

Abstract
In this study, we report a male newborn with severe Zellweger spectrum disorder (ZSDs) presenting asphyxia, hypotonia, poor feeding, and dysmorphic facial features. Despite intensive supportive treatment, the boy’s condition deteriorated progressively. The patient’s diagnosis was made by delayed results after his death. His genetic analysis showed that the boy carried novel compound heterozygous mutation in PEX1 gene (c.2050C > T and c.782_783del). We conducted a literature search and identified 316 patients with ZSD caused by mutations in the PEX1 gene. The p.G843D and p.I700Yfs*42 were the most commonly reported mutations. Among the 316 patients, clinical manifestations were available in 265 patients. The segregation of these patients’ manifestation showed that patients with missense PEX1 mutations have a milder phenotype than those with truncating mutations, while the common p.G843D mutations are milder than other missense mutations. Nearly all truncating mutations in PEX1 except for those with premature stop codons near the end of the gene were associated with a severe disease phenotype. These results indicated that all domains of PEX1 were important in the maintenance of normal peroxisome function. The correlation between severity of the disease and type of mutations in PEX1 can be helpful in predicting prognosis among patients with ZSD caused by mutated PEX1.
Keywords: Case report; neonate; Zellweger spectrum disorder, PEX1
Introduction
Zellweger spectrum disorders (ZSDs) are the most common forms of peroxisome biogenesis disorders (PDBs) and are caused by mutations in the PEX1 gene (~60% in all PBDs) (1). The ZSD is a clinical continuum of overlapping phenotypes including its most severe form, Zellweger syndrome (ZS, MIM 214100); milder phenotypes such as neonatal adrenoleukodystrophy (NALD, MIM 601539), infantile Refsum disease (IRD, MIM 266510) (2-4); and the mildest subgroup, Heimler syndrome (HS; MIM 234580) (5). The PEX1 gene encodes peroxisome biogenesis factor 1, which combine PEX6 to form a heterohexamer and become a membrane attached ATPase that facilitates the recycling of PEX5 and peroxisomal enzyme delivery (6,7). A damaged PEX1 protein impedes the importation of peroxisomal proteins therefore causes dysfunction in the peroxisome organelle (2,3). The ZSD can be diagnosed by a combination of clinical manifestation, biochemical analysis, and genetic testing; however, the currently available treatment methods are mainly supportive or palliative for those with severe forms of the disease (8).
The most common PEX1 mutations are missense c.2528G>A (p.G843D) and frameshift c.2097_2098insT (p.I700Yfs*42) (9,10). The frameshift p.I700Yfs*42 is associated with a more severe phenotype than p.G843D (10). A recent study has shown that peroxisome’s biogenesis, due to misfolded PEX1 proteins caused by p.G843D in the domain linking area, were rescued by in vitro protein stabilizing therapy (11). While the usefulness of therapies (chaperone and many others) in other less frequent and scattered PEX1 mutations are yet to be tested (12), each patient’s accurate diagnosis is undoubtedly fundamental to the initiation of correct treatment.
In the current study, we reported on a neonatal patient with severe ZSD caused by a novel compound heterozygous mutation in PEX1, and conducted a literature review to analyze the possible genotype-phenotype association. We present the following case in accordance with the CARE reporting checklist (available at http://dx.doi.org/10.21037/tp-20-167).
Case presentation
We investigated the case of a male newborn, who was the first child to non-consanguineous healthy parents after an uneventful pregnancy despite prenatal ultrasound detection of mildly dilated cardiac ventricles. The boy was delivered at 41 gestational weeks by cesarean section owing to fetal distress. His birth weight was 2,790 g (−1.4 standard deviation score), and his Apgar scores were 5 at 1 minute and 7 at 5 minutes. He was transferred to our neonatal intensive care unit (NICU) due to suspected asphyxia at birth, significant hypotonia, and poor feeding. The maternal TPAL (i.e., term, preterm, abortions, living children) was 1-0-0-1, and his parents reported no history of miscarriage or premature death in either family.
