

Abstract
Background
Colorectal cancer (CRC) is one of the most common cancers. The aim of our study was to explore its related mutations, identify novel mutation markers, and construct predictive models for postoperative CRC patients, so as to provide evidence for the diagnosis, treatment, and prognosis of CRC.
Methods
A total 50 CRC patients were collected, and the mutations in tissue samples were detected through next-generation sequencing (NGS). Meanwhile, 246 CRC cases with complete mutation data were downloaded from The Cancer Genome Atlas (TCGA) database. Afterwards, the co-mutations in both clinical and TCGA cohorts were identified, and the high-frequency mutation genes were selected. Subsequently, functional enrichment analysis was performed, and overall survival (OS) and progression-free survival (PFS) predictive models were constructed.
Results
In all, 18 out of 238 co-mutation genes mutated in at least 20% of the samples and were selected out as common high-frequency mutation genes. They were significantly enriched in 460 Gene Ontology (GO) terms and 87 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (P<0.05), which were closely related to the occurrence and development of CRC. Among the 18 genes, NOTCH3, histone lysine methyltransferase 2C (KMT2C), and cAMP-response element binding protein-BP (CREBBP) were respectively associated with tumor position, stage, and PFS (P<0.05), and could be considered as potential biomarkers of CRC. Finally, OS and PFS predictive models were constructed and verified using the 50 clinical cases, with both models demonstrating high fitting degrees useful for predicting the OS and PFS of CRC patients.
Conclusions
NOTCH3, KMT2C, and CREBBP were found to be prospective biomarkers for the diagnosis and prognosis of CRC. The prognosis prediction models had high sensitivity and could be used to predict the OS and PFS of CRC patients.
Keywords: Colorectal cancer (CRC), mutation, prognosis, predictive model
Introduction
Colorectal cancer (CRC) is one of the most common cancers in the world. According to the latest global cancer epidemiological statistics, new cases of CRC account for 10.2% of all malignant tumors, ranking third among all cancers, and the total number of deaths account for 9.2%, ranking second, with the proportion of CRC continuing to rise (1,2). In 2020, a projected 150,000 new CRC cases and more than 50,000 CRC-related deaths will occur in the United States (3). According to the current data published in China, in 2015, the number of new cases of CRC reached 388,000, with 225,000 of these cases being men. At present, the goal of CRC screening in China involves improving the screening and detection rates of early CRC and important precancerous lesions (4). Despite the progress in the diagnosis and treatment of the disease, the prognosis of CRC patients is still poor due to the late stage of the initial diagnosis and the high frequency of metastasis and recurrence. Therefore, it is necessary to develop an effective method to improve the diagnosis rate, predict metastasis and recurrence, and monitor the curative effect in real time, so as to improve the overall cure level (5). A thorough understanding of the molecular genetic characteristics of CRC is the key to solving this problem.
CRC develops through a series of differentially expressed or mutated genes which affect the homeostasis of oncogenes or tumor suppressors (6). In recent years, the identification of CRC tumor markers has seen rapid progress. Changes in non-coding RNA (ncRNA) have been confirmed as a key factor in the development of CRC (7). A variety of ncRNAs, including long non-coding RNAs (lncRNAs) and microRNAs (miRNAs), have recently been found to have functional features in the development of CRC (8-11). With the swift emergence of sequencing technology, the somatic mutations of a large number of genes, including TP53, KRAS, PIK3CA, APC, and RNF43, etc., have been proven as drivers in the development of CRC (12). Hence, the current study combined clinical samples and data from The Cancer Genome Atlas (TCGA) database to identify somatic mutations in postoperative CRC patients, and analyzed their correlation with clinical parameters. Finally, a prognostic model was constructed by regression analysis, and the clinical cohort was used as a verification group to further evaluate the prognostic ability of the model in patients with CRC. We present the following article in accordance with the MDAR reporting checklist (available at http://dx.doi.org/10.21037/jgo-21-28).
