

Abstract
Background:
Recent cryo-electron microscopic imaging studies have shown that in addition to binding to the classical extracellular benzodiazepine binding site of the α1β3γ2L γ-aminobutyric acid type A (GABAA) receptor, diazepam also binds to etomidate binding sites located in the transmembrane receptor domain. Because such binding is characterized by low modulatory efficacy, the authors hypothesized that diazepam would act in vitro and in vivo as a competitive etomidate antagonist.
Methods:
The concentration-dependent actions of diazepam on 20 μM etomidate-activated and 6 μM GABA-activated currents were defined (in the absence and presence of flumazenil) in oocyte-expressed α1β3γ2L GABAA receptors using voltage clamp electrophysiology. The ability of diazepam to inhibit receptor labeling of purified α1β3γ2L GABAA receptors by 3[H]azietomidate was assessed in photoaffinity labeling protection studies. The impact of diazepam (in the absence and presence of flumazenil) on the anesthetic potencies of etomidate and ketamine was compared in a zebrafish model.
Results:
At nanomolar concentrations, diazepam comparably potentiated etomidate-activated and GABA-activated GABAA receptor peak current amplitudes in a flumazenil-reversible manner. The half-maximal potentiating concentrations were 39 nM (95% CI, 27 to 55 nM) and 26 nM (95% CI, 16 to 41 nM), respectively. However, at micromolar concentrations, diazepam reduced etomidate-activated, but not GABA-activated, GABAA receptor peak current amplitudes in a concentration-dependent manner with a half-maximal inhibitory concentration of 9.6 μM (95% CI, 7.6 to 12 μM). Diazepam (12.5 to 50 μM) also right-shifted the etomidate-concentration response curve for direct activation without reducing the maximal response and inhibited receptor photoaffinity labeling by 3[H] azietomidate. When administered with flumazenil, 50 μM diazepam shifted the etomidate (but not the ketamine) concentration-response curve for anesthesia rightward, increasing the etomidate EC50 by 18-fold.
Conclusions:
At micromolar concentrations and in the presence of flumazenil to inhibit allosteric modulation via the classical benzodiazepine binding site of the GABAA receptor, diazepam acts as an in vitro and in vivo competitive etomidate antagonist.
For nearly two centuries, general anesthetics have allowed surgical interventions to occur without pain and suffering.1,2 However, recovery from anesthesia still relies on anesthetic redistribution and/or elimination because—unlike many other neuroactive drugs—there are no reversal agents for clinically approved anesthetic agents. In theory, pharmacologic reversal of anesthesia might be achieved by administering an antagonist that competitively displaces anesthetics from their on-target protein binding site(s). Unfortunately, effective strategies for developing competitive antagonists capable of reversing anesthesia have not yet been developed.
The γ-aminobutyric acid type A (GABAA) receptor is the major inhibitory neurotransmitter receptor in the brain and an important target of numerous sedative-hypnotic drugs, including several commonly used general anesthetic agents.3–6 Such agents act as efficacious positive allosteric modulators (i.e., coagonists) of the GABAA receptor. They stabilize the receptor in the open channel state, leading to neuronal hyperpolarization with resultant central nervous system depression. For most anesthetics, the location(s) of their GABAA receptor binding sites remains a subject of debate. However, in the case of etomidate, in vivo and in vitro studies strongly implicate a solvent-accessible hydrophobic cavity located within the transmembrane receptor domain at each of the two β+/α− subunit interfaces as the critical site of action.7–10 This single class of binding sites can quantitatively account for both the agonist-potentiating and the direct activating GABAA receptor actions of etomidate.11
Recent cryo-electron microscopic imaging of the α1β3γ2L GABAA receptor reveals that in addition to binding to the classical high-affinity benzodiazepine binding site located within the extracellular receptor domain at the α+/γ− subunit interface, diazepam can also bind to the etomidate binding sites.12–14 Such binding is thought to occur at micromolar diazepam concentrations, is characterized by low intrinsic efficacy for positively modulating GABAA receptor function, and likely explains why mutations that render the receptor insensitive to etomidate also abolish its sensitivity to high (but not low) concentrations of diazepam.15–17 Together, these findings suggest that diazepam or structurally similar compounds could be developed as competitive antagonists. Such antagonists are predicted to be capable of reversing the anesthetic actions of etomidate and other drugs that act via this class of GABAA receptor binding sites. However, this hypothesis has never been tested. In the current article, we describe studies designed to test this hypothesis and to define the structural features of benzodiazepines that govern their abilities to bind to the etomidate binding site on the GABAA receptor and antagonize etomidate action.
Materials and Methods
Anesthetics and Anesthetic Photoaffinity Labels
Etomidate was purchased from Bachem Americas (USA). Flumazenil was purchased from Fisher Scientific (USA), fludiazepam was purchased from Cayman Chemical (USA), 1-Me (AGN-PC-0JTT8B) from Angene Chemical (United Kingdom), 7-Me (Amb1285986), 1–4’-Me (Amb32896093), and 7-Me (Amb1301849) from Ambinter (France), and 4’–2-ol (Z1352548204) from Enamine (USA), diazenil and hydro-diazeil were custom synthesized by Aberjona Laboratories (USA). All other benzodiazepines and benzodiazepine-like compounds were purchased from Sigma Aldrich (USA). 3[H]azietomidate was synthesized as previously described.18,19
GABAA Receptor Electrophysiology.
Oocytes were harvested from adult female Xenopus laevis frogs under tricaine anesthesia using procedures approved by the Institutional Animal Care and Use Committee of the Massachusetts General Hospital (Boston, Massachusetts) and in accordance with the principles outlined in the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health (Bethesda, Maryland). Human α1β3γ2L GABAA receptors were expressed in harvested oocytes as previously described.20 Whole-cell two-electrode voltage-clamp electrophysiologic experiments were carried out at room temperature using a GeneClamp 500B amplifier (Molecular Devices, USA), and currents were sampled and digitized at a rate of 1 sample per 100 μs. Oocytes were clamped at a holding potential of −50 mV. Solutions were perfused using a gravity fed system with a flow rate of 4 mL/min and controlled by a VC3 8 channel valve commander (ALA Scientific Instruments, USA). Current traces were analyzed in Clampfit 10.6 (Axon Instruments). Adequate receptor expression was confirmed in each oocyte before study with a 10 s application of 1 mM GABA in ND96 buffer (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH = 7.4). To avoid output saturation, oocytes producing 1 mM GABA-evoked peak currents greater than 10 μA were discarded. For all experiments using both diazepam (or another benzodiazepine or benzodiazepine analog) and flumazenil, a test benzodiazepine:flumazenil concentration ratio of at least 1:2 was used to ensure complete competition by flumazenil at the classical benzodiazepine binding site.
Agonist-dependent Effects of Diazepam and Flumazenil on GABAA Receptors.
Each oocyte was perfused with ND96 buffer containing either 20 μM etomidate or 6 μM GABA for 60 s. Following a five-minute washout period with ND96 buffer alone, oocytes were again exposed to etomidate or GABA for 60 s along with the desired concentration of test compound(s) (e.g., diazepam, flumazenil, or both). To correct for oocyte-to-oocyte variability in receptor expression, peak current amplitudes recorded in the presence of test compound(s) were normalized to those elicited in their absence and measured in the same oocyte.
The effects of diazepam on etomidate-activated peak current amplitudes were analyzed with Prism 8 for MacOS software (Graphpad, USA) using their bell-shaped concentration-response equations:
where Dip is the maximal normalized response, EC50 is the diazepam concentration that elicits a normalized response that is half-way between 100% and Dip, and half maximal inhibitory concentration (IC50) is the diazepam concentration that reduces the normalized response to one-half Dip.
The effects of diazepam on peak GABA-activated current amplitudes were analyzed with GraphPad Prism 8.0 for MacOS (USA) using a Hill equation in the form:
where Maximum is the normalized response at infinitely high diazepam concentrations and EC50 is the diazepam concentration that elicits a normalized response that is half-way between 100% and Maximum.
The effects of benzodiazepines or benzodiazepine analogs plus flumazenil on peak etomidate-activated currents were analyzed with GraphPad Prism 8.0 for MacOS using a Hill equation in the form:
where [B] is the benzodiazepines or benzodiazepine analog concentration and IC50 is the benzodiazepine or benzodiazepine analog concentration that reduces the normalized response by one-half.
Impact of Diazepam on the Etomidate Concentration-Response Relationship in GABAA Receptors.
Peak etomidate-activated current amplitudes in the presence of diazepam plus flumazenil were measured and normalized to the peak current amplitude elicited by 1 mM GABA in the same oocyte. The normalized responses were then plotted against the concentration of etomidate and this relationship analyzed with GraphPad Prism 8.0 for MacOS using the Gaddum/Schild method as defined by the following equations:
where Maximum is the peak normalized response at infinitely high etomidate concentrations, EC50 is the etomidate concentration that elicits a normalized response that is one-half of the Maximum, pA2 is the negative logarithm of the diazepam concentration needed to shift the etomidate concentration-response curve by a factor of 2, and n is the Hill slope.
