

Strain-specific differences in respiratory syncytial virus (RSV) isolates are associated with differential pathogenesis in mice. However, the role of RSV genotypes in human infection is incompletely understood. This work demonstrates that one such genotype, 2stop+A4G, present in the RSV attachment (G) gene terminus is associated with greater infant disease severity. The genotype consists of two tandem stop codons preceding an A-to-G point mutation in the 4th position of the G gene end transcription termination signal. Virologically, the 2stop+A4G RSV genotype results in reduced levels of the RSV fusion (F) glycoprotein. A recombinant 2stop+A4G RSV was better able to establish infection in the presence of existing RSV immunity than a virus harboring the common A4G mutation. These data suggest that regulation of G and F expression has implications for virulence and, potentially, immune evasion.
KEYWORDS: respiratory syncytial virus, infant, wheeze, G glycoprotein, gene end, disease severity, transcriptional regulation, A4G, tandem stop codons, gene variant
ABSTRACT
This study identified a genotype of respiratory syncytial virus (RSV) associated with increased acute respiratory disease severity in a cohort of previously healthy term infants. The genotype (2stop+A4G) consists of two components. The A4G component is a prevalent point mutation in the 4th position of the gene end transcription termination signal of the G gene of currently circulating RSV strains. The 2stop component is two tandem stop codons at the G gene terminus, preceding the gene end transcription termination signal. To investigate the biological role of these RSV G gene mutations, recombinant RSV strains harboring either a wild-type A2 strain G gene (one stop codon preceding a wild-type gene end signal), an A4G gene end signal preceded by one stop codon, or the 2stop+A4G virulence-associated combination were generated and characterized. Infection with the recombinant A4G (rA4G) RSV mutant resulted in transcriptional readthrough and lower G and fusion (F) protein levels than for the wild type. Addition of a second stop codon preceding the A4G point mutation (2stop+A4G) restored G protein expression but retained lower F protein levels. These data suggest that RSV G and F glycoprotein expression is regulated by transcriptional and translational readthrough. Notably, while rA4G and r2stop+A4G RSV were attenuated in cells and in naive BALB/c mice compared to that for wild-type RSV, the r2stop+A4G RSV was better able to infect BALB/c mice in the presence of preexisting immunity than rA4G RSV. Together, these factors may contribute to the maintenance and virulence of the 2stop+A4G genotype in currently circulating RSV-A strains.
IMPORTANCE Strain-specific differences in respiratory syncytial virus (RSV) isolates are associated with differential pathogenesis in mice. However, the role of RSV genotypes in human infection is incompletely understood. This work demonstrates that one such genotype, 2stop+A4G, present in the RSV attachment (G) gene terminus is associated with greater infant disease severity. The genotype consists of two tandem stop codons preceding an A-to-G point mutation in the 4th position of the G gene end transcription termination signal. Virologically, the 2stop+A4G RSV genotype results in reduced levels of the RSV fusion (F) glycoprotein. A recombinant 2stop+A4G RSV was better able to establish infection in the presence of existing RSV immunity than a virus harboring the common A4G mutation. These data suggest that regulation of G and F expression has implications for virulence and, potentially, immune evasion.
INTRODUCTION
Respiratory syncytial virus (RSV) is the leading cause of hospitalizations and viral deaths in children under the age of 5 years (1, 2). Currently, no licensed RSV vaccine is available, and prophylaxis is limited to administration of a monoclonal RSV antibody to specific high-risk infants (3).
The single-stranded negative-sense RNA genome of RSV is flanked by noncoding 3′ leader and 5′ trailer regions. Genome transcription is sequential and begins at the 3′ proximal promoter and proceeds in a 3′ to 5′ direction in a “start-stop” manner, mediated by the polymerase to generate 10 monocistronic mRNAs (4, 5). Gene junctions between each gene contain transcription start and end signals that are essential for efficient gene transcription. While gene start signals are highly conserved, variations in transcription end signals are common (6–9).
Variations in the gene end signal of the attachment (G) gene have previously been identified in clinical RSV isolates (10). One such variation harboring a single nucleotide from adenine to guanine at the 4th position (A4G) was shown to result in transcriptional readthrough of the G transcript terminator, a subsequent decrease in mRNA transcripts, and decreased levels of F protein in comparison to that for the prototypic A2 strain in HEp-2 cells. Phylogenetic analysis of those clinical isolates grouped into their own clade separate from the “wild-type” A2-like strains, suggesting that naturally circulating RSV strains vary from the classical laboratory-adapted strains (10).
Moudy et al. demonstrated that laboratory strains might also differ in transcriptional regulation, but they had access to only 53 sequences and no clinical data (11). Alteration in gene expression of the two major surface glycoproteins could theoretically affect clinical disease severity, especially as both the F and G proteins have been implicated in RSV pathogenesis (12, 13) and both F and G are targets for neutralizing antibodies during infection (14, 15). Several studies identified RSV strains that better recapitulate aspects of human RSV disease in mice than commonly used A2 and Long strains (16–18). These strain-specific pathogenicity phenotypes in mice point to RSV disease manifestations in humans being attributable to both host immune responses and viral genetics and highlight RSV genotype as a determinant of disease severity. The impact of RSV strain differences on human disease, however, is variable and incompletely characterized (19–21). Thus, further investigation is needed to assess the impact of RSV genotype on clinical disease.
We sequenced the approximately 270 terminal nucleotides of the RSV G gene in order to identify RSV clades in a term healthy infant cohort (Tennessee Children’s Respiratory Initiative [TCRI] cohort) and assess whether RSV clade differences were associated with infant infection severity. Sequencing was exclusive to the G gene to identify sequences that could play a role in transcriptional readthrough and protein expression of both G and F. We found no differences by clade associated with infant infection severity as measured by an ordinal bronchiolitis severity score (BSS) in the infant cohort. However, a single nucleotide polymorphism (SNP)-like approach identified specific variants in the RSV G gene, A4G and 2stop+A4G genotypes, that were associated with BSS and predominate among RSV-A strains. These gene end variants alter the ratio of G to F protein expression. The implication of this in our BALB/c mouse model is that genotype variants of RSV, such as 2stop+A4G, are able to establish reinfection in the presence of preexisting immunity in mice. Together, these factors may contribute to the predominance and maintenance of the 2stop+A4G genotype in circulating RSV strains.
RESULTS
Distribution and frequency of A4G in glycoprotein G gene end sequences confirms strain A2 sequence is infrequently found in clinical isolates.
The RSV G gene is used to assign clinical isolates to subtypes A or B and to specific clades existing within these subtypes (22). The extracellular domain of G consists of two hypervariable regions, the second of which corresponds to the C-terminal region of the G protein and which reflects the overall gene variability for this gene. The hypervariable C-terminal sequence of G can therefore be used to quantify RSV genetic diversity within a population and has been used extensively in molecular and epidemiological studies (23, 24). To determine the RSV subgroup, we sequenced the last 270 nucleotides of the C-terminal region of the G gene of the 209 RSV-positive nasopharyngeal swabs from infants in the TCRI cohort. We took the approach of sequencing directly from RSV-positive swabs, as data from a small limited sequence study conducted by the CDC showed that no changes occurred between RSV clinical specimens and the isolates obtained from those specimens (T. C. T. Peret, personal communication). We acknowledge that this approach places a potential limitation on our study when comparing sequences obtained directly from clinical samples to those deposited on GenBank, which are likely from culture-adapted virus. However, given the size of the region sequenced in this study (270 nucleotides) and the available data (Peret, personal communication) it is unlikely that sequence changes between clinical specimens and isolates from this specimen will affect our results.
Of the 209 RSV strains, 142 were RSV subgroup A, and the remaining 67 were RSV subgroup B strains (Table 1). Subgroup A contained viruses from clades GA2 and GA5 only, while GB3 was the only clade representative of RSV-B.
TABLE 1.
