

Influenza virus infects a wide range of hosts, resulting in illnesses that vary from asymptomatic cases to severe pneumonia and death. Viral transfer can occur between human and nonhuman hosts, resulting in human and nonhuman origin viruses circulating in novel hosts. In this work, we have identified the first case of a swine-origin influenza A(H1N1)pdm09 virus resulting in a human infection. This shows that these viruses not only circulate in swine hosts, but are continuing to evolve and distinguish themselves from previously circulating human-origin influenza viruses. The development of techniques for distinguishing human-origin and swine-origin viruses are necessary for the continued surveillance of influenza viruses. We show that unique genetic signatures can differentiate circulating swine-associated strains from circulating human-associated strains of influenza A(H1N1)pdm09, and these signatures can be used to enhance surveillance of swine-origin influenza.
KEYWORDS: A(H1N1)pdm09, influenza, pandemic, random forest, swine-origin, zoonotic transmission
ABSTRACT
Human-to-swine transmission of seasonal influenza viruses has led to sustained human-like influenza viruses circulating in the U.S. swine population. While some reverse zoonotic-origin viruses adapt and become enzootic in swine, nascent reverse zoonoses may result in virus detections that are difficult to classify as “swine-origin” or “human-origin” due to the genetic similarity of circulating viruses. This is the case for human-origin influenza A(H1N1) pandemic 2009 (pdm09) viruses detected in pigs following numerous reverse zoonosis events since the 2009 pandemic. We report the identification of two human infections with A(H1N1)pdm09 viruses originating from swine hosts and classify them as “swine-origin” variant influenza viruses based on phylogenetic analysis and sequence comparison methods. Phylogenetic analyses of viral genomes from two cases revealed these viruses were reassortants containing A(H1N1)pdm09 hemagglutinin (HA) and neuraminidase (NA) genes with genetic combinations derived from the triple reassortant internal gene cassette. Follow-up investigations determined that one individual had direct exposure to swine in the week preceding illness onset, while another did not report swine exposure. The swine-origin A(H1N1) variant cases were resolved by full genome sequence comparison of the variant viruses to swine influenza genomes. However, if reassortment does not result in the acquisition of swine-associated genes and swine virus genomic sequences are not available from the exposure source, future cases may not be discernible. We have developed a pipeline that performs maximum likelihood analyses, a k-mer-based set difference algorithm, and random forest algorithms to identify swine-associated sequences in the hemagglutinin gene to differentiate between human-origin and swine-origin A(H1N1)pdm09 viruses.
IMPORTANCE Influenza virus infects a wide range of hosts, resulting in illnesses that vary from asymptomatic cases to severe pneumonia and death. Viral transfer can occur between human and nonhuman hosts, resulting in human and nonhuman origin viruses circulating in novel hosts. In this work, we have identified the first case of a swine-origin influenza A(H1N1)pdm09 virus resulting in a human infection. This shows that these viruses not only circulate in swine hosts, but are continuing to evolve and distinguish themselves from previously circulating human-origin influenza viruses. The development of techniques for distinguishing human-origin and swine-origin viruses are necessary for the continued surveillance of influenza viruses. We show that unique genetic signatures can differentiate circulating swine-associated strains from circulating human-associated strains of influenza A(H1N1)pdm09, and these signatures can be used to enhance surveillance of swine-origin influenza.