Inhibition of R-[3H]azietomidate Photoaffinity Labeling of Purified GABAA Receptors by Diazepam and Flumazenil.
Human embryonic kidney 293 cells transfected to inducibly express α1β3γ2L GABAA receptors containing a FLAG epitope on the N terminus of the α1 subunit were homogenized to form a membrane preparation, which was then further purified using an anti-FLAG affinity purification resin and reconstituted into mixed lipid micelles as previously described.10,21 Receptor aliquots were then photolabled with approximately 2 μM R-[3H]azietomidate in the presence of 1 mM GABA and the desired concentrations of either diazepam or flumazenil using the previously reported approach.22 Receptor subunits were then resolved by polyacrylamide gel electrophoresis. Gels were stained with GelCode Blue Protein Stain (Thermofisher, USA). The gel bands containing GABAA receptor subunits were individually excised, and 3H incorporation into excised gel bands was assayed by liquid scintillation counting. Normalized specific photolabeling was determined by first subtracting counts per minute incorporated into samples photolabeled in the presence of 300 μM nonradioactive etomidate and then dividing the counts per minute measured in samples without competing ligand (i.e., diazepam or flumazenil). Diazepam and flumazenil stock solutions were prepared in ethanol to 60 mM and all photolabeled solutions contained a final concentration of 0.5% (vol/vol) ethanol. Data were analyzed with GraphPad Prism 8.0 for MacOS software using a Hill equation in the form:
where IC50 is the concentration of diazepam required to reduce the normalized specific R-[3H]azietomidate photo-incorporation by one-half.
Anesthetic Concentration-Response Relationships in Zebrafish Larvae.
Zebrafish (Danio rerio) were used in accordance with established protocols approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee. Embryos were collected from adult mating pairs on an as-needed basis and maintained in 140-mm diameter Petri dishes containing E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl, 0.33 mM MgSO4, 2 mM HEPES, pH 7.4) at 28.5°C with a 14/10 h light/dark cycle. The density of embryos and larvae were maintained at fewer than 100 per dish. Larval zebrafish (aged 6 to 7 days postfertilization) and 100 μL of E3 medium were transferred from the Petri dish to a standard 96-well plate using a 1,000-μL pipette fit with a cut and fire-polished tip. Solutions containing etomidate, ketamine, diazepam, flumazenil, and/or dimethyl sulfoxide controls were prepared at twice the desired final concentration in E3 medium and 100 μL was added to the wells to bring the total volume to 200 μL. Final dimethyl sulfoxide concentrations never exceeded 0.7% and were controlled across all conditions.
Immediately after the addition of larvae and test compounds, the 96-well plate was placed inside a Zebrabox (Viewpoint Behavioral Systems, Canada) and incubated in the dark chamber at 28°C for fifteen minutes (etomidate experiments) or thirty minutes (ketamine experiments) before recording. In pilot experiments, these incubation times were determined to produce reproducible steady-state responses. Larvae activity was recorded with an infrared video camera and analyzed using Zebralab v3.2 software (Viewpoint Behavioral Systems). Experiments consisted of four trials each of a 0.2-s exposure to a 500-lux white light separated by 3-min rest periods to avoid acclimation to the light stimulus as previously described.23 Briefly, baseline activity for each fish was established by recording activity in the darkened chamber in the 10 s before each stimulus then averaged across all four prestimulus periods. For each trial, fish were considered to have had an intact photomotor response if their activity score for the 0.2 s during the stimulus or the two 0.2 s epochs after the stimulus exceeded the upper 95% CI of their baseline activity level. In such cases, fish were scored 1, in all other cases where poststimulus activity did not exceed this threshold, fish were scored 0. These scores of 0 or 1 were then averaged across the four trials, giving a probability of response to stimulus for each fish. In each experiment, the mean probability of response was taken for eight fish per drug concentration. To account for cohort variability, each 96-well plate also contained eight control fish in E3 medium and the appropriate concentration of dimethyl sulfoxide. The mean probability of responses for all treatment groups on a single plate were then normalized to this control group. To assure reproducibility, each experiment consisted of five plates containing distinct fish cohorts. Each of these cohorts represent fish from different clutches and mating pairs, collected on different days. Data are reported as the mean ± SEM probability of response derived from the five plates. Data were analyzed and fit with GraphPad Prism 8.0 for MacOS using a Hill equation in the form
where the maximum represented the normalized probability of response in the absence of anesthetic and EC50 is the anesthetic concentration required to obtain a probability of response that is one-half of the Maximum.
Statistical Analysis
Data derived from electrophysiologic experiments are reported as the mean of five individual experiments (using different oocytes) ± SD. Data derived from photoaffinity labeling experiments are reported as results obtained from individual receptor aliquots. The statistical comparisons between the etomidate and ketamine anesthetic EC50s in the presence versus the absence of diazepam plus flumazenil were made using the extra sum-of-squares F test. A correlation between log IC50 and the calculated log of the octanol:water partition coefficient was tested using linear regression. A correlation was assumed if the slope was statistically different from zero. Statistical testing was two-tailed. The assumption of normality around reported mean values was confirmed using the Shapiro-Wilk test with an alpha value of 0.05. There were no lost or missing data, and no outliers appeared in our studies. No statistical power calculations were conducted before the study. All sample sizes were based on our previous experience with these experimental designs. All fitting and statistical tests were performed with GraphPad Prism 8.0 for MacOS. Statistical significance was assumed for P < 0.05.
Results
Diazepam Selectively Inhibits Etomidate-activated GABAA Receptor Currents
In our initial electrophysiologic studies, we sought to obtain functional evidence that diazepam could inhibit etomidate binding to the GABAA receptor by using an in vitro assay in which oocyte-expressed α1β3γ2L GABAA receptors are directly activated by either etomidate or GABA (as a control) in the presence of widely ranging concentrations of diazepam. To facilitate comparisons between experimental results obtained with the two different receptor agonists, we chose concentrations of etomidate (20 μM) and GABA (6 μM) that pilot studies showed comparably activate GABAA receptors (5 to 10% of that activated by 1 mM GABA; Supplemental Digital Content, http://links.lww.com/ALN/C401). We found that at nanomolar concentrations, diazepam comparably potentiated peak etomidate-activated and GABA-activated current amplitudes (fig. 1, A–C). With both agonists, peak current amplitudes increased with diazepam concentration before reaching maximum values by 300 nM diazepam that were 250% to 300% of those measured in the absence of diazepam (fig. 1C). The diazepam EC50 for potentiation of peak current amplitudes was comparable for receptors activated with etomidate versus GABA with values of 39 nM (95% CI, 27 to 55 nM) versus 26 nM (95% CI, 16 to 41 nM), respectively. This is consistent with positive allosteric modulation of agonist-activated currents by diazepam via the classical benzodiazepine binding site. However, at the micromolar concentrations at which diazepam is expected to also bind to the etomidate binding site on the GABAA receptor,15,16 we observed a dramatic diazepam concentration-dependent reduction in peak current amplitudes mediated by receptors that were activated with etomidate. The diazepam IC50 for this antagonist action was 9.6 μM (95% CI, 7.6 to 12 μM). We did not see a similar reduction with receptors that were activated with GABA.
Fig. 1.
