

Summary
Circulating microvesicles (MVs) from patients with systemic lupus erythematosus (SLE) express the type 1 interferon (IFN)‐inducible protein galectin‐3 binding protein (G3BP), which may enhance their deposition in the glomerular basement membrane. The release of G3BP‐expressing MVs from normal peripheral blood mononuclear cells (PBMCs) is induced by Toll‐like receptor 9 (TLR‐9) ligands, and these vesicles contain autoantibody‐accessible double‐stranded DNA (dsDNA). This study compares the release of MVs expressing G3BP and dsDNA from PBMCs derived from SLE patients with or without active lupus nephritis (LN) and from healthy donors, and taps further into the potential dependency on IFN‐α for their generation and impacts of TLR‐7/TLR‐9 co‐stimulation. PBMCs from 10 healthy donors and 12 SLE patients, six of whom had active LN at study inclusion, were stimulated in‐vitro with recombinant human IFN‐α and the TLR‐9 agonists oligodeoxynucleotide (ODN)2216 or ODN2395 alone or in combination with the TLR‐7 agonist gardiquimod. MVs in the supernatants were subsequently isolated by differential centrifugation and their expression of G3BP and dsDNA was quantified by flow cytometry. Stimulation with ODN2395 significantly increased the release of MVs co‐expressing G3BP and dsDNA from PBMCs isolated from healthy donors and SLE patients. The expression of G3BP on individual MVs and the proportion of G3BP and dsDNA double‐positive MVs released were increased in active LN patients. Neither co‐stimulation with gardiquimod nor with the IFN‐α inhibitor IN‐1 had any effect on the MV release induced by ODN2395. In conclusion, the TLR‐9‐mediated inducibility of MVs co‐expressing G3BP and dsDNA is increased in SLE patients with active LN.
Keywords: G3BP, microvesicles, PBMCs, SLE, TLR‐9
Toll‐like receptor‐9 ligation induces the release of microvesicles (MVs) embroidered with the type 1 interferon‐inducible protein galectin‐3 binding protein (G3BP) and double‐stranded DNA that may become targeted with anti‐DNA autoantibodies. Deposition of such MVs in kidney glomeruli expressing G3BP ligands, may cause lupus nephritis.
Introduction
Microvesicles (MVs) are plasma membrane‐derived vesicles with enclosed cytoplasm that range in size from 0·1 to 1 µm and are shed from most cell types constitutively and in response to activation or during apoptosis [1, 2, 3]. Having previously been regarded as inert cell debris [4], evidence now suggests that MVs contain an orchestrated cargo of proteins and nucleic acids involved in inflammation, coagulation and several other physiological processes [3, 5]. In addition to being potential biomarkers, MVs may also be pathogenic players in systemic autoimmune diseases, such as systemic lupus erythematosus (SLE) [6, 7]. Accordingly, the MV‐profile of SLE patients deviates markedly from that of healthy subjects; proteomics show that circulating MVs from SLE patients have an increased content of galectin‐3 binding protein (G3BP), immunoglobulin (Ig)G [8] and several other proteins [9, 10]. These findings have been supplemented by flow cytometric analyses showing increased circulating levels of MVs with surface‐bound IgG [11, 12, 13] or G3BP [14]. MVs containing apoptosis‐modified chromatin are abundant in the blood of SLE patients [15].
The expression of double‐stranded DNA (dsDNA) on the surface renders MVs targets for anti‐dsDNA antibodies in SLE [12], which may implicate the MVs in loss of self‐tolerance and the proinflammatory responses driving lupus disease [15, 16]. A role for vesicular DNA in murine lupus has been indicated, i.e. by the finding that mice deficient in DNase 1L3 develop features characteristic of SLE [17]. In addition, G3BP‐expressing MVs are likely to be glomerulophilic by virtue of G3BP’s affinity for multiple factors comprising the glomerular basement membrane, including nidogen, collagen IV, fibronectin and galectin‐3, which is up‐regulated in patients with lupus nephritis (LN) [18, 19]. Hence, G3BP carried on the surface of MVs may facilitate IgG accumulation in the glomeruli, and glomerular co‐localization of G3BP and IgG was recently demonstrated in LN patients by electron microscopy [14]. Despite their etiopathogenic potential in SLE, G3BP‐expressing MVs remain largely undescribed.
We recently identified Toll‐like receptor 9 (TLR‐9) ligation as a stimulus for release of G3BP‐expressing MVs from peripheral blood mononuclear cells (PBMCs) from healthy donors [20]. The majority of these in‐vitro‐generated vesicles contained dsDNA that proved accessible to an anti‐dsDNA antibody. Moreover, their release depended, at least in part, on interferon (IFN)‐α secretion, compatible with the type I IFN‐inducible nature of G3BP [21] and the increased IFN‐α activity frequently found in SLE patients [21, 22, 23, 24, 25].
TLR‐9 signaling has been linked to the pathogenesis of SLE [26]. The binding of DNA‐rich immune complexes (ICs) to TLR‐9 elicits secretion of IFN‐α from plasmacytoid dendritic cells (pDCs) [27, 28, 29], and synthetic TLR‐9 agonists have been shown to induce the production of anti‐dsDNA antibodies by B cells from SLE patients [30]. Furthermore, high TLR‐9 expression is associated with worsened SLE prognosis [31], and the expression is increased in PBMCs from patients with active LN compared to patients without active LN and healthy individuals [32]. Several murine studies demonstrate that administration of TLR‐9 ligands in lupus‐prone mice triggers or exacerbates renal disease [33, 34, 35, 36]. In these studies, animals injected with cytosine–phosphate–guanosine (CpG) oligodeoxynucleotide (ODN) displayed aggravated IC‐induced glomerulonephritis, severe proteinuria, and elevated serum autoantibodies. Conversely, treatment with TLR‐9‐antagonists reduced serum anti‐dsDNA antibodies, IFN‐α levels and glomerular IgG deposits [37, 38]. Paradoxically, some studies of TLR‐9 knock‐out mice show attenuated renal disease [39, 40], while others show exacerbated renal disease [8, 41, 42, 43, 44, 45, 46, 47, 48].
In the present study, we compare the TLR‐9‐induced MV release by PBMCs derived from healthy blood donors and SLE patients with or without active LN at study inclusion. We further examine the effects of co‐stimulation through TLR‐7 and TLR‐9 and the role of IFN‐α in the TLR‐9‐mediated MV release from SLE PBMCs.
Materials and methods
SLE patients and healthy donors
SLE patients were included from both the inpatient and outpatient rheumatology clinic at Copenhagen University Hospital, Rigshospitalet, and fulfilled the American College of Rheumatology classification criteria for SLE [49]. Patients with histologically confirmed LN and increasing or persistent proteinuria of > 0·5 g/day were considered to have active LN. Blood from anonymous healthy donors was obtained from the Blood Bank at Copenhagen University Hospital, Rigshospitalet. Four of the healthy donors were also included in a previous study [20]. Samples from patients and healthy donors were analyzed in parallel and were sex‐ and age‐matched. The study was approved by the Scientific–Ethical Committee of the Capital Region of Denmark (protocol no. H‐15004075).
Isolation of PBMCs
Blood was collected in ethylenediamine tetraacetic acid (EDTA)‐containing Vacutainer® tubes (Greiner Bio‐one GmbH, Kremsmünster, Austria), and centrifuged at 1800 g for 10 min at 21°C. The plasma was aspirated and discarded, and the PBMCs were poured onto a density gradient medium (LymphoprepTM; Alere Technologies, Oslo, Norway), centrifuged at 1172 g for 30 min at 24°C, washed twice in sterile phosphate‐buffered saline (PBS) (Thermo Fisher Scientific, Waltham, MA, USA), and finally resuspended in sterile medium consisting of RPMI‐1640 GlutaMAX medium (Lonza, Basel, Switzerland) supplemented with 20% heat‐inactivated fetal calf serum (hFCS) (Sigma‐Aldrich, St Louis, MO, USA) and 0·1% gentamicin (BI, Kibbutz Beit Haemek, Israel) (referred to as incubation medium). PBMCs were subsequently counted using the NucleoCounter® NC‐100TM system (ChemoMetec, Allerød, Denmark), according to the manufacturer’s instructions. The purified PBMCs were then divided into 500 µl aliquots in cryotubes (Dacos, Esbjerg, Denmark), followed by addition of 500 µl sterile medium, yielding a final concentration of 25% hFCS and 10% dimethyl sulfoxide (DMSO) (Merck kGaA, Darmstadt, Germany). The cryotubes were inverted, placed in CoolCell® freezing containers (BioCision, San Rafael, CA, USA) and stored at −80°C for at least 24 h before they were cryopreserved.
