

Postsynaptic development requires the Frizzled nuclear import (FNI) pathway entailing the internalization and subsequent cleavage of Frizzled-2. Kim et al. show that Drosophila ALS2 regulates postsynaptic development by directing Frizzled-2 trafficking to late endosomes, where the Frizzled-2 C terminus is cleaved, and also promotes neuronal survival independently of the FNI pathway.
Abstract
Mutations in the human ALS2 gene cause recessive juvenile-onset amyotrophic lateral sclerosis and related motor neuron diseases. Although the ALS2 protein has been identified as a guanine-nucleotide exchange factor for the small GTPase Rab5, its physiological roles remain largely unknown. Here, we demonstrate that the Drosophila homologue of ALS2 (dALS2) promotes postsynaptic development by activating the Frizzled nuclear import (FNI) pathway. dALS2 loss causes structural defects in the postsynaptic subsynaptic reticulum (SSR), recapitulating the phenotypes observed in FNI pathway mutants. Consistently, these developmental phenotypes are rescued by postsynaptic expression of the signaling-competent C-terminal fragment of Drosophila Frizzled-2 (dFz2). We further demonstrate that dALS2 directs early to late endosome trafficking and that the dFz2 C terminus is cleaved in late endosomes. Finally, dALS2 loss causes age-dependent progressive defects resembling ALS, including locomotor impairment and brain neurodegeneration, independently of the FNI pathway. These findings establish novel regulatory roles for dALS2 in endosomal trafficking, synaptic development, and neuronal survival.
Introduction
Mutations in the human ALS2 gene are associated with multiple early-onset motor neuron diseases (MNDs), including juvenile amyotrophic lateral sclerosis 2 (ALS2), juvenile primary lateral sclerosis, and infantile-onset ascending hereditary spastic paraplegia (Chen et al., 2013; Eymard-Pierre et al., 2002; Hadano et al., 2001; Yang et al., 2001). These ALS2-deficient MNDs are commonly characterized by progressive degeneration of motor neurons in the central nervous system, indicating an important role for the ALS2 gene product (ALS2/alsin) in motor neuron survival and maintenance. Consistently, the ALS2 protein is expressed primarily in central nervous system neurons, including motor neurons of the cortex and spinal cord (Devon et al., 2005; Otomo et al., 2003).
A large proportion of ALS2 mutations in MND patients leads to premature termination of protein translation or decreased protein stability (Sato et al., 2018; Yamanaka et al., 2003), implying a loss-of-function disease mechanism. ALS2−/− mice develop mild motor dysfunction and distal axonopathy in the corticospinal tract (Deng et al., 2007; Julien and Kriz, 2006; Yamanaka et al., 2006). Although biochemical and cell biological studies suggest that ALS2 regulates endosomal fusion and trafficking by activating the small GTPase Rab5 (Otomo et al., 2003; Topp et al., 2004), the roles of ALS2 in axon/synaptic maintenance and their potential link to endosomal trafficking are unknown.
Wnt signaling plays key roles in synaptic development, and its dysregulation is increasingly recognized as an important mechanism of synaptic loss in neurodegenerative diseases (Dickins and Salinas, 2013). At the Drosophila glutamatergic neuromuscular junction (NMJ), the Wnt homologue Wingless (Wg) is secreted from presynaptic terminals and binds the Drosophila Frizzled-2 (dFz2) receptor, which is expressed on pre- and postsynaptic membranes (Packard et al., 2002). In postsynaptic muscles, Wg activates the noncanonical Frizzled nuclear import (FNI) pathway by binding to dFz2 and inducing its endocytosis and cleavage (Mathew et al., 2005). The cleaved C-terminal fragment (dFz2-C) is imported into muscle nuclei in an Importin-β11–dependent manner to promote postsynaptic differentiation (Mathew et al., 2005; Mosca and Schwarz, 2010); however, the intracellular compartment in which dFz2 is cleaved to produce dFz2-C remains unknown.
In the present study, we investigate the physiological roles of a Drosophila homologue of human ALS2 (dALS2) in the larval NMJ and adult brain. We demonstrate that loss of dALS2 impairs normal development of the subsynaptic reticulum (SSR), a network of postsynaptic membrane invaginations at the NMJ. Genetic interaction data suggest that dALS2 promotes postsynaptic development by regulating the FNI pathway. We also demonstrate that dALS2 ablation causes a general defect in receptor trafficking from early to late endosomes. Our results also indicate that dFz2-C cleavage occurs in the late endosome/lysosome compartment. Finally, we show that loss of dALS2 causes adult-onset, progressive neurodegeneration resembling ALS in an FNI pathway–independent manner. Together, these findings demonstrate an unexpected role for dALS2-mediated endosomal trafficking in postsynaptic dFz2 signaling and highlight the potential contribution of impaired receptor and endosomal trafficking to the pathogenesis of ALS2-associated MNDs.
Results
dALS2 is required for normal postsynaptic development
To explore the in vivo function of dALS2 protein at synapses, we disrupted the dALS2 gene using two different approaches. First, we imprecisely excised a P-element insertion (G4607) in the first exon of dALS2 and isolated the deletion dALS2Δ73, which deletes a portion of the first exon, including the translation start site (Fig. S1 A). Second, we performed CRISPR/Cas9 mutagenesis to isolate a larger deletion, dALS2Δ1, which removed most of the protein-coding region, including the translation start site (Fig. S1 A). Transheterozygotes of dALS2Δ1 and dALS2Δ73 were null for dALS2 expression, but normally expressed the adjacent gene CG11248 (Fig. S1 B). Homozygotes and transheterozygotes of dALS2Δ1 and dALS2Δ73 were viable and fertile.
Figure S1.

Characterization of dALS2 gene and mutants. (A) Schematic of the genomic dALS2 (CG7158) locus. The position of the transposon G4607 used to generate the dALS2 allele dALS2Δ73 via transposase-mediated excision is indicated by the inverted triangle. The gRNA target sites used to generate the dALS2Δ1 allele via the CRISPR/Cas9 genome editing system are marked by arrows. Exon-intron organization of dALS2 and the neighboring CG11248 gene is shown in the middle. Introns are indicated by horizontal lines, untranslated regions by white boxes, translated regions by black boxes, and translation initiation sites by arrows. Shown below are deletion breakpoints for dALS2Δ1 and dALS2Δ73. (B) RT-PCR analysis of dALS2, CG11248, and rp49 transcripts in WT and dALS2Δ1/dALS2Δ73 third instar larvae. rp49 is used a loading control. (C–H) Multiple aspects of presynaptic development remain unchanged in dALS2 mutants. (C) Confocal images of NMJ 6/7 stained with antibodies against HRP (green) and cysteine string protein (Csp; red) are shown for WT and dALS2Δ1/dALS2Δ73 third instar larvae. (D–F) Quantification of total bouton number (D), bouton size (E), and muscle 6/7 area (F). n = 16 NMJs. (G) Confocal images of anti-Futsch–stained NMJ 6/7 in WT and dALS2Δ1/dALS2Δ73 third instar larvae. Arrowheads indicate Futsch-immunoreactive loops. (H) Quantification of boutons with Futsch loops (n = 18 NMJs). (I and J) Characterization of ghost boutons in dALS2-null mutants. (I) Confocal images of NMJ 6/7 stained with anti-HRP, anti-Brp, and anti-GluRIIC antibodies for WT and dALS2Δ1/dALS2Δ73 third instar larvae. Ghost boutons (arrowheads) are identified as HRP-labeled varicosities lacking Brp and GluRIIC immunoreactivities. (J) Confocal images of NMJ 6/7 labeled with anti-HRP, anti-Synapsin (Syn), and anti-GluRIIC antibodies are shown for WT and dALS2Δ1/dALS2Δ73 third instar larvae. Synapsin is still present in dALS2-induced ghost boutons (arrowheads). (K and L) Postsynaptic, but not presynaptic, dALS2 knockdown results in excessive ghost bouton formation. (K) Confocal images of NMJ 6/7 labeled with anti-HRP and anti-Dlg antibodies are shown for D42-GAL4 /+, UAS-dALS2-RNAi/+; D42-GAL4/+ (dALS2-RNAi-pre), 24B-GAL4/+, and UAS-dALS2-RNAi/+; 24B-GAL4/+ (dALS2-RNAi-post) third instar larvae. Right panels show higher magnification views of areas marked by white boxes. Arrowheads indicate ghost boutons. (L) Quantification of ghost bouton number (n = 30 NMJs). Data are presented as mean ± SEM. Comparisons are with WT (D–F and H) or D42-GAL4 unless otherwise indicated. Scale bars: 20 µm (C, G, and K); 5 µm (I and J). ***, P < 0.001.
We next examined NMJs 6/7 of third instar larvae of WT (w1118) and dALS2Δ1/dALS2Δ73 animals using synaptic markers. Bouton number, bouton size, and muscle size did not differ between WT and dALS2 mutant larvae (Fig. S1, C–F). Furthermore, Futsch-labeled synaptic microtubules were normal in dALS2 mutants (Fig. S1, G and H); however, we observed a 4.2-fold increase of synaptic boutons lacking the SSR marker Discs-large (Dlg; WT: 0.67 ± 0.16; dALS2Δ1/dALS2Δ73: 2.80 ± 0.35; mean ± SEM/NMJ; P < 0.001; Fig. 1, A and B). These aberrant boutons, known as ghost boutons (Ataman et al., 2006), contained the synaptic vesicle marker Synapsin but were devoid of the active zone marker Bruchpilot (Brp) and the glutamate receptor (GluR) subunit GluRIIC (Fig. S1, I and J). Ghost boutons are newly formed, immature boutons sprouting from the main NMJ branches (Ataman et al., 2006; Ataman et al., 2008), so these results suggest that dALS2 is required for maturation of synaptic boutons. To further assess postsynaptic abnormalities of mature boutons located on the main NMJ branch, we analyzed the level and distribution of postsynaptic GluRs. At the Drosophila NMJ, GluRs are present in two types of tetrameric complexes composed of GluRIIC, GluRIID, and GluRIIE, together with either GluRIIA or GluRIIB. GluRIIB, but not GluRIIA, signal intensity relative to HRP immunoreactivity was significantly increased in dALS2 mutant larvae relative WT controls (WT: 100 ± 2.57%; dALS2Δ1/dALS2Δ73: 128.85 ± 2.92%; P < 0.001; Fig. 1, C and D). Consistently, the ratio of GluRIIB to GluRIIA signal intensities in individual GluRII clusters was significantly increased by 19% in dALS2 mutants (P < 0.001; Fig. 1 E); however, the mean size of GluRII clusters and the ratio between GluRIIB domains and Brp puncta remained normal. Finally, overexpression of dALS2 in the WT background did not change synaptic maturation or GluRIIB abundance (Fig. 1, B and D), indicating that dALS2 is not limiting for synaptic development.
Figure 1.

dALS2 regulation of postsynaptic development. (A) Confocal images of NMJ 6/7 stained with anti-HRP and anti-Dlg antibodies are shown for WT (w1118), dALS2Δ1/dALS2Δ73, UAS-HA-dALS2,dALS2Δ1/D42-GAL4,dALS2Δ73 (dALS2 rescue-pre), UAS-HA-dALS2,dALS2Δ1/24B-GAL4,dALS2Δ73 (dALS2 rescue-post), UAS-HA-hALS2/+; dALS2Δ1/24B-GAL4,dALS2Δ73 (hALS2 rescue-post); and UAS-Myc-NLS-dfz2-C/+; dALS2Δ1/24B-GAL4, dALS2Δ73 (NLS-dFz2-C rescue-post) third instar larvae. Arrowheads and arrows mark ghost boutons formed at type Ib and Is terminals, respectively. (B) Quantification of ghost bouton number per NMJ 6/7 in indicated genotypes. dALS2 OE (overexpression)-post indicates UAS-HA-dALS2/24B-GAL4, and NLS-dFz2-C OE-post indicates UAS-Myc-NLS-dfz2-C/+; 24B-GAL4/+. n = 30 NMJs. (C) Confocal images of third instar NMJ 6/7 of the indicated genotypes stained with anti-HRP and anti-GluRIIB antibodies. (D) Quantification of GluRIIA, and GluRIIB immunoreactivities normalized to HRP. n = 18–46 NMJ branches. (E) Quantification of the ratio of mean GluRIIB to GluRIIA fluorescence intensities. n = 42 NMJ branches. Data are presented as mean ± SEM. Comparisons are with WT or 24B-GAL4/+ control unless otherwise indicated. Scale bars: 20 µm (A); 5 µm (C). ***, P < 0.001; *, P < 0.05.