Following admission, it was noticed that the boy had dysmorphic features, including hypertelorism, large anterior fontanelle, low ear position, palmar crease, and micrognathia (Figure 1). His vitals were stable [temperature (T), 36.1 °C; pulse (P), 126 bpm; respiration (R), 45 bpm; blood pressure (BP), 77/50 mmHg; oxygen saturation (SpO2), 95%]. The results of his initial laboratory assessment showed elevated serum transaminases [alanine aminotransferase (ALT) 74 U/L; aspartate aminotransferase (AST) 207 U/L], elevated bilirubin levels (total 114.2 µmol/L; indirect 102.7 µmol/L, at 16 hours after birth), hypoglycemia (1.8 mmol/L), slightly elevated C-reactive protein (9 mg/dL), blood ammonia (102 µmol/L), and lactate (3.6 mmol/L). Complete blood count (CBC) showed a normal white cell count (8.5×109/L), hemoglobin (167 g/L) and slightly decreased platelet count (133×109/L). The arterial blood gas (ABG) results were acceptable: pH 7.28, electrolytes [potassium (K+) 3.0 mmol/L, sodium (Na+) 134 mmol/L, chloride (Cl-) 99 mmol/L, calcium (Ca2+) 2.08 mmol/L, bicarbonate (HCO3−) 21.4 mmol/L, base excess (BE) −4.7 mmol/L]. His chest and abdomen X-ray images (posterior to anterior) did not reveal any obvious abnormality. Transthoracic echocardiogram revealed a small atrial septal defect (1.5 mm), patent ductus arteriosus (2.8 mm), and mild pulmonary hypertension (<40 mmHg). His cranial ultrasound showed mildly dilated ventricles; however, abdominal ultrasound did not detect any deformities in the liver or kidneys.
Figure 1.

The patient at his age 3 days.
The boy was empirically treated with intravenous antibiotics. He received phototherapy and supportive treatments (oxygen cannula, intravenous glucose infusion, and small-volume enteral feeding (30 mL/kg/d) through the nasogastric tube). Because the patient presented dysmorphic features and unusual hypotonicity, we included a tandem mass spectrometry analysis of a dried blood sample and urine in his subsequent laboratory test.
The boy’s condition was not improved over the following days. His blood glucose was maintained within normal ranges by intravenous fluid while feeding volume to 60 mL/kg/d was not tolerated. A second blood test on day 5 showed deterioration in CBC (white cell count: 5.1×109/L, hemoglobin: 142 g/L), ABG (pH 7.19, HCO3− 43.7 mmol/L, BE +11 mmol/L), C-reactive protein (87 mg/L), and procalcitonin (6.3 ng/mL). A sepsis was suspected, and his antibiotic treatment was escalated. After discussing with his parents regarding further management, consent was provided to conduct familial genetic analysis; however, his parents declined further treatment due to diagnostic uncertainty and likely poor prognosis. The boy was discharged against medical advice on his 8th day of life.
His culture results did not reveal bacteria in his bloodstream. Later phone communication with his parents regarding mass spectrometry results (normal results from his dried blood sample, but markedly elevated 4-hydroxyphenyl-lactic acid and 3,6-epoxide tetradecanoic acid in urine sample), the boy’s condition was reported to have deteriorated rapidly after discharge, and he died at age 18 days.
Exome sequencing
Exome sequencing was performed on the boy, and both parents using the protocol reported previously (13). Briefly, the sequencing was performed using genomic DNA extracted from peripheral blood. The DNA library was constructed using Illumina TruSeq DNA Sample Preparation Kit (Illumina Inc., San Diego, CA, USA) and amplified using ligation-mediated polymerase chain reaction (LM-PCR) before hybridization for exon regions (SeqCap EZ Human Exome Kit v2.0; Roche, Basel, Switzerland) and subsequent sequencing on the Illumina HiSeq 2500 platform (Illumina Inc., San Diego, CA, USA) with 150-bp paired-end reads. The sequence data were mapped to the GRCh37 reference and filtered according to categories recommended by the American College of Medical Genetics (ACMG) (14). All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee(s) and with the Helsinki Declaration (as revised in 2013). The study was approved by the ethics committee of Shanghai Children’s Hospital, Shanghai Jiao Tong University (No. 2015RY009-F01). The parents of the patient provided written informed consent.