RSV clinical strain antigenic subgroups and clades
| Years | No. of isolates/RSV season for clade: |
||
|---|---|---|---|
| A (n = 142) |
B (n = 67) |
||
| GA2 (n = 54) | GA5 (n = 88) | GB3 | |
| 2004–2005 | 2 | 0 | 25 |
| 2005–2006 | 13 | 48 | 17 |
| 2006–2007 | 21 | 36 | 9 |
| 2007–2008 | 18 | 4 | 16 |
Further analysis of the subgroup A sequences identified two main categories of gene end variant: wild type (16% [23/142]) and A4G (84% [119/142]) (Fig. 1). Here, we define the wild-type gene end variant as one amber stop codon (UAG) followed by the prototype A2 strain G gene end sequence UUACTTUUUUU (10). Within the A4G gene end category, we identified a second subgroup that we have termed 2stop+A4G (31% [37/119]) due to the presence of 2 tandem stop codons (UGA followed by an amber stop codon) that precede the A4G gene end sequence UCATTUUUUU (Fig. 1). Amber stop codons were present in 58% (82/119) of the A4G gene end variants.
FIG 1.

RSV G gene end variants identified in TCRI clinical samples. Attachment glycoprotein sequence analysis of 142 TCRI clinical samples identified two main categories of gene end variants: wild type (gray circle, 16% [23/142]) and A4G (blue circle, 84% [119/142]). Within the A4G category, a subgroup of gene end variants displaying an additional stop codon was identified, which we termed 2stop+A4G (red circle, 31% [37/119]).
To compare the frequency of the A4G genotype in TCRI sequences with frequency in sequences available in GenBank, we performed maximum-likelihood phylogenetic analyses on the G gene C-terminal region of the 142 subgroup A TCRI RSV sequences and 269 RSV subgroup A GenBank sequences (Fig. 2). Among these, 56.6% of all sequences included A4G. These sequences demonstrate that guanine is the predominant nucleotide found at the fourth position of the RSV G gene end sequence and suggests that previous knowledge regarding glycoprotein gene sequences may not represent extant clinical isolates.
FIG 2.

Comparison of RSV A4G and 2stop+A4G genotype phylogenetics. The C-terminal 270 nucleotides of 411 RSV subgroup A isolates were aligned using ClustalW and phylogenetic reconstruction was performed using MEGA5 software by maximum likelihood (general time reversible model, 1,000 bootstrap replicates). The tree was constructed and annotated using FigTree v1.2.2. Gray nodes (9% prevalence [37/411]) represent strains, such as A2, containing A2-like G gene transcription and translation termination signals containing a single stop codon and an A nucleotide at the 4th position of the gene end transcription terminator. Blue nodes (56.6% prevalence [233/411]) represent A4G genotypes, including strain CH17, and red nodes represent 2stop+A4G genotypes (32% prevalence [132/411]), including strain A2001/2-20. Green nodes (2% prevalence [8/411]) represent wild-type strains, such as MON-5-91, that are distantly related to strains A2 and Long. Branch lengths are measured as number of substitutions per site (scale bar, 0.02).
To determine whether these gene end variants were present in recently circulating strains, particularly in the rapidly spreading ON1 genotype, sequences deposited on GenBank from RSV samples collected between 2009 and 2017 were downloaded. An additional 51 sequences were analyzed, 12 of which were genotype ON1. We found that the 2stop+A4G gene end variant was present in 98% (50/51) of these recently circulating viruses (Fig. 3). Of interest, only 1/12 of the ON1 genotype samples, and the only ON1 sample collected in 2017, displayed a wild-type G gene end signal.
FIG 3.
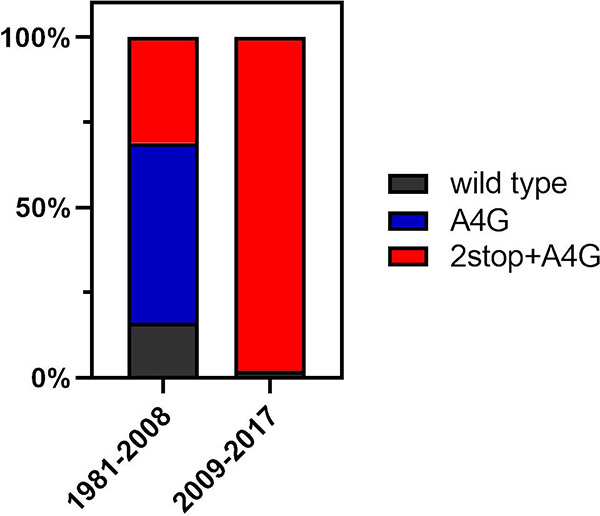
2stop+A4G G gene end variants in recently circulating strains. RSV G sequences from 51 samples collected between 2009 and 2017 were analyzed for the presence of wild-type, A4G, or 2stop+A4G G gene end variations. All additional sequences, bar 1 which had a wild-type gene end signal, displayed the 2stop+A4G gene end variation. In contrast, previously analyzed sequences (samples collected between 1981 and 2008) showed a mix of all three variants, with the 2stop+A4G variation making up a lesser proportion of the gene end variants.
Two tandem stop codons in the gene end terminator of the RSV G gene were associated with increased RSV severity in human infants.
RSV isolates from the Tennessee Children’s Respiratory Initiative (TCRI) cohort were collected from infants during acute respiratory infection. We sequenced the last 270 nucleotides of the C-terminal region of the G gene of 209 RSV-positive nasopharyngeal swabs from these infants, representing isolates from 2004 to 2008. SNP-like analyses of the sequence data of the G gene identified a genetic polymorphism in the C terminus of RSV G (subgroup A), including two tandem stop codons in the gene end terminator of the RSV G gene.
Isolates were categorized by A4G versus wild-type G gene end sequence, and the A4G isolates were further subcategorized according to whether the sequence included 1 (A4G) or 2 stop codons (2stop+A4G). Next, clinical disease severity was statistically evaluated for each of the three genotype categories using the bronchiolitis severity score (BSS) (Fig. 4). In multivariable ordinal logistic regression analysis, the 2stop+A4G genotype was associated with significantly higher BSS scores than both the wild-type and A4G genotypes, with 2stop+A4G having 3-fold increased odds compared to that of the wild type (adjusted odds ratio [aOR] = 3.00; 95% confidence interval [CI], 1.24 to 7.28) and 2-fold increased odds compared to that of A4G (aOR = 2.42; 95% CI, 1.21 to 4.86) for bronchiolitis severity.
FIG 4.
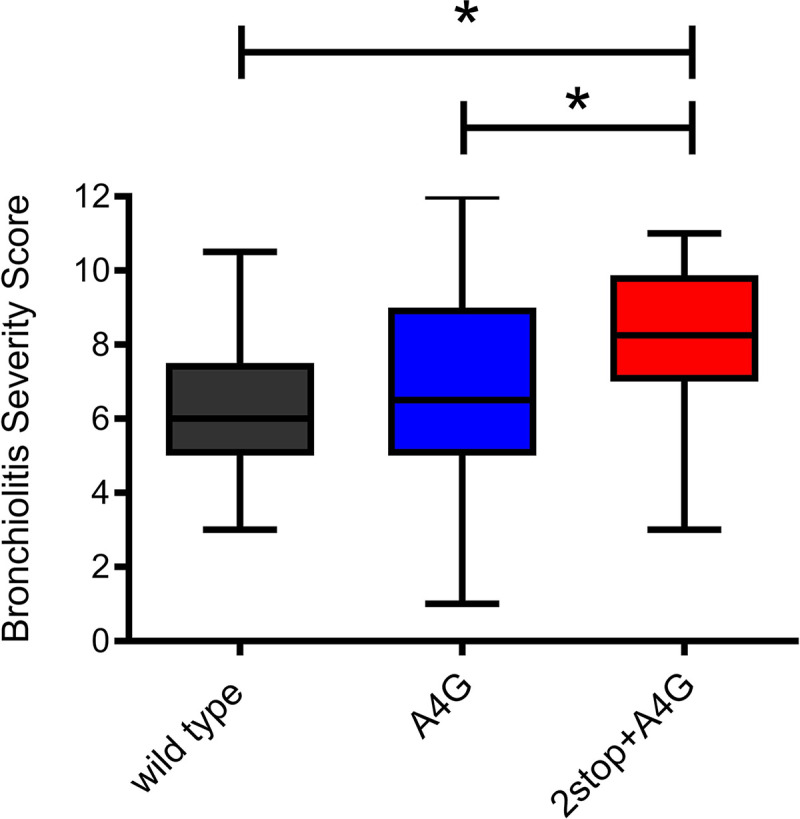
Bronchiolitis severity scores (BSS) associated with RSV genotypes in the TCRI cohort. The BSS (BSS scale 0 to 12, with higher numbers indicating more severe disease) of the 209 infants with RSV infection whose virus was sequenced were compared to identify differences in BSS by RSV genotype (wild type, n = 23; A4G, n = 82, and 2stop+A4G, n = 37). The 2stop+A4G genotype, containing two tandem stop codons in the C-terminal region of the G gene, was associated with clinically significant differences in disease severity in comparison to that for single stop codon (A4G) and wild-type A2-like viruses. Box plots represent median BSS (middle line), interquartile range (length of the box, 25th and 75th), and maximum/minimum values (the whiskers show the extent of the data range). Statistical comparisons indicated on the figure were performed using the Wilcoxon or Kruskal-Wallis nonparametric tests. In multivariable ordinal logistic regression analysis, the 2stop+A4G genotype was associated with significantly increased likelihood of higher bronchiolitis scores than both the wild type (adjusted odds ratio [aOR] = 3.00; 95% CI, 1.23 to 7.28) and A4G genotypes with 2stop (aOR = 2.42; 95% CI, 1.21,4.86). *, P < 0.05.