To examine whether dALS2 function is required in presynaptic neurons or postsynaptic muscles, we expressed HA-tagged dALS2 (HA-dALS2) in dALS2Δ1/dALS2Δ73 mutants using the upstream activating sequence (UAS)–GAL4 system. UAS-HA-dALS2 expression under the muscle-specific 24B-GAL4 driver completely rescued the phenotypes of increased ghost bouton number and increased synaptic GluRIIB in dALS2 mutants (Fig. 1, A–D). Contrastingly, these postsynaptic defects were not rescued by UAS-HA-dALS2 expression under the motor neuron–specific D42-GAL4 driver (Fig. 1, A, B, and D). Notably, muscular expression of HA-tagged human ALS2 (HA-hALS2) almost completely rescued the dALS2 postsynaptic phenotypes (Fig. 1, A, B, and D), confirming that the synaptic function of dALS2 is conserved in its human homologue. The postsynaptic role of dALS2 in NMJ development was further assessed using RNAi-mediated dALS2 depletion. Muscular expression of UAS-dALS2-RNAi induced excessive formation of ghost boutons, while motor neuron expression did not affect postsynaptic differentiation (Fig. S1, K and L).
dALS2 regulates postsynaptic ultrastructure
We next examined the ultrastructure of NMJ synapses in dALS2 mutants using transmission EM. Presynaptic terminals, including synaptic vesicle density and size, number of active zones per bouton area, number of T-bars per active zone, and number of mitochondria per bouton, were normal in dALS2Δ1/dALS2Δ73 mutants (data not shown); however, the SSR, the network of infolded postsynaptic membrane at the NMJ, was structurally defective. Because ghost boutons are completely devoid of postsynaptic structure (Ataman et al., 2006; Packard et al., 2002), our analysis was focused on mature boutons surrounded by SSR. Compared with WT controls, membrane layers in dALS2 mutants were less complex, with lower SSR membrane density (WT: 13.69 ± 1.08; dALS2Δ1/dALS2Δ73: 8.35 ± 0.28; P < 0.001; Fig. 2, A and B). In addition, we observed striking expansion of the postsynaptic pocket (pocket depth; WT: 0.14 ± 0.01 µm; dALS2Δ1/dALS2Δ73: 0.24 ± 0.02 µm; P < 0.001; Fig. 2, A and C), a postsynaptic area immediately apposed to an active zone containing amorphous material (Packard et al., 2002); however, SSR thickness was unchanged (Fig. 2 D). These data suggest that dALS2 is required for normal postsynaptic differentiation at the NMJ.
Figure 2.
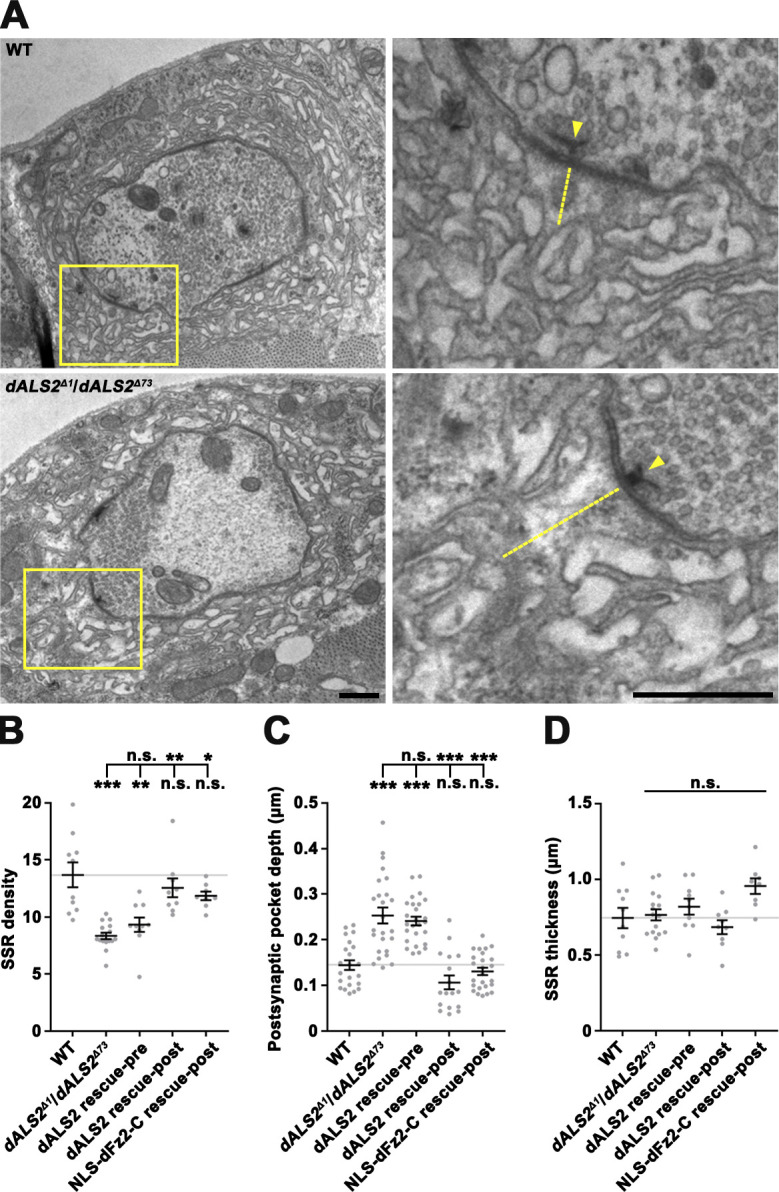
Reduced ultrastructural SSR density in dALS2 mutants. (A) Transmission EMs of type Ib boutons and the surrounding SSR in WT and dALS2Δ1/dALS2Δ73 third instar larvae. Right panels show higher magnification views of areas marked by boxed regions. Labels indicate presynaptic t-bars (arrowheads) and postsynaptic pockets (dotted lines). (B–D) Quantification of SSR density (B; n = 8–16 boutons), calculated as number of SSR layers per micrometer, postsynaptic pocket depth (C; n = 17–25), and SSR thickness (D; n = 8–16) in WT, dALS2Δ1/dALS2Δ73, UAS-HA-dALS2,dALS2Δ1/D42-GAL4,dALS2Δ73 (dALS2 rescue-pre), UAS-HA-dALS2,dALS2Δ1/24B-GAL4,dALS2Δ73 (dALS2 rescue-post), and UAS-Myc-NLS-dfz2-C/+; dALS2Δ1/24B-GAL4, dALS2Δ73 (NLS-dFz2-C rescue-post). Data are presented as mean ± SEM. All comparisons are relative to WT unless otherwise indicated. Scale bars: 0.5 µm. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
dALS2 mutations alter synaptic function
To assess the functional consequence of dALS2 mutations at the NMJ, we performed electrophysiological measurements of excitatory junctional potentials (EJPs). The amplitude of spontaneously occurring miniature EJPs (mEJPs) was significantly reduced in dALS2Δ1/dALS2Δ73 mutants relative to WT (WT: 1.54 ± 0.06 mV; dALS2Δ1/dALS2Δ73: 0.86 ± 0.05 mV; P < 0.001; Fig. 3, A and B), while mEJP frequency was normal (Fig. 3 C). This was consistent with the finding that synaptic levels of GluRIIB relative to GluRIIA are increased in dALS2 mutants, as ion conductivity is decreased in GluRIIB-expressing receptor complexes relative to GluRIIA-expressing receptor complexes (DiAntonio et al., 1999). Decreased mEJP amplitude phenotype was significantly rescued by postsynaptic, but not presynaptic, expression of HA-dALS2 in dALS2 mutants (Fig. 3, A and B). Finally, despite decreased mEJP amplitude, the amplitude of evoked EJPs remained normal in dALS2 mutants due to a compensatory increase in presynaptic neurotransmitter release (quantal content; WT: 11.16 ± 0.84; dALS2Δ1/dALS2Δ73: 19.37 ± 1.05; P < 0.001; Fig. 3 F). The increased quantal content phenotype was also rescued by muscular expression of HA-dALS2, suggesting that a retrograde (post to presynaptic) homeostatic mechanism is involved in this compensatory process.
Figure 3.

Impaired NMJ synaptic transmission in dALS2 mutants. (A) Representative mEJP events from muscle 6 for WT, dALS2Δ1/dALS2Δ73, UAS-HA-dALS2,dALS2Δ1/D42-GAL4,dALS2Δ73 (dALS2 rescue-pre), UAS-HA-dALS2,dALS2Δ1/24B-GAL4,dALS2Δ73 (dALS2 rescue-post), and UAS-Myc-NLS-dfz2-C/+; dALS2Δ1/24B-GAL4, dALS2Δ73 (Myc-NLS-dFz2-C rescue-post) third instar larvae. (B and C) Quantification of mean mEJP amplitude (B) and mEJP frequency (C) for the same genotypes described in A. (D) Representative EJP recordings with 0.2-Hz nerve stimulation are shown for WT and dALS2Δ1/dALS2Δ73 larvae. (E and F) Quantification of mean EJP amplitude (E) and quantal content (F) for the same genotypes described in A. n = 12–15. Data are presented as mean ± SEM. All comparisons are with WT unless otherwise indicated. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
dALS2 regulates the FNI Wg signaling pathway in the muscle
The synaptic defects of dALS2 mutants, including excessive ghost bouton formation and abnormal SSR development, are strikingly similar to those of animals deficient in the postsynaptic FNI Wg pathway or in dGRIP, a PDZ protein required for dFz2 intracellular trafficking (Ataman et al., 2006; Harris et al., 2016; Kamimura et al., 2013; Mosca and Schwarz, 2010; Packard et al., 2002). We therefore hypothesized that the synaptic defects in dALS2-null mutants could be due to dysregulation of the postsynaptic FNI pathway. To test this possibility, we first examined genetic interactions between dALS2 and wg or dGRIP. Heterozygosity for dALS2, wg, or dGRIP did not affect postsynaptic development (Fig. 4, A and B); however, the number of ghost boutons was significantly increased in larvae transheterozygous for dALS2 and wg or dGRIP relative to WT controls (Fig. 4, A and B), suggesting functional linkage between dALS2 and the postsynaptic FNI pathway during synaptic development.
Figure 4.

dALS2 regulation of Wg/dFz2-mediated postsynaptic development. (A) Confocal images of NMJ 6/7 labeled with anti-HRP and anti-Dlg are shown for WT, dALS2Δ1/+, wgI-17/+, dGRIPex36/+, wgI-17/+; dALS2Δ1/+, and dGRIPex36/+; dALS2Δ1/+ third instar larvae. Arrowheads indicate ghost boutons. (B) Quantification of ghost bouton number (n = 30 NMJs). Note transheterozygous interactions between dALS2 and wg or dGRIP during synaptic development. (C) Confocal images of muscle nuclei stained with anti–dFz2-C and DAPI are shown for WT, dALS2Δ1/dALS2Δ73, UAS-HA-dALS2,dALS2Δ1/D42-GAL4,dALS2Δ73 (dALS2 rescue-pre), and UAS-HA-dALS2,dALS2Δ1/24B-GAL4,dALS2Δ73 (dALS2 rescue-post) third instar larvae. Arrowheads indicate dFz2-C nuclear puncta. (D) Quantification of dFz2-C puncta per nucleus (n = 36 nuclei). Data are presented as mean ± SEM. All comparisons are with WT unless otherwise indicated. Scale bars: 20 µm (A); 5 µm (C). ***, P < 0.001; *, P < 0.05.
Next, we determined if loss of dALS2 affects nuclear accumulation of the cleaved dFz2-C. In WT larvae, dFz2-C immunoreactivity was detected in a punctate pattern in muscle nuclei (Fig. 4 C), as previously reported (Mathew et al., 2005). Importantly, the number of nuclear dFz2-C puncta was significantly decreased in dALS2Δ1/dALS2Δ73 mutants (WT: 4.28 ± 0.42; dALS2Δ1/dALS2Δ73: 1.36 ± 0.33; P < 0.001; Fig. 4, C and D). Muscular expression of HA-dALS2 or HA-hALS2 in dALS2 mutants rescued decreased nuclear accumulation of dFz2-C puncta (Fig. 4, C and D), supporting a role for dALS2 in the postsynaptic FNI pathway.
Previous studies have shown that expression of Myc-NLS-dFz2-C, a Myc epitope fused with a nuclear localization signal (NLS) and the C-terminal 88 aa of dFz2, can bypass the requirement of the FNI pathway for postsynaptic development (Mosca and Schwarz, 2010). We therefore tested if Myc-NLS-dFz2-C is able to rescue the synaptic defects of dALS2 mutants. Muscle-specific expression of Myc-NLS-dFz2-C in dALS2Δ1/dALS2Δ73 mutants decreased the number of ghost boutons to that of WT larvae and, furthermore, rescued other key dALS2 phenotypes, including synaptic elevation of GluRIIB expression, decreased SSR complexity, expansion of postsynaptic pockets, and decreased mEJP amplitude (Figs. 1, 2, and 3). These findings are consistent with the model that dALS2 regulates postsynaptic development by modulating the FNI pathway.
dALS2 regulates dFz2 transport from early to late endosomes
The mammalian homologue of dALS2 is localized to Rab5+ early endosomes (Otomo et al., 2003; Topp et al., 2004). To investigate the mechanism by which dALS2 regulates dFz2 signaling, we examined subcellular localization of dALS2 in postsynaptic muscles. Because our polyclonal anti-dALS2 antibody could not detect endogenous dALS2 in immunomicroscopy, we expressed low levels of UAS-HA-dALS2 in muscles by leaky expression from Mhc-GS-GAL4 in the absence of the steroid RU486 (Osterwalder et al., 2001). In muscles, Rab5+ early endosomes preferentially distributed around synaptic boutons, while Rab5+/Rab7+ intermediate endosomes and Rab7+ late endosomes were more common in the perinuclear area (Fig. S2 A). The HA-dALS2 fusion protein primarily appeared in reticular and punctate patterns. HA-dALS2 puncta overlapped with a subset of Rab5+ early endosomes and Rab5+/Rab7+ intermediate endosomes, but rarely colocalized with Rab7+ late endosomes (Fig. S2, A and B).
Figure S2.