Stimulation of PBMCs
PBMCs were stimulated as previously described [20]. In brief, the cryopreserved PBMCs were thawed at room temperature (RT), washed and resuspended in incubation medium. The hFCS used in the incubation medium was filtered through a 0·2 µm filter (Sartorius, Göttingen, Germany). Cell viability was confirmed using the NucleoCounter® NC‐100TM system (ChemoMetec), according to the manufacturer’s instructions. The cells were plated into 48‐well plates with Nunclon® delta surface (Nunc, Roskilde, Denmark) at approximately 600 000 PBMCs per well and rested for 30 min at 37°C and 5% CO2 before incubation for 24 h at 37°C and 5% CO2 with the following components or combinations hereof: the TLR‐7 agonist gardiquimod (Invivogen, San Diego, CA, USA, 1·5 µg/ml); the TLR‐9 agonist ODN2395 (Invivogen, 6 µg/ml); the TLR‐9 agonist ODN2216 (Invivogen, 6 µg/ml) [50]; recombinant human (rh)IFN‐α (Sigma‐Aldrich), 2 µg/ml, ~200 000 U/ml); and the inhibitor of interaction between IFN‐α and the IFN‐α receptor (IFNAR) IFN‐α‐IFNAR‐IN‐1 hydrochloride (IN‐1) (MedchemExpress, Sollentuna, Sweden, 32 µM) [51].
Preparation of culture supernatants
After incubation with stimuli the plates were left at RT for 15 min. Adhered cells were gently loosened and transferred to fluorescence activator cell sorter (FACS) tubes (Corning, New York City, NY, USA). The cell suspensions were centrifuged at 458 g for 10 min at 24°C to pellet PBMCs. The cell‐free supernatants were then harvested, aliquoted into cryotubes and snap‐frozen in liquid nitrogen. Samples were stored at −80°C until analysis.
Staining of PBMCs
After collection of the supernatants, PBMCs were resuspended in 100 µl PBS followed by incubation with 100 µl tetramethylrhodamine ethyl ester (TMRE) solution (Abcam, Cambridge, UK) (100 nM in PBS) for 15 min at 37°C, 5% CO2, washed in PBS containing 2% hFCS (referred to as incubation buffer) and centrifuged at 458 g for 10 min at 24°C. The supernatant was aspirated, and PBMCs were resuspended in 100 µl incubation buffer. Fcγ‐receptors were blocked by addition of 5 µl commercial Fc blocker [Becton Dickinson (BD), Franklin Lakes, NJ, USA] to each FACS tube for 15 min at RT in the dark. Antibody cocktails were prepared containing BV421‐conjugated anti‐human CD3 (BD), peridinin chlorophyll‐cyanin 5·5 (PerCP‐Cy5·5)‐conjugated anti‐human CD19 (BD), allophycocyanin (APC)‐H7‐conjugated anti‐human CD69 (BD) and phycoerythrin‐cyanin 7 (PE‐Cy7)‐conjugated anti‐human CD80 (BD) antibodies, all diluted 1 : 55 in brilliant stain buffer (BD). The cocktails were added to PBMCs to a final dilution of 1 : 110 and the mixtures were incubated for 30 min at RT in the dark, followed by wash in incubation buffer. The cells were resuspended in incubation buffer and left on ice for 5 min before acquisition on a FACSCanto II flow cytometer (BD). TMRE stains cells with active mitochondria, and consequently cells negative for this dye were considered dead/apoptotic and were excluded from analysis.
Isolation of MVs from culture supernatants
The frozen cell‐free supernatants were thawed at RT and transferred to Eppendorf tubes (Corning) (200 µl in each) and centrifuged at 20 000 g for 30 min at 21°C to pellet MVs. The supernatant was subsequently aspired down to 25 µl and discarded, and MVs were resuspended in 175 µl PBS that had been filtered through a 0·2 µm filter (Sartorius, Göttingen, Germany), followed by another ultracentrifugation step. The supernatant was aspirated as before and discarded, and MVs were resuspended in 70 µl filtered PBS to a final volume of 95 µl (MV‐isolate).
Detection and staining of MVs
Calcein was used as a fluorescent marker of MVs and was added to the MV isolates in the form of calcein‐AM; the reliable use of this compound for the general labeling of intact extracellular vesicles has been described previously [52]. Fcγ‐receptors on MVs were blocked by adding 5 µl commercial Fc blocker (BD) to MV‐isolates for 15 min at RT. After this incubation, 5 µl MV‐isolate was pipetted into 22·5‐µl filtered PBS in FACS tubes followed by addition of 5 µl mouse anti‐human G3BP antibody of IgG2b isotype (Proteintech, Manchester, UK) (1 µg/ml in filtered PBS) or an isotype control (Biolegend, San Diego, CA, USA), 5 µl mouse anti‐dsDNA antibody of IgG2a isotype (SSI, Copenhagen, Denmark) (0·5 µg/ml in filtered PBS) or an isotype control (Biolegend). Next, 5 µl APC‐conjugated goat anti‐mouse IgG2b antibody (Southern Biotech, Birmingham, AL, USA) (0·5 µg/ml in filtered PBS), 5 µl BV510‐conjugated rat anti‐mouse IgG2a antibody (BD) (0·5 µg/ml in filtered PBS) and 2·5 µl calcein‐AM (Sigma‐Aldrich) (2·5 µg/ml in filtered PBS) were added, yielding a final volume of 50 µl. Unstained and single‐stained controls were included. The tubes were incubated for 1 h at RT in the dark. After incubation, 100 µl TruCount beads (BD) were added to the tubes and the volume was adjusted to 300 µl with filtered PBS before acquisition on a FACSCanto II flow cytometer (BD) in logarithmic mode, at low flow‐rate, and with lowest SSC threshold (= 200). The TruCount bead solution was prepared by dissolving the lyophilized beads in 500 µl filtered PBS. Each sample was run for 4 min or until a minimum of 1000 TruCount bead events were recorded.
Quantification and size determination of MVs
The absolute count of MVs (MVs/µl) was calculated with TruCount beads as reference by applying the formula:
{[(no. of MV events within gates of interest)/(no. of collected bead events)] × [(total no. of beads)/(test volume)]] × (dilution factor)}.
The MV gate was defined using Megamix‐Plus SSC beads (Biocytex, Marseille, France), as outlined in our previous study [20].
Statistical analysis
Wilcoxon’s rank‐sum test and Wilcoxon’s signed‐rank test (two‐tailed) were used for two‐group comparisons of unpaired and paired continuous data, respectively. Fisher’s exact test (two‐tailed) was applied for two‐group comparisons of categorical data. The tests were carried out using GraphPad Prism software version 8 (GraphPad Software Inc, San Diego, CA, USA). Spearman’s correlations and linear regression analyses (ordinary least squares) were performed using R software version 3.4.3 (R Foundation for Statistical Computing, Vienna, Austria). The validity of the models with regard to linearity, homoscedasticity and normality of error terms was assessed from ‘residuals versus fitted plots’ and ‘normal Q–Q plots of standardized residuals. P‐values less than 0·05 were considered statistically significant.