Methods
Patient enrolment and sample collection
From January 2017 to October 2019, 50 CRC patients were enrolled in this study from Tianjin Medical University Cancer Institute and Hospital, including 23 colon cancer cases and 27 rectal cancer cases. Subsequently, their tumor and paracancerous tissues were collected and sequenced through next-generation sequencing (NGS), and their clinical information was also recorded. This study was approved by the ethics committee of Tianjin Medical University Cancer Institute and Hospital (LLSP2019-016). All participants voluntarily signed informed consent to participate in the study. All procedures performed in this study involving human participants were in accordance with the Declaration of Helsinki (as revised in 2013).
TCGA data screening
Mutation data of CRC patients were downloaded from TCGA database (https://tcga-data.nci.nih.gov/docs/publications/tcga/). The screening criteria were the following: (I) diagnosed as CRC with the pathological type of adenocarcinoma; (II) colon and rectal tumor sites, with a ratio of 23:27; (III) a tissue sample type with complete mutation data; and (IV) complete and detailed clinical information. Finally, a total of 246 cases were enrolled, comprising 110 colon cancer and 136 rectal cancer samples.
Mutation detection
Single-nucleotide variations (SNVs) and insertions/deletions (InDels) were identified with VarScan version 2.4.3, MuTect version 1.1.4, and Genome Analysis Toolkit (GATK) version 2.3.9. CONTRA version 2.0.4 was used for copy number variations (CNVs) detection. An independently developed fusion program was used to detect gene fusion.
The mutation frequency of each genes in 50 clinical cases and 246 TCGA cases was counted by a self-developed Python script, and the common mutation genes were screened. Finally, the genes with a mutation frequency greater than 20% were selected as common high-frequency mutation genes.
Functional enrichment analysis
Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed using clusterProfiler package (v. 3.14.3) in R v.3.6.3 (R Foundation for Statistical Computing, Vienna, Austria), and the database of human reference genome sequence hg19 in org.Hs.eg.db package (v. 3.10.0) was used as the reference data. The P value was corrected by the Benjamin–Hochberg (BH) method, with a P value <0.05 and a q value <0.05 being the cut-off criteria.
Establishment of predictive models and statistical analysis
According to the mutations, overall survival (OS) and progression-free survival (PFS) predictive models were constructed using a multiple linear regression function lm() in R (v. 3.6.3). Statistical analyses were performed using Chi-square test or Fisher’s exact test for categorical variables, and by t-test or Mann–Whitney U test for continuous variables through IBM SPSS Statistics v.21 software (IBM Corp., Armonk, NY, USA). A P value <0.05 was considered statistically significant.
Results
Clinical characteristics
Clinical characteristics of 50 clinical CRC patients are listed in Table 1. In TCGA cases, there were 130 (52.85%) males and 116 (47.15%) females, ranging in age from 31 years to 90 years (65.85±12.58). Furthermore, 129 cases were in stages I–II, 108 were in stages III–IV, and 9 had no recorded stage. In all, 200 survived and 46 died.
Table 1. Clinical characteristics of 50 colorectal cancer patients.
| Parameters | N [%] |
|---|---|
| Sex | |
| Male | 30 [60] |
| Female | 20 [40] |
| Age | |
| <60 years | 27 [54] |
| ≥60 years | 23 [46] |
| Tumor stage | |
| I | 1 [2] |
| II | 12 [24] |
| III | 23 [46] |
| IV | 1 [2] |
| None | 13 [26] |
| Pathological type | |
| Adenocarcinoma | 50 [100] |
| Smoking | |
| Yes | 16 [32] |
| No | 34 [68] |
| Drinking | |
| Yes | 24 [48] |
| No | 26 [52] |
| Tumor size (cm) | |
| <5 | 15 [30] |
| ≥5 | 33 [66] |
| None | 2 [4] |
| Tumor volume (cm2) | |
| <100 | 20 [40] |
| ≥100 | 28 [56] |
| None | 2 [4] |
| Tumor differentiation | |
| Moderate | 33 [66] |
| Low | 6 [12] |
| Middle-low | 6 [12] |