Literature review
We searched for publications of patients with PEX1 mutations from 1997 to 2019 in the Ovid database (all databases) using the following terms: “pex1”, “Zellweger”, and “peroxisome biogenesis disorder”. We conducted a manual search for relevant references in the articles identified in the initial database searches. Information regarding clinical data [gender, symptoms, age at death, diagnosis or disease severity assessed in each report, and PEX1 mutation (in reference to NM_000466.3)] were collected for each reported case. The identification of relevant literature and data extraction were performed independently by two investigators (PL and YS). The data were then reviewed by a third investigator (LM) and conflicts were resolved by consensus. Patients with confirmed rare mutations in PEX1 that were either homozygous or compound heterozygous were included in the analysis. In patients with compound heterozygotic mutations, the mutation with milder impact on the PEX1 protein (12,14) was taken to assess the severity of the disease as evaluated by each publication.
Results
Case genetics
The genetic results of the patients, received 4 weeks after the sampling, showed that the boy carried compound heterozygous mutations in PEX1 (NM_000466.2): a novel c.2050C>T (p.Gln684*) inherited from his father and previously described c.782_783del (p.Gln261Argfs*8) from the mother. These two mutations were predicted to be null variants (nonsense and frame shift) and their frequencies are low in the genome sequencing database (Table 1). The c.782_783del mutation was previously identified in Zellweger syndrome by targeted sequencing using either Sanger termination or parallel sequencing by synthesis (15,16).
Table 1. Mutations in PEX1 identified in the patient with Zellweger syndrome.
| Mutation | Predicted protein change | GRCh37 | gnomAD frequency | Classification | Genotype | ||
|---|---|---|---|---|---|---|---|
| Proband | Father | Mother | |||||
| c.2050C>T | p.Gln684* | chr7:92134067 G>A | 0 | PVS1 | Het | Het | WT |
| c.782_783del | p.Gln261Argfs*8 | chr7:92147045 CTT>C | 0.00004 | PVS1 | Het | WT | Het |
Het, heterozygous; WT, wild type.
Patients with peroxisome biogenesis disorder due to PEX1 mutations
Our literature review retrieved 316 patients (Figure 2) with ZSDs due to either homozygous or compound heterozygous mutations in the PEX1 gene. The most common mutations in the literature were p.G843D and p.I700Yfs*42, which account respectively for 45.4% and 19.3% of the mutated alleles. Among these patients, clinical manifestations and disease severity were available in 265 patients (Tables 2,3). While the disease severity varied in patients caused by missense mutations (Table 2), 84% (47/56) of the patients with truncating mutation presented severe disease phenotypes except for those with premature stop codons near the end of the gene (Table 3). The proportion of patients with severe disease was significantly more in disease caused by truncating mutations than by missense mutations. The disease presentation in patients with the common p.G834D mutation were milder than those of other missense mutations (Table 4).
Figure 2.