Results
This study included 12 SLE patients, six of whom had active LN at study inclusion, and 10 healthy donors. Their clinical and demographical characteristics are shown in Table 1. Patients with active LN were confirmed by renal biopsy; class I (n = 1), class II (n = 1), class III (n = 1), class IV (n = 2) and class IV + V (n = 1). The groups did not differ significantly with regard to sex, age or disease duration nor with regard to leukocyte counts and lymphocyte counts (hematology was only available for SLE patients). Three SLE patients were considered lymphopenic (< 1000/µl) at study inclusion, two of whom had active LN. Notably, the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) scores of patients with active LN were significantly higher than those of patients without active LN at study inclusion (median SLEDAI scores of 8 and 4 for patients with and without active LN, respectively) (P = 0·01). Moreover, both prednisolone treatment and positivity for anti‐dsDNA antibodies was more frequent in the group with active LN (83 and 33% for patients with and without active LN, respectively), but these differences were not statistically significant (P = 0.24).
Table 1.
Characteristics of SLE patients and HDs
| All SLE | Active LN | Not active LN | HD | |
|---|---|---|---|---|
| n = 12 | n = 6 | n = 6 | n = 10 | |
| Sex (female), % (n) | 91·7 (11) | 100 (6) | 83·3 (5) | 90 (9) |
| Age (years), median (IQR) | 32 (23·5–44·8) | 34 (26·8–51·8) | 28 (19–46·8) | 28 (24·5–37) |
| Disease duration (years), median (IQR) | 6 (2–16) | 6 (1–18·5) | 6 (2–19) | – |
| Immunosuppressive medication, a % (n) | 75 (9) | 83·3 (5) | 66·7 (4) | – |
| Prednisolone | 58·3 (7) | 83·3 (5) | 33·3 (2) | – |
| Dose (mg), median (range) | 30 (2–75) | 30 (2–75) | 20 (5–35) | – |
| Hydroxychloroquine | 58·3 (7) | 66·7 (4) | 50 (3) | – |
| Mycophenolate mofetil | 25 (3) | 33·3 (2) | 16·7 (1) | – |
| Cyclophosphamide | 16·7 (1) | 16·7 (1) | 0 (0) | – |
| Disease manifestations, a % (n) | ||||
| Nephritis | 50 (6) | 100 (6) | 0 (0) | – |
| Arthritis | 25 (3) | 16·7 (1) | 33·3 (2) | – |
| Pleuritis | 16·7 (2) | 33·3 (2) | 0 (0) | – |
| CNS | 8·3 (1) | 16·7 (1) | 0 (0) | – |
| APS, thrombotic | 25 (3) | 16·7 (1) | 33·3 (2) | – |
| Low C3/C4 | 66·7 (8) | 100 (6) | 33·3 (2) | – |
| Previous LN, % (n) | 58·3 (7) | 100 (6) | 16·7 (1) | – |
| Leukocytes (103/µl), median (IQR) | 6·5 (4·65–12·2) | 7·9 (4·4–14·95) | 5·7 (3·98–8·7) | – |
| Lymphocytes (103/µl), median (IQR) | 1·5 (0·95–2·38) | 1·7 (0·8–2·5) | 1·5 (1·18–1·98) | – |
| Lymphopenia (< 1000/µl), % (n) | 25 (3) | 33·3 (2) | 16·7 (1) | |
| Anti‐dsDNA antibody positivity, % (n) | 58·3 (7) | 83·3 (5) | 33·3 (2) | – |
| Titer, median (IQR) b | 111 (6·3–380) | 380 (112–380) | 8·1 (0·9–111) | – |
| SLEDAI score, median (IQR) | 6 (4–8) | 8 (6–20) | 4 (0–6·25) | – |
IQR = interquartile range; HD = healthy donors; SLE = systemic lupus erythematosus; LN = lupus nephritis; SLEDAI = Systemic Lupus Erythematosus Disease Activity Index; CNS = central nervous system.
At study inclusion;
n = 10 (five with active LN).
TLR‐9‐mediated release of G3BP‐ and dsDNA‐expressing MVs from mononuclear cells
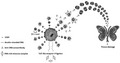
Stimulation of cells from healthy donors and active LN patients with the TLR‐9 agonist ODN2395 caused minor increases in the overall content of MVs in the supernatants (Fig. 1a), but a marked increase in the release of G3BP and dsDNA double‐positive MVs was observed (Fig. 1b), and this was reflected by the number of G3BP‐expressing MVs in toto (Fig. 1c).
Fig. 1.
Release of galectin‐3 binding protein (G3BP)‐ and dsDNA‐expressing microvesicles (MVs) from Toll‐like receptor (TLR)‐9‐stimulated mononuclear cells. Peripheral blood mononuclear cells (PBMCs) from 10 healthy donors (HD; white columns), six systemic lupus erythematosus (SLE) patients without active lupus nephritis (LN) (not active LN; gray columns) and six SLE patients with active LN (active LN; black columns) were left non‐stimulated (NS) or stimulated with the TLR‐9 agonist oligodeoxynucleotide (ODN)2395 for 24 h. MVs released into the culture supernatants were subsequently isolated by ultracentrifugation, quantified and characterized with respect to expression of G3BP and dsDNA by flow cytometry. Columns and error bars represent median values and interquartile range. (a) The total concentration of MVs in culture supernatants. (b) The concentration of G3BP and dsDNA double‐positive MVs and (c) the G3BP‐expressing MV population in toto with (d) corresponding G3BP‐derived median fluorescence intensity (MFI) values. (e) The concentration of the dsDNA‐expressing MV population in toto with (f) corresponding dsDNA‐derived MFI values. Asterisks above horizontal lines represent P‐values for comparisons between non‐stimulated and stimulated samples, whereas values above brackets are P‐values for comparisons between groups. *P < 0·05; **P < 0·01.
In non‐stimulated cultures from all three groups, the median proportion of MVs expressing G3BP was less than 6%, which increased to approximately 40% in the groups of healthy donors and patients without active LN and to approximately 60% in the group of patients with active LN (Table 2). Furthermore, patients with active LN showed higher G3BP expression per MV than the two other groups (Fig. 1d). The differences were even more pronounced for G3BP and dsDNA double‐positive MVs, with median G3BP median fluorescence intensity (MFI)‐values of 182, 145 and 302 for MVs from healthy donors, patients without active LN and patients with active LN, respectively (active LN versus healthy: P = 0·016 and active LN versus not active LN: P = 0·0087; data not shown).
Table 2.
Proportions of G3BP‐ and dsDNA‐expressing MV populations in individual cell cultures
| 1 | 2 | 3 | P‐values a | |||
|---|---|---|---|---|---|---|
| HD (n = 10) | Not active LN (n = 6) | Active LN (n = 6) | 1 versus 2 | 1 versus 3 | 2 versus 3 | |
| MV populations, % of total MVs | ||||||
| Non‐stimulated samples | ||||||
| G3BP‐expressing MVs in toto | 4 | 4 | 6 | 0·96 | 0·43 | 0·82 |
| dsDNA‐expressing MVs in toto | 32 | 32 | 35 | 0·88 | 0·49 | 0·70 |
| G3BP and dsDNA double‐positive MVs | 3 | 2 | 4 | 0·49 | 0·43 | 0·39 |
| ODN2395‐stimulated samples | ||||||
| G3BP‐expressing MVs in toto | 40** | 40* | 58* | 0·96 | 0·001 | 0·026 |
| dsDNA‐expressing MVs in toto | 29 | 25* | 37 | 0·18 | 0·06 | 0·07 |
| G3BP and dsDNA double‐positive MVs | 25* | 21* | 36* | 0·31 | 0·008 | 0·07 |
Median proportions of MVs bearing G3BP and dsDNA released from PBMCs from healthy donors (group 1; HD), patients without ongoing LN (group 2; not active LN), and patients with ongoing LN (group 3; active LN).
G3BP = galectin‐3 binding protein; MV = microvesicle; PBMC = peripheral blood mononuclear cells; HD = healthy donors; ODN = oligodeoxynucleotide; LN = lupus nephritis.
P‐values for comparisons between groups (1–3) with significant P‐values marked in bold type.
P < 0·05 and
P < 0·01 relate to comparisons between stimulated and non‐stimulated samples.