| None | 5 [10] |
| Hypertension | |
| Yes | 11 [22] |
| No | 39 [78] |
| Diabetes | |
| Yes | 1 [2] |
| No | 49 [98] |
| Therapeutic method | |
| Surgery | 28 [56] |
| Chemotherapy | 2 [4] |
| Surgery + chemotherapy | 20 [40] |
| Recurrence | |
| Yes | 5 [10] |
| No | 45 [90] |
| Metastasis | |
| Yes | 12 [24] |
| No | 38 [76] |
| Overall survival (OS) | |
| <6 months | 19 [38] |
| ≥6 months | 31 [62] |
| Progression-free survival (PFS) | |
| <3 months | 13 [26] |
| ≥3 months | 2 [4] |
| None | 35 [70] |
Screening of high-frequency mutation genes
In TCGA cohort, 42,514 mutations were found in 16,378 genes. In clinical cases, a total of 1465 mutations with a frequency ≥ 0.5% in 255 genes were detected, in which TP53 p.Arg342* (22.09%), FBXW7 p.Arg658* (19.71%), BRAF p.Asp594Gly (17.33%), PIK3CA p.His1047Arg (16.67%), and NRAS p.Gly12Val (15.52%) had higher mutation frequencies. A total of 238 co-mutation genes were found in both clinical and TCGA cases. Afterwards, 18 genes with mutation frequency ≥20% were selected as high-frequency mutation genes; among them, TP53, ARID1A, and APC had a mutation frequency over 50%. Figure 1 shows the mutation distribution of 18 genes in all clinical samples, including 191 missense (67.97%), 38 nonsense (13.52%), 32 coding sequence (CDS) InDels (11.39%), 13 frameshift deletions (4.63%), and 7 frameshift insertions (2.49%).
Figure 1.

Mutation landscape of 18 common high frequency mutation genes in The Cancer Genome Atlas (TCGA) and clinical cases. The upper side shows the details of clinical information of each patient. The lower panel illustrates the genetic alterations, with the different colors representing different mutation types. The histogram on the right shows the frequency of mutation genes in 50 clinical patients.
Functional and pathway enrichment of high-frequency mutation genes
After enrichment analysis, the 18 genes were found to be enriched in 460 GO terms and 87 KEGG pathways. The GO terms included 419 biological processes (BPs), 10 cellular components (CCs), and 31 molecular functions (MFs). BPs were mainly enriched in histone modification, covalent chromatin modification, regulation of neuron apoptotic process, and regulation of neuron apoptotic process. CCs were enriched in mixed-lineage leukemia protein 3 (MLL3)/MLL4 complex, nuclear chromatin, lamellipodium, extrinsic component of membrane, etc. MFs were enriched in 1-phosphatidylinositol-3-kinase activity, phosphatidylinositol 3-kinase activity, histone methyltransferase activity (H3-K4 specific), and phosphatidylinositol kinase activity. Figure 2A lists the top 10 BPs, MFs, and all CC terms according to P value. KEGG pathways of 18 high-frequency mutation genes were significantly enriched in pancreatic cancer, breast cancer, CRC, and FoxO signaling pathway, thyroid hormone signaling pathway, cell cycle, Wnt signaling pathway, and others (Figure 2B).
Figure 2.

Enrichment analysis of high-frequency mutation genes. Top 10 enriched BPs, CCs, and MFs in GO terms (A), and the top 30 KEGG pathways (B) of 18 common high-frequency mutation genes among TCGA and clinical cases. The colors of the circles indicate P values of terms and pathways, and the size represents the number of genes enriched in each term and pathway. BP, biological process; CC, cellular component; MF, molecular function; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Clinical significance of the mutation genes
Next, we analyzed the correlation between high-frequency mutation genes and clinical characteristics, including tumor position, stage, recurrence, metastasis, OS, and PFS. Among the 18 genes, NOTCH3 was significantly correlated with the tumor positions of CRC (P=0.021, Figure 3A), histone lysine methyltransferase 2C (KMT2C) with stage (P=0.042, Figure 3B), and cAMP-response element binding protein-BP (CREBBP) with PFS (P=0.015, Figure 3C). The mutations of NOTCH3, KMT2C, and CREBBP might play a potential role to identify the tumor location, cancer stage and PFS
Figure 3.