Characterization of transgenic HA-dALS2 and Flag-dFz2-Myc expression in larval muscles. (A) Single confocal sections of the peribouton area (upper panels) at NMJ 6/7 and the perinuclear area (lower panels) in muscle 6 are shown for Mhc-GS-GAL4/UAS-HA-dALS2 third instar larvae stained with anti-HA, anti-Rab5, and anti-Rab7. Arrowheads indicate endosomal compartments double positive for HA-dALS2 and Rab5, while arrows indicate endosomal compartments triple positive for HA-dALS2, Rab5, and Rab7. Note that HA-dALS2 rarely colocalizes with Rab7. (B) Quantification of the number of HA-dALS2+ puncta associated with Rab5 (red), Rab7 (blue), or Rab5/Rab7 (magenta) in the peribouton or perinuclear area (n = 7 muscles). (C and D) Flag-dFz2-Myc localizes similarly to endogenous dFz2 in the body wall muscles. (C) Confocal image of UAS-Flag-dfz2-Myc/+; 24B-GAL4/+ third instar larval NMJs stained with anti-Myc. (D) Single confocal sections through the equators of muscle nuclei (dashed circles) stained with anti-Myc. Upper panel: Immunoreactivity is not detected in the absence of the UAS-Flag-dfz2-Myc transgene (24B-GAL4/+). Lower panel: Nuclear staining is observed when the UAS-Flag-dfz2-Myc transgene is expressed in body wall muscles (UAS-Flag-dfz2-Myc/+; 24B-GAL4/+). (E and F) Muscle expression of Flag-dFz2-Myc rescues the dfz2 NMJ phenotype. (E) Confocal images of third instar NMJ 6/7 stained with antibodies against HRP (green) and Dlg (red) are shown for WT, dfz2C1/Df(3L)ED4782 (dfz2C1/Df), and UAS-Flag-dfz2-Myc/+; dfz2C1/Df(3L)ED4782,24B-GAL4 (Flag-dFz2-Myc rescue-post) larvae. Right panels show higher magnification views of areas marked by white boxes. Arrowheads indicate ghost boutons. (F) Quantification of ghost bouton number per NMJ 6/7 (n = 15–19 NMJs). Data are presented as mean ± SEM. Comparisons are relative to WT unless otherwise indicated. Scale bars: 5 µm (A and D); 20 µm (C and E). **, P < 0.01. n, muscle nucleus.
The endosomal localization of dALS2 led us to hypothesize that dALS2 regulates the FNI pathway by facilitating dFz2 endocytic trafficking. To test this possibility, we first generated a UAS transgene of a Flag-dfz2-Myc construct containing a Flag epitope directly following the signal peptide and a Myc epitope fused to the C terminus of full-length dFz2. When this transgene was expressed in postsynaptic muscles, anti-Myc signals were detected in the postsynaptic region of larval NMJs as well as punctate structures in the cytoplasm and nuclei (Fig. S2, C and D). Furthermore, muscular expression of Flag-dFz2-Myc completely suppressed increased ghost bouton numbers in dfz2 mutants (Fig. S2, E and F), suggesting that Flag-dFz2-Myc behaves similarly to endogenous dFz2 protein in vivo.
Subsequently, we used the UAS-Flag-dfz2-Myc transgene to assess the role of dALS2 in dFz2 internalization and endosomal trafficking. In this assay, filets of WT and dALS2Δ1/dALS2Δ73 larvae expressing UAS-Flag-dfz2-Myc in postsynaptic muscles were prelabeled with an anti-Flag antibody at 4°C, a temperature that blocks endocytosis. Trafficking was then initiated by incubating filets at RT. After permeabilization, three-color immunofluorescence was performed to monitor colocalization of internalized Flag-dFz2-Myc receptors with Rab5 and Rab7. In WT muscles, 10 min after prelabeling, most internalized Flag-dFz2-Myc receptors localized to Rab5+ puncta closely associated with synaptic boutons (Fig. 5, A and C). At this time point, only a few receptors were associated with perinuclear Rab5+/Rab7+ and Rab7+ endosomes (Fig. 5, B and C). Thirty minutes after prelabeling WT muscles, internalized Flag-dFz2-Myc was slightly increased in periboutonal Rab5+ puncta, but substantially increased in perinuclear Rab7+ puncta (Fig. 5, A–C), demonstrating successful transport of Flag-dFz2-Myc from periboutonal early endosomes to perinuclear late endosomes. In dALS2 mutant muscles imaged 10 min after prelabeling, the amount and distribution of internalized Flag-dFz2-Myc were similar to those of WT muscles (Fig. 5, A–C). By sharp contrast, 30 min after prelabeling, the level of Flag-dFz2-Myc in periboutonal Rab5+ puncta was significantly higher, and the level of Flag-dFz2-Myc in perinuclear Rab7+ puncta was significantly lower, in dALS2 mutant larvae relative to WT (Fig. 5, A–C). These results indicate that dALS2 loss selectively impairs dFz2 trafficking to late endosomes, which is associated with transport from NMJ synapses to the perinuclear area.
Figure 5.

dALS2 loss impairs dFz2 transport from early to late endosomes in postsynaptic muscles. Filets of UAS-Flag-dfz2-Myc/+; 24B-GAL4/+ (WT) and UAS-Flag-dfz2-Myc/+; dALS2Δ1/dALS2Δ73,24B-GAL4 (dALS2Δ1/dALS2Δ73) larvae expressing Flag-dFz2-Myc in muscles were prelabeled with anti-Flag antibody at 4°C and subjected to a Flag-dFz2-Myc trafficking assay. (A) Single confocal sections of NMJs 6/7 in WT and dALS2Δ1/dALS2Δ73 larvae at 10 or 30 min after anti-Flag prelabeling. Arrowheads indicate endosomal compartments double positive for internalized Flag-dFz2-Myc and Rab5 in the postsynaptic peribouton area, defined as an area of 1 µm around anti-HRP–stained synaptic boutons (dashed line). (B) Single confocal sections of muscle 6 in WT and dALS2Δ1/dALS2Δ73 larvae at 10 or 30 min after anti-Flag prelabeling. Arrows indicate endosomal compartments triple positive for internalized Flag-dFz2-Myc, Rab5, and Rab7 in the perinuclear area defined as an area 5 µm around the nucleus (dashed line). Arrowheads indicate endosomal compartments double positive for internalized Flag-dFz2-Myc and Rab7. (C) Quantification of the number of internalized Flag-dFz2-Myc puncta associated with Rab5, Rab7, or Rab5/Rab7 in the peribouton and perinuclear area (n = 7 larvae). Scale bars: 2 µm.
To further solidify these findings, we tested the impact of dALS2 knockdown on endosomal trafficking of Wg in BG2-c2 Drosophila neuronal cells. We found that transient expression of Flag-dFz2-Myc in BG2-c2 cells potently induced Wg uptake and that depletion of dALS2 did not affect dFz2-mediated Wg internalization (Fig. S3 A–C). We then assessed trafficking of internalized Wg in control and dALS2 knockdown cells coexpressing Flag-dFz2-Myc and GFP-Rab5 or GFP-Rab7. In control cells, 10 min after Wg treatment, a majority (∼66%) of intracellular Wg puncta were labeled by GFP-Rab5, while only ∼34% of puncta overlapped with GFP-Rab7 (Fig. S3, D, E, and H). In control cells, after 30 min of Wg treatment, Wg puncta preferentially colocalized with GFP-Rab7 rather than GFP-Rab5 (Fig. S3, F–H), suggesting the progression of Wg from early to late endosomes. However, in dALS2 knockdown cells at the same time point, Wg puncta remained highly associated with GFP-Rab5, and Wg colocalization with GFP-Rab7 was significantly decreased relative to control cells (Fig. S3, F–H). The sizes of GFP-Rab5+ and GFP-Rab7+ endosomes remained unchanged by dALS2 knockdown (Fig. S3 I). Collectively, these results confirm that dALS2 loss impairs dFz2 trafficking from early to late endosomes without grossly affecting endosomal dynamics.
Figure S3.

dALS2 is required for Wg trafficking to Rab7+ late endosomes in BG2-c2 cells. (A) RT-PCR analysis of dALS2 expression in control and dALS2 dsRNA BG2-c2 cells using rp49 as a loading control. (B and C) Depletion of dALS2 in BG2-c2 cells does not impair dFz2-mediated Wg internalization. (B) Representative confocal images for control and dALS2 knockdown cells allowed to uptake Wg for 10 min. Cells were transfected with pAc-Flag-dfz2-Myc in the presence or absence of dALS2 dsRNA, serum starved for 6 h, and pulsed with Wg-conditioned medium (100 µg/ml Wg) at 4°C for 30 min. Cells were further incubated in serum-free M3 medium at RT for 10 min and fixed for immunostaining with anti-Wg antibody. Notably, Wg internalization into BG2-c2 cells occurs in a receptor-mediated manner. (C) Quantification of the amount of internalized Wg per cell (n = 15 cells). (D–H) Depletion of dALS2 impairs Wg trafficking from Rab5+ early endosomes to Rab7+ late endosomes. (D–G) Representative confocal images for control and dALS2 knockdown cells allowed to uptake Wg for 10 (D and E) or 30 min (F and G). Cells were cotransfected with pAc-Flag-dfz2-Myc together with either pAc-GFP-Rab5 (D and F) or pAc-GFP-Rab7 (E and G) in the presence or absence of dALS2 dsRNA were treated with Wg-conditioned medium as in B. Cells were further incubated in serum-free medium for 10 or 30 min at RT before immunostaining with anti-Wg and anti-GFP antibodies. Arrowheads indicate Wg puncta overlapping with GFP-Rab5 (D and F) or GFP-Rab7 (E and G). (H) Quantification of Wg-GFP-Rab5 and Wg-GFP-Rab7 colocalization control and dALS2 knockdown cells (n = 15 cells). (I) Quantification of the average areas of Rab5-GFP and Rab7-GFP puncta per cell. Data are presented as mean ± SEM. Comparisons are with the mock-treated control. Scale bars: 5 µm. ***, P < 0.001.
Internalization of malelylated BSA (mBSA) mediated by the class C scavenger receptor SR-CI occurs via clathrin-mediated endocytosis (Gupta et al., 2009), while the BMP receptor Thickveins (Tkv) is internalized via macropinocytosis upon stimulation by the BMP ligand Glass bottom boat (Gbb; Kim et al., 2019). To examine the general role of dALS2 in intracellular receptor trafficking, we addressed the effect of dALS2 knockdown on endosomal trafficking of mBSA and Tkv. First, we pulsed SR-CI-expressing BG2-c2 cells for 1 min with fluorescently labeled mBSA and assessed mBSA trafficking at different chase time points. As assessed at the 4-min chase time point, dALS2 knockdown did not impair internalization of mBSA into Rab5+ early endosomes (Fig. S4, A and D); however, mBSA did not efficiently progress to Rab7+ late endosomes in dALS2 knockdown cells during a longer chase time (19 min; Fig. S4, B–D). Next, we performed an Myc-Tkv trafficking assay in BG2-c2 cells. dALS2 knockdown did not affect Gbb-induced Myc-Tkv enrichment on Rab5+ early endosomes (Fig. S4, E and H), but strongly impaired its progression to Rab7+ late endosomes (Fig. S4, F–H). Thus, dALS2 is required for early to late endosome trafficking of multiple endocytosed receptors, regardless of their internalization routes.
Figure S4.

Depletion of dALS2 disrupts the transport of mBSA and the BMP receptor Tkv from early to late endosomes. (A–D) Knockdown of dALS2 impairs progression of mBSA from early to late endosomes. (A–C) BG2-c2 cells were transfected with a Sr-CI construct in the absence (control) or presence of dALS2 dsRNA and pulsed with Cy3-mBSA for 1 min in serum-free M3 medium containing 1.5 mg/ml BSA. The pulse was chased for 4 min (A) and 19 min (B and C) in serum-free medium. After fixation, cells were immunostained for endogenous Rab5 (A and B) or Rab7 (C). Single confocal sections through the middles of cells are shown. Arrowheads indicate mBSA puncta colocalizing with Rab5 (A and B) or Rab7 (C). (D) Quantification of mBSA-Rab5 or mBSA-Rab7 colocalization in control and dALS2 knockdown cells (n = 15 cells). (E–H) dALS2 knockdown impairs progression of Myc-Tkv from early to late endosomes. (E–G) BG2-c2 cells were transfected with a Myc-Tkv construct alone (control) or together with dALS2 dsRNA. Cells were serum starved for 6 h, prelabeled with anti-Myc antibody at 4°C for 30 min, and subsequently stimulated with Gbb-conditioned medium (50 ng/ml Gbb) at RT for 1 min. After further incubation in serum-free M3 medium for 4 min (E) or 19 min (F and G), cells were immunostained for endogenous Rab5 (E and F) or Rab7 (G). Single confocal sections through the middles of cells are shown. Arrowheads indicate Myc-Tkv puncta colocalizing with Rab5 (E and F) or Rab7 (G). (H) Quantification of Myc-Tkv-Rab5 and Myc-Tkv-Rab7 colocalization (n = 10 cells). Data are presented as mean ± SEM. Comparisons are with mock-treated control. Scale bars: 5 µm. ***, P < 0.001.
dALS2-mediated transport of dFz2 to late endosomes is required for cleavage of its C terminus
To determine the significance of dALS2-mediated endosomal transport in the FNI pathway, we examined the effect of dALS2 deficiency on cleavage of the dFz2-C using the Flag-dfz2-Myc transgene. Western blots of body wall lysates from WT and dALS2Δ1/dALS2Δ73 larvae overexpressing Flag-dFz2-Myc specifically in muscles were probed with anti-Myc antibody, and two protein bands were detected: a ∼75-kD band corresponding to full-length Flag-dFz2-Myc and a ∼9.9-kD band corresponding to the cleaved C-terminal fragment (Fig. 6 A). The ratio of cleaved dFz2-C-Myc to full-length Flag-dFz2-Myc was significantly reduced in dALS2 mutant larvae relative to WT controls (Fig. 6 B). Thus, defective transport of dFz2 from Rab5+ early endosomes to Rab7+ late endosomes in dALS2-null larvae is accompanied by decreased cleavage of the dFz2 receptor.
Figure 6.

