome using yeasts, fibroblasts and cybrid models of the disease. Br. J. Pharmacol. 2012;167(6):1311–1328. doi: 10.1111/j.1476-5381.2012.02086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jeong M.H. β-Lapachone attenuates mitochondrial dysfunction in MELAS cybrid cells. Biochem. Biophys. Res. Commun. 2014;454(3):417–422. doi: 10.1016/j.bbrc.2014.10.093. [DOI] [PubMed] [Google Scholar]
- 67.Gammage P.A. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 2014;6(4):458–466. doi: 10.1002/emmm.201303672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reddy P. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell. 2015;161(3):459–469. doi: 10.1016/j.cell.2015.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bacman S.R. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013;19(9):1111–1113. doi: 10.1038/nm.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yahata N. TALEN-mediated shift of mitochondrial DNA heteroplasmy in MELAS-iPSCs with m.13513G>A mutation. Sci. Rep. 2017;7(1):15557. doi: 10.1038/s41598-017-15871-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Antón Z. Mitochondrial import, health and mtDNA copy number variability using type II and type V CRISPR effectors. J. Cell Sci. 2020;133(18) doi: 10.1242/jcs.248468. [DOI] [PubMed] [Google Scholar]
- 72.Bian W.P. Knock-in strategy for editing human and zebrafish mitochondrial DNA using mito-CRISPR/cas9 system. ACS Synth. Biol. 2019;8(4):621–632. doi: 10.1021/acssynbio.8b00411. [DOI] [PubMed] [Google Scholar]
- 73.Gammage P.A., Moraes C.T., Minczuk M. Mitochondrial genome engineering: the revolution may not Be CRISPR-ized. Trends Genet. 2018;34(2):101–110. doi: 10.1016/j.tig.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mok B.Y. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020;583(7817):631–637. doi: 10.1038/s41586-020-2477-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lorenz C. Human iPSC-derived neural progenitors are an effective drug discovery model for neurological mtDNA disorders. Cell Stem Cell. 2017;20(5):659–674. doi: 10.1016/j.stem.2016.12.013. e9. [DOI] [PubMed] [Google Scholar]
- 76.Hämäläinen R.H. Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model. Proc. Natl. Acad. Sci. U. S. A. 2013;110(38):E3622–E3630. doi: 10.1073/pnas.1311660110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Winanto Organoid cultures of MELAS neural cells reveal hyperactive Notch signaling that impacts neurodevelopment. Cell Death Dis. 2020;11(3):182. doi: 10.1038/s41419-020-2383-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Respiratory enzymes and substrate transporters in MELAS iPSCs. mRNA levels of hexokinase 1 (HK1), hexokinase 2 (HK2), 3-phosphoinositide-dependent protein kinase 1 (PDK1), nicotinamide phosphoribosyltransferase (NAMPT), glucose transporters SLC2A1 and SLC2A3, and lactate transporters SLC16A1 and SLC16A3 in the control (454E2) and MELAS (A24#1–3) iPSCs (n = 3). *P < 0.05.
Drug screening system and effects of PDK inhibitors that activate pyruvate dehydrogenase (A, B) A drug screening system based on the effects of 2-deoxy-D-glucose (2DG). (C–E) The relative areas occupied by live cells stained by calcein and treated with 3-phosphoinositide-dependent protein kinase 1 (PDK1) inhibitors JX06 (1, 15, 30 mM, C), DAP (10, 30, 60 μM, D), or DCA (10, 50, 100 μM, E) were compared with those of the DMSO-treated cells in the control (454E) and MELAS (A24#1-3) iPSCs.
Enzymes for GSH generation in MELAS iPSCs. mRNA levels of glutamate-cysteine ligase catalytic subunit (GCLC), glutathione synthetase (GSS), cysteine dioxygenase type 1 (CDO1), cysteine sulfinic acid decarboxylase (CSAD), and 2-aminoethanthiol dioxygenase (ADO) in the control (454E2) and MELAS (A24#1–3) iPSCs (n = 3). Enzymatic cascades for GSH generation are schematically shown. *P < 0.05 for comparisons between the control and MELAS iPSCs; #P < 0.05 for comparisons of data obtained in the presence and absence of the treatment with taurine in MELAS iPSCs (ANOVA followed by the post hoc Tukey’s test in both cases).
Glucose metabolism in MELAS iPSCs. Results of the metabolome assay measuring the levels of the components of glycolysis, pentose phosphate pathway (PPP), and TCA cycle are shown (N = 6). The nitrogen shift pathway that contributes to nucleotide production by activating PPP and glutamine metabolism is schematically shown. mRNA levels of pyruvate kinases M1 and M2 (PKM1 and PKM2), phosphoribosyl pyrophosphate amidotransferase (PPAT), as well as of glutaminases 1 and 2 (GLS1 and GLS2) in the control and MELAS iPSCs are illustrated (n = 3). *P < 0.05 for comparisons between control and MELAS iPSCs; #P < 0.05 for comparisons of data obtained in the presence and absence of the treatment with taurine in MELAS iPSCs (ANOVA followed by the post hoc Tukey’s test in both cases).
Metabolism of nucleotides and amino acids in MELAS iPSCs. Results of the metabolome assay measuring the levels of nucleic acids and amino acids are shown (N = 6). *P < 0.05 for comparisons between control and MELAS iPSCs; #P < 0.05 for comparisons of data obtained in the presence and absence of the treatment with taurine in MELAS iPSCs (ANOVA followed by the post hoc Tukey’s test in both cases).
Expression levels of an autophagy marker and amino acid transporters. mRNA levels of the genes encoding the autophagy marker LC3A, LC3B, the amino acid transporters SLC7A5, SLC7A8, SLC43A1, SL43A2, SLC7A11, and SLC3A2, were measured in each iPSC line in the presence or absence of the treatment with taurine (n = 3). The role of each molecule is schematically shown. *P < 0.05 for comparisons between the control and MELAS iPSCs; #P < 0.05 for comparisons of data obtained in the presence and absence of the treatment with taurine in iPSCs (ANOVA followed by the post hoc Tukey’s test in both cases).
Urea cycle products in MELAS iPSCs. Results of the metabolome assay, measuring the levels of the urea cycle products are shown (N = 6). *P < 0.05 for comparisons between the control and MELAS iPSCs; #P < 0.05 for comparisons of data obtained in the presence and absence of the treatment with taurine in MELAS iPSCs (ANOVA followed by the post hoc Tukey’s test in both cases).