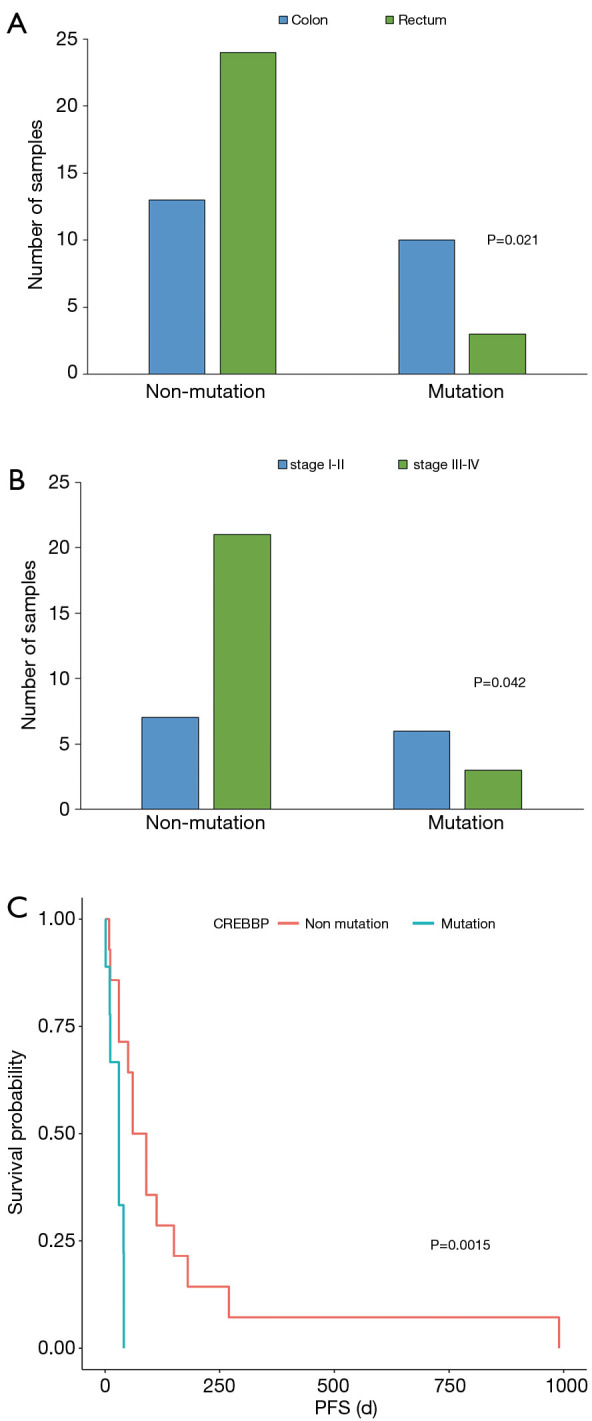
Correlation analysis between gene mutations and clinical characteristics. NOTCH3 (A), KMT2C (B), and CREBBP (C) mutations were respectively correlated with the tumor position, stage, and progression-free survival (PFS) of colorectal cancer (CRC) patients.
Predictive models of prognostic index
According to the 18 mutated genes, OS and PFS prediction models were constructed by multiple regression analysis, and the significance test of regression equations were examined by F test. The P values of OS and PFS predictive models were 0.006 and 0.013 respectively, indicating that the test of regression equations was significant. Comparing the OS and PFS of clinical patients respectively with the predictive models, both of the predictive models had high fitting degrees (Figure 4A,B), and could be used to predict the OS and PFS of CRC patients.
Figure 4.

The predictive ability of 18-gene prognostic models for colorectal cancer (CRC). Predictive models of the overall survival (OS) (A) and progression-free survival (PFS) (B) of CRC patients were constructed based on the 18 high-frequency mutation genes. The X-axis represents the predictive days, and the Y-axis represents the real days.
Discussion
The ability to predict the prognosis of patients has great clinical value, as it can prolong the survival time and improve the quality of life of patients, and can be used to adjust follow-up management. In CRC, the combined application of multiple markers can improve the accuracy of CRC screening and diagnosis, thus improving the diagnostic efficiency of gene detection (13). In this study, we found 18 high-frequency mutation genes (mutation frequency ≥20%) in clinical CRC patients and TCGA patients, including two common cancer related genes, TP53 and KRAS. Combined with clinical characteristics, we further screened out NOTCH3, KMT2C, and CREBBP as candidate markers for the diagnosis and prognosis of CRC patients. Meanwhile, the predictive models based on the mutation genes were proven, through comparison with the OS and PFS of clinical cases, to have high reliability in predicting the OS and PFS of CRC patients.