Late endosomal dFz2 trafficking is required for Wnt-dependent postsynaptic development. (A) Western blots of body wall muscle extracts from 24B-GAL4/+ (Ab control), UAS-Flag-dfz2-Myc/+; 24B-GAL4/+ (WT), and UAS-Flag-dfz2-Myc/+; dALS2Δ1/dALS2Δ73,24B-GAL4 (dALS2Δ1/dALS2Δ73) larvae, probed with anti-Myc (top), anti-Flag (middle), and anti–β-actin (bottom). Markers are shown in kilodaltons. Note that the level of a ∼9.9-kD band, corresponding to the cleaved C-terminal peptide of Flag-dFz2-Myc (dFz2-C-Myc), is decreased in dALS2Δ1/dALS2Δ73 larvae relative to WT controls. (B) Quantification of cleaved dFz2-C-Myc relative to full-length Flag-dFz2-Myc by densitometric measurements (n = 5 lysates). (C) Western blot analysis of body wall muscle extracts from third instar larvae carrying BG57-GAL4/+ alone (Ab control), or BG57-GAL4 and UAS-Flag-dfz2-Myc with or without (WT) the indicated UAS-RNAi transgene, probed with anti-Myc (top), anti-Flag (middle), and anti–β-actin (bottom) antibodies. (D) Quantification of cleaved dFz2-C-Myc relative to full-length Flag-dFz2-Myc by densitometric measurements (n = 3 lysates). (E and F) Depletion of Rab7, Mon1, Hrs, or VhaAC39-1 causes excessive ghost bouton formation. (E) Confocal images of NMJ 6/7 labeled with anti-HRP and anti-Dlg antibodies are shown for third instar larvae carrying 24B-GAL4 alone (WT) or together with the indicated UAS-RNAi transgene. Right panels show higher magnification views of areas marked by white boxes. Arrowheads indicate ghost boutons. (F) Quantification of ghost bouton numbers in third instar larvae carrying 24B-GAL4 (white background)/D42-GAL4 (gray background) alone (WT) or together with the indicated UAS-RNAi transgene. n = 30 NMJs. Data are presented as mean ± SEM. Scale bar: 20 µm. ***, P < 0.001; **, P < 0.01.
To directly test whether dFz2-C cleavage requires progression of dFz2 to late endosomal compartments, we knocked down Rab7 or Mon1, a critical component of guanine-nucleotide exchange factor for late endosomal Rab7 (Nordmann et al., 2010), in Flag-dFz2-Myc–overexpressing postsynaptic muscles and measured the effect on steady-state levels of dFz2-C-Myc by Western blot. Muscle-specific expression of a UAS-RNAi construct targeting Rab7 or Mon1 using BG57-GAL4 significantly reduced the level of dFz2-C in postsynaptic muscles (Fig. 6, C and D), indicating that C-terminal cleavage of dFz2 requires its trafficking to Rab7+ late endosomes. In addition to the Rab5-to-Rab7 switch, endosomal maturation involves the formation of intraluminal vesicles, which is initiated by the endosomal sorting complex required for transport-0 complex, and endosomal acidification by vacuolar H+-ATPase (V-ATPase; Huotari and Helenius, 2011). Expression of a UAS-RNAi construct targeting the endosomal sorting complex required for transport-0 component Hrs or the V-ATPase subunit VhaAC39-1 in Flag-dFz2-Myc–overexpressing postsynaptic muscles decreased dFz2-C-Myc levels (Fig. 6, C and D), supporting that dFz2-C cleavage requires progression of dFz2 to late endosomal compartments. Consistently, presynaptic but not postsynaptic knockdown of Rab7, Mon1, Hrs, and VhaAC39-1 aberrantly increased the formation of ghost boutons (Fig. 6, E and F), as in dALS2-null mutants.
To further confirm dFz2-C cleavage in late endosomal compartments, Flag-dFz2-Myc–expressing BG2-c2 cells were subjected to a receptor trafficking assay in which the two epitope tags of internalized Flag-dFz2-Myc receptors in comparison with endosomal markers were monitored by immunofluorescence. Ten minutes after Wg treatment, anti-Flag and anti-Myc signals were fully overlapped on Rab5+ early endosomes, but to a much lesser extent on Rab7+ late endosomes (Fig. 7, A and D). At later times (30 and 90 min), overlapping anti-Myc and anti-Flag signals dissociated from Rab5+ early endosomes, colocalizing instead with Rab7+ late endosomes (Fig. 7, B and D), indicating progression of Flag-dFz2-Myc to the late endosomal compartment. Even 90 min after Wg treatment, anti-Flag and anti-Myc signals remained associated on Rab5+ early endosomes. By sharp contrast, Rab7+ late endosomes also contained anti-Myc signal, but progressively lost anti-Flag signal, generating Flag−Myc+ puncta (Fig. 7, B and D). Importantly, a Flag-dFz2-Myc mutant—Flag-dFz2ΔSGKTLESW-Myc—with deletion at the site of predicted dFz2-C cleavage by glutamyl-endopeptidase (Mathew et al., 2005) generated Flag+Myc+Rab7+ endosomes but failed to form Flag−Myc+Rab7+ endosomes (Fig. 7, C and D), suggesting progressive formation of Flag−Myc+Rab7+ late endosomes results from physiologically relevant dFz2-C cleavage. Flag−Myc+ puncta also colocalized with the lysosomal marker Lysotracker (Fig. 7 E). Finally, Flag−Myc+ puncta, but not Flag+Myc+ puncta, colocalized with Importin-β11 (Fig. 7 F), which mediates nuclear import of dFz2-C (Mosca and Schwarz, 2010). Collectively, these data strongly suggest that the C terminus of dFz2 is cleaved in late endosome/lysosome compartments.
Figure 7.
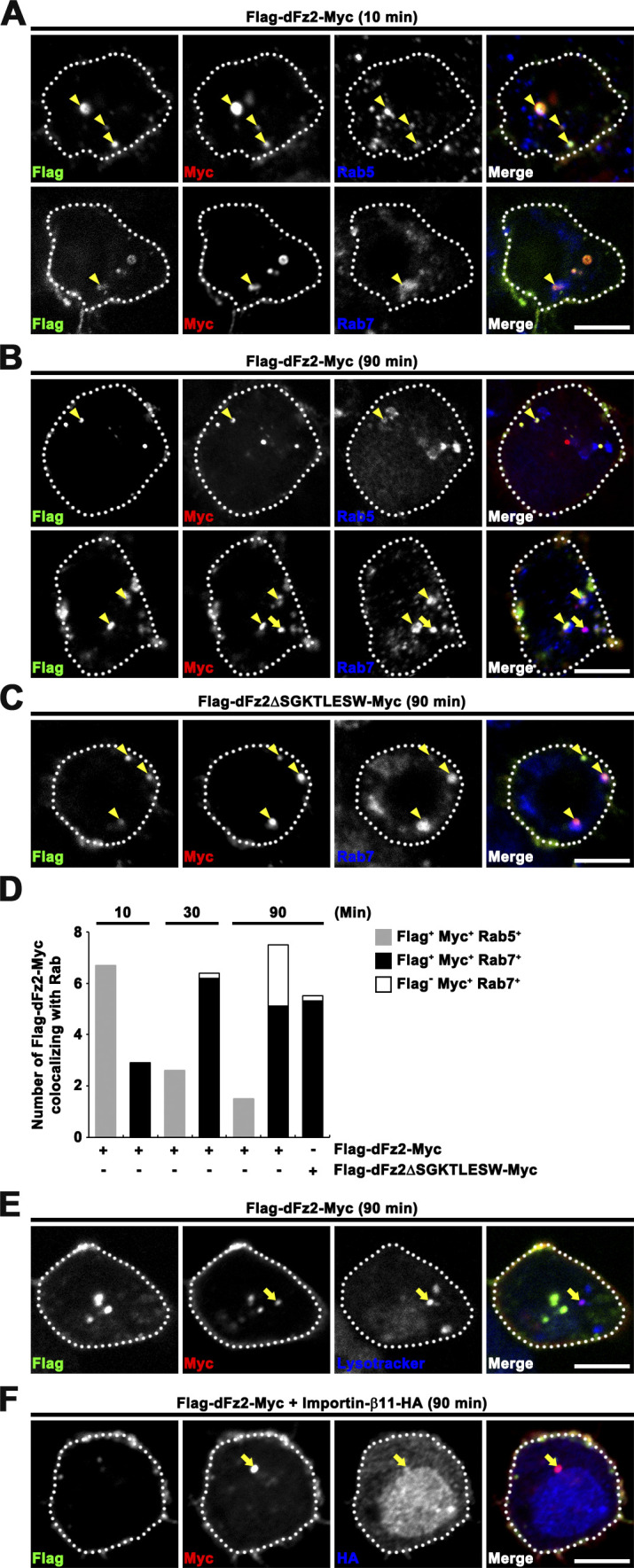
The dFz2 C-terminal fragment is cleaved in late endocytic compartments. (A–C) Single confocal slices through the middle of BG2-c2 cells. Surface Flag-dFz2-Myc (A and B) or Flag-dFz2ΔSGKTLESW-Myc (C) receptors in live BG2-c2 cells were prelabeled with anti-Flag antibody in Wg-conditioned medium (100 ng/ml Wg) at 4°C for 30 min, returned to RT to allow for receptor internalization, and fixed after 10 min (A) or 90 min (B and C). After permeabilization, cells were stained with anti-Myc and anti-Rab5 or anti-Rab7 and subsequently with fluorescent secondary antibodies. Arrowheads indicate Flag+Myc+Rab+ endosomal structures, and arrows indicate Flag−Myc+Rab+ endosomal structures. (D) Quantification of the number of Flag+Myc+ and Flag−Myc+ structures associated with Rab5 or Rab7 for cells shown in A–C. n = 12 cells. Notably, internalized Flag-dFz2-Myc receptors lost anti-Flag signals on Rab7+ late endosomes in a manner dependent on the sequence SGKTLESW. (E) Single confocal slice of Flag-dFz2-Myc–transfected BG2-c2 cells from experiments performed as in B. Cells were labeled with 1 µM Lysotracker for 5 min before cell fixation and subsequently immunostained for Flag and Myc. Flag−Myc+ structures also colocalize with the lysosomal marker Lysotracker (arrow). (F) Single confocal slice of Flag-dFz2-Myc/Importin-β11-HA–transfected BG2-c2 cells from experiments performed as in B. Flag−Myc+ puncta colocalize with Importin-β11-HA (arrow). Scale bar: 5 µm.
Subsequently, we tested if mammalian ALS2 has a conserved role in promoting the FNI pathway. We focused on Frizzled-5 (Fz5), the closest mammalian homologue of dFz2 that contains a conserved sequence (KTLES) for glutamyl-endopeptidase cleavage in the intracellular domain (Mathew et al., 2005). We produced a human Fz5 construct with a GFP tag fused to the Fz5 C terminus (Fz5-GFP) and transfected mouse motor neuron–like NSC-34 cells with this construct in the presence or absence of ALS2 siRNA (Fig. S5 A). In unstimulated control cells, Fz5-GFP was detected predominantly in the cytoplasm as diffuse and punctate patterns (Fig. S5 B). The Fz5 ligand, Wnt5a, stimulated accumulation of Fz5-GFP in nuclear puncta (Fig. S5 B). The number of cells with nuclear Fz5-GFP+ puncta was significantly increased by 5.9-fold upon Wnt5a stimulation relative to control (Fig. S5 C). Importantly, ALS2 knockdown decreased the number of nuclear Fz5-GFP+ cells by 47% in the presence of Wnt5a stimulation (Fig. S5 C). Western blot analysis of whole-cell lysates probed with anti-GFP antibody confirmed that, upon Wnt5a treatment, Fz5-GFP was cleaved to produce a band of ∼32 kD (Fig. S5 D), corresponding to the cleaved dFz5 C terminus fused to GFP. This band was also detected in the nuclear fraction of cells stimulated with Wnt5a, but not in that of unstimulated control cells (Fig. S5 E). Finally, ALS2 knockdown impaired Wnt5a-induced nuclear accumulation of the 32-kD band (Fig. S5, E and F). Together, these results imply that mammalian ALS2 plays a critical role in promoting the FNI pathway.
Figure S5.

Depletion of mammalian ALS2 impairs Wnt5a-induced nuclear translocation of the Fz5 receptor in motor neuron–like NSC-34 cells. (A) RT-PCR analysis of ALS2 expression in NSC-34 cells treated with control or ALS2 siRNA using GAPDH as a loading control. (B–F) NSC-34 cells were transfected with pEGFP-Fz5 and empty vector (−Wnt5a) or pcDNA-Wnt5a (+Wnt5a) in the presence of control or ALS2 siRNA. (B) Representative confocal images for Fz5-GFP–expressing cells under indicated conditions. Notably, Wnt5a-induced nuclear localization of Fz5-GFP is abrogated by ALS2 knockdown. (C) Quantification of the percentage of cells with nuclear GFP signal among all GFP+ cells. Approximately 50 cells were counted per genotype in each of three replicate experiments. (D) Western blot analysis of whole-cell lysates using anti-GFP and anti-GAPDH (loading control). Markers are shown in kilodaltons. Notably, a ∼34-kD band corresponding to the cleaved C-terminal peptide of Fz5-GFP is decreased in ALS2 knockdown cells relative to controls. (E) Western blot analysis of nuclear extracts using anti-GFP, anti–Lamin B1, and anti-GAPDH. The purity of nuclear fractions was confirmed by detection of Lamin B1 (a nuclear fraction marker), but not GAPDH (a cytoplasmic fraction marker). (F) Quantification of cleaved Fz5-GFP levels by densitometric measurements from three independent experiments. Notably, Wnt5a-induced nuclear accumulation of cleaved Fz5-GFP is abrogated by ALS2 knockdown. Data are presented as mean ± SEM. Scale bar: 10 µm. ***, P < 0.001; **, P < 0.01; *, < 0.05.
dALS2 regulates locomotor activity and neuronal survival
The hallmark of ALS is progressive degeneration of cortical and spinal motor neurons, leading to locomotor defects (Taylor et al., 2016). To ascertain the role of dALS2 in locomotor performance, we performed a negative-geotaxis climbing assay in adult flies (20 d). dALS2Δ1/dALS2Δ73 mutant flies exhibited significantly decreased climbing ability relative to WT and revertant controls (P < 0.001; Fig. 8, A and B). This locomotion deficit in mutant animals was rescued by neuronal expression of dALS2 or hALS2, but not NLS-dFz2-C.