To further address whether these differences relate to active LN specifically, or to disease activity in general, linear regressions were performed with the proportions of G3BP‐expressing MVs in toto as dependent variable (DV) and with LN and SLEDAI scores as independent variables (Table 3a; models 1–3). The group size (n = 12) was considered too small for multivariable analyses. Nevertheless, in these simple models, active LN associated positively with the proportions of G3BP‐expressing MVs in toto (MV percentage change in the presence of LN) (B = 19·3, P = 0·02, model 1), whereas no significant relationship was found between SLEDAI scores and this DV (MV percentage change per SLEDAI score unit increment) (B = 0·4, P = 0·53, model 2). Moreover, excluding renal manifestations from the SLEDAI (i.e. subtracting four points for each patient with active LN) resulted in even higher P‐values and further weakened the beta coefficients (model 3). Similar pictures were seen with the corresponding G3BP MFI‐values as DV (Table 3b, models 1–3). These findings support that disease activity, in general, is not confounding the association between active LN and G3BP‐expressing MVs and that the differences are related directly to renal disease. In the same manner, we also examined potential connections between the titers of anti‐dsDNA antibodies measured in patients and the inducibility of these vesicles (Table 3a,b, model 4). From these analyses we found that the antibody titers were positively associated with the G3BP expression by individual MVs but did not associate significantly with the proportions of G3BP‐expressing MVs in toto (P = 0·04 and 0·11, respectively).
Table 3.
Linear regression analyses of galectin‐3 binding protein‐expressing microvesicles by clinical and serological features of SLE
| Independent variables | Coefficients (B) | 95% CI | P‐values | |
|---|---|---|---|---|
| G3BP‐expressing MVs in % of total MV count | ||||
| LN (active) | 19·3 | 4·5 to 34·2 | 0·02 | Model 1 |
| SLEDAI score | 0·4 | −0·1 to 1·8 | 0·53 | Model 2 |
| SLEDAI score (excluding LN) | 0·02 | −1·6 to 1·7 | 0·98 | Model 3 |
| Anti‐dsDNA antibodies (titer) a | 0·05 | −0·01 to 0·12 | 0·11 | Model 4 |
| MFI of G3BP‐expressing MVs | ||||
| LN (active) | 69·3 | 16·6 to 122 | 0·02 | Model 1 |
| SLEDAI score | 2·4 | −2·4 to 7·2 | 0·3 | Model 2 |
| SLEDAI score (excluding LN) | 1·3 | −4·5 to 7·2 | 0·62 | Model 3 |
| Anti‐dsDNA antibodies (titer) a | 0·2 | 0·01 to 0·41 | 0·04 | Model 4 |
Simple linear regressions were performed for SLE patients (n = 12), with (a) proportions of G3BP‐expressing MVs in toto and (b) the corresponding G3BP MFIs as dependent variables (after ODN2395‐stimulation), and with LN, SLEDAI scores and anti‐dsDNA antibody titers as independent variables (models 1–4). Model 3 in both (a) and (b) depicts the regression coefficients and P‐values for SLEDAI scores when excluding renal manifestations from the index (i.e. subtracting four points for each patient with active LN). Significant P‐values are marked in bold type.
SLE = systemic lupus erythematosus; MV = microvesicle; G3BP = galectin‐3 binding protein; MFI = median fluorescence intensity; ODN = oligodeoxynucleotide; LN = lupus nephritis; SLEDAI = Systemic Lupus Erythematosus Disease Activity Index.
n = 10 (five with active LN).
In contrast to the findings for G3BP, the proportion of MVs bearing dsDNA on the surface was approximately one‐third before stimulation (Table 2), and stimulation with ODN2395 did not affect the total number of dsDNA‐expressing MVs (Fig. 1e), nor did it affect the proportion of MVs bearing dsDNA (Table 2). The majority of the dsDNA‐expressing MVs also carried G3BP, but not vice versa. Without stimulation, MVs in cultures from healthy donors expressed higher amounts of dsDNA than MVs from either patient group, but this difference was not observed in cultures stimulated with ODN2395 (Fig. 1f).
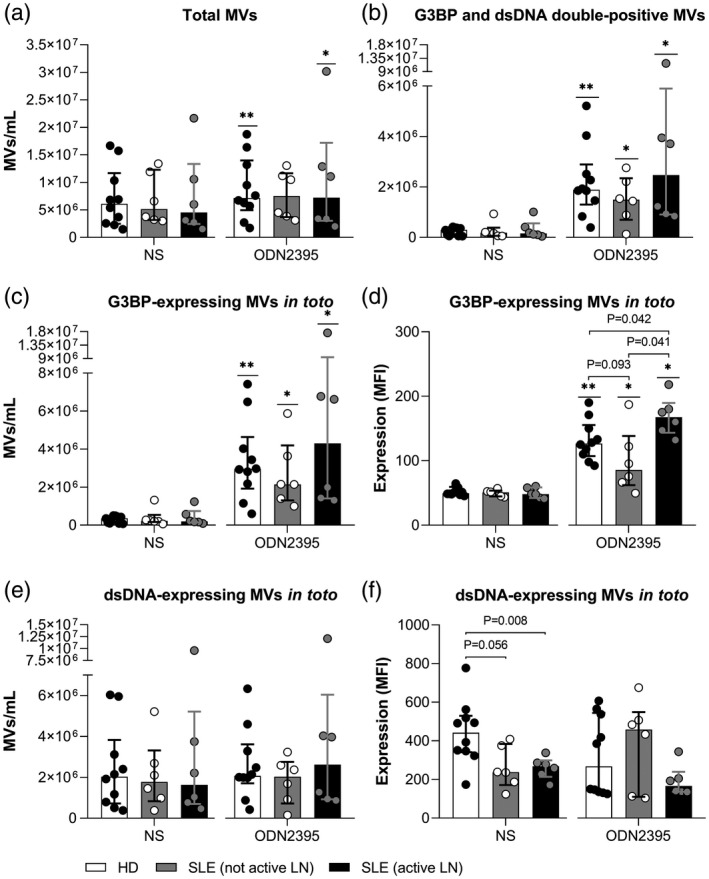
The specificity of the signals derived from the anti‐G3BP and anti‐dsDNA antibodies was validated with isotype controls (Fig. 2) and DNase treatment (not shown).
Fig. 2.
Validation of signal‐specificity. Representative fluorescence scatter‐plots from a healthy donor, showing the staining of microvesicles (MVs) from non‐stimulated (NS) and oligodeoxynucleotide (ODN)2395‐stimulated peripheral blood mononuclear cells (PBMCs) with calcein, as a general MV marker, and with (a) anti‐G3BP and anti‐dsDNA antibodies or (b) corresponding isotype controls.
The fraction of dead/apoptotic B cells was generally decreased in the ODN2395‐stimulated cell cultures but was significantly higher in the active LN group relative to the other two groups after 24 h incubation with this TLR‐9 agonist (data not shown). However, using Spearman’s rho for both healthy donors and SLE patients (non‐stratified), the fraction of dead/apoptotic B cells or T cells did not correlate with the proportions of dsDNA‐expressing MVs in toto or G3BP and dsDNA double‐positive MVs, nor with the amount of dsDNA and G3BP on individual MVs (data not shown). Thus, the proportional differences of dead/apoptotic lymphocytes between the groups are unlikely to explain our findings.
TLR‐9‐mediated expression of CD69 and CD80 on B cells
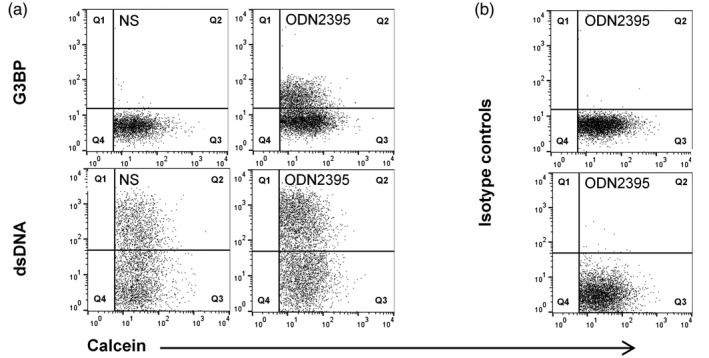
In humans, the cells responsive to the ODN2395 stimulus used in the experiments described above are mainly B cells and pDCs [53, 54]. In particular, B cells frequently show aberrant responsiveness to TLR‐9 agonists in SLE [55, 56, 57]. The B cell and the T cell responses to TLR‐9 stimulation were therefore compared between healthy donors and patients with and without ongoing LN using CD69 and CD80 as activation markers (Fig. 3). Circulating pDCs were considered too sparse to be analyzed in this set‐up [58].