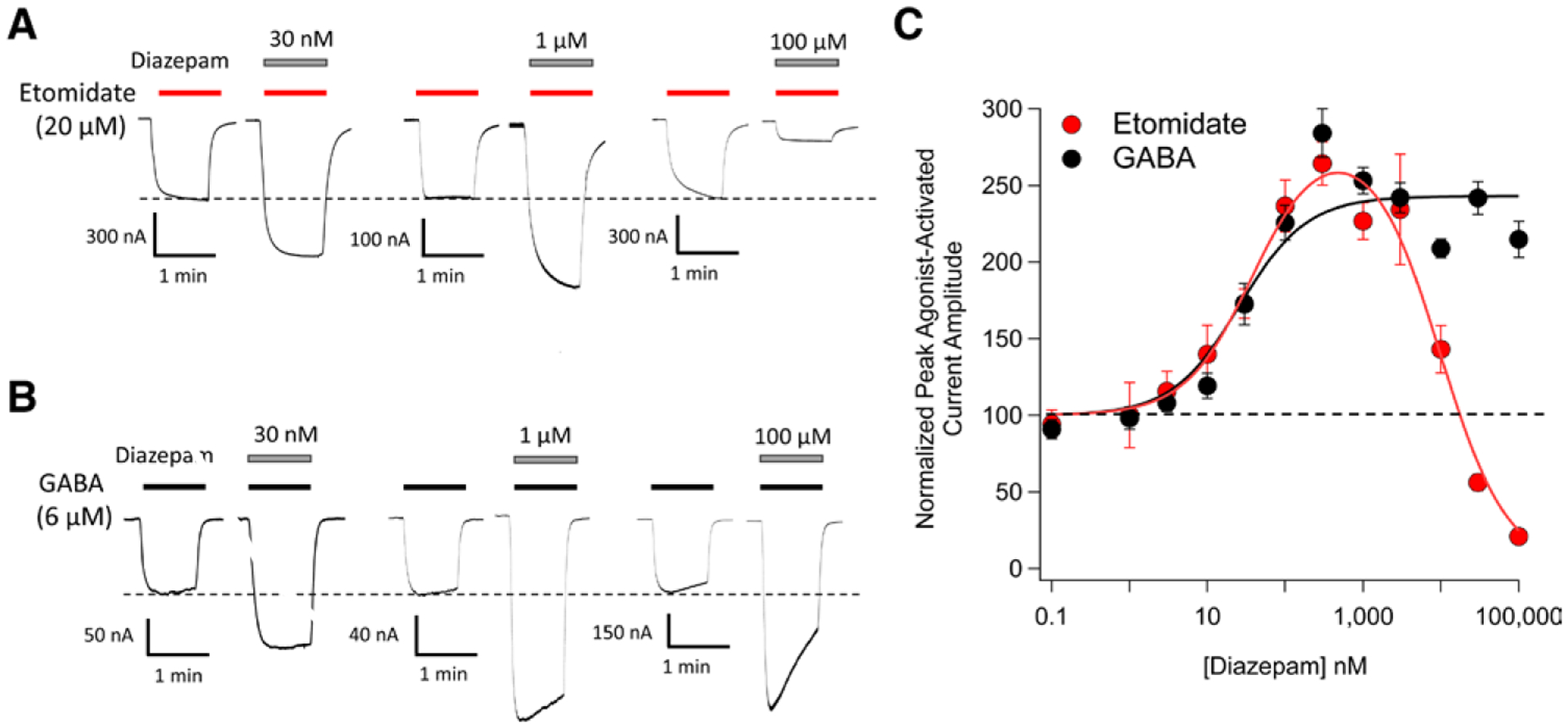
Diazepam modulation of 20 μM etomidate-activated and 6 μM γ-aminobutyric acid (GABA)-activated α1β3γ2L GABAA receptor currents. (A) Representative traces showing the impact of diazepam at the indicated concentrations on etomidate-activated currents. (B) Representative traces showing the impact of diazepam at the indicated concentrations on GABA-activated currents. (C) Diazepam concentration–response curves for etomidate-activated and GABA-activated peak current amplitudes. Each data point represents the mean ± SD derived from five different oocytes. Each curve is a nonlinear least-squares fit of the dataset to either a bell-shaped equation (etomidate-activated currents) or Hill equation (GABA-activated currents). For etomidate-activated peak currents, the diazepam EC50 was 39 nM (95% CI, 27 to 55 nM) and IC50 was 9.6 μM (95% CI, 7.6 to 12 μM). For GABA-activated peak currents, the diazepam EC50 was 26 nM (95% CI, 16 to 41 nM).
We then repeated these experiments in the presence of flumazenil, a competitive antagonist that binds to the classical benzodiazepine binding site and prevents diazepam from acting via that site.10,22,24 Although flumazenil had no impact on either GABA-activated or etomidate-activated currents in the absence of diazepam (data not shown), it completely abolished the nanomolar potentiating actions of diazepam on both etomidate-activated and GABA-activated currents (compare fig. 2, A–C with fig. 1, A–C). This confirmed our assumption that such potentiation is mediated via the classical benzodiazepine binding site regardless of whether the activating agonist is etomidate or GABA. Flumazenil had a negligible effect on the micromolar antagonist actions of diazepam on etomidate-activated currents as the IC50 of diazepam in the presence of flumazenil was 13 μM (95% CI, 10 to 16 μM).
Fig. 2.
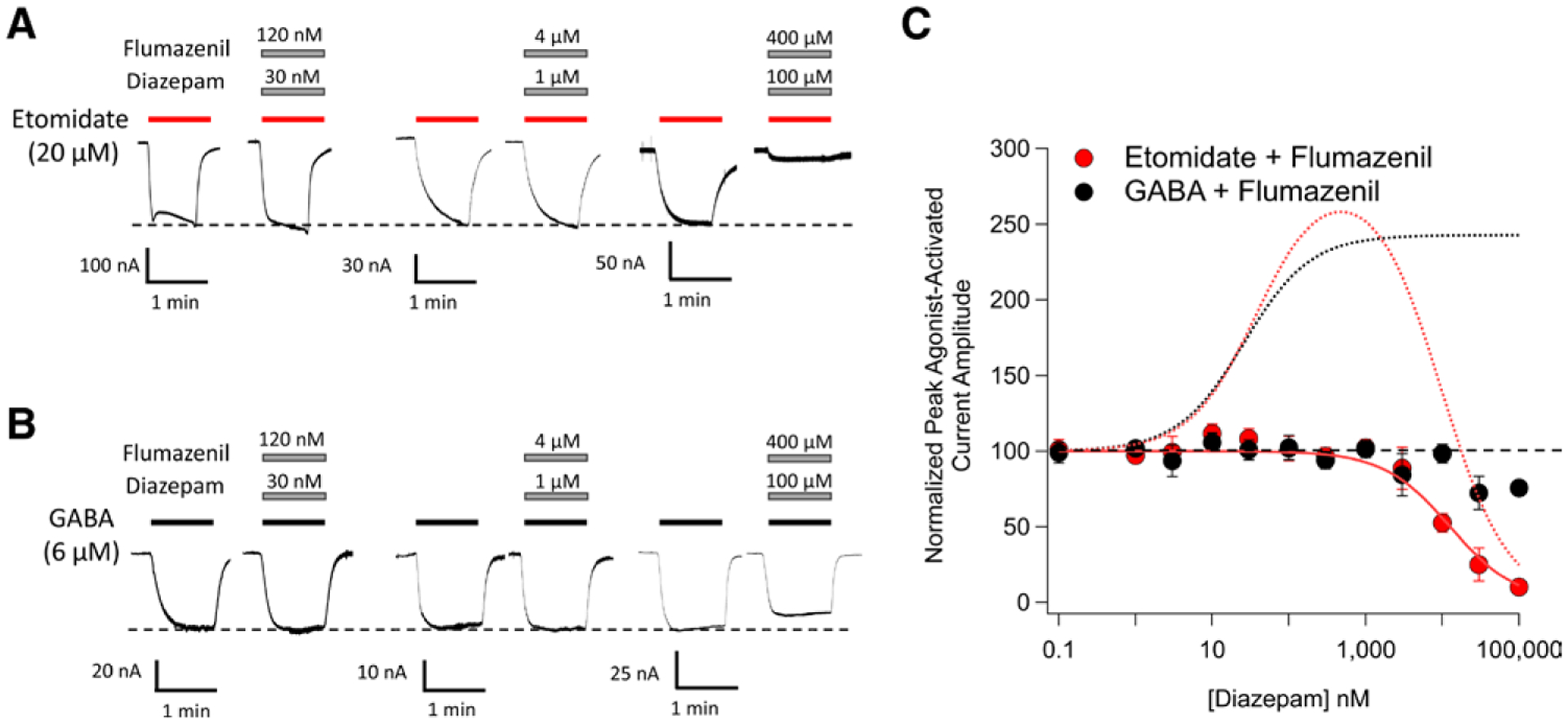
The impact of flumazenil on diazepam modulation of 20 μM etomidate-activated and 6 μM γ-aminobutyric acid (GABA)-activated α1β3γ2L GABAA receptor currents. (A) Representative traces showing the impact of diazepam on etomidate-activated currents in the presence of flumazenil. (B) Representative traces showing the impact of diazepam on GABA-activated currents in the presence of flumazenil. (C) Diazepam concentration–response curves for etomidate-activated and GABA-activated peak current amplitudes in the presence of flumazenil. Each data point represents the mean ± SD derived from five different oocytes. The solid red curve is a fit of the etomidate-activated dataset to a Hill equation yielding a diazepam IC50 of 13 μM (95% CI, 10 to 16 μM). The dotted curves reproduce the fits of the figure 1 data obtained in the absence of flumazenil to illustrate that for both etomidate-activated currents (red dotted curve) and GABA-activated currents (black dotted curve), flumazenil abolishes the nanomolar potentiating action of diazepam. The diazepam:flumazenil concentration ratio was 1:4.