INTRODUCTION
Human-origin influenza A(H1N1) pandemic 2009 (pdm09) viruses are regularly isolated from swine in North America and Europe (1, 2). The global seasonal spread of A(H1N1)pdm09 influenza viruses in humans and their reintroduction into swine resulted in the generation of circulating swine-associated genetic clades (3, 4). The reintroduction of human viruses into swine was estimated to have occurred at least 133 times globally, leading to the circulation and reassortment of these viruses with other circulating swine influenza A viruses (1, 5). Reassortment of human A(H1N1)pdm09 viruses with North American swine influenza viruses containing the triple reassortant internal gene (TRIG) cassette was also identified, resulting in numerous genome constellations found in pigs since the 2009 pandemic (6, 7). This onward reassortment of swine-origin A(H1N1)pdm09 influenza viruses has greatly increased their genetic variability and generated novel genotypes of unknown phenotypic and epidemiologic consequences. Swine-origin A(H1N1)pdm09 virus detection in pigs in France also resulted in the likely human-to-swine-to-human transmission of these viruses to swine workers and exemplifies the real-time nature by which these cross-species transmission events may occur (8). Onward transmission of these genetically diverse swine influenza viruses into humans could lead to unpredictable outcomes, including further human-to-human spread.
This report details the first two human cases of infection with swine-origin A(H1N1)pdm09 influenza viruses in the United States. National public health surveillance platforms, diagnostic sequence analyses, and phylogenetic comparisons to circulating swine influenza viruses allowed for their detection and further characterization. To assess their genetic relationships to A(H1N1)pdm09 vaccine strains, the hemagglutinin (HA) protein sequences were analyzed. Viruses isolated from samples obtained from each case were also tested for antigenic cross-reactivity with ferret antisera raised to previous A(H1N1)pdm09 vaccine strains, as well as postvaccination human antisera obtained from immunized children and adults. Identification of these cases highlighted the potential complexity of distinguishing swine-origin viruses from typical human, seasonal A(H1N1)pdm09 viruses. Recognition of host-specific genetic signatures is critical to this process when (i) reassortment does not result in acquisition of clearly defined swine-origin gene segments and (ii) if swine virus genomic sequences from potential sources of exposure are not readily available for comparison. We therefore developed HA-gene-sequence-based methodologies to assess the host-origin of these viruses using three independent analyses: (i) maximum likelihood (ML) HA gene phylogenies; (ii) k-mer set comparisons, and (iii) random forest algorithms. A combination of these three approaches was used to assess the first confirmed human case of a swine-associated A(H1N1)pdm09 virus infection in the United States and to identify another suspected human infection of swine-associated A(H1N1)pdm09 virus containing molecular signatures suggestive of a swine origin.
RESULTS
Identification of human infections with swine-associated A(H1N1)pdm09 virus.
In October 2017, a female with no underlying medical conditions but with a history of swine exposure developed an influenza-like illness in Iowa. The patient was not hospitalized and completely recovered. No human-to-human transmission was identified. Real-time PCR (RT-PCR) testing of the specimen indicated infection with a seasonal influenza A(H1N1)pdm09 virus. However, results from next-generation sequencing suggested infection with an influenza A(H1N1) variant (A[H1N1]v) virus based on the similarity of several internal genes to those of swine influenza viruses circulating in North America. Subsequently, a virus isolate, A/Iowa/33/2017, was cultured in MDCK cells following a 48 h incubation at 35°C. The specimen and isolate were forwarded to the U.S. Centers for Disease Control and Prevention (CDC) for additional testing and sequences were deposited in the GISAID EpiFlu database (A/Iowa/33/2017; accession number EPI_ISL_329966). Follow-up investigation by state veterinarians determined the patient had direct exposure to swine in the week preceding illness onset and specimens collected from swine at the patient’s farm were sent to the National Veterinary Services Laboratories (Ames, Iowa) for sequencing (A/swine/Iowa/A01104106/2017; accession number EPI_ISL_296456).
The second case, not epidemiologically linked to the first, was detected in Michigan in May 2019, for which the patient recovered from illness following oseltamivir treatment prior to being discharged. The patient reported no recent travel history, nor exposure to swine or other livestock, in the weeks preceding illness onset. The follow-up investigation of close contacts excluded further human-to-human transmission. Virus was isolated in MDCK cells following 48 h incubation and Illumina MiSeq sequences were deposited in the GISAID EpiFlu database (A/Michigan/288/2019; accession number EPI_ISL_381463). The specimen and isolate were forwarded to the U.S. CDC for additional testing.