NOTCH3 is one of the important members of the NOTCH family, and is involved in the occurrence and development of various cancers by regulating tumor microenvironment, and promoting tumor formation, progression, angiogenesis, migration, and invasion (14,15). Its overexpression activates Notch signaling pathway, and promotes tumor cell growth and migration (16). The abnormal activation of Notch pathway has been detected in acute lymphoblastic leukemia, gliomas, CRC and other tumors, and has been significantly correlated with prognosis (16,17). Although few studies on NOTCH3 mutation in cancers have been published, 199 mutations of NOTCH3 gene are reported to be present in malignant tumors of the lung, breast, gastric system, prostate, and lymphoma in the COSMIC database (https://cancer.sanger.ac.uk/cell_lines/search?q=NOTCH3#muts). Despite not applied in clinical by now, Notch3 targeting has been proved as an effective way against cancer, such as anti-NOTCH3 and targeted miRNAs (18-20). Therefore, the mutations of NOTCH3 gene might play a role in the occurrence and development of these tumors.
Histone lysine methyltransferase 2C (KMT2C), also known as myeloid/lymphoid or mixed-lineage leukemia protein 3 (MLL3), encodes a nuclear protein with histone methylation activity and participates in transcriptional synergistic activation. As an important regulator of epigenetics, KMT2C participates in the methylation of various histone amino acid sites, changing the structure of chromatin and affecting the transcription process of target genes, and is thus an attractive drug target for cancer treatment (21-23). KMT2C mutates in a variety of human cancers and is considered to be crucial to the occurrence and development of cancers. However, the research on its mutation function is still limited, which may be related to the lack of a mutation hotspot and mutation domain in KMT2C (22). Interestingly, KMT2C has been previously reported to have a well-established hotspot mutation, S338L (31%), in CRC, which may lead to new epigenetic insights into the carcinogenesis of CRC (24,25). In this study, although no KMT2C hotspot mutation was found in CRC samples, nine KMT2C mutation sites were screened out with a frequency more than 1%, and the KMT2C gene was enriched in the GO term of histone modification. More in-depth study is necessary to understand the mechanism and function of these KMT2C mutations in CRC. The cAMP-response element binding protein-BP (CREBBP) gene encodes the CREBBP protein that binds to the cAMP response element (CRE). CREBBP acts as a transcription factor and plays a role in transcription by participating in chromatin remodeling and helping RNA aggregation, which is known to underlie general cancer pathogenesis (26,27). Recent research has found that mutations in CREBBP are associated with poor prognosis in head and neck squamous cell carcinoma (HNSCC), and synthetic cytotoxicity has been identified in CREBBP mutant tumors (28). Kim et al. (29) found a frameshift mutation of CREBBP in microsatellite instability-high (MSI-H) gastric cancer, which could lead to the premature stop of amino acid synthesis in CREBBP protein; however, they also found that the mutation rate of CREBBP was very low (1.4%) in gastric cancer and CRC patients with MSI-H. In our study, CREBBP mutation occurred in 43.14% of clinical patients, which was very different from that of TCGA patients (6.91%) and of previous research. These differences can perhaps be attributed to a few factors. (I) The above study by Kim et al. only analyzed the gene mutation frequency in microsatellite instability-high (MSI-H) patients, while our study included all CRC patients and ignored MSI status, and thus might have found a higher mutation frequency. Additionally, (II) most of the TCGA data are from Caucasians and African Americans, while our study included Asians, and thus the variability in ethnicity, environment, and lifestyle might also have resulted in a difference of mutation frequency. Therefore, this issue needs to clarified by further research.
Finally, we developed predictive OS and a PFS models based on the mutated genes, and the predictive ability of the models were validated in the validation cohort, with a good predictive accuracy for OS and PFS in CRC patients. The predictive models represent a significant advance in the prognostic monitoring of CRC. Indeed, another recent study has reported that the prediction of CRC using genetic markers is feasible (30). Although there have not been appropriate drugs targeted to NOTCH3, KMT2C, and CREBBP, the three-gene model still become a useful tool to predict CRC patients prognosis.