Fig. 3.
Effect of Toll‐like receptor (TLR)‐9 stimulation on the expression of CD69 and CD80 by B cells and T cells. Peripheral blood mononuclear cells (PBMCs) from 10 healthy donors (HD; white columns), six systemic lupus erythematosus (SLE) patients without active lupus nephritis (LN) (not active LN; grey columns) and six SLE patients with active LN (black columns; active LN) were stimulated with the TLR‐9 agonist oligodeoxynucleotide (ODN)2395 or left non‐stimulated (NS) for 24 h, and the expression of the activation markers (a,b) CD69 and (c) CD80 by CD19‐positive B cells and CD3‐positive T cells were measured by flow cytometry (CD19 expression was undetectable on B cells from one SLE patient without active LN). (d) Comparisons of the T/B cell ratios and the proportions of T cells and B cells among healthy donors and SLE patients with and without active LN in the non‐stimulated cell cultures after 24 h incubation. (e) Representative example of CD3 and CD19 staining of PBMCs from a healthy donor, after exclusion of doublets. (f) Forward‐/side‐scatter (FSC/SSC) plot showing the gates for dead/apoptotic cells, lymphocytes and monocytes. The black events represent CD19 and tetramethylrhodamine ethyl ester (TMRE) double‐positive (viable) cells, backgated from the CD19 gate in (e). (g) Corresponding FSC/SSC plot but with the black events representing CD19‐positive and TMRE‐negative (dead/apoptotic) cells – mainly localizing within the dead/apoptotic gate. Columns and error bars represent median values and interquartile range. Asterisks above horizontal lines represent P‐values for comparisons between non‐stimulated and stimulated samples, whereas values above brackets are P‐values for comparisons between groups. (*)P < 0·1; *P < 0·05; **P < 0·01.
As expected, T cells were non‐responsive to CpG ODN (Fig. 3a). Conversely, B cells from patients with active LN and B cells from healthy donors significantly up‐regulated the expression of CD69 after stimulation with ODN2395, while the expression of this activation marker was significantly lower on B cells from patients without active LN (Fig. 3b). Similarly, ODN2395 induced a significant or borderline‐significant up‐regulation of CD80 on B cells from healthy donors and on B cells from disease‐active LN patients, respectively, but not on B cells from SLE patients without active LN (Fig. 3c). In a simple linear regression analysis involving all 12 SLE patients, this lower responsiveness seemed unlinked to treatment with hydroxychloroquine, which is known to inhibit TLR‐9 signalling [59], at study inclusion (data not shown). Similarly, stratifying patients into those with and without active LN did not reveal any notable intragroup differences in the induced MFIs for CD69/CD80 between individuals receiving and not receiving hydroxychloroquine (data not shown).
Importantly, no marked intergroup differences were observed in the T/B cell ratios or in the proportions of T and B cells in the non‐stimulated cell cultures (Fig. 3d), favoring that variations in the cellular responses as well as the MV responses among groups are unrelated to baseline deviations in the lymphocytic cell distribution.
Effect of TLR‐7 and TLR‐9 co‐stimulation on the release of MVs from TLR‐stimulated mononuclear cells
Previous studies have shown that TLR‐7 ligands interfere with TLR‐9‐mediated chemokine and cytokine production, including that of IFN‐α [60, 61]. We therefore wanted to investigate whether such interference also extends to the TLR‐9‐induced MV response.
Co‐stimulation of PBMCs through TLR‐7 with gardiquimod had no effect on the TLR‐9‐induced release of G3BP‐expressing MVs in toto or of G3BP and dsDNA double‐positive MVs specifically (Fig. 4a,b); nor did it affect the expression of G3BP or dsDNA by individual MVs (data not shown). Notably, co‐stimulation with gardiquimod did inhibit the ODN2395‐induced up‐regulation of CD69 and CD80 by B cells from healthy donors and tended to inhibit the up‐regulation of CD69 on B cells from SLE patients (Fig. 4c,d).
Fig. 4.
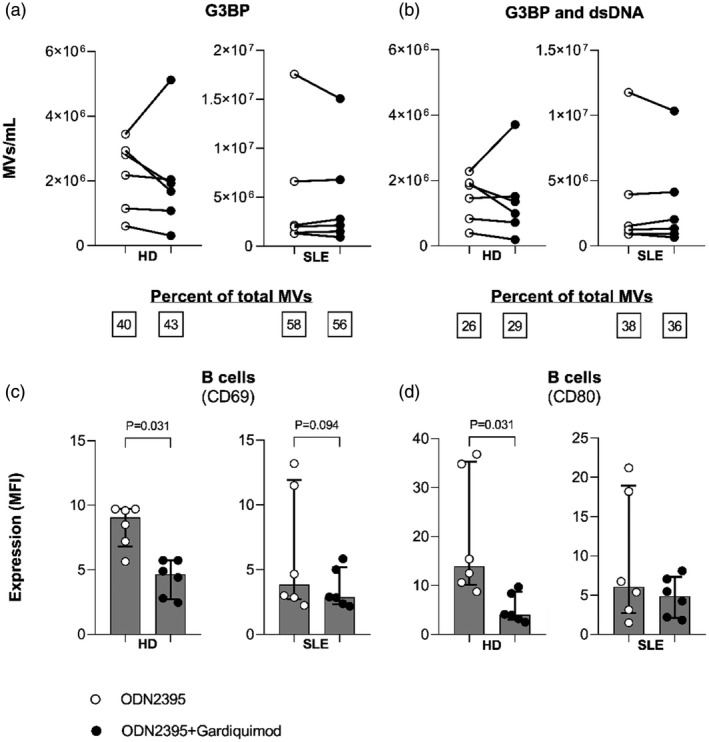
Interplay between Toll‐like receptor (TLR)‐9 and TLR‐7 in induction of microvesicle (MV) release and B cell activation. Peripheral blood mononuclear cells (PBMCs) from six healthy donors (HD) and six systemic lupus erythematosus (SLE) patients [four of whom had active lupus nephritis (LN) at study inclusion] were incubated for 24 h with the TLR‐9 agonist oligodeoxynucleotide (ODN)2395 alone (white events; ODN2395) or in combination with the TLR‐7 agonist gardiquimod (black events; ODN2395+gardiquimod). MVs released into the culture supernatants were subsequently isolated by ultracentrifugation, quantified and characterized with respect to expression of G3BP and dsDNA by flow cytometry. (a) The concentrations of G3BP‐expressing MVs in toto and (b) G3BP and dsDNA double‐positive MVs released from healthy and SLE PBMCs in culture supernatants are shown together with median proportions of MVs bearing these markers (inserted boxes below). (c) Expression of the activation markers CD69 and (d) CD80 by B cells was measured in the corresponding cell fractions by flow cytometry and is shown as median fluorescence intensity (MFI) values. Columns and error bars represent median values and interquartile range.
Role of IFN‐α in the release of MVs from TLR‐stimulated mononuclear cells
Given the prominent role of IFN‐α in the pathogenesis of SLE [62], we examined the influence of this cytokine on the release of G3BP‐expressing MVs from PBMCs derived from SLE patients (Fig. 5). The IFN‐α inhibitor IN‐1 showed no influence on the release of G3BP‐expressing MVs as a whole or G3BP and dsDNA double‐positive MVs (Fig. 5a,b), nor did it affect the expression of G3BP and dsDNA by individual MVs (data not shown).
Fig. 5.