Diazepam Shifts the Etomidate Concentration-Response Curve for GABAA Receptor Activation Rightward
The observation that micromolar concentrations of diazepam reduce the magnitude of peak currents activated by etomidate but not GABA is most consistent with an underlying mechanism that is competitive rather than noncompetitive because the latter is generally expected to be independent of agonist identity. We further tested this inference by assessing the impact of diazepam on the etomidate-concentration response curve for direct activation. We did these studies in the presence of flumazenil to prevent any confounding potentiating actions arising from diazepam binding to the classical benzodiazepine binding site. We found that increasing concentrations of diazepam progressively shift the etomidate-concentration response rightward without significantly altering the maximum response evoked by high etomidate concentrations (fig. 3A). Such behavior typifies competitive antagonism and is consistent with the structural data.12 A Schild analysis of the data yielded an etomidate EC50 in the absence of diazepam of 76 μM (95% CI, 58 to 100 μM), a pA2 of 4.70 (95% CI, 4.4 to 4.9), a Schild slope of 1.1 (95% CI, 0.57 to 1.6), a Hill slope of 2.1 (95% CI, 1.5 to 2.7), and a KB of 20 μM (95% CI, 11 to 36 μM). Because the Schild slope was not significantly different from 1, we refit the data with the Schild slope constrained to 1 to obtain an estimated diazepam dissociation constant of 18 μM (95% CI, 12 to 29 μM).
Fig. 3.
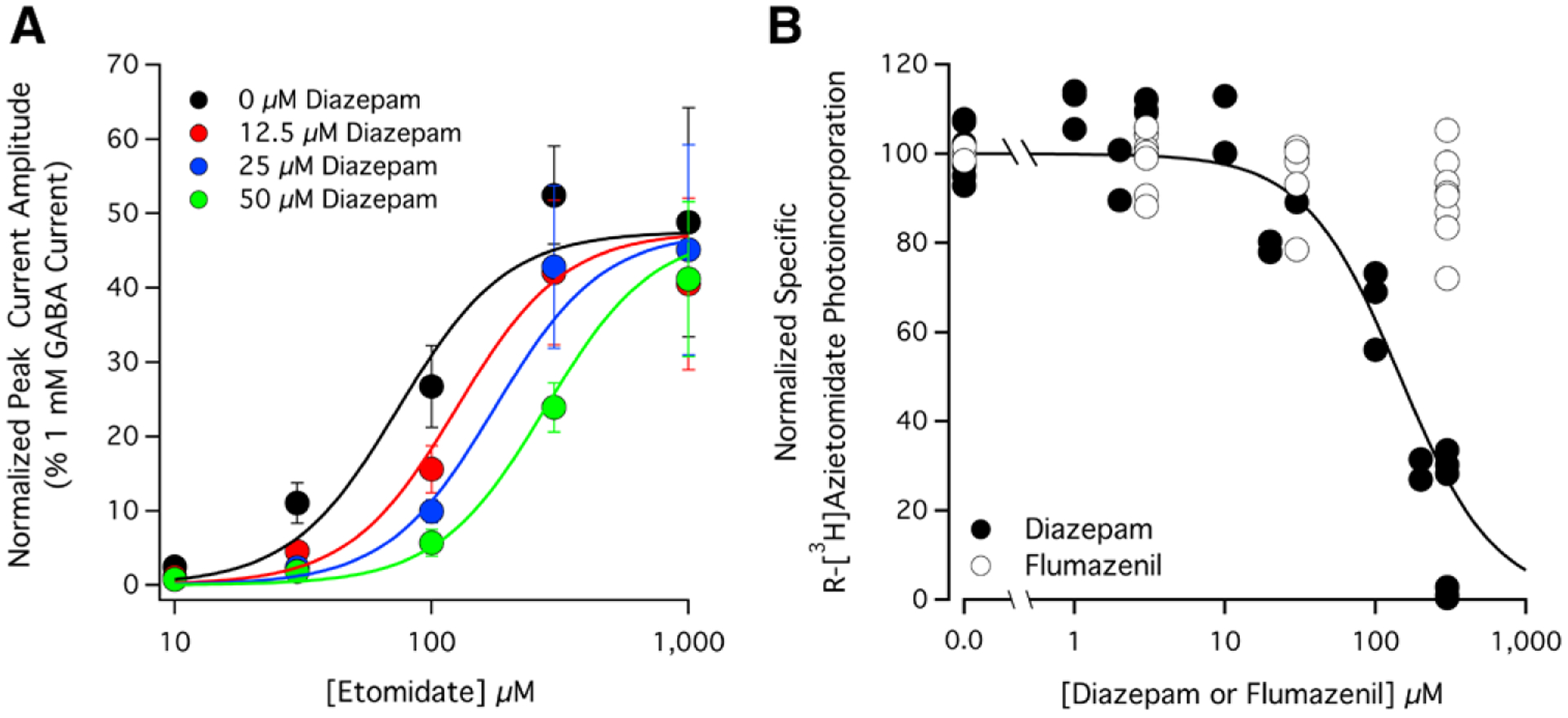
Competitive interactions between diazepam and (A) etomidate or (B) the photoreactive etomidate analog R-[3H]azietomidate at α1β3γ2L γ-aminobutyric acid type A (GABAA) receptors. (A) The impact of diazepam on the etomidate concentration-response curve for direct activation of GABAA receptors in the presence of flumazenil. Each data point represents the mean ± SD derived from five different oocytes. Flumazenil was present to prevent modulation from the classical benzodiazepine binding site. The diazepam:flumazenil concentration ratio was 1:4. The curves are derived from a Schild analysis of the data and yielded an etomidate EC50 in the absence of diazepam of 76 μM (95% CI, 58 to 100 μM), a pA2 (in Molar) of 4.70 (95% CI, 4.4 to 4.9), a Schild slope of 1.1 (95% CI, 0.57 to 1.6), a Hill slope of 2.1 (95% CI, 1.5 to 2.7), and a KB of 20 μM (95% CI, 11 to 36 μM). Because the Schild slope was not significantly different from 1, we refit the data with the Schild slope constrained to 1 to obtain an estimated diazepam dissociation constant of 18 μM (95% CI, 12 to 29 μM). (B) Diazepam and flumazenil concentration-response curves for inhibition of specific R-[3H]azietomidate photoaffinity labeling of purified GABAA receptors. Data were normalized to the specific counts per minute measured in the absence of competing ligand (i.e., diazepam or flumazenil). Each data point represents data from a single receptor aliquot. The curve is a nonlinear least squares fit of the diazepam dataset to a Hill equation yielding an IC50 of 150 μM (95% CI, 120 to 200 μM).
Diazepam Inhibits Photoaffinity Labeling of GABAA Receptors by Azietomidate
As a final test of the ability of diazepam to bind to the etomidate binding sites on the GABAA receptor and act in a competitive manner, we used a photoaffinity label protection assay. This assay uses purified α1β3γ2L GABAA receptors and an etomidate photoaffinity label (R-[3H] azietomidate) that selectively photo-incorporates into the receptor at the etomidate binding site.9,10 This photo-incorporation is inhibited by other ligands that also bind to this site, providing a way to monitor competitive interactions.10,22 Figure 3B shows that diazepam reduced photo-incorporation of R-[3H]azietomidate in a concentration-dependent manner. The diazepam IC50 defined by this assay was 150 μM (95% CI, 120 to 200 μM). This value is substantially higher than those defined in our electrophysiology experiments (10 to 20 μM), which we believe largely reflects a reduction in the free aqueous diazepam concentration due to partitioning of the drug into the mixed lipid micelles into which the GABAA receptors are purified. We previously saw a quantitatively similar reduction in the apparent affinity of flunitrazepam (sevenfold) when using similarly purified GABAA receptors as part of a functional assay.21 An additional factor in the photolabeling experiments that may contribute to the higher IC50 is the inherent nonequilibrium nature of the assay as binding of diazepam is reversible but photolabeling by R-[3H]azietomidate is not. Flumazenil at up to 300 μM had little inhibitory effect on R-[3H]azietomidate photo-incorporation, which is consistent with our electrophysiologic studies suggesting that it binds to the etomidate binding site with much lower affinity than diazepam.
Other Benzodiazepines and Benzodiazepine-like Compounds Inhibit GABAA Receptor Activation by Etomidate
We then evaluated 22 additional benzodiazepines and benzodiazepine-like compounds to see whether they could similarly antagonize etomidate action on the GABAA receptor. The structures of these compounds are shown in figure 4. We found that 18 of these compounds measurably reduced etomidate activated currents. Their inhibitory potencies ranged by 30-fold, and none had a higher potency than diazepam (fig. 5A and table 1). The remaining four compounds produced little or no inhibition of etomidate-activated currents (less than 10%) even at the highest concentration studied (100 μM), implying that they have IC50s that are at least 1,000 μM.
Fig. 4.