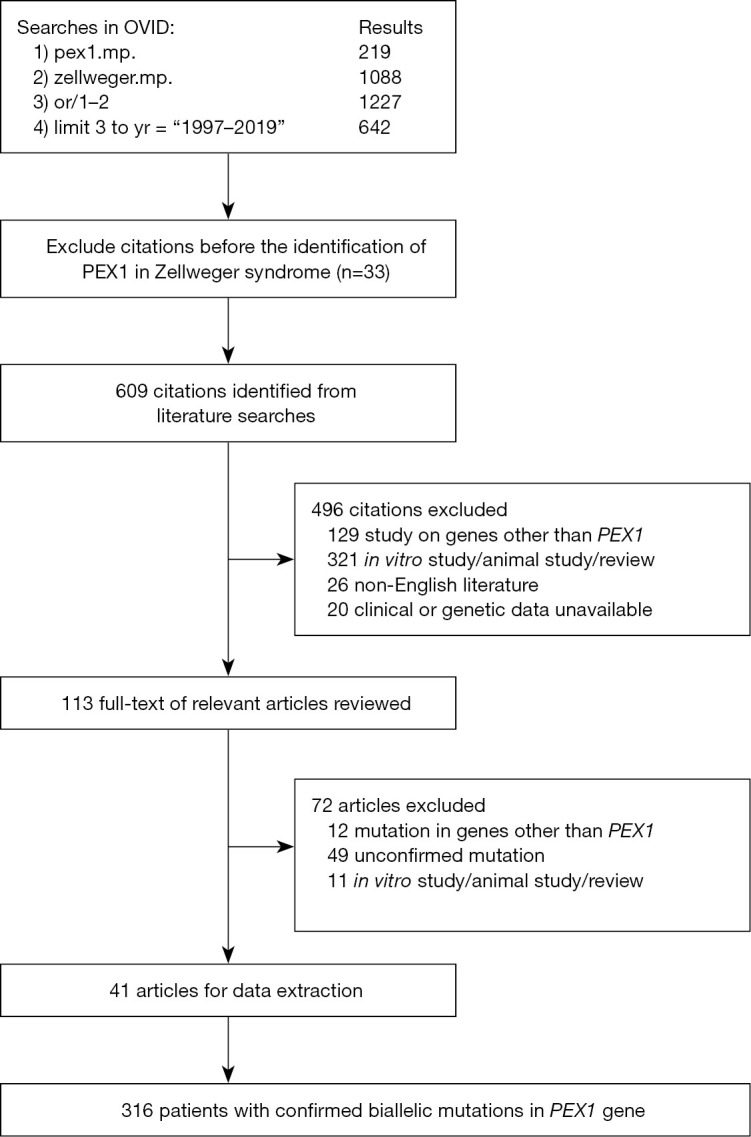
Flow chart of literature review for confirmed ZSDs (due to mutated PEX1). OVID, Ovid database (http://ovidsp.ovid.com/); mp, ‘multi-purpose’; yr, year; n, number.
Table 2. List of PEX1 missense mutations in patients with Zellweger spectrum disorder.
| DNA level | Protein level | Total | Mild | Intermediate | Severe |
|---|---|---|---|---|---|
| c.130-2A>G | p.54_91del | 1 | 1 | ||
| c.274G>C | p.V92L | 3 | 3 | ||
| c.475G>C | p.A159P | 1 | 1 | ||
| c.1240_1359del | p.414_453del | 4 | 4 | ||
| c.1742G>C | p.R581P | 1 | 1 | ||
| c.1777G>A | p.G593R | 5 | 5 | ||
| c.1865_1866insCAGTGTGGA | p.A622_H623insSVD | 2 | 2 | ||
| c.2008C>A | p.L670M | 1 | 1 | ||
| 9bp insertion in exon12 | inframe ins 633_690 | 1 | 1 | ||
| c.1991T>C | p.L664P | 1 | 1 | ||
| c.2083_2085delATG | p.M695del | 2 | 2 | ||
| c.2114T>G | p.L705W | 2 | 2 | ||
| c.2387T>C | p.L796P | 2 | 1 | 1 | |
| c.2528G>A | p.G843D | 162 | 40 | 101 | 21 |
| c.2636T>C | p.L879S | 2 | 1 | 1 | |
| c.2845C>T | p.R949W | 1 | 1 | ||
| c.2966T>C | p.I989T | 1 | 1 | ||
| c.2993G>A | p.R998Q | 1 | 1 | ||
| c.3037C>G | p.R1013G | 2 | 1 | 1 | |
| c.3037C>T | p.R1013C | 5 | 4 | 1 | |
| c.3038G>A | p.R1013H | 3 | 1 | 2 | |
| c.3077T>C | p.L1026P | 2 | 1 | 1 | |
| c.3124A>C | p.F1042L | 1 | 1 | ||
| c.3140T>C | p.L1047P | 1 | 1 | ||
| NA | p.L1256_A1283del | 1 | 1 | ||
| c.3850T>C | Stop lost | 1 | 1 | ||
| Total | 209 | 50 | 121 | 38 |
DNA, deoxyribonucleic acid; NA, not available.