Effects of A4G and 2stop+A4G mutations on viral transcripts and proteins.
Using a bicistronic minigenome reporter system, Moudy and colleagues demonstrated that the A4G gene end mutation results in reduced efficiency of transcription termination and generation of readthrough transcripts (10). In order to compare directly the differences between the genotypes (Fig. 5b) in the context of viral infection, we generated two recombinant (r)RSV mutants based on the strain rA2-K-Line19F, which was chosen because its pathogenesis has been characterized in BALB/c mice (17). For the first virus, we changed the fourth position in the G gene end from an A to a G (A4G) to generate rA2-K-Line19F-A4G (termed rA4G henceforth). For the second mutant virus, in addition to the A4G mutation, we inserted a second stop codon in tandem with the existing stop codon in the G gene end to generate mutant rA2-K-Line19F-2stop+A4G (called r2stop+A4G henceforth).
FIG 5.

Effects of A4G and 2stop+A4G genotypes on RSV glycoprotein transcription and expression levels. (a) Hypothesized mRNA transcription and protein expression by wild-type A2-like, A4G, and 2stop+A4G genotypes. Each horizontal line indicates a predicted mRNA transcript. Light gray dots indicate stop codons present on single G transcripts or on G/F cotranscripts. The size of the black circles under “predicted protein levels” indicate the expected relative levels of each glycoprotein. (b) Comparison of RSV wild type, A4G and 2stop+A4G genotype G gene end sequences. (c) Representative Northern blots performed using probes specific for RSV G, F, M2, or mammalian actin mRNA as a control. Two virus clones of recombinant rA4G and r2stop+A4G RSV were tested. G, F, and G/F cotranscripts were quantified as a percentage of total transcripts detected with G- or F-specific probes. rA2-K-Line19F was used as the wild-type A2-like control. Data from both clones of rA4G and r2stop+A4G viruses were combined and presented as means plus standard deviation (SD). (d) Representative RSV N-, G-, and F-specific Western blots of rA4G, r2stop+A4G, and wild-type (rA2-K-Line19F) viruses. Quantification of three replicates of Western blots were calculated for rA4G, r2stop+A4G, and wild type rA2-K-Line19F viruses by densitometry. G and F levels were normalized to N levels for each virus prior to comparison with wild-type rA2-K-Line19F. Data represent means from 3 experimental replicates plus SD. *, P < 0.05 by one-way ANOVA with Tukey’s multiple-comparison test.
Following the rescue of these recombinant viruses, mRNA transcript and protein expression levels of G and F were investigated. We hypothesized that translational readthrough of the amber codon in RSV G occurs in the presence of A4G and that this results in reduced G protein expression levels due to the generation of a G-F cotranscript. The predicted mRNA species and relative glycoprotein levels of the recombinant RSV strains based on this hypothesis are shown in Fig. 5a. We hypothesized that the A4G genotype would have reduced F and G protein expression levels compared to that of the wild type, while viruses with a 2stop+A4G genotype would produce reduced F levels but wild-type G levels (Fig. 5a).
We tested this model by measuring mRNA transcripts during RSV infection in HEp-2 cells (Fig. 5c) as well as glycoprotein levels in virus preparations (Fig. 5d). Northern blots using a G-specific probe revealed the presence of a G/F cotranscript during infection with the rA4G or r2stop+A4G RSV mutants (Fig. 5c). The same G/F cotranscripts were visible using an F-specific probe (Fig. 5c). In the F-specific Northern blot, there was also a faint band approximately the size of the G/F cotranscript in the lane corresponding to wild-type RNA (Fig. 5c). This may have been attributable to transcriptional readthrough at the F-M2 gene junction, which has previously been reported for RSV and results in approximately the same size cotranscript as the G/F cotranscript (11). Alternatively, there may have been transcriptional readthrough at the G/F junction in wild-type RSV that resulted in a small but detectable amount of G/F cotranscription. An M2-specific probe revealed the presence of an F/M2 transcript in wild-type and mutant virus samples (Fig. 5c), as has been previously reported (7, 25, 26). The RSV M2 gene and cellular actin mRNA were used as the viral RNA loading control and total RNA loading control, respectively. By analyzing densitometry from the Northern blots, it was determined, as expected, that approximately 30% to 40% of the total G transcripts resulted from readthrough of the A4G gene end for the rA4G and r2stop+A4G RSV mutants, while approximately 20% of the total F transcripts were a result of this readthrough (Fig. 5c). Together, these findings support the previously reported bicistronic minigenome results (10) and support our model of transcriptional readthrough in the context of RSV infection.
The relative glycoprotein abundance present during RSV infection was quantified by performing Western blotting of virus stocks specific for the RSV G and F proteins (Fig. 5d). The intensities of the G- or F-specific bands were normalized to the intensity of the nucleoprotein (N)-specific signal for each virus. The rA4G RSV mutant exhibited lower levels of both G and F glycoproteins than wild-type RSV, with the G protein expression being 76% that of the wild type, and the F protein level approximately 61% that of the wild type (Fig. 5d). These values were reflective of reduced mRNA transcription seen in Fig. 5c, although they did not completely correlate with reduction in protein expression levels.
The r2stop+A4G RSV mutant, however, had G levels similar to that of wild-type virus (95%) but F levels similar to that of the rA4G RSV mutant (65%) (Fig. 5d). Although protein expression was reduced for both the rA4G and r2stop+A4G RSV mutants as expected, this reduction did not directly correlate with G and F mRNA transcript levels. In an attempt to probe for translational readthrough during viral infection, an epitope tag was introduced downstream of the stop codon(s); however, those mutants were not able to be rescued. Nevertheless, the Northern and Western blotting results presented here support our model of both transcriptional and translational readthrough occurring during RSV infection.
Attenuation of A4G and 2stop+A4G viruses in vitro and in vivo.
We hypothesized that the A4G mutation confers some advantage to the virus, because it was the predominant gene end sequence in our data set. We also tested whether the 2stop+A4G genotype, associated with virulence in infants, was more pathogenic in a mouse model. We compared the in vitro growth kinetics of the rA4G and r2stop+A4G RSV mutants to those of the wild-type strain, rA2-K-Line19F, in a human bronchial epithelial immortalized cell line (BEAS-2B). In these cells, both the rA4G and r2stop+A4G RSV mutants exhibited delayed replication and significantly lower titers than the wild type (Fig. 6, left). As the rA4G RSV mutant exhibited lower G protein expression in virus preparations than r2stop+A4G RSV and the wild type (Fig. 5d), the corresponding decrease in cell binding activity was tested using an RSV binding assay we previously employed (27). However, no difference in binding activity was observed between wild-type, rA4G, and r2stop+A4G viruses (data not shown).
FIG 6.