Figure 8.

Loss of dALS2 function in neurons causes locomotor dysfunction and progressive brain neurodegeneration. (A) Distance climbed by 20-d-old WT, dALS2Δ1/dALS2Δ73, and C155-GAL4/+; UAS-HA-dALS2,dALS2Δ1/dALS2Δ73 (dALS2 rescue) flies over a 30-s period (n = 9 trials, 100 flies/trial). (B) Quantification of average distance climbed by WT, revertant, dALS2Δ1/dALS2Δ73, C155-GAL4/+; UAS-HA-dALS2,dALS2Δ1/dALS2Δ73 (dALS2 rescue), C155-GAL4/+; UAS-HA-hALS2/+; dALS2Δ1/dALS2Δ73 (hALS2 rescue), and C155-GAL4/UAS-Myc-NLS-dfz2-C; dALS2Δ1/dALS2Δ73 (NLS-dFz2-C rescue) flies (n = 9 trials). (C) Frontal sections (5-µm thickness) of brains from 20-d-old WT and dALS2Δ1/dALS2Δ73 flies were H&E stained. Vacuoles are marked by arrowheads. (D) Quantification of vacuoles with diameter greater than 5 µm in WT and dALS2Δ1/dALS2Δ73 brains at indicated ages (n = 10 brains). (E) Quantification of brain vacuolization in 20-d-old flies of indicated genotypes (n = 10 brains). (F) Confocal images of TUNEL-stained 20-d-old WT and dALS2Δ1/dALS2Δ73 brains. Arrowheads indicate TUNEL+ cells. (G) Quantification of TUNEL+ cells in brains of 20-d-old flies of the indicated genotypes (n = 12 brains). (H) Confocal single slices of 20-d-old dALS2Δ1/dALS2Δ73 brains costained with TUNEL or anti-cleaved Dcp-1, anti-Elav, and anti-Repo. TUNEL+ or cleaved Dcp-1+ cells coexpress the neuronal cell marker Elav (arrowheads) but not the glial cell marker Repo. (I) Single confocal slice of TUNEL-stained 20-d-old OK6-GAL4/UAS-NLS-mCherry; dALS2Δ1/dALS2Δ73 VNCs. Arrowheads indicate a TUNEL+mCherry+ motor neuron. (J) Quantification of TUNEL+mCherry+ cell number per VNC (n = 9 VNCs). Data are presented as mean ± SEM. Comparisons are with WT unless otherwise indicated. Scale bars: 50 µm (C and F); 10 µm (H and I). ***, P < 0.001; **, P < 0.01; *, P < 0.05.
To determine if the locomotion deficit of dALS2 mutants was associated with neurodegeneration, we analyzed fly brain morphology by performing hematoxylin and eosin (H&E) staining. Brains of newly eclosed dALS2Δ1/dALS2Δ73 mutants were anatomically and histologically normal; however, aged dALS2 mutants exhibited numerous vacuoles in the cortex and neuropils (Fig. 8 C), a hallmark of degeneration in the Drosophila brain (Muqit and Feany, 2002). At 10 and 20 d of age, the average number of brain vacuoles in dALS2 mutant flies was significantly increased by 2.4-fold and 2.9-fold, respectively, relative to WT controls (P < 0.001; Fig. 8 D). The brains of 30- and 40-d-old dALS2 flies exhibited further increased vacuolization, while age-matched controls were well preserved (Fig. 8 D). To characterize age-related degeneration in dALS2 mutants, we assessed apoptotic cell death in 20-d-old brains. The number of TUNEL+ cells per brain was significantly increased in dALS2 mutant flies relative to WT (WT: 12.92 ± 1.44; dALS2Δ1/dALS2Δ73: 25.50 ± 1.64; P < 0.001; Fig. 8, F and G). To determine the nature of apoptotic cells, dALS2 mutant brains were stained with TUNEL or anti-cleaved death caspase-1 (Dcp-1) in combination with antibodies against the neuronal marker anti-Elav and the glial marker Repo. TUNEL+ and Dcp-1+ cells expressed Elav, but not Repo (Fig. 8 H), suggesting that apoptotic cells are predominantly neurons. Notably, dALS2 deficiency also induced apoptotic cell death in OK6-GAL4/UAS-NLS-mCherry–labeled motor neurons residing in the ventral nerve cord (VNC; Fig. 8, I and J). Finally, neuronal expression of dALS2 or hALS2, but not NLS-dFz2-C, in dALS2 mutants rescued the phenotypes of brain vacuolization and neuronal cell death (Fig. 8, E and G). This finding suggests that dALS2 regulates neuronal cell survival in a dFz2-independent manner. Together, these results demonstrate a causal relationship between locomotor deficits and neurodegeneration in aged dALS2 mutants.
Discussion
In the present study, we establish a role for dALS2 in synaptic maturation at the NMJ, a model of glutamatergic synapses. We demonstrate that dALS2 regulates postsynaptic development by activating the FNI pathway of noncanonical Wg signaling. First, we showed that loss of dALS2 led to postsynaptic phenotypes, including excess ghost boutons and a structurally abnormal SSR, which have previously been associated with disruption of the postsynaptic FNI pathway (Ataman et al., 2006; Harris et al., 2016; Mathew et al., 2005; Mosca and Schwarz, 2010). Second, dALS2 exhibited transheterozygous interactions with wg and dGRIP, resulting in excess ghost bouton formation, and decreased nuclear levels of dFz2-C. Third, an NLS-tagged dFz2-C completely rescued the postsynaptic phenotypes of dALS2 mutants. Together, these findings suggest that impairment of the FNI pathway is responsible for postsynaptic defects observed in dALS2 mutants.
dALS2 mutants exhibit normal evoked EJP amplitude, despite decreased mEJP amplitude, indicating a homeostatic increase in presynaptic neurotransmitter release (quantal content). The balance between GluRIIA and GluRIIB and the association of GluR with synaptic homeostasis at NMJ terminals have recently been further elucidated (Hong et al., 2020; Zhao et al., 2020). GluRIIB overexpression reduces the level of GluRIIA (Zhao et al., 2020), while dALS2 loss selectively increases GluRIIB levels without affecting GluRIIA. Moreover, T-bar clustering elicited by reducing the GluRIIC level (Hong et al., 2020) is not present in dALS2 mutants, suggesting that canonical homeostatic control may be also perturbed by dALS2 loss.
How might dALS2 regulate the FNI pathway? Studies have shown that mammalian ALS2 regulates early endosomal fusion and trafficking by activating Rab5 (Devon et al., 2006; Otomo et al., 2003; Topp et al., 2004). We thus assessed the potential role of dALS2 in endocytic dFz2 trafficking. Although dFz2 transport from synapses to the nucleus is essential for activation of the postsynaptic FNI pathway (Ataman et al., 2006), the exact nature of this retrograde transport remains unclear. In the present study, we demonstrate that retrograde dFz2 transport occurs via trafficking from Rab5+ early endosomes to Rab7+ late endosomes. We further demonstrate that dALS2 is required for Rab5-to-Rab7 conversion of dFz2 endosomes.
The above findings raise a key question as to how the Rab5-to-Rab7 conversion of dFz2 endosomes influences the FNI pathway. Our data suggest that cleavage of the signaling-competent C-terminal fragment of dFz2 depends on its trafficking to Rab7+ late endosomes. First, muscle-specific depletion of central regulators of late endosome formation and maturation (Hrs, Rab7, Mon1, and VhaAC39-1) inhibited dFz2-C cleavage and increased ghost bouton formation at the NMJ. Second, in a receptor trafficking assay, Flag-dFz2-Myc receptors internalized in response to Wg stimulation, progressively inducing formation of Flag−Myc+ dFz2-C puncta colocalizing with Rab7 or Lysotracker, but not Rab5, implying physical separation of the N and C termini of dFz2 in the late endosome/lysosome compartment. However, a Flag-dFz2-Myc mutant with deletion at the site of dFz2-C cleavage formed Flag+Myc+Rab7+ endosomes but did not form Flag−Myc+Rab7+ endosomes, indicating that Flag−Myc+ dFz2-C puncta arose from physiologically relevant cleavage of Flag-dFz2-Myc. Finally, Flag−Myc+ dFz2-C puncta overlapped with Importin-β11, which mediates nuclear import of dFz2-C (Mosca and Schwarz, 2010). We thus propose that C-terminal cleavage of dFz2 occurs in late endocytic compartments, such as late endosomes and lysosomes.
It is presently unclear why C-terminal cleavage of dFz2 requires progression of dFz2 to late endocytic compartments; however, the necessity of V-ATPase implies that acidic luminal pH in late endosomes and lysosomes creates an environment that is favorable for proteolytic dFz2 cleavage. V-ATPase–mediated acidification is thought to promote γ-secretase proteolysis (S3 cleavage) of Notch by facilitating ectodomain shedding, which can be accomplished by inducing denaturation or acid hydrolase–dependent degradation of the Notch extracellular domain (Schnute et al., 2018). Future investigations will determine whether ectodomain shedding is required for C-terminal cleavage of dFz2.
We found that dALS2 mutant flies recapitulated some key phenotypes of ALS, including adult-onset motor impairment and brain neurodegeneration. Intriguingly, these defects were suppressed by neuronal expression of dALS2, but not NLS-dFz2-C, indicating that dALS2 regulates neuronal cell survival in a dFz2-independent manner. These findings raise an important question as to the cellular mechanisms of neuronal cell death in dALS2 mutant flies and ALS2 patients. Increasing evidence suggests a central role for impaired receptor and endosomal trafficking in the pathogenesis of ALS (Burk and Pasterkamp, 2019). For example, modulation of endosomal trafficking by constitutively active Rab5 or chemical modulators of Rab5 effectors ameliorates neurodegeneration caused by an intronic GGGGCC hexanucleotide repeat expansion in the C9ORF72 gene (Shi et al., 2018), which is the most common cause of ALS and frontotemporal dementia (Renton et al., 2014). Moreover, loss-of-function mutations in charged multivesicular body protein 2B and the valsolin-containing protein (VCP or ALS14) gene, both of which are involved in endosome maturation, have also been linked to ALS (Cox et al., 2010; Johnson et al., 2010), further supporting the potential connection between endosomal trafficking defects and neurodegeneration in ALS. Given the generalized role of dALS2 in controlling endosomal trafficking, defining the detailed mechanism by which endosomal trafficking defects trigger or contribute to neuronal cell death in ALS2 is of paramount importance.
Materials and methods
Drosophila stocks
All Drosophila stocks were maintained at 25°C on normal food. The WT flies were w1118. The wgI-17, Df(3L)ED4782 (a deficiency uncovering the dfz2 locus), UAS-NLS-mCherry, UAS-Rab7-RNAi, UAS-Hrs-RNAi, and UAS-VhaAC39-1-RNAi flies were obtained from the Bloomington Drosophila Stock Center. UAS-dALS2-RNAi and UAS-Mon1-RNAi flies were obtained from the Vienna Drosophila Resources Center. The UAS-myc-NLS-dfz2-C flies were a kind gift from Thomas Schwarz (Boston Children’s Hospital, Boston, MA). The dfz2C1 flies were obtained from Makoto Sato (Kanazawa University, Kanazawa, Japan). Stephan Sigrist (Freie Universität Berlin, Berlin, Germany) provided the dGRIPex36 flies. Transgenic expression was driven by the following GAL4 drivers: D42-GAL4, OK6-GAL4, 24B-GAL4, BG57-GAL4, and Mhc-GS-GAL4 (Brand and Perrimon, 1993; Budnik et al., 1996; Osterwalder et al., 2001; Sanyal, 2009; Yeh et al., 1995).
A dALS2-null allele, dALS2Δ1, was generated as described previously via a CRISPR/Cas9-assisted nonhomologous end-joining strategy (Gratz et al., 2013). Briefly, two pU6-BbsI-chiRNA plasmids encoding gRNA sequences targeting the first and fourth exon sequences of dALS2 (5′-GGTGCCAGTAGTGCCTAGCGTGG-3′ and 5′-GCCTACGAACACGCCATGGCAGG-3′, respectively) were generated using a standard protocol (Gratz et al., 2013), and a mixture of these plasmids was injected into y2,cho2,v1; nos-Cas9 Drosophila embryos (BestGene). Flies reared from the injected embryos were individually crossed to the TM6B balancer line, and all isolated engineered alleles were analyzed by PCR across the target sites for the two gRNAs. An additional dALS2-null allele, dALS2Δ73, was generated via Δ2-3 transposase-mediated imprecise excision of G4607, an enhanced promoter insertion in the dALS2 locus (GenExel). The molecular lesions in dALS2Δ1 and dALS2Δ73 were characterized by sequencing across the deletions.
Transgenic flies carrying UAS-HA-dALS2, UAS-HA-hALS2, or UAS-Flag-dfz2-Myc were generated in the w1118 background using standard procedures.