Table 3. List of PEX1 truncating mutations in patients with Zellweger spectrum disorder.
| DNA level | Position of last wild type amino acid | Total | Mild | Intermediate | Severe |
|---|---|---|---|---|---|
| c.657_660delGTCA | 220 | 1 | 1 | ||
| c.1670+5G>T | 556 | 1 | 1 | ||
| c.1727dupG | 577 | 1 | 1 | ||
| del exon 12/13 | 633 | 2 | 2 | ||
| del exon 13/14 | 690 | 1 | 1 | ||
| c.2097insT | 700 | 20 | 1 | 1 | 18 |
| c.2176C>T | 726 | 2 | 1 | 1 | |
| c.2226+2T>C | 742 | 1 | 1 | ||
| c.2227-2416del | 743 | 3 | 1 | 2 | |
| c.2391_2392delTC | 798 | 1 | 1 | ||
| c.2416+1G>T | 830 | 1 | 1 | ||
| c.2584-2A>G | 861 | 1 | 1 | ||
| c.2614C>T | 872 | 3 | 3 | ||
| c.2730delA | 910 | 2 | 2 | ||
| c.2814-2818del | 938 | 1 | 1 | ||
| c.2875C>T | 959 | 1 | 1 | ||
| c.2916delA | 973 | 6 | 1 | 5 | |
| c.2926+2T>C | 975 | 2 | 2 | ||
| C.3691_3694delCAGT | 1,231 | 2 | 2 | ||
| c.3750G>A | 1,250 | 2 | 2 | ||
| del exon24 | 1,255 | 2 | 2 | ||
| Total | 56 | 3 | 6 | 47 |
DNA, deoxyribonucleic acid; NA, not available.
Table 4. Severity of patients with peroxisome biogenesis disorder due to mutations in PEX1.
| Disease severity | Missense | All (N=209) | Truncation (N=56) | P | ||
|---|---|---|---|---|---|---|
| p.G834D (N=162) | Other (N=47) | P | ||||
| Mild | 40 (25%) | 10 (21%) | 0.7 | 50 (24%) | 3 (5%) | 0.001 |
| Intermediate | 101 (62%) | 20 (43%) | 0.006 | 121 (58%) | 6 (11%) | <0.0001 |
| Severe | 21 (13%) | 17 (36%) | 0.0009 | 38 (31%) | 47 (84%) | <0.0001 |
Discussion
The patient we studied had a constellation of clinical findings that fulfilled the diagnosis, however delayed, of Zellweger syndrome: hypotonia, poor feeding, dysmorphic face, brain malformations, hepatic dysfunction, and increased excretion of 3,6-epoxydicarboxylic acid and 4-hydroxyphenyl-lactic acid that are among the pattern of disease manifestations. The clinical characteristics and genetics of patients with PEX1 mutations in the literature indicated that the function of the PEX1 gene could hardly be maintained after an interruption in any of its C-terminal domains. The identification of compound heterozygotic truncating PEX1 mutations in our patient, confirmed the belated diagnosis of a severe form of ZSDs.
The rarity of ZSDs increased the difficulty in the development of clinicians’ awareness of the disease, and screening protocols for ZSDs are not commonly available in clinics (17). Also, severe forms of ZSDs can barely allow physicians to implement a correct etiological treatment during intensive care before establishing a correct diagnosis (18). Furthermore, there is currently no known effective treatment for ZSDs, the value of genetic diagnosis is limited to prognostic estimation and prenatal genetic diagnosis for future pregnancies in the same family (19). However, the identification of temperature-sensitive peroxisomal defects in p.G843D fibroblasts has suggested that increased protein instability is among the underlying mechanisms for ZSD (20), and indeed there are evolving in vitro evidence showing that ZSDs could benefit from chaperone therapy that rescues molecular misfolding (11,21). It was proposed that ZSDs due to mutated PEX1 might benefit differently from molecular stabilizing treatment due to variations in each mutation’s actual functional impairment (12). Nevertheless, aside from the current paradigm where truncating mutation causes a more severe phenotype than missense mutations, the aggregation of current cases with PEX1 mutations did not show an apparent association between certain mutated PEX1 protein domains and disease severity. Future studies on the structure of human PEX1 protein would add to the body of understand to determine correct etiological treatment for each patient.