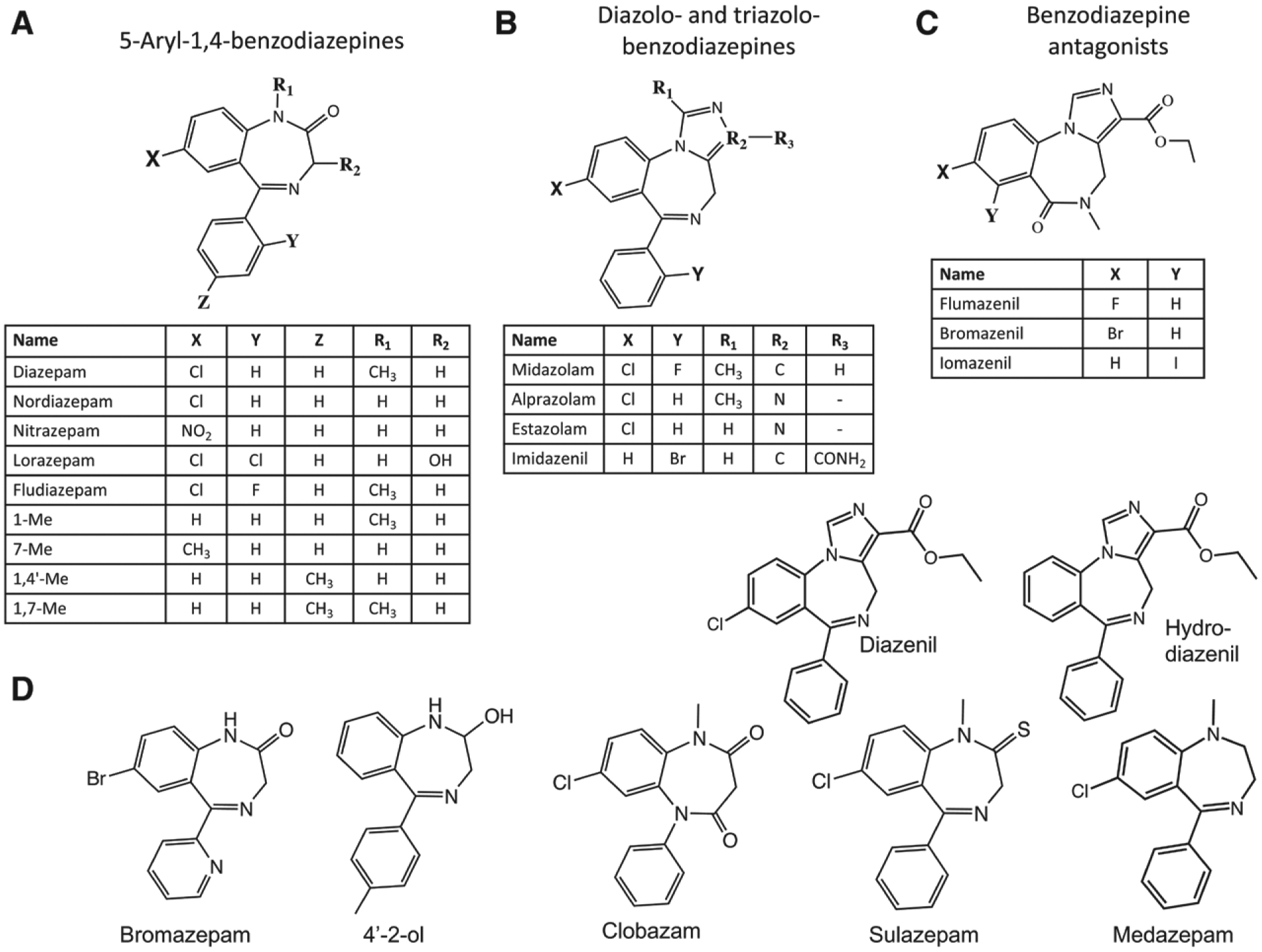
Structures of the 23 benzodiazepines and benzodiazepine-like compounds that we evaluated grouped by class. (A) 5-arl-1,4-benzodiazepines, (B) diazolo-benzodiazepines and triazolo-benzodiazepines, (C) benzodiazepines antagonists, and (D) compounds that do not belong to the three previous groups.
Fig. 5.
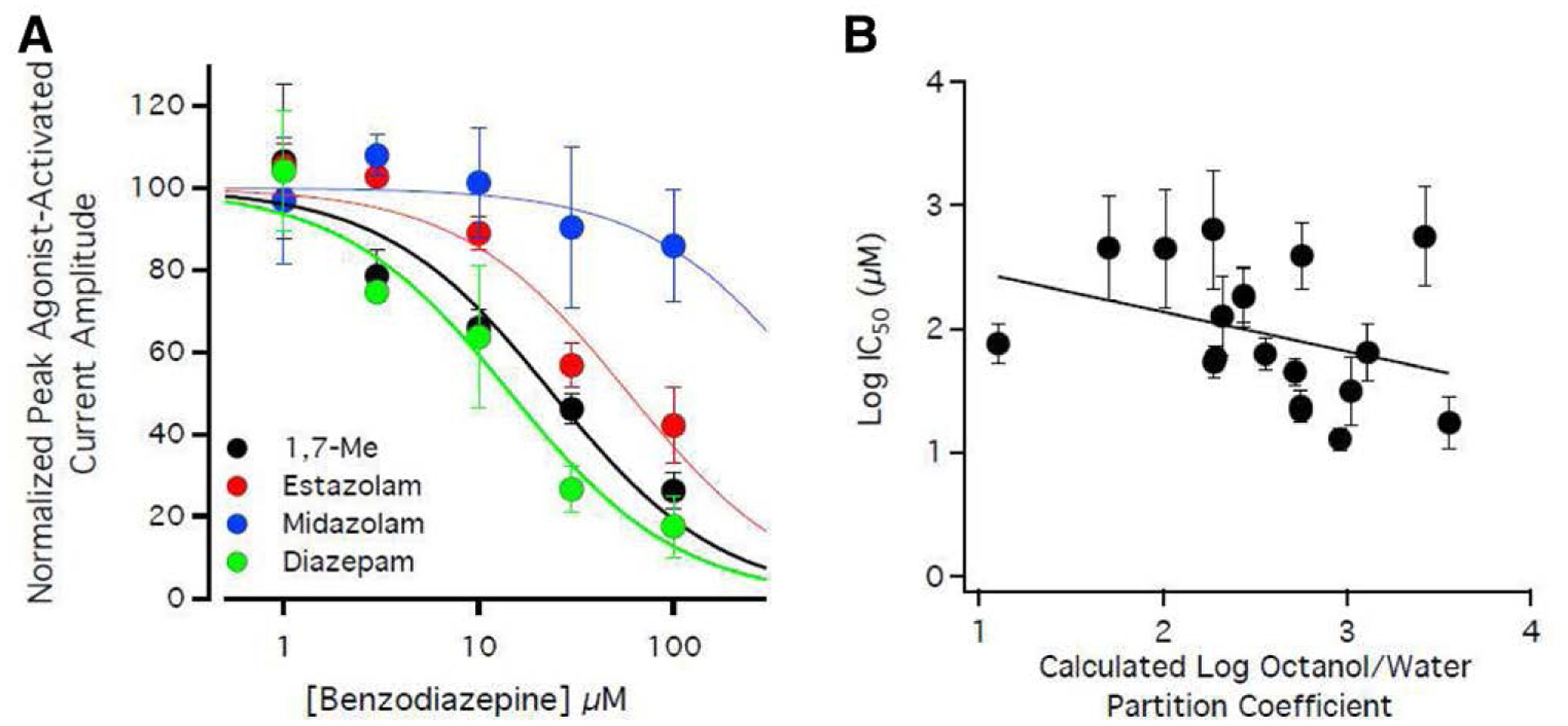
Inhibition of 20 μM etomidate-activated currents by representative benzodiazepines and benzodiazepine-like compounds in the presence of flumazenil. The benzodiazepine:flumazenil concentration ratio was 1:2. (A) 1,7-Me, estazolam, midazolam, and diazepam concentration-response curves for inhibition of etomidate-activated peak current amplitudes. Each data point represents the mean ± SD derived from five different oocytes. The curves are fits of the datasets to a Hill equation yielding 1,7-Me, estazolam, midazolam, and diazepam IC50s of 24 μM (95% CI, 18 to 32 μM), 59 μM (95% CI, 47 to 74 μM), 560 μM (95% CI, 220 to 1400 μM), and 15 μM (95% CI, 11 to 20 μM) respectively. (B) Relationship between the LogIC50 values of the nineteen compounds studied for whom this value could be quantified and their calculated log octanol:water partition coefficient values. A linear fit of this relationship yielded a slope of −0.31 (95% CI, −0.75 to 0.13), which was not significantly different from zero (P = 0.157), and an r2 of 0.114. Error bars indicate the 95% CI. Some symbols overlap.