Attenuation of rA4G and r2stop+A4G RSV in vitro and in BALB/c mice. (Left) Replication kinetics of recombinant viruses in BEAS-2B cells infected at an MOI of 0.01. Supernatant samples were collected at indicated time points. The t0 indicates the input titer, and the dotted line indicates the limit of detection of the assay. Data represent the means from three experimental replicates ± standard errors of the means (SEMs). *, P < 0.05 when comparing wild type to both mutants (rA4G and r2stop+A4G RSV) by two-way ANOVA and Bonferroni’s multiple-comparison test. FFU, fluorescent focus units. (Right) Lung viral load in BALB/c mice shown as FFU per gram of lung homogenate quantified on the indicated days postinfection. Groups of 5 BALB/c mice were infected intranasally with 105 PFU of wild-type (black), rA4G (blue), and r2stop+A4G (red). On the indicated days postinfection, the left lung was harvested and homogenized, and viral load was determined using a fluorescent focus assay. Data represent the means from two experimental replicates ± SEMs. *, P < 0.05 when comparing wild type (rRSV-A2-K-Line19F) to both mutants (rA4G and r2stop+A4G) by two-way ANOVA test with Bonferroni’s correction for multiple comparisons.
Next, the effect of each of the recombinant viruses on viral load in the lungs of BALB/c mice was determined. Both rA4G and r2stop+A4G RSV mutants were modestly attenuated relative to wild-type strain rA2-K-Line19F on days 4 and 6 postinfection (p.i.), which is the period of peak viral load in this mouse model (Fig. 6, right). These results were suspected to be attributable to the reduced F protein expression in both mutant viruses (Fig. 5c) and suggested that in the immunologically naive mouse model, improved virus replication is not likely to be responsible for the association of disease severity with the 2stop+A4G genotype or the prevalence of the A4G genotype.
Furthermore, despite the attenuation in replication observed in vivo, neither the rA4G or r2stop+A4G RSV mutants exhibited significantly altered pathogenesis or histopathology in the BALB/c mouse model compared to that of rA2-K-Line19F, as measured by histopathology parameters, lung cytokine levels, induction of neutralizing antibodies, CD8+ T-cell numbers, or RSV F protein-specific (major histocompatibility complex [MHC] tetramers) CD8+ T-cell numbers (data not shown). The absence of altered pathogenesis, other than mild attenuation, in vivo in naive mice led us to consider whether the 2stop+A4G genotype modulates evasion of RSV-specific antibody, thus resulting in increased disease severity observed in infants who may have various levels of waning maternal antibody or antibody resulting from previous exposure. Finally, we hypothesized that more efficient reinfection might explain its higher prevalence among circulating RSV strains and studied this possibility in mice.
In vitro and in vivo neutralization of wild-type, A4G, and 2stop+A4G RSVs.
To determine whether clinical isolates harboring the 2stop+A4G genotype were more difficult to neutralize than the A4G or wild-type A2-like genotypes, a plaque reduction assay was performed using MPE8, a potent pre-F-specific monoclonal antibody (MAb) (15, 28). The neutralization titers of polyclonal antisera (BEI reference serum) or monoclonal antibodies MPE8 or palivizumab were each equivalent against rA2-K-Line19F, rA4G, and r2stop+A4G recombinant viruses, suggesting the G gene mutations had no effect on in vitro neutralization (data not shown). This result led us to investigate neutralization in vivo, because in vitro neutralization assays do not recapitulate all of the mechanisms of antiviral neutralizing antibodies (29).
We used a BALB/c mouse model of reinfection following low-dose primary infection to assess in vivo neutralization (30). Mice were first infected with 2 × 102 PFU of rRSV A2 and, 2 months later, reinfected with either wild-type (rA2-K-Line19F), rA4G, or r2stop+A4G RSV. The mean serum neutralizing antibody (nAb) titer of A2-infected mice at the time of secondary infection was 1:4, similar to our previous study (30). Viral titers were measured in lung homogenates 4 days post-secondary infection to determine the ability of each respective virus to reinfect BALB/c mice in the presence of RSV-specific immunity (Fig. 7). We found that, in contrast to results obtained in immunologically naive animals, r2stop+A4G RSV tended to better reinfect/reestablish infection in mice upon secondary infection than rA4G RSV (Fig. 7). Thus, the A4G genotype was more easily neutralized in vivo than the 2stop+A4G and wild-type A2-like genotypes. These data suggest that the increase in relative G protein abundance associated with addition of the 2stop mutation restores the A4G virus’s ability to infect immune mice to wild-type levels. It does not, however, suggest a mechanism for a selective advantage to explain the predominance of the 2stop+A4G mutations in circulating viruses.
FIG 7.

Reinfection of mice. Groups of five 6-week-old BALB/c mice were infected with strain rRSV-A2 to initiate a primary RSV infection in these animals. We confirmed that rRSV-A2 generated high-titer infections in all three groups by measuring lung viral titers (data not shown). To determine whether the mutant viruses generated in this study were able to establish infection in the presence of RSV-specific antibody, previously infected mice were subsequently inoculated with either the wild-type (rA2-K-Line19F), rA4G, or r2stop+A4G viruses as a secondary infection 56 days post-primary infection. Four days post-secondary infection (60 days overall), viral load was determined in lung homogenates of these animals. In comparison to wild-type and r2stop+A4G viruses, the rA4G mutant was unable to establish reinfection in the majority of mice. Primary infection is shown as gray circles for each virus (gray); secondary infection is shown as wild type (black), rA4G (blue), and r2stop+A4G (red). Each dot represents a single animal. Experiments were performed in duplicates, results are presented as means ± SDs, and statistical comparisons were performed using Tukey’s multiple-comparison test.
DISCUSSION
The association between RSV genotype and disease presentation is poorly understood (31). In the present study, we investigated viral genetic differences associated with clinical disease among an otherwise low-risk group of infants using a SNP-like analysis. This analysis identified G gene sequences, i.e., 2stop+A4G, significantly associated with RSV clinical disease severity. We then investigated the mechanisms behind the observed clinically significant increased disease severity (32) with RSV genotype 2stop+A4G (Fig. 4). The 2stop+A4G genotype variants were named as such due to the presence of an additional stop codon directly preceding the naturally occurring amber (UAG) codon in a subset of isolates containing a previously described G gene end variant, A4G (Fig. 5b). A4G has been demonstrated to generate a leaky stop codon between the F and G glycoprotein sequences that results in “wild-type” or A2-like levels of G transcript but lower levels of F transcript (10). Lower expression of F could be a mechanism for immune evasion by presenting less F to bind antibody, increasing virus replication in the setting of preexisting immunity and resulting in increased disease severity in children and increased fitness for circulation. We found that two G gene end variations (A4G and 2stop+A4G) were common among the RSV-A strains identified in our cohort. They were also common among globally isolated strains (Fig. 2 and 3), suggesting that these G gene end variations may be advantageous to RSV.
We hypothesized that concomitant transcriptional and translational readthrough associated with the A4G genotype would decrease G and F protein levels, but tandem stop codons, i.e., the 2stop+A4G genotype, would restore G protein levels (Fig. 5a). As suggested by this model, we found an increase in transcriptional readthrough and reduced expression levels of G and F with the rA4G virus, whereas the r2stop+A4G virus restored levels of G protein expression to that of the “wild type” while maintaining low F protein expression levels (translational readthrough) (Fig. 5c and d).
The aforementioned amber (UAG) stop codon in G is intrinsic in most naturally occurring RSV isolates, including those in this study, and is read through by ribosomes. Amber suppression has been described for tobacco mosaic virus and murine leukemia virus, whereby an amino acid insertion in a growing polypeptide chain quashes translation termination (33, 34). In the case of the 2stop+A4G genotype, reduction in F expression is likely due to the inability of ribosomes to reinitiate translation of F in the middle of a transcript postdissociation following G translation (35, 36).
Thus, one effect of the A4G mutation is to alter the G-to-F ratio, which in turn might affect virus infection in previously infected persons. It is well known that antibodies toward the RSV F and G glycoproteins are responsible for virus neutralization during infection (37), likely by different mechanisms. F antibodies neutralize virus by blocking fusion required for cell entry (38). Anti-G antibodies block binding to the cell surface, which is required for fusion and entry, likely through two mechanisms: blocking binding to glycosaminoglycans on the cell surface (39) and, in humans, also blocking binding to CX3CR1 on the cell surface (40, 41). Studies in mice suggest that G induces host immune responses that contribute to disease pathogenesis (12, 42, 43). Coimmunoprecipitation and cryo-electron tomography (cryo-ET) studies suggest that G and F interact; in vitro studies suggest G affects F binding and fusion, and in vivo mouse studies suggest both contribute to lung mucus production (42, 44–47). Therefore, altering the G/F ratio could potentially alter virus neutralizability as well as disease severity. Levels of G independent of levels of F could also affect its ability to act as an antigen decoy and antagonize the effectiveness of neutralizing antibodies (13).