Molecular biology
Full-length cDNA for dALS2 was obtained from the Drosophila Genomic Resources Center (DGRC; clone ID: LD33266). The entire open reading frame of hALS2 was amplified from human HeLa cell cDNA by PCR and subcloned into pGEM-T easy vector (Promega). For transgenic rescue experiments, dALS2 and hALS2 cDNA inserts were moved into the pUAST-HA vector (Nahm et al., 2010a), termed UAS-HA-dALS2 and UAS-HA-hALS2, respectively. Full-length cDNA for dfz2 was obtained from the DGRC (clone ID: LD10629). For transgenic expression of Flag-dFz2-Myc in animals, dfz2 cDNA with an N-terminal Flag tag inserted immediately after the signal peptide was generated by two-stage PCR-based mutagenesis and introduced into the pGEM-T vector. The Flag-dfz2 insert was reamplified by PCR to introduce a C-terminal Myc tag and subsequently ligated to the pUAST vector, termed UAS-Flag-dfz2-Myc. For transient expression in Drosophila BG2-c2 neuronal cells, the UAS-Flag-dfz2-Myc insert was moved into the pAc5.1 vector (Invitrogen), termed pAc-Flag-dfz2-Myc. This construct was subsequently used to generate pAc-Flag-dfz2 ΔSGKTLESW-Myc via PCR-based mutagenesis. Full-length cDNAs for Sr-CI and Importin-β11 were amplified by RT-PCR of total RNA extracted from S2R+ cells and cloned into the pGEM-T Easy vector. The Sr-CI cDNA insert was subcloned into pAc5.1 to produce pAc-Sr-CI, and the Importin-β11 insert was inserted into pAc5.1-HA to produce pAc-Importin-β11-HA. For transient expression in mouse motor neuron–like NSC-34 cells, full-length cDNAs for human Wnt5a and Fz5 (Addgene) were subcloned into the pcDNA3.1 (Invitrogen) and pEGFP-N2 (Clontech) vectors to generate pcDNA-Wnt5a and pEGFP-Fz5, respectively.
For dALS2 depletion in BG2-c2 cells, double-stranded RNA (dsRNA) for dALS2 was synthesized as described previously (Kim et al., 2019). To generate a DNA template for in vitro transcription, primers containing the T7 promoter sequence upstream of the following dALS2 sequences were used: 5′-TGGCGGATACACACTTCAAAACCA-3′ and 5′-CCACCGCCTCTCCAAATCCATTGA-3′. For ALS2 depletion in NSC-34 cells, the following duplex siRNA sequences for mouse ALS2 were used: 5′-GAGAGACAAGAGGACCAU-3′ and 5′-AUGGUCCUCUUUGUCUGUC-3′.
For molecular characterization of dALS2 deletions, the following primers were used for genomic PCR: 5′-CACGTGCTCATATGCGGCCTTCAG-3′ and 5′-CGTCACTTTTACCCGAACACCCTTAC-3′. To assess the effect of dALS2 deletions on the expression of dALS2, CG11248, and rp49, total RNA isolated from larvae was reverse transcribed into cDNA. cDNAs were quantified by PCR using the following primers: dALS2: 5′-CGCATCACTCCATGGCCG-3′ and 5′-GATGCGCAAGGTGTGGCT-3′; CG11248: 5′-CAAAGGCTGCAAGGCCAG-3′ and 5′-GGCGGAAATACCCTCGCC-3′; and rp49: 5′-CACCAGTCGGATCGATATGC-3′ and 5′-CACGTTGTGCACCAGGAACT-3′.
Cell transfection and production of Wg-conditioned medium
BG2-c2 cells (DGRC) were maintained at 25°C in Shields and Sang M3 medium (Sigma-Aldrich) supplemented with 10% heat-inactivated FBS, 1 g/liter yeast extract, and 2.5 g/liter bactopeptone, 10 µg/ml insulin, 100 U/ml penicillin, and 100 µg/ml streptomycin (Gibco). BG2-c2 cells were transfected in serum-free M3 medium using Cellfectin II according to the manufacturer’s protocol (Invitrogen). NSC-34 cells were grown in DMEM (Gibco) supplemented with 10% FBS (Gibco), 100 U/ml penicillin, and 100 µg/ml streptomycin. Cells were transfected in serum-free DMEM using Lipofectamine 2000 (Thermo Fisher Scientific).
To produce Wg-conditioned medium, S2 cells stably expressing Wg (S2-Tub-wg; DGRC) were maintained in M3 medium supplemented with 10% FBS (vol/vol), 1 g/liter yeast extract, 2.5 g/liter bactopeptone, 100 U/ml penicillin, 100 µg/ml streptomycin, and 125 µg/ml hygromycin (Sigma-Aldrich). The conditioned medium was produced in serum-free M3 medium and centrifuged at 10,000 g for 5 min and the supernatant collected. The supernatant concentration of Wg was determined by immunoblotting using a mouse anti-Wg antibody (1:500; Cat# 4D4; Developmental Studies Hybridoma Bank [DSHB]), and Wg-HA–conditioned medium was used as a protein concentration standard. Multiple Tag Fusion (GenScript) was used to determine Wg-HA concentration by immunoblotting using rabbit anti-HA (1:1,000; Cat# 3724; Cell Signaling Technology). BG2-c2 cells were stimulated with conditioned medium containing 100 ng/ml Wg.
Generation of GluRIIB and GluRIIC antibodies
Rabbit anti-Drosophila GluRIIB and GluRIIC antibodies were generated by Afrontier against the C-terminal residues of GluRIIB (ASSAKKKKKTRRIEK) and GluRIIC (QGSGSSSGSNNAGRGEKEARV), respectively. Immune sera were purified using IgG affinity chromatography.
Western blotting
To characterize the transgenic Flag-dFz2-Myc receptor, body wall muscles were collected from third instar larvae overexpressing UAS-Flag-dfz2-Myc under the control of the 24B-GAL4 muscle driver. To assess the effects of dALS2 or endosome maturation on Flag-dFz2-Myc cleavage, UAS-Flag-dfz2-Myc expression was driven in the dALS2Δ1/dALS2Δ73 background under the control of 24B-GAL4 or together with UAS-Rab7-RNAi, UAS-Mon1-RNAi, UAS-Hrs-RNAi, or UAS-VhaAC39-1-RNAi under the control of BG57-GAL4. Samples were homogenized in SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, 2.88 mM β-mercaptoethanol, and 0.02% bromophenol blue), boiled for 5 min, and centrifuged at 13,000 g for 5 min. Supernatant samples were subjected to 12% SDS-PAGE and transferred to polyvinylidene fluoride membrane (Merck Millipore). Blots were incubated overnight at 4°C with mouse anti-Flag (1:2,000; Cat# F1804; Sigma-Aldrich), rabbit anti-Myc (1:1,000; Cat# 2278; Cell Signaling Technology), or rabbit anti–β-actin (1:1,000; Cat# A2066; Sigma-Aldrich) antibodies, and then for 1 h at RT with HRP-conjugated anti-rabbit or anti-mouse secondary antibody (1:4,000; Cat# 111–035-144 or 715–035-150; Jackson ImmunoResearch Laboratories) in blocking solution (5% skim milk/0.1% Tween-20/TBS). Protein bands were visualized by ECL reagents (iNtRON Biotechnology).
NSC-34 cells were transfected with pEGFP-Fz5 and either pcDNA-Wnt5a or empty vector in the presence of control or ALS2 siRNA. For whole-cell lysates, cells were lysed in lysis buffer (50 mM Tris-HCl, pH 7.4, 0.5% Triton X-100, 150 mM NaCl, and protease inhibitor mixture). Nuclear fractions were isolated using a NE-PER nuclear and cytoplasmic extraction reagent kit (Thermo Scientific Scientific). Whole-cell lysates and nuclear fractions were analyzed by Western blotting using rabbit anti-GFP (1:1,000; Cat# A11122; Invitrogen), rabbit anti-Lamin B1 (1:1,000; Cat# ab16048; Abcam), and rabbit anti-GAPDH (1:1,000; Cat# sc-25778; Santa Cruz Biotechnology).
Larval NMJ immunohistochemistry and quantification
Wandering third instar larvae were dissected in Ca2+-free HL3 saline (70 mM NaCl, 5 mM KCl, 20 mM MgCl2, 10 mM NaHCO3, 5 mM trehalose, 115 mM sucrose, 5 mM Hepes, pH 7.2) and fixed for 30 min in 4% formaldehyde/PBS or for 7 min in Bouin’s fixative (Sigma-Aldrich). Fixed larvae were washed in 0.1% Triton X-100/PBS and then sequentially incubated with primary and secondary antibodies in 0.1% Triton X-100/PBS containing 0.2% BSA as described previously (Kim et al., 2019). The following primary antibodies were used: mouse anti-Dlg (1:500; Cat# 4F3; DSHB), mouse anti-Brp (1:10; Cat# nc82; DSHB), mouse anti-GluRIIA (1:50; Cat# 8B4D2; DSHB), mouse anti-Futsch (1:5; Cat# 22C10; DSHB), mouse anti-Synapsin (1:25; Cat# 3C11; DSHB), mouse anti-Csp (1:50; Cat# 1G12; DSHB), rabbit anti-Rab5 (1:200; Cat# ab31261; Abcam), mouse anti-Rab7 (1:50; Cat# Rab7; DSHB), rat anti-HA (1:200; Cat# 11867423001; Roche), mouse anti-Myc (1:200; Cat# 551101; BD Biosciences), rabbit anti-GluRIIB (1:2,000; custom-produced by Afrontier), rabbit anti-GluRIIC (1:2,000; custom-produced by Afrontier), and rabbit anti–dFz2-C (1:200; a kind gift from Vivian Budnik, University of Massachusetts, Worcester, MA). The following secondary antibodies from Jackson ImmunoResearch Laboratories were used at 1:200: FITC-, Cy3-, and Cy5-conjugated donkey anti-mouse (Cat# 715–095-151, 715–165-151, and 715–175-151, respectively); FITC-, Cy3-, and Cy5-conjugated donkey anti-rabbit (Cat# 711–095-152, 711–165-152, and 711–175-152, respectively); FITC- and Cy3-conjugated donkey anti-rat (Cat# 712–095-153 and 712–165-153, respectively); and FITC-, Cy5-, and DyLight405-conjugated goat anti-HRP (Cat# 123–195-021, 123–175-021, and 123–475-021, respectively). Labeled larvae were mounted in SlowFade mounting medium (Molecular Probes), and images of NMJs and muscle nuclei were acquired as confocal z stacks with an LSM 800 confocal microscope (Carl Zeiss) with a C-Apo 40× 1.2 NA W, Plan-Apo 63× 1.4 NA Oil, or Plan-Apo 100× 1.4 NA Oil objective at RT. Images were processed with the Zen microscopy software and Adobe Photoshop. For comparison between genotypes, samples were processed simultaneously and imaged using identical acquisition settings.
For quantification of NMJ morphology, NMJs on muscles 6/7 from segment A3 were selected for analysis. To quantify bouton numbers, ghost bouton numbers, and bouton size, maximum intensity projection images of NMJs 6/7 costained with anti-Dlg or anti-Csp and anti-HRP were produced from confocal z stacks collected at 1-µm intervals. Larval muscle size was not significantly different among the genotypes evaluated in this study. Ghost boutons were identified by the presence of anti-HRP–stained boutons lacking postsynaptic Dlg staining. Bouton size was quantified by measuring the bouton diameter for one terminal and four adjacent nonterminal Ib boutons on two different NMJ 6/7 branches per animal using ImageJ. The proportion of Ib boutons with Futsch-immunoreactive loops was quantified using anti-HRP and anti-Futsch costaining. Synaptic levels of GluR receptors were quantified using anti-HRP and anti-GluRIIA or anti-GluRIIB costaining. The synaptic area was delimited by HRP immunoreactivity, and the average GluRIIA or GluRIIB signal intensity within the synaptic area was normalized to the average HRP signal intensity. Nuclear dFz2-C was quantified in muscle 6 by counting distinct dFz2-C immunoreactive spots in the three nuclei closest to the NMJ, as described previously (Mathew et al., 2005). The nuclear boundary was defined by DAPI staining.
Fluorescence-based internalization and trafficking studies
The Flag-dFz2-Myc trafficking assay in dissected third instar larvae was performed as described previously (Ataman et al., 2006; Mathew et al., 2005). Briefly, WT and dALS2Δ1/dALS2Δ73 larvae expressing Flag-dFz2-Myc specifically in muscles were dissected in HL3 solution containing 0.1 mM Ca2+ and incubated at 4°C in 0.1 mM Ca2+/HL3 containing 5 µg/ml rat anti-Flag (Cat# 637302; BioLegend) for 1 h to label surface Flag-dFz2-Myc receptors. Samples were then washed with 0.1 mM Ca2+/HL3 to remove unbound antibody and incubated at RT for 10 or 30 min to allow internalization of the labeled receptors. Samples were fixed in 4% formaldehyde/PBS for 30 min and incubated with 20 µg/ml anti-rat IgG secondary antibody (Cat# 712–005-150; Jackson ImmunoResearch Laboratories) under nonpermeant conditions to mask external Flag-dFz2-Myc. Samples were permeabilized in 0.1% Triton X-100/PBS and incubated with rabbit anti-Rab5 (1:200; Cat# ab31261; Abcam) and mouse anti-Rab7 (1:50; Cat# Rab7; DSHB), followed by incubation with FITC-conjugated donkey anti-rat, Cy3-conjugated donkey anti-rabbit, and Cy5-conjugated donkey anti-mouse secondary antibodies, together with DyLight405-conjugated goat anti-HRP antibody (1:200; Cat# 712–095-153, 711–165-152, 715–175-151, and 123–475-021, respectively; Jackson ImmunoResearch Laboratories). After 10 and 30 min of internalization, the majority of internalized Flag-dFz2-Myc was detected in discrete punctate structures. For each sample, images were captured with an LSM 800 confocal microscope using a Plan-Apo 63× 1.4 NA Oil objective at RT. To quantitatively monitor endosomal trafficking of internalized Flag-dFz2-Myc, the number of Flag-immunoreactive puncta (green) containing Rab5 (red) and/or Rab7 (blue) was manually counted in a 1-µm area surrounding anti-HRP–stained synaptic branches (peribouton area) and a 5-µm area surrounding the nucleus (perinuclear area) using ImageJ.