Table 1.
Half-maximal Concentration (IC50) for Antagonizing Direct Activation by 20 μM Etomidate (in the Presence of Flumazenil) and Calculated Log Octanol/Water Partition Coefficient Values
| Class | Compound Name | IC50 (μM) | 95% Confidence Interval (μM) | Calculated Log Octanol/Water Partition Coefficien† |
|---|---|---|---|---|
| 5-Aryl-1,4-benzodiazepines | Diazepam | 13* | 10–16* | 2.96 |
| 15 | 11–20 | |||
| Nordiazepam | 32 | 17–60 | 3.02 | |
| Nitrazepam | 130 | 61–260 | 2.32 | |
| Lorazepam | ≥ 1,000 | — | 2.37 | |
| Fludiazepam | 390 | 210–730 | 2.75 | |
| 1-Me | 76 | 53–110 | 1.10 | |
| 7-Me | 45 | 35–58 | 2.72 | |
| 1,4’-Me | 22 | 18–27 | 2.75 | |
| 1,7-Me | 24 | 18–32 | 2.75 | |
| Diazolo- and triazolo- benzodiazepines | Midazolam | 560 | 220–1,400 | 3.42 |
| Alprazolam | 63 | 46–85 | 2.55 | |
| Estazolam | 59 | 47–74 | 2.29 | |
| Imidazenil | 190 | 110–310 | 2.44 | |
| Benzodiazepine antagonists | Flumazenil | ≥ 1,000 | — | 1.29 |
| Bromazenil | 450 | 150–1,400 | 2.01 | |
| Iomazenil | 640 | 210–1,900 | 2.27 | |
| Unclassified | Bromazepam | 460 | 170–1,200 | 1.70 |
| 4’–2-ol | 54 | 41–73 | 2.28 | |
| Clobazam | 180 | 100–320 | 2.44 | |
| Sulazepam | 64 | 38–110 | 3.11 | |
| Medazepam | ≥ 1,000 | — | 4.12 | |
| Diazenil | 18 | 11–29 | 3.56 | |
| Hydro-diazenil | ≥ 1,000 | — | 2.84 |
[Benzodiazepine]:[Flumazenil] = 1:2 unless otherwise indicated.
[Diazepam]:[Flumazenil] = 1:4.
Calculated by ChemBioDraw Ultra 12.0.3 (Cambridgesoft, USA).
Although the etomidate binding site is located in a relatively hydrophobic protein domain (i.e., the transmembrane domain of the GABAA receptor), figure 5B shows that the correlation between the inhibitory potencies of all 23 compounds and their hydrophobicities (as reflected by their cLogP values) is weak. A linear fit of the relationship between LogIC50 versus cLogP yielded a slope that is not significantly different from zero (−0.31; 95% CI, −0.75 to 0.13; P = 0.157), with an r2 of only 0.114.
Diazepam Shifts the Etomidate Concentration-Response Curve for Anesthesia Rightward
If diazepam can competitively antagonize the GABAA receptor actions of etomidate in vitro, could it do the same in vivo with resultant reversal of general anesthesia? Such an action would manifest itself as an increase in the etomidate concentration required to inhibit behavioral responses to strong stimuli. To answer this question, we used a validated zebrafish larva assay that measures the motor responses of these aquatic animals when subjected to strong light stimuli.23 We used larvae that were 6 to 7 days postfertilization, because by this stage of development there has been sufficient upregulation of the NKCC2 chloride transporter to establish a mature chloride gradient and thus render GABAergic action inhibitory.25,26
We first defined the potencies of etomidate and ketamine, an anesthetic that acts primarily via a glutaminergic rather than a GABAergic mechanism and thus serves as a mechanistic control.27–29 We found that both anesthetics produce a concentration-dependent reduction in zebrafish larvae motor responses to the light pulse (fig. 6, A and B). The etomidate and ketamine EC50s for abolishing these responses were 0.66 μM (95% CI, 0.42 to 1.0 μM) and 170 μM (95% CI, 110 to 270 μM), respectively. We also found that at a concentration of 50 μM, diazepam alone essentially abolishes such responses. This diazepam action was completely flumazenil-reversible, indicating that positive modulation via the classical benzodiazepine site (perhaps together with some weak positive modulation via the etomidate binding site) is necessary for diazepam to produce anesthesia in this animal model. Finally, we tested whether 50 μM diazepam—when given along with 200 μM flumazenil to prevent its action at the classical benzodiazepine binding site—could antagonize the action of etomidate in zebrafish as it had done in the GABAA receptor studies. We found that it did, shifting the etomidate concentration-response curve rightward and statistically significantly (P < 0.0001) increasing the etomidate EC50 by 18-fold to 12 μM (95% CI, 4.7 to 31 μM; fig. 6C). This antagonistic action was selective for etomidate anesthesia as no rightward shift was seen in parallel studies using zebrafish larvae anesthetized with ketamine. In fact, we observed the opposite as the combination of 50 μM diazepam and 200 μM flumazenil statistically significantly (P < 0.0001) shifted the ketamine concentration–response curve leftward by 19-fold to 9 μM (95% CI, 6 to 13 μM; fig. 6D).
Fig. 6.
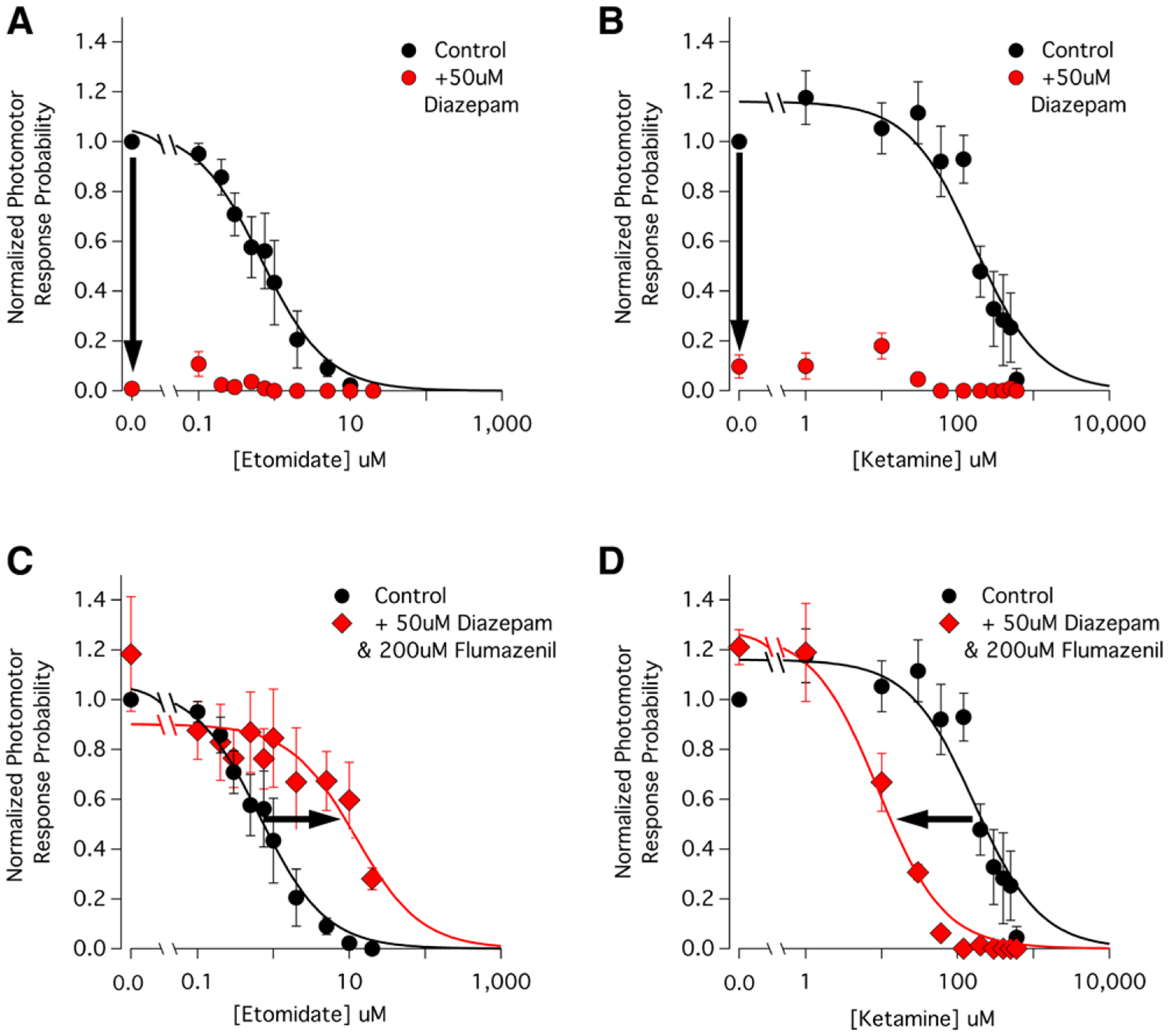
Contrasting effects of diazepam (50 μM)/flumazenil (200 μM) on the etomidate and ketamine concentration–response curves for abolishing zebrafish larvae motor responses to a light stimulus. Each data point represents the mean ± SEM normalized photomotor response derived from 40 larvae. The curves are nonlinear least squares fits of the datasets to a Hill equation. (A) Etomidate concentration-response curve for abolishing zebrafish larvae motor responses to a light stimulus in the absence (control) and presence of 50 μM diazepam. Etomidate abolishes responsiveness in a concentration-dependent manner with an EC50 of 0.66 μM (95% CI, 0.42 to 1.0 μM). Diazepam (50 μM) alone essentially abolishes all responsiveness even in the absence of etomidate (downward arrow). (B) Ketamine concentration-response curve for abolishing zebrafish larvae motor responses to a light stimulus in the absence (control) and presence of 50 μM diazepam. Ketamine abolishes responsiveness in a concentration-dependent manner with an EC50 of 170 μM (95% CI, 110 to 270 μM), whereas 50 μM diazepam alone essentially abolishes all responsiveness even in the absence of ketamine (downward arrow). (C) The combination of diazepam and flumazenil increases the etomidate EC50 for abolishing zebrafish larvae motor responses to a light stimulus from a control value of 0.66 μM to 12 μM (95% CI, 4.7 to 31 μM). The resulting rightward shift is highlighted by the arrow. (D) The combination of diazepam and flumazenil reduces the ketamine EC50 for abolishing zebrafish larvae motor responses to a light stimulus from a control value of 170 μM to 9 μM (95% CI, 6 to 13 μM). The resulting leftward shift is highlighted by the arrow.