Our in vitro studies show that altering the G/F ratio with A4G mutation significantly decreased virus replication relative to that of the wild-type virus either without or with the 2stop mutation both in vitro and in primary infection in mice (Fig. 6). In the rechallenge studies in mice, the infection rates for 2stop+A4G virus and wild-type viruses, 7 of 10 and 6 of 9, respectively, were higher than for the A4G virus, 2 of 9, though not significantly so (Fig. 7). Thus, we did not identify a selective advantage as indicated by an increase in virus replication for the A4G mutation over that of the wild-type virus in either in vitro or in vivo studies. Our data do, however, suggest that the addition of the 2stop mutation to the A4G mutation increases replication in immune mice, presumably due to the increased levels of G protein. This suggests that altering relative levels of gene transcription and translation is another site and mechanism for virus evolution.
One limitation in these results is the fact that in vitro and animal model studies do not necessarily apply to human infection. Though the virus we chose for these studies causes more-human-like RSV disease in BALB/c mice (i.e., RSV rA2-K-Line19F) than the usually studied viruses (17, 48), we still do not know if our findings apply to human infection.
In summary, we did not identify a replication advantage with the rA4G mutation that might explain its emergence as a common circulating strain but did show that a mutation such as the 2stop mutation in the A4G virus affects relative levels of transcription and translation, which appeared to increase replication in immune mice, i.e., a selective advantage in this RSV model and another site where mutations might affect virus fitness in humans.
MATERIALS AND METHODS
Infant study population and clinical sample collection.
The Tennessee Children’s Respiratory Initiative (TCRI) is a longitudinal prospective investigation of previously healthy term infants and their biological mothers, who were enrolled at the time of acute respiratory tract infection during the winter virus seasons from 2004 to 2008. TCRI objectives and study population have been previously described (32). The protocol and informed consent documents were approved by the Institutional Review Board at Vanderbilt University Medical Center. One parent of each participant provided written informed consent for participation in this study. During acute infection, nasal wash and throat swab samples were collected from each infant, placed in viral transport medium, and stored at −80°C. Following viral RNA extraction from each nasal wash sample, a 96-well plate-based single-step reverse transcription-PCR (RT-PCR) was used to amplify the hypervariable C-terminal region of the G gene for sequencing and phylogenetic analysis, as described previously (24).
RSV infection severity by RSV genotype.
To determine the clinical disease severity and collect the components of the bronchiolitis severity score (BSS), a structured clinical assessment and chart abstraction were performed for each infant (49). This ordinal score assigns 0 to 3 points for each of 4 categories, representing severity of wheezing, degree of oxygen desaturation, respiratory rate, and chest retractions (50). The BSS is a 13-point scale of 0 (least severe disease) to 12 (most severe disease) previously shown to reproducibly reflect a clinically statistically significant difference in clinical disease for each whole unit on the scale and correlates with level of health care utilization (hospitalization, emergency department visit, outpatient visit, or no care) (32, 51, 52). The BSS distribution values were compared between RSV sequences in the analysis detailed below.
Cells and mice.
Human bronchial epithelial cells (BEAS-2B) and HEp-2 cells were propagated as described (18). BSR-T7/5 cells were a gift from Karl-Klaus Conzelmann and Ursula Buchholz and were propagated in Glasgow’s minimal essential medium (GMEM) containing 10% fetal bovine serum (FBS), 1 μg/ml penicillin G-streptomycin sulfate-amphotericin B (PSA) solution, 2% minimal essential amino acids, and supplemented with 1 mg/ml Geneticin every second passage.
Six-week-old female BALB/c mice were obtained from the Jackson Laboratories (Bar Harbor, ME). Mice were housed under specific-pathogen-free conditions, and all experiments were conducted using protocols approved by the Emory University Institutional Animal Care and Use Committee (IACUC).
Rescue of recombinant viruses.
Using recombination-mediated mutagenesis, a bacterial artificial chromosome (BAC) containing the antigenome of RSV strain rA2-K-Line19F was altered in the G gene end fourth position to contain the A4G variation, generating the A4G antigenome (53). In a separate recombination mediated-mutagenesis, the rA2-K-Line19F antigenome-BAC was mutated to encode two tandem stop codons in the last two G amino acid positions as well as the A4G gene end variation, generating the 2stop+A4G antigenome. Each antigenomic BAC was transfected into confluent BSR-T7/5 cells in 6-well plates using Lipofectamine 2000 (Life Technologies, Grand Island, NY) with cotransfection of human codon bias-optimized helper plasmids expressing RSV N, P, M2-1, and L as described previously (53). Two independent BAC clones were used to rescue rA4G and r2stop+A4G viruses in duplicates. Transfected BSR-T7/5 cells were passed until a cytopathic effect (CPE) was present in approximately 70% of the culture, at which time, the cells were scraped into the medium, snap-frozen in liquid nitrogen, and stored at −80°C until further use. Viral stocks were propagated on HEp-2 cells to make master and working stocks as previously described (18). Viral working stock sequences were confirmed to harbor the desired mutation.
Northern blotting.
To obtain viral RNA, subconfluent HEp-2 cells in T-75 flasks were mock infected or infected with a multiplicity of infection (MOI) of 1 for each virus. Twenty-four hours postinfection (p.i.), total RNA was extracted using Trizol (Life Technologies) according to the manufacturer’s instructions. mRNA was isolated from total RNA using the Dynabeads mRNA purification kit (Ambion, Life Technologies). Equal amounts of mRNA from each isolation were separated in a 2% formaldehyde-1% agarose gel and transferred to a nylon membrane (54). RNAs were UV cross-linked to the membrane using a Spectrolinker (Spectronics Corporation, Westbury, NY). Digoxigenin (DIG)-labeled oligonucleotide probes were hybridized to the membrane, and signal was detected using CDP-Star (Roche Applied Science, Indianapolis, IN). Hybridization and detection steps were carried out according to Roche supplied protocols. Following developing, probes were stripped from the membrane, and membranes were reprobed for different mRNA species. Four DIG-labeled probes were generated and used for Northern blotting: three probes specific to RSV mRNAs, including G (G-1, 5′-GTATATTGGGGTTGTGTTCTTGATC-3′), F (F700r, 5′-GCATTAACACTAAATTCCCTGGT-3′), and M2 (8486r, 5′-AATGGGATCCATTTTGTCCCACCAC-3′), and one probe specific for mammalian actin mRNA (Act-1, 5′-AGGGTGTAACGCAACTAAGTCATAG-3′).
Western blotting.
Virus stocks were diluted to equal PFU in radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich, St. Louis, MO), mixed with Laemmli gel sample buffer, loaded onto a 10% SDS-PAGE gel, and separated by electrophoresis. Proteins were transferred to a polyvinylidene fluoride (PVDF) membrane (Bio-Rad, Hercules, CA). Membranes were probed using anti-RSV G antibody 131-2G (EMD Millipore, Billerica, MA) followed by a horseradish peroxidase (HRP)-conjugated donkey anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA). Signal was detected using SuperSignal West Femto substrate (Thermo Scientific, Waltham, MA) and a ChemiDoc Analyzer (Bio-Rad). Following developing, antibodies were stripped from the membrane using Restore Plus Western stripping buffer (Thermo Scientific). Membranes were then probed using the anti-RSV F antibody motavizumab (a gift from Nancy Ulbrandt, MedImmune, Gaithersburg, MD) and an HRP-conjugated goat anti-human secondary antibody (Jackson ImmunoResearch Laboratories). Signal was detected as described above, and antibodies were again stripped from the membrane. Membranes were then probed using the anti-RSV N antibody clone D14 (a gift from Edward Walsh, University of Rochester) and an HRP-conjugated donkey anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories), and signal was detected as described above. Densitometry was performed using Image Lab software (Bio-Rad).
Multistep growth analysis.