This study has some limitations which should be addressed. First, our current research was retrospective in nature. Although the OS and PFS models were verified in the validation cohort, the bias inherent in this study could not be completely eliminated. Second, the sample size used for this analysis was relatively small (fewer than 100 cases), which might have led to deviation. Finally, this study included only CRC patients in our hospital, whose baseline characteristics may differ from those in western countries. Therefore, it is not clear whether our current prognosis models are directly applicable to populations with different ethnic composition. In the further study, we will focus on the Asian populations and collect more multi-center CRC cases to improve the predictive model.
Conclusions
Our study identified 18 common high frequency mutations in clinical and TCGA CRC cases, with NOTCH3, KMT2C, and CREBBP constituting a potential novel signature for the diagnosis and prognosis of CRC. Based on this signature, we constructed predictive OS and PFS models which were preliminary proven to be highly reliable. However, the findings of our study and the related mechanisms need to be thoroughly validated and explored in a larger cohort study.
Acknowledgments
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The study was approved by Tianjin Medical University Cancer Institute and Hospital (LLSP2019-016). All participants voluntarily signed the informed consent before inclusion into this study. All procedures performed in this study involving human participants were in accordance with the Declaration of Helsinki (as revised in 2013).
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Reporting Checklist: The authors have completed the MDAR reporting checklist. Available at http://dx.doi.org/10.21037/jgo-21-28
Data Sharing Statement: Available at http://dx.doi.org/10.21037/jgo-21-28
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/jgo-21-28). The authors have no conflicts of interest to declare.
(English Language Editor: J. Gray)
References
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394-424. 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7-30. 10.3322/caac.21590 [DOI] [PubMed] [Google Scholar]
- 3.Sun YL, Zheng RS, Zhang SW, et al. Report of Cancer Incidence and Mortality in Different Areas of China, 2015. China Cancer 2019;20:1-11.31892356 [Google Scholar]
- 4.National Clinical Research Center for Digestive Diseases (Shanghai) , National Early Gastrointestinal-Cancer Prevention & Treatment Center Alliance (GECA), Chinese Society of Digestive Endoscopy, et al. Chinese Consensus of Early Colorectal Cancer Screening (2019, Shanghai). Chin J Intern Med 2019;58:736-44. [DOI] [PubMed] [Google Scholar]
- 5.Cheng R, Zhang ST. Accurate diagnosis of colorectal cancer and precancerous diseases. Zhonghua Nei Ke Za Zhi 2020;59:145-7. [DOI] [PubMed] [Google Scholar]
- 6.Ma R, Jing CW, Zhang Y, et al. The somatic mutation landscape of Chinese Colorectal Cancer. J Cancer 2020;11:1038-46. 10.7150/jca.37017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ling H, Vincent K, Pichler M, et al. Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene 2015;34:5003-11. 10.1038/onc.2014.456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carotenuto P, Fassan M, Pandolfo R, et al. Wnt signalling modulates transcribed-ultraconserved regions in hepatobiliary cancers. Gut 2017;66:1268-77. 10.1136/gutjnl-2016-312278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Almeida MI, Nicoloso MS, Zeng L, et al. Strand-Specific miR-28-5p and miR-28-3p have distinct effects in colorectal cancer cells. Gastroenterology 2012;142:886-896.e9. 10.1053/j.gastro.2011.12.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ling H, Pickard K, Ivan C, et al. The clinical and biological significance of miR-224 expression in colorectal cancer metastasis. Gut 2016;65:977-89. 10.1136/gutjnl-2015-309372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dragomir MP, Knutsen E, Calin GA. Snapshot: unconventional miRNA functions. Cell 2018;174:1038-1038.e1. 10.1016/j.cell.2018.07.040 [DOI] [PubMed] [Google Scholar]