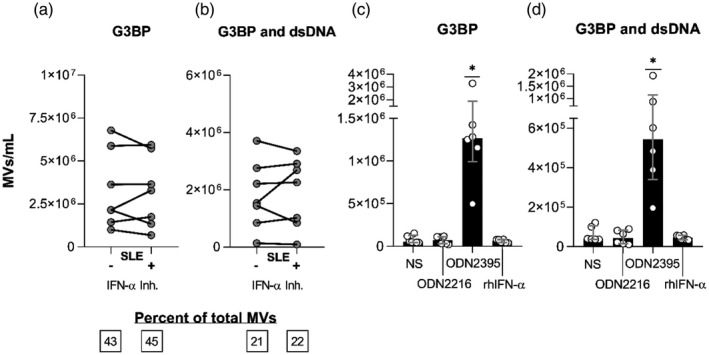
Interferon (IFN)‐α’s role in the Toll‐like receptor (TLR)‐9‐induced release of microvesicles (MVs) from systemic lupus erythematosus (SLE) mononuclear cells. Peripheral blood mononuclear cells (PBMCs) from seven SLE patients [two of whom had active lupus nephritis (LN) at study inclusion] were incubated for 24 h with the TLR‐9 agonist oligodeoxynucleotide (ODN)2395 alone (−) or in combination with the IFN‐α inhibitor IN‐1 (+). MVs released into the culture supernatants were subsequently isolated by ultracentrifugation, quantified and characterized with respect to expression of G3BP and dsDNA by flow cytometry. (a) Concentrations of G3BP‐expressing MVs in toto and (b) G3BP and dsDNA double‐positive MVs in the culture supernatants. Median proportions of MVs bearing these markers are inserted in the boxes below. (c,d) PBMCs from six healthy donors were left non‐stimulated (NS) or were incubated for 24 h with the TLR‐9‐agonist ODN2216, a weak B cell activator and strong IFN‐α inducer; with the TLR‐9 agonist ODN2395, a potent B cell activator and strong IFN‐α inducer; or with recombinant human (rh)IFN‐α. (c) The concentrations of G3BP‐expressing MVs in toto and (d) MVs double‐positive for G3BP and dsDNA in the culture supernatants from non‐stimulated (NS) and stimulated cells. Columns and error bars represent median values and interquartile range. Asterisks above horizontal lines represent P‐values for comparisons between non‐stimulated and stimulated samples. *P < 0·05.
To further assess the dependency on IFN‐α for the TLR‐9‐induced MV response, PBMCs from six healthy donors were stimulated with rhIFN‐α, the TLR‐9 agonists ODN2216 (CpG ODN class A) or with ODN2395 (CpG ODN class C). ODN2216 mainly engages pDCs to secrete large amounts of IFN‐α but only weakly activates B cells, whereas ODN2395 is considered a strong IFN‐α inducer and potent B cell activator [50]. Evidently, ODN2395 but not rhIFN‐α or ODN2216 induced the release of G3BP‐expressing MVs (Fig. 5c,d). Similarly, stimulation of PBMCs from one healthy donor with lower concentrations of rhIFN‐α [0·01 µg/ml (~1000 U/ml) and 0·5 µg/ml (~50 000 U/ml)] had no effect either on the release of these vesicles (data not shown). None of the stimuli affected the release of MVs expressing dsDNA in toto (data not shown).
Discussion
This study confirms our previous findings that stimulation of PBMCs with the class C TLR‐9 agonist ODN2395 induces release of MVs co‐expressing G3BP and dsDNA in healthy donors [20] and extend this finding to also include SLE patients. This is a relevant finding, as co‐expression of G3BP and dsDNA may affect the antigenic and glomerulophilic properties of MVs, and MVs from SLE patients more frequently contain apoptosis‐modified chromatin or G3BP than MVs from healthy donors [9, 10, 12, 14, 15, 16]. However, the class A TLR‐9 agonist ODN2216 failed to induce such MV release. Both TLR‐9 agonists stimulate pDCs, but ODN2395 also potently stimulates B cells [50], suggesting that B cells are involved in the generation of G3BP‐expressing MVs, either as primary source or as critical contributors to the local cytokine environment. We have previously shown that also the class B TLR‐9 agonist ODN2006 fails to induce the release of G3BP‐expressing MVs from PBMCs [20]. These differences may relate to the effects of ODN2395 as both a potent B cell activator and strong IFN‐α inducer [50]. Hence, TLR‐9 ligation in B cells combined with IFN‐α secretion from pDCs may be necessary for such MV release to occur. Moreover, this would also explain the lack of effect when incubating healthy donor PBMCs with rhIFN‐α alone.
Following stimulation with ODN2395, MVs in cell cultures from patients with active LN contained a higher proportion of MVs bearing G3BP (approximately 60%) than MVs in cultures from patients without LN or healthy donors (both approximately 40%), and the patients with LN also showed increased amounts of surface‐bound G3BP per MV. The over‐expression of G3BP in this group may be linked to an increased TLR‐9 expression by mononuclear cells from SLE patients with active disease [30, 63], and particularly from patients with ongoing LN [32]. The expression of TLR‐9 in naive B cells is generally low, but it can be induced by BCR triggering, whereas TLR‐9 is expressed constitutively at high levels in memory B cells [64]. Hence, differences in B cell activity and differentiation between groups may, in part, explain the differences observed in MV release and composition. In our data set, the titers of anti‐dsDNA antibodies measured in patients associated positively with the in‐vitro‐provoked expression of microvesicular G3BP. Potentially, pre‐exposure of cells to ICs rich in dsDNA may accentuate subsequent TLR‐9‐induced MV‐responses, perhaps partly by up‐regulating TLR‐9. However, an increased expression of TLR‐9 does not necessarily correlate with higher responsiveness to TLR‐9 ligands, at least with regard to cytokine production [56].
Another relevant aspect to consider is the potential influence of the high‐mobility group box 1 protein (HMGB1) on the TLR‐9‐induced MV response. HMGB1 facilitates the interaction between TLR‐9 and its ligands and is elevated in serum from SLE patients [65, 66], which may contribute to the higher concentration of MVs expressing G3BP in the circulation of these individuals [14].
Approximately one‐third of MVs expressed dsDNA on their surface in all three groups of subjects, irrespective of whether or not the cell cultures were stimulated. Thus, generation of dsDNA‐expressing MVs is not subject to control by TLR‐9 and is not limited to SLE patients. It appears from our data that the dsDNA‐bearing part of the MVs is largely the same part that acquires G3BP after stimulation of the originator cells with TLR‐9 ligands.
The TLR‐9 agonist, ODN2395, used as stimulus in this study, is primarily a stimulus for B cells and pDCs [53, 54]. Concomitantly with the release of G3BP‐bearing MVs, ODN2395 induced up‐regulation of the activation markers CD69 and CD80 on B cells from patients with active LN and on healthy donor B cells. However, the ODN2395‐induced MV‐release from patients without active LN was not accompanied by such a co‐increase. These data suggest that B cells from patients without active LN were generally less responsive to TLR‐9 stimulation, which was paralleled by lower G3BP expression in the corresponding MV‐fraction compared to the two other groups.
We also addressed the interplay between TLR‐9 and TLR‐7 in the induction of MV release. TLR‐7 ligands have been reported to suppress TLR‐9 signalling and thereby reduce production of chemokines and cytokines, including IFN‐α [60, 61]. Although co‐stimulation with the TLR‐7 agonist gardiquimod down‐regulated the expression of CD69 and CD80 on healthy donor B cells, it did not counteract the TLR‐9‐mediated release of G3BP‐expressing MVs, confirming that this release was not a reflection of cell activation per se. The weaker inhibitory effect on CD69 and CD80, seen for the patient‐derived PBMCs relative to healthy donors, probably results from a markedly lower inductional‐capacity of these activation markers in response to ODN2395. Regrettably, we did not select PBMCs for this specific experiment based on their responsiveness. In fact, cells from four of the six subjects in the SLE group were among the low responders also observed in Fig. 3b,c.
Supplementation of ODN2395‐stimulated PBMCs from SLE patients with IN‐1, which neutralizes secreted IFN‐α, did not affect the release of MVs expressing G3BP from these cells, thus contrasting with our previous findings for healthy donor PBMCs [20]. The patient cells may have already been primed by IFN‐α prior to isolation due to the excess IFN‐α often found in SLE [21, 22, 23, 24, 25]. However, it could also suggest that paracrine or autocrine secretion of IFN‐α is not responsible for release of these MVs, although ODN2395 is a strong IFN‐α inducer [50].