Subconfluent BEAS-2B cells were infected at an MOI of 0.01 for 1 h at room temperature. Following infection, inoculum was removed and cells were washed in phosphate-buffered saline (PBS) before complete Eagle minimal essential medium (EMEM) was added back to cells. Supernatant samples were collected at 12, 24, 48, 72, and 96 h p.i. and snap-frozen in liquid nitrogen. Virus titer in the samples was later determined by fluorescent focus unit (FFU) assay on HEp-2 cells. Briefly, samples were serially diluted and used to infect HEp-2 cells in duplicates in 96-well plates. Infection proceeded for 1 h at room temperature, at which time, the cells were overlaid with complete medium containing 7.5% methylcellulose as described above. FFUs were counted 2 days later to determine viral titer.
In vitro viral load determination.
To quantify lung viral load, 5 mice per group per time point were infected intranasally with 1 × 105 PFU virus. On days 1, 2, 4, 6, and 8 p.i., mice were euthanized and the left lung was removed for viral load quantification. Lungs were homogenized as described previously (18), and viral titers were determined by FFU assay on HEp-2 cells as described above.
Plaque reduction assays on clinical isolates.
MPE8, a pre-F-specific MAb (28), was serially diluted in PBS, and equal volumes of each dilution were mixed with approximately 2 FFU/μl of each virus (for a final concentration of 1 FFU/μl). Virus-antibody mixtures were incubated for 1 h at 37°C. Following incubation, 100 μl of each mixture was used to infect HEp-2 cells in duplicates in 24-well plates and left to rock for 1 h at room temperature. Next, complete growth medium containing 7.5% methylcellulose was added to the wells, the plates were incubated at 37°C, and PFU was counted 6 days p.i. To determine the concentration at which 50% of virus was neutralized (EC50), PFU present in each antibody dilution treatment well was compared to that in the negative control of virus mixed with PBS alone.
Reinfection of mice.
For the primary infection, 15 6-week-old female BALB/c mice were infected intranasally with 2 × 102 PFU/mouse of rRSV-A2. On day 56 post-primary infection, groups of 5 mice were infected with either wild-type (rRSV-A2-K-Line19F), rA4G, or r2stop+A4G viruses at 1 × 106 PFU/mouse. As a control, three groups of 5 naive mice were also infected with each of the above-mentioned recombinant viruses. On day 4 post-secondary infection (day 60 overall), mice were euthanized and the left lung collected for quantification of viral load by plaque assay as described previously (18, 55).
Data analysis.
The C-terminal 270 nucleotides of the RSV G protein of subgroup A strains were aligned using ClustalW, and phylogenetic reconstruction was performed using MEGA5 software by maximum likelihood using a bootstrap cutoff of 1,000 replicates. Trees were constructed and annotated using FigTree v1.2.2. The association of the identified RSV genotypes (A2-like, A4G, and 2stop+A4G) with clinical severity outcomes (the BSS) in the TCRI cohort was assessed using Kruskal-Wallis nonparametric tests and figuratively reported using box plots. To assess the association between RSV sequence and increased bronchiolitis severity outcome, we used the ordinal logistic regression and calculated the adjusted odds ratios and 95% CIs. The model included the phenotype of interest as the outcome variable and viral genotype as an explanatory variable. Adjustment covariates included infant age, gestational age, sex, and race. A P value of <0.05 was considered statistically significant. Multivariable regression analyses were performed using R version 3.4.0 (https://cran.r-project.org/).
All experimental in vitro data sets contain a minimum of three biological replicates. Statistical analysis was performed using either one-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test or a two-way ANOVA with Bonferroni’s multiple comparisons (P < 0.05) within the GraphPad Prism file.
In vivo mouse studies were carried out in duplicates, and each group consisted of 5 BALB/c mice. Statistical analysis was performed within the GraphPad Prism file using either a two-way ANOVA with Bonferroni’s multiple comparisons (P < 0.05) for viral kinetics or a one-way ANOVA with Tukey’s multiple-comparison test for the reinfection experiments.
Data availability.
The RSV sequences used in the analyses in this study encompassed the C-terminal hypervariable domain of the G gene (region <649.918; RSV strain A2 used as reference; GenBank KT992094). The sequences are available in GenBank under accession numbers MW088172 to MW088313.
ACKNOWLEDGMENTS
This work was supported by NIH grants R01AI087798 and U19AI095227 awarded to R.S.P., T.V.H., and M.L.M. and the Emory Children's Center for Childhood Infections and Vaccines (CCIV).
We thank Edward Walsh (University of Rochester) for the anti-N antibody, Ursula Buchholz and Karl-Klaus Conzelmann for the BSR-T7/5 cells used for virus rescue, and Nancy Ulbrandt for providing the anti-RSV F antibody motavizumab. We also thank all the families who participated in this study, the clinical staff at Vanderbilt Children’s Hospital and clinics for their support and help in recruiting mothers and children who participated in this study, and the entire Center for Asthma Research Study team for their assistance with the conduct of the study and data collection. We also thank Zhouwen Liu for his assistance in database building and management, research study nurses Patricia Minton and Kimberly Woodward for their assistance in the clinical study visits, Sarah Reiss and Kaitlin Costello for their assistance with biospecimen handling and analyses, Paul Moore and William Cooper for providing expert clinical case reviews, William Dupont for his biostatistical expertise, and Teresa Chipps for her administrative support.
Conceptualization: A. L. Hotard, S. Human, M. L. Moore, and T. V. Hartert; Data curation: A. L. Hotard, S. Human, T. V. Hartert, K. Carroll, S. Lee, T. C. T. Peret, J. Jorbe, J. V. Williams, M. Bloodworth, M. Stier; Formal analysis: A. L. Hotard, S. Human, T. Gebretsadik, E. K. Larkin; Funding acquisition: M. L. Moore, T. V. Hartert, S. Peebles, Jr; Investigation: A. L. Hotard, S. Human, E. K. Larkin, T. Gebretsadik, K. Carroll, S. Lee, T. C. T. Peret, M. Bloodworth, M. Stier; Methodology: A. L. Hotard, S. Human, C. A. Rostad, T. V. Hartert, T. Gebretsadik; Project administration: A. L. Hotard, S. Human, M. L. Moore, T. V. Hartert; Resources: M. L. Moore, T. V. Hartert, J. Lanzone; Supervision: M. L. Moore, T. V. Hartert, L. McCormick, L. J. Anderson; Writing – original draft: A. L. Hotard, T. V. Hartert, M. L. Moore; Writing – redrafting, review and editing: S. Human, T. V. Hartert, M. L. Moore, L. McCormick, L. J. Anderson.