Phylogenetic analysis and genetic comparisons to vaccine strains.
A phylogenetic tree of human and swine influenza A(H1N1)pdm09 HA genes was built to visualize and understand the larger phylogenetic context of the distribution of the swine-associated and human-associated strains within the influenza A(H1N1)pdm09 lineage through both time and sequence divergence (Fig. 1A). The initial assessment of the location of A/Iowa/33/2017 indicated that it grouped in a phylogenetic clade primarily made up of swine-associated viruses, with a small set of human-associated strains isolated in 2016 that were closely related to the most recent common ancestor of this clade. Phylogenetic analysis of each viral gene segment indicated the virus was a reassortant containing PB2, PB1, NP, and NS genes derived from the TRIG cassette, but with HA, NA, PA, and M genes closely related to A(H1N1)pdm09 viruses circulating in swine (Fig. 1 and 2; Fig. S1 to S6 in the supplemental material). No amino acid residues were identified in the NA or PA proteins to indicate reduced susceptibility to antiviral drugs. An influenza virus collected from swine being reared on the farm where the patient also resided, A/swine/Iowa/A01104106/2017, was found to be nearly identical (99.9% identity) at the nucleotide level for each gene segment compared to the virus isolated from the patient. Strains belonging to the HA clade have continued to circulate in the swine population and were isolated in 2018 (Fig. 1A). Isolate A/Iowa/33/2017 had six HA1 amino acid changes relative to the nearest A(H1N1)pdm09 vaccine strain in use at the time of detection, A/Michigan/45/2015. Genetic analysis revealed that there were 6 potential amino acid differences in the H1 region of the HA, including mixed bases, compared to candidate vaccine virus A/Michigan/45/2015 (Table S1A). Four of these were identified at putative antigenic sites: site E (A48A/T), site A (D/E127D), and site B (N156I/N and I/L191L) (9). A/Iowa/33/2017 contained no HA amino acid differences relative to the swine influenza virus isolated from sampling swine on the patient’s farm.
FIG 1.


Evolutionary relationships of the hemagglutinin (A) and neuraminidase (B) nucleotide sequences. The evolutionary history was inferred using the neighbor-joining method (28). (A and B) The optimal trees with the sums of branch length equaling 1.65521810 (A) and 2.16241627 (B). The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are shown next to the branches (29). The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Jukes-Cantor method (30) and are in the units of the number of base substitutions per site. The analysis involved 203 (A) and 209 (B) nucleotide sequences. All ambiguous positions were removed for each sequence pair. There were a total of 1,795 (A) and 1,489 (B) positions in the final data set. Evolutionary analyses were conducted in MEGA7 (19).
FIG 2.

Genome constellations identified in A(H1N1) variant viruses.
A/Michigan/288/2019 was an atypical A(H1N1)pdm09 subtype virus with swine-lineage internal genes, indicating swine origin. While the HA and neuraminidase (NA) genes clustered with contemporary human A(H1N1)pdm09 viruses (clade 6B.1A) and a small number of swine influenza viruses from the United States, all of the internal protein-coding vRNAs were similar to swine viruses circulating in the United States (Fig. 1 and 2; Fig. S1 to S6). The PB1 and PA genes clustered with those of the A/swine/Texas/4199-2/1998 virus used in a commercial, live-attenuated swine vaccine in the United States (Fig. 2; Fig. S2 and S3) (10). No amino acid residues were identified in the NA or PA proteins to indicate reduced susceptibility to antiviral drugs. There were six amino acid substitutions in HA1 of A/Michigan/288/2019 compared to the A/Idaho/07/2018 cell-based seasonal influenza vaccine component and seven amino acid substitutions in the HA1 compared to the A/Brisbane/02/2018 egg-based vaccine component (Table S1B). Of these differences, only one amino acid residue was located in putative antigenic site A (K142R).