The Flag-dFz2-Myc trafficking assay in BG2-c2 cells was performed to determine the intracellular location of dFz2-C cleavage. BG2-c2 cells were transiently transfected with pAc-Flag-dFz2-Myc or pAc-Flag-dFz2ΔSGKTLESW-Myc—a dFz2 mutant deleting the coding sequences for the glutamyl-endopeptidase cleavage site—in the absence or presence of pAc-Importin-β11-HA. At 48 h post-transfection, cells were incubated for 6 h in serum-free M3 medium containing 100 µg/ml cycloheximide (Sigma-Aldrich), a protein synthesis inhibitor. Cells were incubated in Wg-conditioned medium (100 ng/ml Wg) containing 5 µg/ml rat anti-Flag (Cat# 637302; BioLegend) at 4°C for 30 min to label surface Flag-dFz2-Myc receptors. Cells were then rinsed in M3 medium and incubated at RT for 10, 30, or 90 min to allow for internalization of labeled receptors. In some experiments, Lysotracker (Molecular probe) was added to a final concentration of 1 µM, 85 min after Wg stimulation. Cells were fixed in 4% formaldehyde/PBS for 10 min and subsequently permeabilized with 0.2% Triton X-100/PBS for 10 min. After blocking with 0.2% BSA/PBS for 10 min, cells were incubated overnight with the following combinations of primary antibodies in 0.2% BSA/PBS at 4°C: mouse anti-Myc (1:100; Cat# 551101; BD Biosciences); mouse anti-Myc (1:100; Cat# 551101; BD Biosciences) and rabbit anti-Rab5 (1:200; Cat# ab31261; Abcam); rabbit anti-Myc (1:1,000; Cat# 2278; Cell Signaling Technology) and mouse anti-Rab7 (1:50; Cat# Rab7; DSHB); and mouse anti-Myc (1:100; Cat# 551101; BD Biosciences) and rabbit anti-HA (1:200; Cat# 3724; Cell Signaling Technology). After washing with PBS, cells were incubated with FITC-conjugated donkey anti-rat, Cy3- or Cy5-conjugated donkey anti-rabbit, and Cy3- or Cy5-conjugated donkey anti-mouse secondary antibodies (1:200; Cat# 712–095-153, 711–165-152, 711–175-152, 715–165-151, and 715–175-151, respectively; Jackson ImmunoResearch Laboratories) in 0.2% BSA/PBS for 1 h. For each cell, a z stack of an optical section (0.23-µm thickness) was captured with an LSM 800 confocal microscope using a Plan-Apo 63× 1.4 NA Oil objective at RT. The numbers of intracellular Flag+Myc+Rab5+, Flag+Myc+Rab7+, or Flag−Myc+Rab7+ puncta per cell were manually counted using ImageJ.
For the Wg trafficking assay, BG2-c2 cells were transfected with pAc-Flag-dfz2-Myc in the presence or absence of dALS2 dsRNA. Cells at 48 h post-transfection were serum starved in M3 medium for 6 h and subsequently incubated at 4°C in Wg-conditioned medium (∼100 ng/ml Wg) for 30 min. After removing unbound Wg with ice-cold M3 medium, cells were incubated in M3 medium at RT for 10 min to allow Wg internalization. Cells were fixed in 4% formaldehyde/PBS for 10 min, washed three times with PBS, and permeabilized with 0.2% Triton X-100/PBS for 10 min. Cells were incubated with mouse anti-Wg (1:5; Cat# 4D4; DSHB) and rabbit anti-Flag (1:200; Cat# F7425; Sigma-Aldrich), and then with Cy3-conjugated donkey anti-rabbit and FITC-conjugated donkey anti-mouse secondary antibodies (1:200; Cat# 711–165-152 and 715–095-151, respectively; Jackson ImmunoResearch Laboratories). For each cell, a z stack of 2D images (0.23-µm thickness) was acquired with a Plan-Apo 63× 1.4 NA Oil objective at RT. For quantification of Wg internalization, total green fluorescence per cell was determined by integrating green fluorescence on all optical sections after correcting for background fluorescence. Transfected cells were identified by the presence of anti-Flag signals (red). To monitor endosomal Wg trafficking, a Wg internalization assay was performed in BG2-c2 cells transfected with pAc-Flag-dfz2-Myc and pAc-GFP-Rab5 or pAc-GFP-Rab7. After 10 or 30 min of Wg internalization, cells were processed for immunostaining for GFP-Rab5/GFP-Rab7, Wg, and Flag-dFz2-Myc. For quantitative analysis of endocytic Wg trafficking, the number of GFP-Rab5+/GFP-Rab7+ Wg puncta was manually counted in Flag-dFz2-Myc–expressing cells using ImageJ.
For the mBSA trafficking assay, BG2-c2 cells were transfected with pAc-Sr-CI in the presence or absence of dALS2 dsRNA. Cells at 48 h post-transfection were treated with 10 µg/ml Cy3-mBSA in serum-free M3 medium supplemented with 1.5 mg/ml BSA at RT for 1 min (pulse). The pulse was chased for 4 or 19 min in serum-free M3 medium. Cells were fixed in 4% formaldehyde/PBS for 10 min and subsequently permeabilized with 0.1% saponin/PBS for 10 min. Cells were stained for Rab5 and Rab7 and imaged for quantification of colocalization between mBSA and Rab5 or Rab7, as described for the Wg trafficking assay.
For the Myc-Tkv receptor trafficking assay, BG2-c2 cells were transfected with pAc-Myc-Tkv in the presence or absence of dALS2 dsRNA. Cells at 48 h post-transfection were serum starved in M3 medium for 6 h and subsequently incubated at 4°C with 2.5 µg/ml mouse anti-Myc antibody or rabbit anti-Myc antibody for 30 min to label surface Myc-Tkv. Cells were treated at RT for 1 min with Gbb-conditioned medium (50 ng/ml Gbb) and further incubated for 4 or 19 min in serum-free M3 medium. Cells were stained for Myc and Rab5 or Rab7 and imaged for quantification of colocalization between Myc-Tkv and Rab5 or Rab7, as described for the Wg trafficking assay.
For quantitative analysis of nuclear Fz5-GFP, NSC-34 cells were transfected with pEGFP-Fz5 and empty vector or pcDNA-Wnt5a in the presence of control or ALS2 siRNA. Cells at 72 h post-transfection were fixed, stained for nuclei (1:10,000; Hoechst; Thermo Fisher Scientific), and imaged for quantification of cells with nuclear GFP signal among all GFP+ cells. Images of NSC-34 cells were acquired as confocal z stacks with a Leica TCS SP8 confocal microscope (Leica) with a HCX Apo 63× 0.9 NA W objective at RT. Images were processed with the Leica LASX software and Adobe Photoshop.
EM
Wandering third instar larvae were dissected in Ca2+-free HL3 saline at RT. Larval fillets were fixed in 1% glutaraldehyde/4% paraformaldehyde/0.1 M cacodylic acid (pH 7.2) solution at 4°C for 12 h. The samples were then washed with 0.1 M cacodylic acid (pH 7.2) solution three times, post-fixed in 1% OsO4/0.1 M cacodylic acid (pH 7.2) solution at RT for 3 h and subjected to serial dehydration from 30% to 100% ethanol. Samples were subsequently incubated in propylene oxide, a mixture of propylene oxide and resin, and pure resin before being embedded in 100% resin. Type Ib boutons at NMJ 6/7 were imaged with Tecnai G2 Spirit TWIN (FEI Company) and a Gatan CCD Camera (794.10.BP2 MultiScan) at ≥4,400× magnification. We analyzed the following parameters as described previously (Dani et al., 2014; Nahm et al., 2010b): SSR density (number of SSR layers/µm), postsynaptic pocket depth (μm), SSR thickness (μm), vesicle density (number of vesicles/µm2), vesicle diameter (nm), active zone density (number of active zones/µm2), and number of mitochondria per bouton.
NMJ electrophysiology
Wandering third instar larvae were dissected in ice-cold Ca2+-free HL3 saline and subsequently incubated with 0.5 mM Ca2+ HL3 for 5–10 min. Whole-muscle recording was performed on muscle 6 in segment A3 as described previously (Yao et al., 2017). Intracellular electrodes with a resistance of ∼40 MΩ filled with 3 M KCl solution were used for recording. We analyzed recordings from muscles with resting membrane potential less than −60 mV and input resistance more than 5 MΩ. EJPs were elicited by 0.2-Hz stimulation. Stimulus pulses were fixed at 0.5-ms duration using pClamp 10.6 software (Axon Instruments). Spontaneous mEJPs were recorded for 3 min. Both EJPs and mEJPs were amplified with an Axoclamp 900A amplifier (Axon Instruments) under bridge mode and filtered at 10 kHz. The mean amplitude of EJPs and the mean amplitude and frequency of mEJPs were analyzed using Clampfit software (Molecular Devices).
Histology, TUNEL staining, and immunostaining of adult brains
Paraffin sectioning and H&E staining of adult brains were performed as previously described (Heo et al., 2017). Heads from adult flies at 2, 10, 20, 30, and 40 d posteclosion were fixed overnight in 4% formaldehyde/PBS and embedded in paraffin. Frontal sections (5-µm thickness) were cut through the entire brain on an RM2145 microtome (Leica), placed on a single glass slide, and stained with H&E using a standard protocol. To quantify brain degeneration, vacuoles greater than 5 µm in diameter were counted through all serial sections of the entire brain.
TUNEL staining of adult brains (Fig. 8, F and I) was performed using the In Situ Cell Death Detection Kit (Roche) as previously described (Ojelade et al., 2019). Briefly, 20-d-old adult fly brains and VNCs were dissected in PBS and fixed in 4% formaldehyde/PBS for 20 min. Samples were blocked overnight in 0.3% Triton X-100/PBS containing 5% normal goat serum at 4°C, incubated in 10% Triton X-100/PBS containing 100 mM sodium citrate for 30 min at 65°C, and washed three times with 0.3% Triton X-100/PBS. Samples were then equilibrated in labeling solution for 15 min and incubated in a 1:9 mixture of terminal deoxynucleotidyl transferase enzyme and labeling solution for 3 h at 37°C. Samples were washed three times in 0.3% Triton X-100/PBS for 15 min/wash and stained with DAPI for 5 min. Z stack (1-µm thickness) images of brain or VNC samples were acquired with a Plan/Apo 20× 0.8 M27 objective lens at RT. Only TUNEL+ cells greater than 2 µm in diameter were counted.
For immunostaining of adult brains (Fig. 8 H), 20-d-old adult fly brains were stained as previously described (Nahm et al., 2013). Briefly, dissected brains were fixed in 4% formaldehyde/PBS for 20 min at RT. Brains were then permeabilized in 0.3% Triton X-100/PBS, blocked with 5% BSA in 0.3% Triton X-100/PBS at RT for 1 h, and incubated with primary antibodies in blocking buffer for 2 d at 4°C. Samples were then incubated in 0.3% Triton X-100/PBS containing 5% BSA with secondary antibodies overnight at 4°C. To stain brains with TUNEL and antibodies, brains were incubated in 0.1% Triton X-100/PBS containing 100 mM sodium citrate for 30 min at RT and incubated with TUNEL reaction mixture in the dark for 1 h at 37°C before incubation with primary antibodies. The following antibodies were used in this study: rat anti-Elav (1:10; Cat# 7E8A10; DSHB), mouse anti-Repo (1:10; Cat# 8D12; DSHB), rabbit anti–cleaved Dcp-1 (1:100; Cat# 9578; Cell Signaling Technology), and donkey secondary antibodies (1:200) from Jackson ImmunoResearch Laboratories, including FITC-conjugated anti-rabbit (Cat# 711–095-152), Cy3-conjugated anti-rat (Cat# 712–095-153), and Cy5-conjugated anti-mouse (Cat# 715–175-151).
Adult behavioral analysis
Adult locomotor function was assessed using a climbing ability test (Nahm et al., 2013). For each genotype, ∼100 flies were collected within 1 d after eclosion and aged for 20 d. Aged flies were placed into an empty glass cylinder, allowed to acclimate for 5 min, and gently tapped to the bottom. The distance climbed by individual flies after 30 s was measured.
Statistical analysis
Data are presented as mean ± SEM. Statistical significance was calculated using a one-way ANOVA followed by pairwise Tukey’s tests (for multiple group comparisons) or using a two-tailed Student’s t test (for two-group comparisons).
Online supplemental material
Fig. S1 shows characterization of dALS2 mutants and extends Fig. 1. Fig. S2 shows the subcellular localization of HA-dALS2 in larval muscles and demonstrates that Flag-dFz2-Myc behaves similarly to endogenous dFz2 protein in vivo. Fig. S3 shows the impact of dALS2 depletion on intracellular trafficking of Wg in BG2-c2 cells. Fig. S4 shows the impact of dALS2 depletion on intracellular trafficking of mBSA and Tkv in BG2-c2 cells. Fig. S5 shows that depletion of mammalian ALS2 impairs Wnt5a-induced nuclear translocation of the Fz5 receptor in mouse motor neuron–like NSC-34 cells.
Acknowledgments
This work was supported by Ministry of Science and Technology, Taiwan grants 107-2311-B-001-003-MY3, 106-0210-01-15-02, and 107-0210-01-19-01 (T.-N. Li, H.-C. Lin, and C.-K. Yao) and National Research Foundation of Korea grants BK21+ program (J. Lee) and 2017M3C7A1025368 and 2019R1A2C2089437 (S. Lee).
The authors declare no competing financial interests.
Author contributions: Conceptualization: J. Kim, J. Lee, C.-K. Yao, and S. Lee; investigation: J. Kim, S. Kim, M. Nahm, T.-N. Li, H.-C. Lin, and Y.D. Kim; formal analysis: J. Kim, S. Kim, M. Nahm, J. Lee, C.-K. Yao, and S. Lee; writing - original draft: J. Kim and S. Lee; writing - review and editing: S. Lee; supervision: C.-K. Yao and S. Lee; funding acquisition: C.-K. Yao and S. Lee.