The cellular origin of G3BP‐expressing MVs was not specifically addressed in this study, although the specificity of ODN2395 suggests that B cells may be involved. Efforts should be made to identify general cell surface markers on these vesicles. Moreover, both depletion and enrichment of B cells could further assist to disclose their potential B cell dependency or origin. Other major limitations to our study include the relatively small sample sizes which may cause type II errors and the exclusive use of frozen material. Moreover, it cannot be ruled out that the observed intergroup differences in cell and MV responses, at least in part, relate to treatment or variations in non‐lymphocytic cell distributions. For instance, even though administration of hydroxychloroquine did not seem to affect the cellular responsiveness to ODN2395, we did not consider the individual doses which may have been higher among patients without ongoing LN. Furthermore, as monocytes are pivotal for the clearance of MVs, differential frequencies of these cells between patients and controls and/or between patients with and without active LN may affect the final recovery of G3BP‐expressing MVs in the culture supernatants. Besides, in‐vitro findings may not readily be translated into more complex in‐vivo conditions.
In vivo, release of MVs expressing G3BP may be induced by circulating dsDNA‐rich ICs through TLR‐9, in accordance with the higher concentration of G3BP‐expressing MVs in the circulation of SLE patients than in healthy individuals [14]. The expression of G3BP is likely to facilitate glomerular deposition of the MVs in galectin‐3‐expressing glomeruli, while their content of dsDNA makes the MVs accessible for anti‐dsDNA antibody binding and subsequent complement activation. Thus, the propensity to generate and deposit MVs expressing G3BP as well as dsDNA may mediate potentiated tissue damage that may manifest as LN.
In conclusion, our study shows that stimulation of PBMCs by the class C TLR‐9 agonist ODN2395 induces release of G3BP‐expressing MVs, particularly in SLE patients with active LN. The glomerulophilic and pathogenic potential of MVs co‐expressing G3BP and dsDNA in LN deserves further investigation.
Disclosures
The authors declare no conflicts of interest.
Acknowledgements
This study was supported by grants to Søren Jacobsen from the Lundbeck Foundation (R208‐2015‐4018&4019) and the Danish Rheumatism Association (A3865).
Data Availability Statement
Not applicable.
References
- 1. Arraud N, Linares R, Tan S et al. Extracellular vesicles from blood plasma: determination of their morphology, size, phenotype and concentration. J Thromb Haemost 2014; 12:614–27. [DOI] [PubMed] [Google Scholar]
- 2. Lötvall J, Hill AF, Hochberg F et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 2014; 3:26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Inal JM, Ansa‐Addo EA, Stratton D et al. Microvesicles in health and disease. Arch Immunol Ther Exp 2012; 60:107–21. [DOI] [PubMed] [Google Scholar]
- 4. Wolf P. The nature and significance of platelet products in human plasma. Br J Haematol 1967; 13:269–88. [DOI] [PubMed] [Google Scholar]
- 5. Distler JH, Pisetsky DS, Huber LC, Kalden JR, Gay S, Distler O. Microparticles as regulators of inflammation: novel players of cellular crosstalk in the rheumatic diseases. Arthritis Rheum 2005; 52:3337–48. [DOI] [PubMed] [Google Scholar]
- 6. Perez‐Hernandez J, Cortes R. Extracellular vesicles as biomarkers of systemic lupus erythematosus. Dis Markers 2015; 2015:613536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turpin D, Truchetet ME, Faustin B et al. Role of extracellular vesicles in autoimmune diseases. Autoimmun Rev 2016; 15:174–83. [DOI] [PubMed] [Google Scholar]
- 8. Jackson SW, Scharping NE, Kolhatkar NS et al. Opposing impact of B cell‐intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol 2014; 192:4525–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Østergaard O, Nielsen CT, Iversen LV et al. Unique protein signature of circulating microparticles in systemic lupus erythematosus. Arthritis Rheum 2013; 65:2680–90. [DOI] [PubMed] [Google Scholar]
- 10. Østergaard O, Nielsen CT, Tanassi JT, Iversen LV, Jacobsen S, Heegaard NHH. Distinct proteome pathology of circulating microparticles in systemic lupus erythematosus. Clin Proteomics 2017; 14:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nielsen CT, Østergaard O, Stener L et al. Increased IgG on cell‐derived plasma microparticles in systemic lupus erythematosus is associated with autoantibodies and complement activation. Arthritis Rheum 2012; 64:1227–36. [DOI] [PubMed] [Google Scholar]
- 12. Ullal AJ, Reich CF, Clowse M et al. Microparticles as antigenic targets of antibodies to DNA and nucleosomes in systemic lupus erythematosus. J Autoimmun 2011; 36:173–80. [DOI] [PubMed] [Google Scholar]
- 13. Mobarrez F, Fuzzi E, Gunnarsson I et al. Microparticles in the blood of patients with SLE: Size, content of mitochondria and role in circulating immune complexes. J Autoimmun 2019; 102:142–9. [DOI] [PubMed] [Google Scholar]
- 14. Nielsen CT, Østergaard O, Rekvig OP, Sturfelt G, Jacobsen S, Heegaard NH. Galectin‐3 binding protein links circulating microparticles with electron dense glomerular deposits in lupus nephritis. Lupus 2015; 24:1150–60. [DOI] [PubMed] [Google Scholar]
- 15. Dieker J, Tel J, Pieterse E et al. Circulating apoptotic microparticles in systemic lupus erythematosus patients drive the activation of dendritic cell subsets and prime neutrophils for NETosis. Arthritis Rheum 2016; 68:462–72. [DOI] [PubMed] [Google Scholar]
- 16. Kaul A, Gordon C, Crow MK et al. Systemic lupus erythematosus. Nat Rev Dis Primers 2016; 2:16039. [DOI] [PubMed] [Google Scholar]
- 17. Sisirak V, Sally B, D'Agati V et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell 2016; 166:88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sasaki T, Brakebusch C, Engel J, Timpl R. Mac‐2 binding protein is a cell‐adhesive protein of the extracellular matrix which self‐assembles into ring‐like structures and binds beta1 integrins, collagens and fibronectin. EMBO J 1998; 17:1606–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kang EH, Moon KC, Lee EY et al. Renal expression of galectin‐3 in systemic lupus erythematosus patients with nephritis. Lupus 2009; 18:22–8. [DOI] [PubMed] [Google Scholar]
- 20. Rasmussen NS, Nielsen CT, Jacobsen S, Nielsen CH. Stimulation of mononuclear cells through Toll‐like receptor 9 induces release of microvesicles expressing double‐stranded DNA and galectin 3‐binding protein in an interferon‐α‐dependent manner. Front Immunol 2019; 10. 10.3389/fimmu.2019.02391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baechler EC, Batliwalla FM, Karypis G et al. Interferon‐inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA 2003; 100:2610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bennett L, Palucka AK, Arce E et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 2003; 197:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crow MK, Kirou KA, Wohlgemuth J. Microarray analysis of interferon‐regulated genes in SLE. Autoimmunity 2003; 36:481–90. [DOI] [PubMed] [Google Scholar]
- 24. Crow MK, Wohlgemuth J. Microarray analysis of gene expression in lupus. Arthritis Res Ther 2003; 5:279–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han GM, Chen SL, Shen N, Ye S, Bao CD, Gu YY. Analysis of gene expression profiles in human systemic lupus erythematosus using oligonucleotide microarray. Genes Immun 2003; 4:177–86. [DOI] [PubMed] [Google Scholar]
- 26. Celhar T, Fairhurst AM. Toll‐like receptors in systemic lupus erythematosus: potential for personalized treatment. Front Pharmacol 2014; 5:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody‐DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 2005; 115:407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vallin H, Perers A, Alm GV, Rönnblom L. Anti‐double‐stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN‐alpha inducer in systemic lupus erythematosus. J Immunol 1999; 163:6306–13. [PubMed] [Google Scholar]