Differentiating swine-associated from human-associated A(H1N1)pdm09 viruses using k-mer set differences.
K-mers generated in different data sets were compared to eliminate matches and select only matching values. By using the k-mers generated from an inclusive and exclusive group, we defined a set difference that identified only k-mers unique to one group. In addition, this process could identify multibase insertions or deletions that may be present in any selected set of A(H1N1)pdm09 reference strains or vaccine strains. As described in the methods, the inclusive group was selected as the circulating swine influenza viruses, while the exclusive group was set as the human-associated strains of A(H1N1)pdm09 viruses based on both phylogeny and host of isolation. Differentiating sequences based on this technique uses all possible k-mers present in the sequence data and resulted in the identification of several k-mers associated with only the circulating swine-associated A(H1N1)pdm09 viruses. There were four 11-mers identified as unique to the circulating swine influenza viruses. Each of these k-mers contained a site with a single base pair change that differentiated these strains at positions 78, 375, 966, and 1,413. In total, 121 of 149 circulating swine-associated influenza viruses with an A(H1N1)pdm09 HA gene contained 1 or more of these sites. A/Iowa/33/2017 and A/Michigan/288/2019 had all four positions described in Table 1. This is relative to 94 of 3,188 unique human-associated strains collected after 2015 containing one or more of the identified sites.
TABLE 1.
Sites identified by the k-mer analysis representing the circulating swine-associated strains
| Sequencea | Position in HA nucleotide coding sequenceb | Frequency in circulating swine (%)c | Frequency in circulating human (%)d |
|---|---|---|---|
| CATGCAAACAA | 78 | 67/149 (45.0) | 57/3188 (1.8) |
| TCAGTATCATC | 375 | 59/149 (39.6) | 50/3188 (1.6) |
| CCAAAATATGT | 966 | 105/149 (70.5) | 33/3188 (1.0) |
| TTAAAGAACAA | 1413 | 60/149 (40.2) | 31/3188 (1.0) |
Nucleotides defining each k-mer’s unique portion are underlined.
Nucleotide positions are relative to the HA nucleotide sequences.
Fraction of swine-associated influenza A virus strains observed with unique k-mer.
Fraction of human-associated influenza A virus strains observed with unique k-mer.
Identification of swine-associated predictive patterns in A(H1N1) pdm09 viruses by random forest algorithms.
The random forest model classified strains as either predicted swine or predicted human, and used those differences to identify sites in the HA gene segment to differentiate circulating swine-associated influenza viruses with A(H1N1)pdm09 HA genes from circulating human-associated influenza A(H1N1)pdm09 viruses. The training set consisted of 149 strains identified as circulating swine-associated A(H1N1)pdm09 strains and 149 human-associated strains of A(H1N1)pdm09 collected after 2015 using the previously described criteria (4). The results of the training set identified three noncoding nucleotide sites as highly predictive of host status (Table 2). Compared to the coding region sequence, including the signal peptide of the reference A(H1N1)pdm09 virus A/California/07/2009, the nucleotide sites were identified at positions 954, 966, and 1,359 (Fig. 3). Using this model resulted in human-associated viruses and swine-associated viruses being predicted correctly 85.7% and 100% of the time using the test data set (Table 1), respectively. Using the entire data set, including all collected human-associated and swine-associated A(H1N1)pdm09 influenza A viruses, the predictions were accurate for 98.0% of human-associated and 97.3% of swine-associated viruses. In addition, using the full deduplicated human-associated data set containing 3,188 viruses resulted in correct identification of 97.4% (3,038 of 3,188) of the human-associated viruses. The model differentiated circulating human-associated viruses based on position 954 containing a guanine, position 966 containing a guanine, and position 1,359 containing a cysteine. A collection of 3,421 influenza A(H1) virus sequences downloaded from the Influenza Resource Database and identified as isolated from swine hosts across all swine-associated lineages were analyzed for the occurrence of the marker set (11). The human-associated markers of 954G and 966G were identified in only 4.7% (161/3,421) of the swine-isolated influenza A(H1) viruses.