Discussion
Our studies show that at micromolar concentrations and in the presence of flumazenil (to inhibit its action at the classical extracellular benzodiazepine site), diazepam exhibits the pharmacology of a competitive etomidate antagonist. Specifically, diazepam antagonizes the allosteric agonist actions of etomidate on the GABAA receptor in a concentration-dependent manner, rightward shifts the etomidate concentration-response curve for direct activation without reducing the maximal response at high etomidate concentrations, and inhibits photoaffinity labeling of the GABAA receptor by the etomidate analog R-[3H]azietomidate. Additionally and most notably, it increases the etomidate concentration required to produce immobility in an animal model of anesthesia. As expected for an antagonist acting competitively at the etomidate binding site, this behavior is selective for etomidate as diazepam failed to reverse the orthosteric agonist actions of GABA on the GABAA receptor or increase the ketamine concentration necessary to produce anesthesia. In fact, diazepam (in the presence of flumazenil) had the opposite effect on ketamine anesthesia, reducing the ketamine concentration needed to induce anesthesia. We believe that this reflects an additive or synergistic interaction between the inhibitory action of ketamine at glutaminergic synapses and a weak positive modulatory action of diazepam at the etomidate binding sites of the GABAA receptor.
Our studies also show that this antagonism is not specific to diazepam because many other benzodiazepines and benzodiazepine-like compounds similarly inhibit the agonist actions of etomidate on the GABAA receptor. However, the inhibitory potencies of the 22 other compounds that we studied were all lower than that of diazepam, in some cases by orders of magnitude. Such selective binding likely explains why diazepam was visualized within the etomidate binding sites of the GABAA receptor in cryo-electron microscopic imaging studies whereas flumazenil and alprazolam were not.14 Although the etomidate binding sites are located within the hydrophobic transmembrane receptor domain, we observed no statistically significant correlation between the hydrophobicity of these compounds and their apparent affinities for the etomidate binding site as reflected by their IC50 values. This suggests that the specific structural features of these compounds govern their binding affinities. As a class, we found that the benzodiazepine antagonists (i.e., flumazenil, bromazenil, and iomazenil) possessed the lowest etomidate inhibitory potencies. The distinguishing structural feature of these antagonists is the absence of a pendent phenyl ring, suggesting that attractive π–π stacking interactions between such rings and an aromatic ring in the etomidate binding site (e.g., β3F289, which has also been proposed to form such interactions with the aromatic ring in propofol30) enhance binding affinity to the etomidate binding site. This interpretation is supported by the observation that the presence of a halogen substituent on this ring—which withdraws electrons from the ring thus reducing π-π stacking interaction strength-reduces inhibitory potency. For example, although fludiazepam differs from diazepam only by the presence of a fluorine atom at the 2′ position of the pendent phenyl ring, it possesses an etomidate inhibitory potency that is 30× lower. In fact, of the thirteen 5-aryl-1,4-benzodiazepines, diazolo-benzodiazepines, and triazolo-benzodiazepines that we studied, the four with the lowest etomidate inhibitory potencies all have a halogen substituent on the pendent phenyl ring.
A key feature of benzodiazepines that allows them to antagonize etomidate action is their relatively low intrinsic efficacies when bound to the etomidate binding site of the GABAA receptor. In the case of diazepam, for example, such binding is insufficiently efficacious to produce direct activation and potentiates currents activated by low concentrations of GABA by no more than two- to threefold.15 In comparison, high concentrations of etomidate can produce significant direct activation and potentiate currents activated by low concentrations of GABA by more than an order of magnitude.11 The ideal competitive antagonist would bind specifically and with high affinity to the etomidate binding site—thus eliminating the need for flumazenil—while retaining low intrinsic efficacy. Additional benzodiazepine analog design, synthesis, and experimental studies would be necessary to determine whether such an agent can be developed.
Previous groups have worked to develop strategies to pharmacologically reverse anesthetic actions. For example, Solt et al.31 found that methylphenidate could induce a shift in electroencephalogram power of rats anesthetized with isoflurane from delta to theta and reduce the time to emergence after isoflurane administration was terminated. This group similarly found that dextroamphetamine could reduce the emergence time after propofol administration.32 However, the underlying mechanism used to achieve such reversal is quite different from that demonstrated in the current studies as methylphenidate and dextroamphetamine are believed to act at the network level by increasing arousal rather than by directly competing with the anesthetic at its molecular site of action. We have previously shown that similar to diazepam, the novel etomidate analog naphthalene-etomidate could bind with low efficacy to the etomidate binding sites on the GABAA receptor and partially reverse etomidate action.33 However, the relatively low apparent binding affinity of this analog for the etomidate binding site (dissociation constant: less than 300 μM compared with 10 to 20 μM for diazepam) along with its low aqueous solubility limited its ability to compete with etomidate.
Our observation that diazepam can act in a competitive manner at the etomidate binding site of the GABAA receptor has potentially important implications for anesthetic mechanisms research because this phenomenon may be useful for assessing the extent to which other anesthetic agents act by binding to this site. In our zebrafish model of anesthesia, etomidate and ketamine likely define the opposite ends of that mechanistic spectrum with the actions of etomidate being completely mediated by this site and those of ketamine mediated elsewhere. As a result, 50 μM diazepam (in the presence of 200 μM flumazenil) had opposite effects on the etomidate and ketamine concentration-response curves for anesthesia, inducing an 18-fold rightward shift when the anesthetic is etomidate (reflecting competitive antagonism) and a 19-fold leftward shift when it is ketamine (reflecting additivity/synergy). Other anesthetics whose actions are thought to be mediated only in part by this site (e.g. propofol and thiopental) are thus predicted to produce concentration-response curve shifts that are quantitatively in between these two extremes. Future studies will test this prediction.
Our results may also have implications for the clinical development of selective anesthetic reversal agents. Much as opioid actions can be pharmacologically reversed by the competitive antagonist naloxone, our studies demonstrate that anesthetic actions can similarly be reversed by a compound that acts as a competitive antagonist at the on-target binding site. However, a limitation of the competitive antagonist approach to anesthetic reversal is that some anesthetic agents likely act (in whole or in part) via different GABAA receptor sites or (as exemplified by ketamine) on non-GABAergic targets. In such cases, an antagonist acting selectively at the etomidate binding site would not be expected to produce complete reversal of all anesthetic actions.
In conclusion, our studies show that on binding to the etomidate binding sites of the GABAA receptor, diazepam acts as a competitive etomidate antagonist capable of selectively reversing the agonist actions of etomidate. This antagonism manifests itself both in vitro and in vivo as a reduction in etomidate potency and provides proof-of-concept for the development of competitive anesthetic antagonists capable to reversing anesthetic actions.
EDITOR’S PERSPECTIVE.