- 12.Dienstmann R, Vermeulen L, Guinney J, et al. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer 2017;17:79-92. 10.1038/nrc.2016.126 [DOI] [PubMed] [Google Scholar]
- 13.Bustos García de Castro A, Ferreirós Domínguez J, Delgado Bolton R, et al. PET-CT in presurgical lymph node staging in non-small cell lung cancer: the importance of false-negative and false-positive findings. Radiologia 2017;59:147-58. [DOI] [PubMed] [Google Scholar]
- 14.Huang Q, Li J, Zheng J, et al. The carcinogenic role of the notch signaling pathway in the development of hepatocellular carcinoma. J Cancer 2019;10:1570-9. 10.7150/jca.26847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X, Shi HL, Yao JN, et al. FAM225A facilitates colorectal cancer progression by sponging miR-613 to regulate NOTCH3. Cancer Med 2020;9:4339-49. 10.1002/cam4.3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furukawa S, Kawasaki Y, Miyamoto M, et al. The miR-1-NOTCH3-Asef pathway is important for colorectal tumor cell migration. PLoS One 2013;8:e80609. 10.1371/journal.pone.0080609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu L, Xue F, Shao M, et al. Aberrant expression of Notch3 predicts poor survival for hepatocellular carcinomas. Biosci Trends 2013;7:152-6. [PubMed] [Google Scholar]
- 18.Rosen LS, Wesolowski R, Baffa R, et al. A phase I, dose-escalation study of PF-06650808, an anti-Notch3 antibody-drug conjugate, in patients with breast cancer and other advanced solid tumors. Invest New Drugs 2020;38:120-30. 10.1007/s10637-019-00754-y [DOI] [PubMed] [Google Scholar]
- 19.Song G, Zhang Y, Wang L. MicroRNA-206 targets notch3, activates apoptosis, and inhibits tumor cell migration and focus formation. J Biol Chem 2009;284:31921-7. 10.1074/jbc.M109.046862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang XW, Xi XQ, Wu J, et al. MicroRNA-206 attenuates tumor proliferation and migration involving the downregulation of NOTCH3 in colorectal cancer. Oncol Rep 2015;33:1402-10. 10.3892/or.2015.3731 [DOI] [PubMed] [Google Scholar]
- 21.McGrath J, Trojer P. Targeting histone lysine methylation in cancer. Pharmacol Ther 2015;150:1-22. 10.1016/j.pharmthera.2015.01.002 [DOI] [PubMed] [Google Scholar]
- 22.Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer 2015;15:334-46. 10.1038/nrc3929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawrence M, Daujat S, Schneider R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet 2016;32:42-56. 10.1016/j.tig.2015.10.007 [DOI] [PubMed] [Google Scholar]
- 24.Lu YW, Zhang HF, Liang R, et al. Colorectal cancer genetic heterogeneity delineated by multi-region sequencing. PLoS One 2016;11:e0152673. 10.1371/journal.pone.0152673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X. Association between histone lysine methyltransferase KMT2C mutation and clinicopathological factors in breast cancer. Biomed Pharmacother 2019;116:108997. 10.1016/j.biopha.2019.108997 [DOI] [PubMed] [Google Scholar]
- 26.Sakamoto KM, Frank DA. CREB in the pathophysiology of cancer: implications for targeting transcription factors for cancer therapy. Clin Cancer Res 2009;15:2583-7. 10.1158/1078-0432.CCR-08-1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang H, Guo J, Linpeng SY, et al. Next generation sequencing identified two novel mutations in NIPBL and a frame shift mutation in CREBBP in three Chinese children. Orphanet J Rare Dis 2019;14:45. 10.1186/s13023-019-1022-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar M, Molkentine D, Molkentine J, et al. CREBBP/EP300 mutation is associated with poor outcome in HNSCC and targetable with synthetic cytotoxicity. BioRxiv 2020. doi: 10.1101/2020.04.10.028217 [DOI] [Google Scholar]
- 29.Kim MS, Yoo NJ, Lee SH. Expressional and Mutational Analysis of CREBBP Gene in Gastric and Colorectal Cancers with Microsatellite Instability. Pathol Oncol Res 2014;20:221-2. 10.1007/s12253-013-9668-3 [DOI] [PubMed] [Google Scholar]
- 30.Zheng P, Liang C, Ren L, et al. Additional Biomarkers beyond RAS That Impact the Efficacy of Cetuximab plus Chemotherapy in mCRC: A Retrospective Biomarker Analysis. J Oncol 2018;2018:5072987. 10.1155/2018/5072987 [DOI] [PMC free article] [PubMed] [Google Scholar]