TABLE 2.
Random forest algorithm results for training, testing, and full data
| True host | Test dataseta
|
True host | Training and test datasetb
|
True host | Complete datasetc
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Predicted host |
Predicted host |
Predicted host |
|||||||||
| Human | Swine | Percent | Human | Swine | Percent | Human | Swine | Percent | |||
| Human | 12 | 2 | 85.7 | Human | 146 | 3 | 97.9 | Human | 3,038 | 150 | 95.3 |
| Swine | 0 | 15 | 100.0 | Swine | 4 | 145 | 97.3 | Swine | 4 | 145 | 97.3 |
The test data represents a randomly selected 10% of data separated from the initial random forest subset. These data were not used until after training had been performed.
The training and test data sets were combined and then rerun through the determined random forest algorithm to determine the accuracy of the model given the data.
The complete data set represents 100% of available data.
FIG 3.
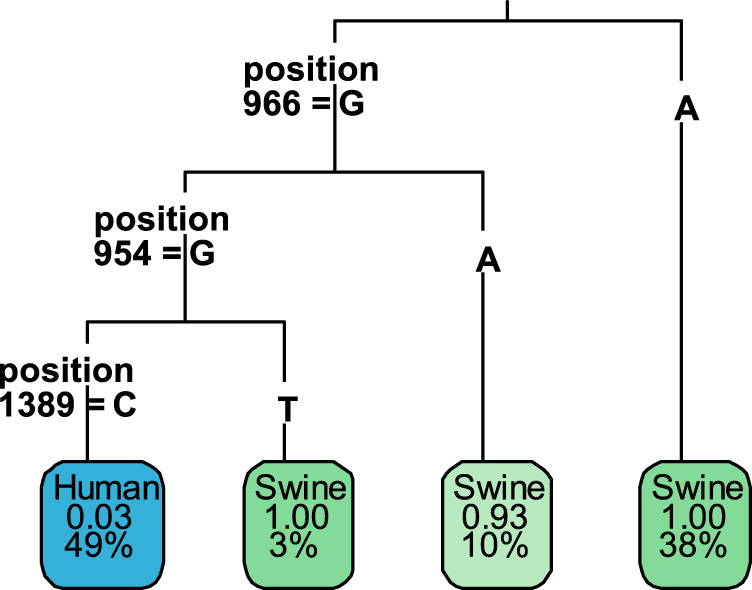
Random forest tree output. Positions represented in the random forest tree represent the human-associated nucleotide. The green colored rectangles represent the fraction of strains in the collection that are swine associated. The blue colored rectangles represent the human-associated endpoint for the categorization of human-associated strains. Percentage values represent the fraction of the data set represented by in that endpoint. (i.e., 49% represents that human-associated strains make up ∼50% of the data set).
Antigenic characterization.
A/Iowa/33/2017 was well inhibited by ferret antisera raised against A/California/7/2009 and A/Michigan/45/2015 viruses (Table 3). Compared to the adult serum pool, the child pool had generally lower HI titers with each antigen tested and a 16-fold reduced titer relative to the adult antisera when tested with A/Iowa/33/2017 antigen. Ferret antisera raised against A(H1N1)pdm09 vaccine viruses (i.e., A/Michigan/45/2015, A/Brisbane/02/2018, and A/Idaho/7/2018) inhibited A/Michigan/288/2019 and A/Iowa/33/2017 with titers that were equivalent to homologous virus titers. Sera from vaccinated children and adults also inhibited the viruses at titers comparable to those with 2016 to 2017 and 2018 to 2019 vaccine virus antigens for A/Iowa/33/2017 and A/Michigan/288/2019, respectively (Tables 3 and 4).
TABLE 3.