REFERENCES
- 1.Scheltema NM, Gentile A, Lucion F, Nokes DJ, Munywoki PK, Madhi SA, Groome MJ, Cohen C, Moyes J, Thorburn K, Thamthitiwat S, Oshitani H, Lupisan SP, Gordon A, Sanchez JF, O'Brien KL, PERCH Study Group, Gessner BD, Sutanto A, Mejias A, Ramilo O, Khuri-Bulos N, Halasa N, de-Paris F, Pires MR, Spaeder MC, Paes BA, Simoes EAF, Leung TF, da Costa Oliveira MT, de Freitas Lazaro Emediato CC, Bassat Q, Butt W, Chi H, Aamir UB, Ali A, Lucero MG, Fasce RA, Lopez O, Rath BA, Polack FP, Papenburg J, Roglic S, Ito H, Goka EA, Grobbee DE, Nair H, Bont LJ. 2017. Global respiratory syncytial virus-associated mortality in young children (RSV GOLD): a retrospective case series. Lancet Glob Health 5:e984–e991. doi: 10.1016/S2214-109X(17)30344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi T, McAllister DA, O'Brien KL, Simoes EAF, Madhi SA, Gessner BD, Polack FP, Balsells E, Acacio S, Aguayo C, Alassani I, Ali A, Antonio M, Awasthi S, Awori JO, Azziz-Baumgartner E, Baggett HC, Baillie VL, Balmaseda A, Barahona A, Basnet S, Bassat Q, Basualdo W, Bigogo G, Bont L, Breiman RF, Brooks WA, Broor S, Bruce N, Bruden D, Buchy P, Campbell S, Carosone-Link P, Chadha M, Chipeta J, Chou M, Clara W, Cohen C, de Cuellar E, Dang DA, Dash-Yandag B, Deloria-Knoll M, Dherani M, Eap T, Ebruke BE, Echavarria M, de Freitas Lazaro Emediato CC, Fasce RA, Feikin DR, et al. 2017. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet 390:946–958. doi: 10.1016/S0140-6736(17)30938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meissner HC, Long SS, American Academy of Pediatrics Committee on Infectious Diseases, and Committee on Fetus and Newborn. 2003. Revised indications for the use of palivizumab and respiratory syncytial virus immune globulin intravenous for the prevention of respiratory syncytial virus infections. Pediatrics 112:1447–1452. doi: 10.1542/peds.112.6.1447. [DOI] [PubMed] [Google Scholar]
- 4.Collins PL, Melero JA. 2011. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res 162:80–99. doi: 10.1016/j.virusres.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dickens LE, Collins PL, Wertz GW. 1984. Transcriptional mapping of human respiratory syncytial virus. J Virol 52:364–369. doi: 10.1128/JVI.52.2.364-369.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harmon SB, Megaw AG, Wertz GW. 2001. RNA sequences involved in transcriptional termination of respiratory syncytial virus. J Virol 75:36–44. doi: 10.1128/JVI.75.1.36-44.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harmon SB, Wertz GW. 2002. Transcriptional termination modulated by nucleotides outside the characterized gene end sequence of respiratory syncytial virus. Virology 300:304–315. doi: 10.1006/viro.2002.1541. [DOI] [PubMed] [Google Scholar]
- 8.Kuo L, Fearns R, Collins PL. 1997. Analysis of the gene start and gene end signals of human respiratory syncytial virus: quasi-templated initiation at position 1 of the encoded mRNA. J Virol 71:4944–4953. doi: 10.1128/JVI.71.7.4944-4953.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuo L, Grosfeld H, Cristina J, Hill MG, Collins PL. 1996. Effects of mutations in the gene-start and gene-end sequence motifs on transcription of monocistronic and dicistronic minigenomes of respiratory syncytial virus. J Virol 70:6892–6901. doi: 10.1128/JVI.70.10.6892-6901.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moudy RM, Harmon SB, Sullender WM, Wertz GW. 2003. Variations in transcription termination signals of human respiratory syncytial virus clinical isolates affect gene expression. Virology 313:250–260. doi: 10.1016/s0042-6822(03)00299-x. [DOI] [PubMed] [Google Scholar]
- 11.Moudy RM, Sullender WM, Wertz GW. 2004. Variations in intergenic region sequences of human respiratory syncytial virus clinical isolates: analysis of effects on transcriptional regulation. Virology 327:121–133. doi: 10.1016/j.virol.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 12.Boyoglu-Barnum S, Todd SO, Chirkova T, Barnum TR, Gaston KA, Haynes LM, Tripp RA, Moore ML, Anderson LJ. 2015. An anti-G protein monoclonal antibody treats RSV disease more effectively than an anti-F monoclonal antibody in BALB/c mice. Virology 483:117–125. doi: 10.1016/j.virol.2015.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bukreyev A, Yang L, Collins PL. 2012. The secreted G protein of human respiratory syncytial virus antagonizes antibody-mediated restriction of replication involving macrophages and complement. J Virol 86:10880–10884. doi: 10.1128/JVI.01162-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson LJ, Bingham P, Hierholzer JC. 1988. Neutralization of respiratory syncytial virus by individual and mixtures of F and G protein monoclonal antibodies. J Virol 62:4232–4238. doi: 10.1128/JVI.62.11.4232-4238.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ngwuta JO, Chen M, Modjarrad K, Joyce MG, Kanekiyo M, Kumar A, Yassine HM, Moin SM, Killikelly AM, Chuang GY, Druz A, Georgiev IS, Rundlet EJ, Sastry M, Stewart-Jones GB, Yang Y, Zhang B, Nason MC, Capella C, Peeples ME, Ledgerwood JE, McLellan JS, Kwong PD, Graham BS. 2015. Prefusion F-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci Transl Med 7:309ra162. doi: 10.1126/scitranslmed.aac4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lukacs NW, Moore ML, Rudd BD, Berlin AA, Collins RD, Olson SJ, Ho SB, Peebles RS, Jr. 2006. Differential immune responses and pulmonary pathophysiology are induced by two different strains of respiratory syncytial virus. Am J Pathol 169:977–986. doi: 10.2353/ajpath.2006.051055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore ML, Chi MH, Luongo C, Lukacs NW, Polosukhin VV, Huckabee MM, Newcomb DC, Buchholz UJ, Crowe JE, Jr, Goleniewska K, Williams JV, Collins PL, Peebles RS, Jr. 2009. A chimeric A2 strain of respiratory syncytial virus (RSV) with the fusion protein of RSV strain line 19 exhibits enhanced viral load, mucus, and airway dysfunction. J Virol 83:4185–4194. doi: 10.1128/JVI.01853-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stokes KL, Chi MH, Sakamoto K, Newcomb DC, Currier MG, Huckabee MM, Lee S, Goleniewska K, Pretto C, Williams JV, Hotard A, Sherrill TP, Peebles RS, Jr, Moore ML. 2011. Differential pathogenesis of respiratory syncytial virus clinical isolates in BALB/c mice. J Virol 85:5782–5793. doi: 10.1128/JVI.01693-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilca R, De Serres G, Tremblay M, Vachon ML, Leblanc E, Bergeron MG, Dery P, Boivin G. 2006. Distribution and clinical impact of human respiratory syncytial virus genotypes in hospitalized children over 2 winter seasons. J Infect Dis 193:54–58. doi: 10.1086/498526. [DOI] [PubMed] [Google Scholar]
- 20.Martinello RA, Chen MD, Weibel C, Kahn JS. 2002. Correlation between respiratory syncytial virus genotype and severity of illness. J Infect Dis 186:839–842. doi: 10.1086/342414. [DOI] [PubMed] [Google Scholar]
- 21.Anderson LJ, Peret TC, Piedra PA. 2019. RSV strains and disease severity. J Infect Dis 219:514–516. doi: 10.1093/infdis/jiy498. [DOI] [PubMed] [Google Scholar]
- 22.Johnson PR, Spriggs MK, Olmsted RA, Collins PL. 1987. The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: extensive sequence divergence between antigenically related proteins. Proc Natl Acad Sci U S A 84:5625–5629. doi: 10.1073/pnas.84.16.5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng H, Storch GA, Zang C, Peret TC, Park CS, Anderson LJ. 1999. Genetic variability in envelope-associated protein genes of closely related group A strains of respiratory syncytial virus. Virus Res 59:89–99. doi: 10.1016/S0168-1702(98)00132-4. [DOI] [PubMed] [Google Scholar]
- 24.Peret TC, Hall CB, Schnabel KC, Golub JA, Anderson LJ. 1998. Circulation patterns of genetically distinct group A and B strains of human respiratory syncytial virus in a community. J Gen Virol 79:2221–2229. doi: 10.1099/0022-1317-79-9-2221. [DOI] [PubMed] [Google Scholar]
- 25.Cartee TL, Megaw AG, Oomens AG, Wertz GW. 2003. Identification of a single amino acid change in the human respiratory syncytial virus L protein that affects transcriptional termination. J Virol 77:7352–7360. doi: 10.1128/jvi.77.13.7352-7360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sutherland KA, Collins PL, Peeples ME. 2001. Synergistic effects of gene-end signal mutations and the M2-1 protein on transcription termination by respiratory syncytial virus. Virology 288:295–307. doi: 10.1006/viro.2001.1105. [DOI] [PubMed] [Google Scholar]