What We Already Know about This Topic
Diazepam binds to the γ-aminobutyric acid type A (GABAA) receptor high-affinity extracellular benzodiazepine site
Diazepam can also bind to the GABAA receptor transmembrane etomidate site
It is unknown whether diazepam or similar compounds can antagonize etomidate
What This Article Tells Us That Is New
In vitro and in vivo zebrafish studies show that diazepam and other like compounds can competitively antagonize etomidate at the GABAA receptor etomidate binding site
This provides proof-of-concept for development of competitive anesthetic antagonists
Research Support
This work was funded by grants Nos. GM122806, GM128989, and GM058448 from the National Institutes of Health (Bethesda, Maryland) and the Department of Anesthesia, Critical Care, and Pain Medicine, Massachusetts General Hospital (Boston, Massachusetts).
Competing Interests
Dr. Raines has served as a paid consultant to The Medicines Company (Parsippany, New Jersey) on an unrelated project. The remaining authors declare no competing interests.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are available in both the HTML and PDF versions of this article. Links to the digital files are provided in the HTML text of this article on the Journal’s Web site (www.anesthesiology.org).
References
- 1.Moore FD: John Collins Warren and his act of conscience: A brief narrative of the trial and triumph of a great surgeon. Ann Surg 1999; 229:187–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walker AJ, Mashour GA: A brief history of sleep and anesthesia. Int Anesthesiol Clin 2008; 46:1–10 [DOI] [PubMed] [Google Scholar]
- 3.Orser BA: Extrasynaptic GABAA receptors are critical targets for sedative-hypnotic drugs. J Clin Sleep Med 2006; 2:S12–8 [PubMed] [Google Scholar]
- 4.Franks NP: General anaesthesia: From molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci 2008; 9:370–86 [DOI] [PubMed] [Google Scholar]
- 5.Antkowiak B: Closing the gap between the molecular and systemic actions of anesthetic agents. Adv Pharmacol 2015; 72:229–62 [DOI] [PubMed] [Google Scholar]
- 6.Weir CJ, Mitchell SJ, Lambert JJ: Role of GABAA receptor subtypes in the behavioural effects of intravenous general anaesthetics. Br J Anaesth 2017; 119(suppl_1):i167–75 [DOI] [PubMed] [Google Scholar]
- 7.Siegwart R, Jurd R, Rudolph U: Molecular determinants for the action of general anesthetics at recombinant alpha(2)beta(3)gamma(2)gamma-aminobutyric acid(A) receptors. J Neurochem 2002; 80:140–8 [DOI] [PubMed] [Google Scholar]
- 8.Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U: General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J 2003; 17:250–2 [DOI] [PubMed] [Google Scholar]
- 9.Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB: Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci 2006; 26:11599–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiara DC, Jayakar SS, Zhou X, Zhang X, Savechenkov PY, Bruzik KS, Miller KW, Cohen JB: Specificity of intersubunit general anesthetic-binding sites in the transmembrane domain of the human α1β3γ2 γ-aminobutyric acid type A (GABAA) receptor. J Biol Chem 2013; 288:19343–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rüsch D, Zhong H, Forman SA: Gating allosterism at a single class of etomidate sites on alpha1beta2gamma2L GABA A receptors accounts for both direct activation and agonist modulation. J Biol Chem 2004; 279:20982–92 [DOI] [PubMed] [Google Scholar]
- 12.Sigel E, Buhr A: The benzodiazepine binding site of GABAA receptors. Trends Pharmacol Sci 1997; 18:425–9 [DOI] [PubMed] [Google Scholar]
- 13.Sigel E, Lüscher BP: A closer look at the high affinity benzodiazepine binding site on GABAA receptors. Curr Top Med Chem 2011; 11:241–6 [DOI] [PubMed] [Google Scholar]
- 14.Masiulis S, Desai R, Uchański T, Serna Martin I, Laverty D, Karia D, Malinauskas T, Zivanov J, Pardon E, Kotecha A, Steyaert J, Miller KW, Aricescu AR: GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 2019; 565:454–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walters RJ, Hadley SH, Morris KD, Amin J: Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nat Neurosci 2000; 3:1274–81 [DOI] [PubMed] [Google Scholar]
- 16.Drexler B, Zinser S, Hentschke H, Antkowiak B: Diazepam decreases action potential firing of neocortical neurons via two distinct mechanisms. Anesth Analg 2010; 111:1394–9 [DOI] [PubMed] [Google Scholar]
- 17.Olsen RW: GABAA receptor: Positive and negative allosteric modulators. Neuropharmacology 2018; 136(Pt A):10–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Husain SS, Ziebell MR, Ruesch D, Hong F, Arevalo E, Kosterlitz JA, Olsen RW, Forman SA, Cohen JB, Miller KW: 2-(3-Methyl-3H-diaziren-3-yl)ethyl 1-(1-phenylethyl)-1H-imidazole-5-carboxylate: A derivative of the stereoselective general anesthetic etomidate for photolabeling ligand-gated ion channels. J Med Chem 2003; 46:1257–65 [DOI] [PubMed] [Google Scholar]
- 19.Savechenkov PY, Zhang X, Chiara DC, Stewart DS, Ge R, Zhou X, Raines DE, Cohen JB, Forman SA, Miller KW, Bruzik KS: Allyl m-trifluoromethyldiazirine mephobarbital: An unusually potent enantioselective and photoreactive barbiturate general anesthetic. J Med Chem 2012; 55:6554–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGrath M, Yu Z, Jayakar SS, Ma C, Tolia M, Zhou X, Miller KW, Cohen JB, Raines DE: Etomidate and etomidate analog binding and positive modulation of γ-aminobutyric acid type A receptors: Evidence for a state-dependent cutoff effect. Anesthesiology 2018; 129:959–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dostalova Z, Zhou X, Liu A, Zhang X, Zhang Y, Desai R, Forman SA, Miller KW: Human α1β3γ2L gamma-aminobutyric acid type A receptors: High-level production and purification in a functional state. Protein Sci 2014; 23:157–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jayakar SS, Zhou X, Chiara DC, Jarava-Barrera C, Savechenkov PY, Bruzik KS, Tortosa M, Miller KW, Cohen JB: Identifying drugs that bind selectively to intersubunit general anesthetic sites in the α1β3γ2 GABAAR transmembrane domain. Mol Pharmacol 2019; 95:615–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang X, Jounaidi Y, Dai JB, Marte-Oquendo F, Halpin ES, Brown LE, Trilles R, Xu W, Daigle R, Yu B, Schaus SE, Porco JA Jr, Forman SA: High-throughput screening in larval zebrafish identifies novel potent sedative-hypnotics. Anesthesiology 2018; 129:459–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Philip BK: Drug reversal: Benzodiazepine receptors and antagonists. J Clin Anesth 1993; 5(6 Suppl 1):46S–51S [DOI] [PubMed] [Google Scholar]
- 25.Reynolds A, Brustein E, Liao M, Mercado A, Babilonia E, Mount DB, Drapeau P: Neurogenic role of the depolarizing chloride gradient revealed by global overexpression of KCC2 from the onset of development. J Neurosci 2008; 28:1588–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang RW, Wei HP, Xia YM, Du JL: Development of light response and GABAergic excitation-to-inhibition switch in zebrafish retinal ganglion cells. J Physiol 2010; 588(Pt 14):2557–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lodge D, Anis NA, Burton NR: Effects of optical isomers of ketamine on excitation of cat and rat spinal neurones by amino acids and acetylcholine. Neurosci Lett 1982; 29:281–6 [DOI] [PubMed] [Google Scholar]
- 28.Anis NA, Berry SC, Burton NR, Lodge D: The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br J Pharmacol 1983; 79:565–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Solt K, Forman SA: Correlating the clinical actions and molecular mechanisms of general anesthetics. Curr Opin Anaesthesiol 2007; 20:300–6 [DOI] [PubMed] [Google Scholar]
- 30.Fahrenbach VS, Bertaccini EJ: Insights into receptor-based anesthetic pharmacophores and anesthetic-protein interactions. Methods Enzymol 2018; 602:77–95 [DOI] [PubMed] [Google Scholar]
- 31.Solt K, Cotten JF, Cimenser A, Wong KF, Chemali JJ, Brown EN: Methylphenidate actively induces emergence from general anesthesia. Anesthesiology 2011; 115:791–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kenny JD, Taylor NE, Brown EN, Solt K: Dextroamphetamine (but not atomoxetine) induces reanimation from general anesthesia: Implications for the roles of dopamine and norepinephrine in active emergence. PLoS One 2015; 10:e0131914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma C, Pejo E, McGrath M, Jayakar SS, Zhou X, Miller KW, Cohen JB, Raines DE: Competitive antagonism of anesthetic action at the γ-aminobutyric acid type A receptor by a novel etomidate analog with low intrinsic efficacy. Anesthesiology 2017; 127:824–37 [DOI] [PMC free article] [PubMed] [Google Scholar]