Hemagglutination inhibition assay of A(H1N1)v viruses (2016 to 2017)
| Antigen | Subtype (lineage) | Ferret antisera |
||||||
|---|---|---|---|---|---|---|---|---|
| CA/07 | MI/45 | OH/9 | MN/19 | WI/71 | Childa | Adultb | ||
| Reference antigens | ||||||||
| A/California/7/09× PR8 (X-179) | H1N1 (pdm09) | 160 | 160 | 160 | <10 | 20 | 320 | 320 |
| A/Michigan/45/2015 | H1N1 (pdm09) | 1280 | 5120 | 80 | <10 | 80 | 80 | 640 |
| A/Ohio/9/2015 IDCDC-RG48A | H1N1v (gamma) | <10 | <10 | 5120 | <10 | <10 | 10 | 320 |
| A/Minnesota/19/2011 | H1N2v (delta1) | <10 | <10 | 10 | 640 | 640 | 20 | 80 |
| A/Wisconsin/71/2016 | H1N2v (delta1) | <10 | <10 | 20 | 320 | 5120 | 80 | 80 |
| Test antigens | ||||||||
| A/Iowa/33/2017 | H1N1v (pdm09) | 640 | 5120 | 160 | <10 | 10 | 40 | 640 |
2016–2017 postvaccine immune serum pool from child (0 to 3 yrs) vaccinees (A/California/7/2009 vaccine).
2016–2017 postvaccine immune serum pool from adult (19 to 49 yrs) vaccinees (A/California/7/2009 vaccine).
TABLE 4.
Hemagglutination inhibition assay of A(H1N1)v viruses (2018 to 2019)
| Antigen | Subtype (lineage) | Ferret antisera |
||||||
|---|---|---|---|---|---|---|---|---|
| MI/45 | ID/07 | WI/505 | RG48A | IA/32 | Childa | Adultb | ||
| Reference antigens | ||||||||
| A/Michigan/45/2015 | H1N1 (pdm09) | 5120 | 5120 | 5120 | 20 | <10 | 1280 | 2560 |
| A/Idaho/7/2018 | H1N1 (pdm09) | 5120 | 5120 | 2560 | 10 | <10 | 320 | 640 |
| A/Wisconsin/505/2018 | H1N1 (pdm09) | 5120 | 5120 | 5120 | <10 | <10 | 640 | 1280 |
| A/Ohio/9/2015 IDCDC-RG48A | H1N1v (gamma) | 10 | 10 | 10 | 1280 | <10 | 20 | 160 |
| A/Iowa/32/2016 | H1N2v (delta1) | <10 | <10 | <10 | <10 | 2560 | <10 | 80 |
| Test antigens | ||||||||
| A/Michigan/288/2019 | H1N1v (pdm09) | 5120 | 5120 | 5120 | <10 | <10 | 640 | 640 |
2018–2019 postvaccine immune serum pool from child (0 to 3 yrs) vaccinees (A/Michigan/45/2015 vaccine).
2018–2019 postvaccine immune serum pool from adult (19 to 49 yrs) vaccinees (A/Michigan/45/2015 vaccine).