Recent reports have shown that exome sequencing can help diagnose ZSDs, both in neonates and older patients with milder forms of the disease (17,22). It can be anticipated that implementation sequencing could also enable identifying sporadic PEX1 mutations and phenotypes (17). Nevertheless, despite advances in clinical sequencing, the patient reported herein with severe ZSD died before the genetic diagnosis was achieved; therefore, no management specific to the disease could be initiated. The future development of rapid exome sequencing (~one week) would certainly facilitate differential diagnosis and improve the management of such rare diseases (23).
There are several limitations of our current study, firstly the assessment of mutation’s impact on the PEX1 protein was based on homology modeling which might deviated from the actual effect of each mutation. Secondly, assessment on severity of the disease was based on previous case reports rather than standardized analysis of each individual patient. Therefore, the current genotype-phenotype relation can be biased. Nevertheless, the rarity of the disease could make it difficult to accumulate large number of patients in a single center. The report and summarization of cases in the future is most likely to provide further insight of the disease.
In conclusion, we identified a Chinese newborn with a severe form of ZSD due to novel compound heterozygous mutations in the PEX1 gene. The segregation of cases with PEX1 mutations showed that all domains of PEX1 were important in the maintenance of normal peroxisome function. A future increase in metabolite analysis and sequencing speed might be of life-saving benefit to patients and families with ZSDs.
Supplementary
The article’s supplementary files as
Acknowledgments
We thank the family that participated in our study.
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee(s) and with the Helsinki Declaration (as revised in 2013). The study was approved by the ethics committee of Shanghai Children’s Hospital, Shanghai Jiao Tong University (No. 2015RY009-F01). The parents of the patient provided written informed consent.
Footnotes
Reporting Checklist: The authors have completed the CARE reporting checklist. Available at http://dx.doi.org/10.21037/tp-20-167
Peer Review File: Available at http://dx.doi.org/10.21037/tp-20-167
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/tp-20-167). The authors have no conflicts of interest to declare.
References
- 1.Poll-The BT, Saudubray JM, Ogier HA, et al. Infantile Refsum disease: an inherited peroxisomal disorder. Comparison with Zellweger syndrome and neonatal adrenoleukodystrophy. Eur J Pediatr 1987;146:477-83. 10.1007/BF00441598 [DOI] [PubMed] [Google Scholar]
- 2.Reuber BE, Germain-Lee E, Collins CS, et al. Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat Genet 1997;17:445-8. 10.1038/ng1297-445 [DOI] [PubMed] [Google Scholar]
- 3.Portsteffen H, Beyer A, Becker E, et al. Human PEX1 is mutated in complementation group 1 of the peroxisome biogenesis disorders. Nat Genet 1997;17:449-52. 10.1038/ng1297-449 [DOI] [PubMed] [Google Scholar]
- 4.Moser AB, Rasmussen M, Naidu S, et al. Phenotype of patients with peroxisomal disorders subdivided into sixteen complementation groups. J Pediatr 1995;127:13-22. 10.1016/S0022-3476(95)70250-4 [DOI] [PubMed] [Google Scholar]
- 5.Ratbi I, Falkenberg KD, Sommen M, et al. Heimler Syndrome Is Caused by Hypomorphic Mutations in the Peroxisome-Biogenesis Genes PEX1 and PEX6. Am J Hum Genet 2015;97:535-45. 10.1016/j.ajhg.2015.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Francisco T, Rodrigues TA, Dias AF, et al. Protein transport into peroxisomes: Knowns and unknowns. Bioessays 2017;39. doi: . 10.1002/bies.201700047 [DOI] [PubMed] [Google Scholar]