References
- Ataman, B., Ashley J., Gorczyca D., Gorczyca M., Mathew D., Wichmann C., Sigrist S.J., and Budnik V.. 2006. Nuclear trafficking of Drosophila Frizzled-2 during synapse development requires the PDZ protein dGRIP. Proc. Natl. Acad. Sci. USA. 103:7841–7846. 10.1073/pnas.0600387103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataman, B., Ashley J., Gorczyca M., Ramachandran P., Fouquet W., Sigrist S.J., and Budnik V.. 2008. Rapid activity-dependent modifications in synaptic structure and function require bidirectional Wnt signaling. Neuron. 57:705–718. 10.1016/j.neuron.2008.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand, A.H., and Perrimon N.. 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 118:401–415. [DOI] [PubMed] [Google Scholar]
- Budnik, V., Koh Y.H., Guan B., Hartmann B., Hough C., Woods D., and Gorczyca M.. 1996. Regulation of synapse structure and function by the Drosophila tumor suppressor gene dlg. Neuron. 17:627–640. 10.1016/S0896-6273(00)80196-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk, K., and Pasterkamp R.J.. 2019. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 137:859–877. 10.1007/s00401-019-01964-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S., Sayana P., Zhang X., and Le W.. 2013. Genetics of amyotrophic lateral sclerosis: an update. Mol. Neurodegener. 8:28. 10.1186/1750-1326-8-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, L.E., Ferraiuolo L., Goodall E.F., Heath P.R., Higginbottom A., Mortiboys H., Hollinger H.C., Hartley J.A., Brockington A., Burness C.E., et al. 2010. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One. 5:e9872. 10.1371/journal.pone.0009872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani, N., Zhu H., and Broadie K.. 2014. Two protein N-acetylgalactosaminyl transferases regulate synaptic plasticity by activity-dependent regulation of integrin signaling. J. Neurosci. 34:13047–13065. 10.1523/JNEUROSCI.1484-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, H.X., Zhai H., Fu R., Shi Y., Gorrie G.H., Yang Y., Liu E., Dal Canto M.C., Mugnaini E., and Siddique T.. 2007. Distal axonopathy in an alsin-deficient mouse model. Hum. Mol. Genet. 16:2911–2920. 10.1093/hmg/ddm251 [DOI] [PubMed] [Google Scholar]
- Devon, R.S., Schwab C., Topp J.D., Orban P.C., Yang Y.Z., Pape T.D., Helm J.R., Davidson T.L., Rogers D.A., Gros-Louis F., et al. 2005. Cross-species characterization of the ALS2 gene and analysis of its pattern of expression in development and adulthood. Neurobiol. Dis. 18:243–257. 10.1016/j.nbd.2004.10.002 [DOI] [PubMed] [Google Scholar]
- Devon, R.S., Orban P.C., Gerrow K., Barbieri M.A., Schwab C., Cao L.P., Helm J.R., Bissada N., Cruz-Aguado R., Davidson T.L., et al. 2006. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc. Natl. Acad. Sci. USA. 103:9595–9600. 10.1073/pnas.0510197103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAntonio, A., Petersen S.A., Heckmann M., and Goodman C.S.. 1999. Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J. Neurosci. 19:3023–3032. 10.1523/JNEUROSCI.19-08-03023.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickins, E.M., and Salinas P.C.. 2013. Wnts in action: from synapse formation to synaptic maintenance. Front. Cell. Neurosci. 7:162. 10.3389/fncel.2013.00162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eymard-Pierre, E., Lesca G., Dollet S., Santorelli F.M., di Capua M., Bertini E., and Boespflug-Tanguy O.. 2002. Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. Am. J. Hum. Genet. 71:518–527. 10.1086/342359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz, S.J., Cummings A.M., Nguyen J.N., Hamm D.C., Donohue L.K., Harrison M.M., Wildonger J., and O’Connor-Giles K.M.. 2013. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 194:1029–1035. 10.1534/genetics.113.152710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, G.D., Swetha M.G., Kumari S., Lakshminarayan R., Dey G., and Mayor S.. 2009. Analysis of endocytic pathways in Drosophila cells reveals a conserved role for GBF1 in internalization via GEECs. PLoS One. 4:e6768. 10.1371/journal.pone.0006768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadano, S., Hand C.K., Osuga H., Yanagisawa Y., Otomo A., Devon R.S., Miyamoto N., Showguchi-Miyata J., Okada Y., Singaraja R., et al. 2001. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 29:166–173. 10.1038/ng1001-166 [DOI] [PubMed] [Google Scholar]
- Harris, K.P., Akbergenova Y., Cho R.W., Baas-Thomas M.S., and Littleton J.T.. 2016. Shank Modulates Postsynaptic Wnt Signaling to Regulate Synaptic Development. J. Neurosci. 36:5820–5832. 10.1523/JNEUROSCI.4279-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo, K., Nahm M., Lee M.J., Kim Y.E., Ki C.S., Kim S.H., and Lee S.. 2017. The Rap activator Gef26 regulates synaptic growth and neuronal survival via inhibition of BMP signaling. Mol. Brain. 10:62. 10.1186/s13041-017-0342-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, H., Zhao K., Huang S., Huang S., Yao A., Jiang Y., Sigrist S., Zhao L., and Zhang Y.Q.. 2020. Structural Remodeling of Active Zones Is Associated with Synaptic Homeostasis. J. Neurosci. 40:2817–2827. 10.1523/JNEUROSCI.2002-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari, J., and Helenius A.. 2011. Endosome maturation. EMBO J. 30:3481–3500. 10.1038/emboj.2011.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, J.O., Mandrioli J., Benatar M., Abramzon Y., Van Deerlin V.M., Trojanowski J.Q., Gibbs J.R., Brunetti M., Gronka S., Wuu J., et al. ITALSGEN Consortium . 2010. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 68:857–864. 10.1016/j.neuron.2010.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien, J.P., and Kriz J.. 2006. Transgenic mouse models of amyotrophic lateral sclerosis. Biochim. Biophys. Acta. 1762:1013–1024. 10.1016/j.bbadis.2006.03.006 [DOI] [PubMed] [Google Scholar]
- Kamimura, K., Ueno K., Nakagawa J., Hamada R., Saitoe M., and Maeda N.. 2013. Perlecan regulates bidirectional Wnt signaling at the Drosophila neuromuscular junction. J. Cell Biol. 200:219–233. 10.1083/jcb.201207036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, N., Kim S., Nahm M., Kopke D., Kim J., Cho E., Lee M.J., Lee M., Kim S.H., Broadie K., and Lee S.. 2019. BMP-dependent synaptic development requires Abi-Abl-Rac signaling of BMP receptor macropinocytosis. Nat. Commun. 10:684. 10.1038/s41467-019-08533-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew, D., Ataman B., Chen J., Zhang Y., Cumberledge S., and Budnik V.. 2005. Wingless signaling at synapses is through cleavage and nuclear import of receptor DFrizzled2. Science. 310:1344–1347. 10.1126/science.1117051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca, T.J., and Schwarz T.L.. 2010. The nuclear import of Frizzled2-C by Importins-beta11 and alpha2 promotes postsynaptic development. Nat. Neurosci. 13:935–943. 10.1038/nn.2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muqit, M.M., and Feany M.B.. 2002. Modelling neurodegenerative diseases in Drosophila: a fruitful approach? Nat. Rev. Neurosci. 3:237–243. 10.1038/nrn751 [DOI] [PubMed] [Google Scholar]
- Nahm, M., Kim S., Paik S.K., Lee M., Lee S., Lee Z.H., Kim J., Lee D., Bae Y.C., and Lee S.. 2010a. dCIP4 (Drosophila Cdc42-interacting protein 4) restrains synaptic growth by inhibiting the secretion of the retrograde Glass bottom boat signal. J. Neurosci. 30:8138–8150. 10.1523/JNEUROSCI.0256-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahm, M., Long A.A., Paik S.K., Kim S., Bae Y.C., Broadie K., and Lee S.. 2010b. The Cdc42-selective GAP rich regulates postsynaptic development and retrograde BMP transsynaptic signaling. J. Cell Biol. 191:661–675. 10.1083/jcb.201007086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahm, M., Lee M.J., Parkinson W., Lee M., Kim H., Kim Y.J., Kim S., Cho Y.S., Min B.M., Bae Y.C., et al. 2013. Spartin regulates synaptic growth and neuronal survival by inhibiting BMP-mediated microtubule stabilization. Neuron. 77:680–695. 10.1016/j.neuron.2012.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordmann, M., Cabrera M., Perz A., Bröcker C., Ostrowicz C., Engelbrecht-Vandré S., and Ungermann C.. 2010. The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr. Biol. 20:1654–1659. 10.1016/j.cub.2010.08.002 [DOI] [PubMed] [Google Scholar]
- Ojelade, S.A., Lee T.V., Giagtzoglou N., Yu L., Ugur B., Li Y., Duraine L., Zuo Z., Petyuk V., De Jager P.L., et al. 2019. cindr, the Drosophila Homolog of the CD2AP Alzheimer’s Disease Risk Gene, Is Required for Synaptic Transmission and Proteostasis. Cell Rep. 28:1799–1813.e5. 10.1016/j.celrep.2019.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder, T., Yoon K.S., White B.H., and Keshishian H.. 2001. A conditional tissue-specific transgene expression system using inducible GAL4. Proc. Natl. Acad. Sci. USA. 98:12596–12601. 10.1073/pnas.221303298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otomo, A., Hadano S., Okada T., Mizumura H., Kunita R., Nishijima H., Showguchi-Miyata J., Yanagisawa Y., Kohiki E., Suga E., et al. 2003. ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum. Mol. Genet. 12:1671–1687. 10.1093/hmg/ddg184 [DOI] [PubMed] [Google Scholar]
- Packard, M., Koo E.S., Gorczyca M., Sharpe J., Cumberledge S., and Budnik V.. 2002. The Drosophila Wnt, wingless, provides an essential signal for pre- and postsynaptic differentiation. Cell. 111:319–330. 10.1016/S0092-8674(02)01047-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton, A.E., Chiò A., and Traynor B.J.. 2014. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17:17–23. 10.1038/nn.3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal, S. 2009. Genomic mapping and expression patterns of C380, OK6 and D42 enhancer trap lines in the larval nervous system of Drosophila. Gene Expr. Patterns. 9:371–380. 10.1016/j.gep.2009.01.002 [DOI] [PubMed] [Google Scholar]
- Sato, K., Otomo A., Ueda M.T., Hiratsuka Y., Suzuki-Utsunomiya K., Sugiyama J., Murakoshi S., Mitsui S., Ono S., Nakagawa S., et al. 2018. Altered oligomeric states in pathogenic ALS2 variants associated with juvenile motor neuron diseases cause loss of ALS2-mediated endosomal function. J. Biol. Chem. 293:17135–17153. 10.1074/jbc.RA118.003849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnute, B., Troost T., and Klein T.. 2018. Endocytic Trafficking of the Notch Receptor. Adv. Exp. Med. Biol. 1066:99–122. 10.1007/978-3-319-89512-3_6 [DOI] [PubMed] [Google Scholar]
- Shi, Y., Lin S., Staats K.A., Li Y., Chang W.H., Hung S.T., Hendricks E., Linares G.R., Wang Y., Son E.Y., et al. 2018. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24:313–325. 10.1038/nm.4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, J.P., Brown R.H. Jr., and Cleveland D.W.. 2016. Decoding ALS: from genes to mechanism. Nature. 539:197–206. 10.1038/nature20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topp, J.D., Gray N.W., Gerard R.D., and Horazdovsky B.F.. 2004. Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J. Biol. Chem. 279:24612–24623. 10.1074/jbc.M313504200 [DOI] [PubMed] [Google Scholar]
- Yamanaka, K., Vande Velde C., Eymard-Pierre E., Bertini E., Boespflug-Tanguy O., and Cleveland D.W.. 2003. Unstable mutants in the peripheral endosomal membrane component ALS2 cause early-onset motor neuron disease. Proc. Natl. Acad. Sci. USA. 100:16041–16046. 10.1073/pnas.2635267100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka, K., Miller T.M., McAlonis-Downes M., Chun S.J., and Cleveland D.W.. 2006. Progressive spinal axonal degeneration and slowness in ALS2-deficient mice. Ann. Neurol. 60:95–104. 10.1002/ana.20888 [DOI] [PubMed] [Google Scholar]
- Yang, Y., Hentati A., Deng H.X., Dabbagh O., Sasaki T., Hirano M., Hung W.Y., Ouahchi K., Yan J., Azim A.C., et al. 2001. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 29:160–165. 10.1038/ng1001-160 [DOI] [PubMed] [Google Scholar]
- Yao, C.K., Liu Y.T., Lee I.C., Wang Y.T., and Wu P.Y.. 2017. A Ca2+ channel differentially regulates Clathrin-mediated and activity-dependent bulk endocytosis. PLoS Biol. 15:e2000931. 10.1371/journal.pbio.2000931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh, E., Gustafson K., and Boulianne G.L.. 1995. Green fluorescent protein as a vital marker and reporter of gene expression in Drosophila. Proc. Natl. Acad. Sci. USA. 92:7036–7040. 10.1073/pnas.92.15.7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, K., Hong H., Zhao L., Huang S., Gao Y., Metwally E., Jiang Y., Sigrist S.J., and Zhang Y.Q.. 2020. Postsynaptic cAMP signalling regulates the antagonistic balance of Drosophila glutamate receptor subtypes. Development. 147:dev191874. 10.1242/dev.191874 [DOI] [PMC free article] [PubMed] [Google Scholar]