DISCUSSION
Since reporting of novel influenza A viruses became nationally notifiable in 2005, 22 human infections with A(H1N1)v viruses, including the two cases reported here, have been confirmed in the United States. While A(H1)v viruses of multiple HA lineages (i.e., 1A.1 [alpha], 1A.2 [beta], 1A.3.1 [gamma], 1B.2.1 [delta 2], and 1B.2.2 [delta 1]) have been sporadically detected, these two cases represent the first identification of swine-origin A(H1N1)pdm09-like viruses (1A.3.3.2) detected in humans in the United States. Human-to-swine transmission (reverse zoonosis) of seasonal influenza viruses has led to sustained circulation of human-like influenza viruses in swine in the United States (12). While these viruses evolve and eventually become adapted in swine, reverse zoonoses can result in virus detections that are difficult to distinguish as “swine-origin” versus “human-origin” due to the genetic similarity of viruses circulating in both hosts. In October 2017, A/Iowa/33/2017 was identified as a A(H1N1)pdm09 reassortant virus with HA, NA, PA, and M gene segments grouping phylogenetically with swine-origin A(H1N1)pdm09 viruses and the remaining genes derived from the TRIG cassette, making this the first identification of a swine-associated influenza A(H1N1)pdm09-like variant virus identified in the United States. Epidemiologic investigation identified direct exposure to swine, and samples collected from swine at the household of the case confirmed the presence of the same virus that infected the individual. A second A(H1N1)v virus infection originating from circulating swine-origin A(H1N1)pdm09-like viruses was detected in 2019 and, like A/Iowa/33/2017, was also originally diagnosed as a human seasonal A(H1N1)pdm09 virus based on correctly interpreted rRT-PCR results. Subsequent genomic sequence analysis confirmed the swine source and revealed that both viruses were reassortants containing a combination of A(H1N1)pdm09 genes and those detected in swine influenza viruses. Of note was the detection of PB1 and PA genes in A/Michigan/288/2019 that clustered with genes from a live-attenuated swine influenza vaccine used commercially in the United States. Although these genes were also identified in circulating swine influenza viruses, reassortment of live-attenuated influenza vaccine (LAIV) virus genes in pigs and their eventual detection in an infected person is of concern, given the unknown virologic and epidemiologic consequences. A report from Sharma et al. demonstrated detection of LAIV HA gene segments in 58 of 1,116 viruses sampled in a period from January to October 2018 collected from 11 different U.S. states (10). Although only a subset of these were selected for whole-genome sequencing, GenBank analysis of swine influenza viruses collected beginning in 2018 revealed 5.6% of viruses with both PB1 and PA gene segments related to the LAIV virus, indicating low levels of this combination of genes in total U.S. swine influenza viruses. While both cases completely recovered from illness, follow-up investigations of the case in Michigan failed to confirm exposure to swine. Neither case, however, resulted in detection of human-to-human transmission based on follow-up of close contacts and no other human A(H1N1)pdm09 or A(H3N2) viruses have been detected to date with PB1 or PA genes similar to the swine live-attenuated influenza vaccine.
While the source of the viruses collected from these cases were resolved by codon complete genome sequence comparison of the zoonotic viruses to available swine influenza virus genomes, future cases may not be as readily discernible if (i) reassortment does not result in acquisition of well-defined swine influenza virus gene segments or (ii) if related swine influenza virus genomic sequences are not available. Therefore, we sought to identify host-specific molecular markers that distinguished human, seasonal A(H1N1)pdm09 influenza viruses from circulating swine A(H1N1)pdm09 influenza viruses that have infected a human host. Initially, the determination of viruses as swine-associated or human-associated was based on the phylogenetic analysis of the HA gene tree using inferences described by Nelson et al. (4). Specifically, the inference of onward transmission in swine (i.e., swine-associated viruses) was based on the overwhelmingly higher level of influenza virus surveillance in humans compared with swine and the low probability that long branch lengths in the HA tree arose from unsampled human viruses (4). Analysis of A/Iowa/33/2017 along with other human- and swine-associated viruses were then used in a random forest analysis and a unique k-mer analysis to identify molecular markers in the HA gene that differentiated human- and swine-associated A(H1N1)pdm09 viruses. The k-mer analysis and the random forest analysis both identified site 966 as highly predictive for differentiating between swine-associated and human-associated viruses. The initial viruses detected at the start of the 2009 pandemic maintained the swine-associated nucleotides at positions 954, 966, and 1,359. However, using a timescale phylogenetic tree generated in Nextstrain (https://nextstrain.org) (data not shown) from between late 2010 and early 2011, a single nucleotide polymorphism at position 966 occurred in humans, mutating the original nucleotide from adenosine (A) to guanine (G) in the HA sequence. After this occurred, the circulating human-associated viruses maintained guanines in both nucleotide positions 954 and 966.