- 7.Pedrosa AG, Francisco T, Ferreira MJ, et al. A Mechanistic Perspective on PEX1 and PEX6, Two AAA+ Proteins of the Peroxisomal Protein Import Machinery. Int J Mol Sci 2019;20:5246. 10.3390/ijms20215246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braverman NE, Raymond GV, Rizzo WB, et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab 2016;117:313-21. 10.1016/j.ymgme.2015.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ebberink MS, Mooijer PA, Gootjes J, et al. Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder. Hum Mutat 2011;32:59-69. 10.1002/humu.21388 [DOI] [PubMed] [Google Scholar]
- 10.Poll-The BT, Gootjes J, Duran M, et al. Peroxisome biogenesis disorders with prolonged survival: phenotypic expression in a cohort of 31 patients. Am J Med Genet A 2004;126A:333-8. 10.1002/ajmg.a.20664 [DOI] [PubMed] [Google Scholar]
- 11.Zhang R, Chen L, Jiralerspong S, et al. Recovery of PEX1-Gly843Asp peroxisome dysfunction by small-molecule compounds. Proc Natl Acad Sci U S A 2010;107:5569-74. 10.1073/pnas.0914960107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schieferdecker A, Wendler P. Structural Mapping of Missense Mutations in the Pex1/Pex6 Complex. Int J Mol Sci 2019;20:3756. 10.3390/ijms20153756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Wang C, Yang K, et al. A novel compound heterozygous mutation of the L2HGDH gene in a Chinese boy with L-2-hydroxyglutaric aciduria: case report and literature review. Neurol Sci 2018;39:1697-703. 10.1007/s10072-018-3483-2 [DOI] [PubMed] [Google Scholar]
- 14.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405-24. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yik WY, Steinberg SJ, Moser AB, et al. Identification of novel mutations and sequence variation in the Zellweger syndrome spectrum of peroxisome biogenesis disorders. Hum Mutat 2009;30:E467-80. 10.1002/humu.20932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun Y, Wang L, Wei X, et al. Analysis of a Chinese pedigree with Zellweger syndrome reveals a novel PEX1 mutation by next-generation sequencing. Clin Chim Acta 2013;417:57-61. 10.1016/j.cca.2012.12.005 [DOI] [PubMed] [Google Scholar]
- 17.Ge MM, Hu L, Li Z, et al. Novel compound heterozygous mutations in the PEX1 gene in two Chinese newborns with Zellweger syndrome based on whole exome sequencing. Clin Chim Acta 2017;470:24-8. 10.1016/j.cca.2017.04.016 [DOI] [PubMed] [Google Scholar]
- 18.Klouwer FC, Berendse K, Ferdinandusse S, et al. Zellweger spectrum disorders: clinical overview and management approach. Orphanet J Rare Dis 2015;10:151. 10.1186/s13023-015-0368-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Sayed M, Al-Hassan S, Rashed M, et al. Preimplantation genetic diagnosis for Zellweger syndrome. Fertil Steril 2007;87:1468.e1-3. 10.1016/j.fertnstert.2006.09.014 [DOI] [PubMed] [Google Scholar]
- 20.Imamura A, Tamura S, Shimozawa N, et al. Temperature-sensitive mutation in PEX1 moderates the phenotypes of peroxisome deficiency disorders. Hum Mol Genet 1998;7:2089-94. 10.1093/hmg/7.13.2089 [DOI] [PubMed] [Google Scholar]
- 21.MacLean GE, Argyriou C, Di Pietro E, et al. Zellweger spectrum disorder patient-derived fibroblasts with the PEX1-Gly843Asp allele recover peroxisome functions in response to flavonoids. J Cell Biochem 2019;120:3243-58. 10.1002/jcb.27591 [DOI] [PubMed] [Google Scholar]
- 22.Ventura MJ, Wheaton D, Xu M, et al. Diagnosis of a mild peroxisomal phenotype with next-generation sequencing. Mol Genet Metab Rep 2016;9:75-8. 10.1016/j.ymgmr.2016.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clowes Candadai SV, Sikes MC, Thies JM, et al. Rapid clinical exome sequencing in a pediatric ICU: Genetic counselor impacts and challenges. J Genet Couns 2019;28:283-91. 10.1002/jgc4.1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as