- 27.Hotard AL, Laikhter E, Brooks K, Hartert TV, Moore ML. 2015. Functional analysis of the 60-nucleotide duplication in the respiratory syncytial virus Buenos Aires strain attachment glycoprotein. J Virol 89:8258–8266. doi: 10.1128/JVI.01045-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corti D, Bianchi S, Vanzetta F, Minola A, Perez L, Agatic G, Guarino B, Silacci C, Marcandalli J, Marsland BJ, Piralla A, Percivalle E, Sallusto F, Baldanti F, Lanzavecchia A. 2013. Cross-neutralization of four paramyxoviruses by a human monoclonal antibody. Nature 501:439–443. doi: 10.1038/nature12442. [DOI] [PubMed] [Google Scholar]
- 29.Alter G, Barouch DH. 2015. Natural evolution of broadly neutralizing antibodies. Cell 161:427–428. doi: 10.1016/j.cell.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng J, Lee S, Hotard AL, Moore ML. 2014. Refining the balance of attenuation and immunogenicity of respiratory syncytial virus by targeted codon deoptimization of virulence genes. mBio 5:e01704-14. doi: 10.1128/mBio.01704-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vandini S, Biagi C, Lanari M. 2017. Respiratory syncytial virus: the influence of serotype and genotype variability on clinical course of infection. Int J Mol Sci 18:1717. doi: 10.3390/ijms18081717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartert TV, Carroll K, Gebretsadik T, Woodward K, Minton P, Vanderbilt Center for Asthma and Environmental Health Research Investigators and Collaborators. 2010. The Tennessee Children's Respiratory Initiative: objectives, design and recruitment results of a prospective cohort study investigating infant viral respiratory illness and the development of asthma and allergic diseases. Respirology 15:691–699. doi: 10.1111/j.1440-1843.2010.01743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beier H, Barciszewska M, Krupp G, Mitnacht R, Gross HJ. 1984. UAG readthrough during TMV RNA translation: isolation and sequence of two tRNAs with suppressor activity from tobacco plants. EMBO J 3:351–356. doi: 10.1002/j.1460-2075.1984.tb01810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshinaka Y, Katoh I, Copeland TD, Oroszlan S. 1985. Murine leukemia virus protease is encoded by the gag-pol gene and is synthesized through suppression of an amber termination codon. Proc Natl Acad Sci U S A 82:1618–1622. doi: 10.1073/pnas.82.6.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cattaneo R, Rebmann G, Schmid A, Baczko K, ter Meulen V, Billeter MA. 1987. Altered transcription of a defective measles virus genome derived from a diseased human brain. EMBO J 6:681–688. doi: 10.1002/j.1460-2075.1987.tb04808.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sourimant J, Plemper RK. 2016. Organization, function, and therapeutic targeting of the Morbillivirus RNA-dependent RNA polymerase complex. Viruses 8:251. doi: 10.3390/v8090251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLellan JS, Yang Y, Graham BS, Kwong PD. 2011. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J Virol 85:7788–7796. doi: 10.1128/JVI.00555-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLellan JS, Ray WC, Peeples ME. 2013. Structure and function of respiratory syncytial virus surface glycoproteins. Curr Top Microbiol Immunol 372:83–104. doi: 10.1007/978-3-642-38919-1_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feldman SA, Hendry RM, Beeler JA. 1999. Identification of a linear heparin binding domain for human respiratory syncytial virus attachment glycoprotein G. J Virol 73:6610–6617. doi: 10.1128/JVI.73.8.6610-6617.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chirkova T, Lin S, Oomens AGP, Gaston KA, Boyoglu-Barnum S, Meng J, Stobart CC, Cotton CU, Hartert TV, Moore ML, Ziady AG, Anderson LJ. 2015. CX3CR1 is an important surface molecule for respiratory syncytial virus infection in human airway epithelial cells. J Gen Virol 96:2543–2556. doi: 10.1099/vir.0.000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson SM, McNally BA, Ioannidis I, Flano E, Teng MN, Oomens AG, Walsh EE, Peeples ME. 2015. Respiratory syncytial virus uses CX3CR1 as a receptor on primary human airway epithelial cultures. PLoS Pathog 11:e1005318. doi: 10.1371/journal.ppat.1005318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boyoglu-Barnum S, Gaston KA, Todd SO, Boyoglu C, Chirkova T, Barnum TR, Jorquera P, Haynes LM, Tripp RA, Moore ML, Anderson LJ. 2013. A respiratory syncytial virus (RSV) anti-G protein F(ab')2 monoclonal antibody suppresses mucous production and breathing effort in RSV rA2-line19F-infected BALB/c mice. J Virol 87:10955–10967. doi: 10.1128/JVI.01164-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radu GU, Caidi H, Miao C, Tripp RA, Anderson LJ, Haynes LM. 2010. Prophylactic treatment with a G glycoprotein monoclonal antibody reduces pulmonary inflammation in respiratory syncytial virus (RSV)-challenged naive and formalin-inactivated RSV-immunized BALB/c mice. J Virol 84:9632–9636. doi: 10.1128/JVI.00451-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stokes KL, Currier MG, Sakamoto K, Lee S, Collins PL, Plemper RK, Moore ML. 2013. The respiratory syncytial virus fusion protein and neutrophils mediate the airway mucin response to pathogenic respiratory syncytial virus infection. J Virol 87:10070–10082. doi: 10.1128/JVI.01347-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meng J, Hotard AL, Currier MG, Lee S, Stobart CC, Moore ML. 2016. Respiratory syncytial virus attachment glycoprotein contribution to infection depends on the specific fusion protein. J Virol 90:245–253. doi: 10.1128/JVI.02140-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kiss G, Holl JM, Williams GM, Alonas E, Vanover D, Lifland AW, Gudheti M, Guerrero-Ferreira RC, Nair V, Yi H, Graham BS, Santangelo PJ, Wright ER. 2014. Structural analysis of respiratory syncytial virus reveals the position of M2-1 between the matrix protein and the ribonucleoprotein complex. J Virol 88:7602–7617. doi: 10.1128/JVI.00256-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yi H, Strauss JD, Ke Z, Alonas E, Dillard RS, Hampton CM, Lamb KM, Hammonds JE, Santangelo PJ, Spearman PW, Wright ER. 2015. Native immunogold labeling of cell surface proteins and viral glycoproteins for cryo-electron microscopy and cryo-electron tomography applications. J Histochem Cytochem 63:780–792. doi: 10.1369/0022155415593323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sacco RE, Durbin RK, Durbin JE. 2015. Animal models of respiratory syncytial virus infection and disease. Curr Opin Virol 13:117–122. doi: 10.1016/j.coviro.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller EK, Gebretsadik T, Carroll KN, Dupont WD, Mohamed YA, Morin LL, Heil L, Minton PA, Woodward K, Liu Z, Hartert TV, Williams JV. 2013. Viral etiologies of infant bronchiolitis, croup and upper respiratory illness during 4 consecutive years. Pediatr Infect Dis J 32:950–955. doi: 10.1097/INF.0b013e31829b7e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feldman AS, Hartert TV, Gebretsadik T, Carroll KN, Minton PA, Woodward KB, Larkin EK, Miller EK, Valet RS. 2015. Respiratory severity score separates upper versus lower respiratory tract infections and predicts measures of disease severity. Pediatr Allergy Immunol Pulmonol 28:117–120. doi: 10.1089/ped.2014.0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feldman AS, He Y, Moore ML, Hershenson MB, Hartert TV. 2015. Toward primary prevention of asthma. Reviewing the evidence for early-life respiratory viral infections as modifiable risk factors to prevent childhood asthma. Am J Respir Crit Care Med 191:34–44. doi: 10.1164/rccm.201405-0901PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez H, Hartert TV, Gebretsadik T, Carroll KN, Larkin EK. 2016. A simple respiratory severity score that may be used in evaluation of acute respiratory infection. BMC Res Notes 9:85. doi: 10.1186/s13104-016-1899-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hotard AL, Shaikh FY, Lee S, Yan D, Teng MN, Plemper RK, Crowe JE, Jr, Moore ML. 2012. A stabilized respiratory syncytial virus reverse genetics system amenable to recombination-mediated mutagenesis. Virology 434:129–136. doi: 10.1016/j.virol.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moore ML, McKissic EL, Brown CC, Wilkinson JE, Spindler KR. 2004. Fatal disseminated mouse adenovirus type 1 infection in mice lacking B cells or Bruton's tyrosine kinase. J Virol 78:5584–5590. doi: 10.1128/JVI.78.11.5584-5590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller AL, Bowlin TL, Lukacs NW. 2004. Respiratory syncytial virus-induced chemokine production: linking viral replication to chemokine production in vitro and in vivo. J Infect Dis 189:1419–1430. doi: 10.1086/382958. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The RSV sequences used in the analyses in this study encompassed the C-terminal hypervariable domain of the G gene (region <649.918; RSV strain A2 used as reference; GenBank KT992094). The sequences are available in GenBank under accession numbers MW088172 to MW088313.