- 29. Yasuda K, Richez C, Uccellini MB et al. Requirement for DNA CpG content in TLR9‐dependent dendritic cell activation induced by DNA‐containing immune complexes. J Immunol 2009; 183:3109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakano S, Morimoto S, Suzuki J et al. Role of pathogenic auto‐antibody production by Toll‐like receptor 9 of B cells in active systemic lupus erythematosus. Rheumatology 2008; 47:145–9. [DOI] [PubMed] [Google Scholar]
- 31. Yuan Y, Zhao L, Ye Z, Ma H, Wang X, Jiang Z. Association of toll‐like receptor 9 expression with prognosis of systemic lupus erythematosus. Exp Ther Med 2019; 17:3247–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chauhan SK, Singh VV, Rai R, Rai G. Distinct autoantibody profiles in systemic lupus erythematosus patients are selectively associated with TLR7 and TLR9 upregulation. J Clin Immunol 2013; 33:954–64. [DOI] [PubMed] [Google Scholar]
- 33. Anders HJ, Vielhauer V, Eis V et al. Activation of toll‐like receptor‐9 induces progression of renal disease in MRL‐Fas(lpr) mice. FASEB J 2004; 18:534–6. [DOI] [PubMed] [Google Scholar]
- 34. Anders HJ, Banas B, Linde Y et al. Bacterial CpG‐DNA aggravates immune complex glomerulonephritis: role of TLR9‐mediated expression of chemokines and chemokine receptors. J Am Soc Nephrol 2003; 14:317–26. [DOI] [PubMed] [Google Scholar]
- 35. Pawar RD, Patole PS, Ellwart A et al. Ligands to nucleic acid‐specific toll‐like receptors and the onset of lupus nephritis. J Am Soc Nephrol 2006; 17:3365–73. [DOI] [PubMed] [Google Scholar]
- 36. Hasegawa K, Hayashi T. Synthetic CpG oligodeoxynucleotides accelerate the development of lupus nephritis during preactive phase in NZB x NZWF1 mice. Lupus 2003; 12:838–45. [DOI] [PubMed] [Google Scholar]
- 37. Patole PS, Zecher D, Pawar RD, Gröne HJ, Schlöndorff D, Anders HJ. G‐rich DNA suppresses systemic lupus. J Am Soc Nephrol 2005; 16:3273–80. [DOI] [PubMed] [Google Scholar]
- 38. Dong L, Ito S, Ishii KJ, Klinman DM. Suppressive oligodeoxynucleotides delay the onset of glomerulonephritis and prolong survival in lupus‐prone NZB x NZW mice. Arthritis Rheum 2005; 52:651–8. [DOI] [PubMed] [Google Scholar]
- 39. Summers SA, Hoi A, Steinmetz OM et al. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J Autoimmun 2010; 35:291–8. [DOI] [PubMed] [Google Scholar]
- 40. Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med 2006; 203:553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bossaller L, Christ A, Pelka K et al. TLR9 deficiency leads to accelerated renal disease and myeloid lineage abnormalities in pristane‐induced murine lupus. J Immunol 2016; 197:1044–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nickerson KM, Cullen JL, Kashgarian M, Shlomchik MJ. Exacerbated autoimmunity in the absence of TLR9 in MRL.Fas(lpr) mice depends on Ifnar1. J Immunol 2013; 190:3889–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nickerson KM, Wang Y, Bastacky S, Shlomchik MJ. Toll‐like receptor 9 suppresses lupus disease in Fas‐sufficient MRL mice. PLOS ONE 2017; 12:e0173471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Celhar T, Yasuga H, Lee HY et al. Toll‐like receptor 9 deficiency breaks tolerance to RNA‐associated antigens and up‐regulates Toll‐like receptor 7 protein in Sle1 mice. Arthritis Rheum 2018; 70:1597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nickerson KM, Christensen SR, Shupe J et al. TLR9 regulates TLR7‐ and MyD88‐dependent autoantibody production and disease in a murine model of lupus. J Immunol 2010; 184:1840–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu P, Wellmann U, Kunder S et al. Toll‐like receptor 9‐independent aggravation of glomerulonephritis in a novel model of SLE. Int Immunol 2006; 18:1211–9. [DOI] [PubMed] [Google Scholar]
- 47. Wu X, Peng SL. Toll‐like receptor 9 signaling protects against murine lupus. Arthritis Rheum 2006; 54:336–42. [DOI] [PubMed] [Google Scholar]
- 48. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll‐like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006; 25:417–28. [DOI] [PubMed] [Google Scholar]
- 49. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997; 40:1725. [DOI] [PubMed] [Google Scholar]
- 50. Vollmer J, Weeratna RD, Jurk M et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur J Immunol 2004; 34:251–62. [DOI] [PubMed] [Google Scholar]
- 51. Geppert T, Bauer S, Hiss JA, Conrad E, Reutlinger M, Schneider P. Immunosuppressive small molecule discovered by structure‐based virtual screening for inhibitors of protein‐protein interactions. Angew Chem Int Ed Engl 2012; 51:258–61. [DOI] [PubMed] [Google Scholar]
- 52. Gray WD, Mitchell AJ, Searles CD. An accurate, precise method for general labeling of extracellular vesicles. MethodsX 2015; 10:360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bauer S, Kirschning CJ, Hacker H et al. Human TLR9 confers responsiveness to bacterial DNA via species‐specific CpG motif recognition. Proc Natl Acad Sci USA 2001; 98:9237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hornung V, Rothenfusser S, Britsch S et al. Quantitative expression of toll‐like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol 2002; 168:4531–7. [DOI] [PubMed] [Google Scholar]
- 55. Gies V, Schickel JN, Jung S et al. Impaired TLR9 responses in B cells from patients with systemic lupus erythematosus. JCI Insight 2018; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zorro S, Arias M, Riaño F et al. Response to ODN‐CpG by B cells from patients with systemic lupus erythematosus correlates with disease activity. Lupus 2009; 18:718–26. [DOI] [PubMed] [Google Scholar]
- 57. Sieber J, Daridon C, Fleischer SJ et al. Active systemic lupus erythematosus is associated with a reduced cytokine production by B cells in response to TLR9 stimulation. Arthritis Res Ther 2014; 16. 10.1186/s13075-014-0477-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rovati B, Mariucci S, Manzoni M, Bencardino K, Danova M. Flow cytometric detection of circulating dendritic cells in healthy subjects. Eur J Histochem 2008; 52:45–52. [DOI] [PubMed] [Google Scholar]
- 59. Kuznik A, Bencina M, Svajger U, Jeras M, Rozman B, Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J Immunol 2011; 186:4794–804. [DOI] [PubMed] [Google Scholar]
- 60. Berghöfer B, Haley G, Frommer T, Bein G, Hackstein H. Natural and synthetic TLR7 ligands inhibit CpG‐A‐ and CpG‐C‐oligodeoxynucleotide‐induced IFN‐alpha production. J Immunol 2007; 178:4072–9. [DOI] [PubMed] [Google Scholar]
- 61. Butchi NB, Du M, Peterson KE. Interactions between TLR7 and TLR9 agonists and receptors regulate innate immune responses by astrocytes and microglia. Glia 2010; 58:650–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Crow MK. Type I interferon in the pathogenesis of lupus. J Immunol 2014; 192:5459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Papadimitraki ED, Choulaki C, Koutala E et al. Expansion of Toll‐like receptor 9‐expressing B cells in active systemic lupus erythematosus: implications for the induction and maintenance of the autoimmune process. Arthritis Rheum 2006; 54:3601–11. [DOI] [PubMed] [Google Scholar]
- 64. Bernasconi N, Onai N, Lanzavecchia A. A role for Toll‐like receptors in acquired immunity: up‐regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood 2003; 101:4500–4. [DOI] [PubMed] [Google Scholar]
- 65. Ivanov S, Dragoi A, Wang X et al. A novel role for HMGB1 in TLR9‐mediated inflammatory responses to CpG‐DNA. Blood 2007; 110:1970–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu T, Son M, Diamond B. HMGB1 in systemic lupus erythematosus. Front Immunol 2020; 11. 10.3389/fimmu.2020.01057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.