In the human population, these molecular markers have remained stable and continue to be identified in seasonal A(H1N1)pdm09 viruses. As time has progressed, human-associated viruses have reentered the swine population (1) and, based on the results from the phylogenetic trees and their isolation dates, the originally circulating human viruses that maintained circulation in U.S. swine populations also gained the swine-associated mutations at either or both position 954 or 966, mutating the human-associated G at those sites to the swine-associated A. This observation was supported by the identification of the second pdm09-like A(H1N1)v virus, A/Michigan/288/2019, which contained an HA with an A at position 966, despite this virus being closely related (percent identity of 99.49%) to the currently circulating human-associated A(H1N1)pdm09 virus A/Idaho/07/2018. As described above, full genome phylogenetic analysis further confirmed the swine origin of A/Michigan/288/2019 based on the presence of internal gene segments from circulating swine influenza A viruses. This virus would have been categorized as circulating human cases of seasonal A(H1N1)pdm09 without further investigation of the internal gene segments or lack of molecular markers to suggest otherwise.
The k-mer analysis alone found that strains representing the circulating swine-associated populations were 70.5 times more likely to be identified with the G966A mutation than the circulating human-associated strains. The random forest model identified additional nucleotide positions that increased the predictability of using molecular markers to differentiate the host association. While the primary G966A mutation occurred in circulating swine-associated viruses, 10% of the circulating swine-associated strains were identified as having G at position 966, but were also identified as having A at position 954. This less-frequent mutation may shed light on the region itself and the usefulness of multidimensional analysis of nucleotide sequences to identify these patterns. Overall, the random forest analysis differentiated circulating swine-associated from circulating human-associated A(H1N1)pdm09 influenza viruses using the test and training data with an accuracy of 97.3% (Table 2). In addition, the random forest model identified a significantly lower percentage of circulating swine viruses containing the human-associated positions 954G and 966G, but was able to differentiate host using a third mutation at nucleotide position C1389T. The additional site may not represent a large enough percentage of the swine population to properly differentiate based on this site alone but, combined with other genetic and epidemiologic factors, may also support additional sequence analysis and follow-up investigation. Analysis of a collection of 3,421 swine influenza A(H1) virus HA sequences from the Influenza Research Database (IRD) representing all lineages of swine-associated viruses identified the human-associated marker of 954G and 966G in 161 of 3,421 (4.7%), suggesting that these markers are not unique to only swine influenza A(H1N1)pdm09 lineage viruses and may represent a region of significance for adaptation of A(H1) viruses in the swine host.
Based on these results, A(H1N1)pdm09 viruses detected in a human host identified with A at position 954 and/or 966 should be flagged for additional analysis, especially full-genome characterization of swine-origin internal genes. A major limitation of this work, however, is understanding the rate at which these mutations occur following a cross-species transmission event and how quickly they become fixed in a host population. This is critical, as fixation in the host population would ultimately determine the predictability of these markers. Experimental studies in a swine infection model may elucidate this further. Further work will also be needed to determine other factors that may contribute to the accumulation of these mutations and whether these sites are the result of fitness advantages in human or swine hosts and/or the potential mechanisms driving the nucleotide site polymorphism.
Antigenic testing of both A(H1N1)v viruses by hemagglutination inhibition (HI) assay demonstrated that ferret antisera raised to the A(H1N1)pdm09 components of previous seasonal influenza vaccines inhibited the viruses well with titers that were equivalent to homologous virus titers. Additionally, postvaccination immune sera obtained from seasonal influenza-vaccinated children (0 to 3 years) and adults (19 to 49 years) also inhibited the viruses at titers comparable to those of 2016 to 2017 and 2018 to 2019 A(H1N1)pdm09 vaccine viruses. The antiserum titers of children against the viruses were overall lower than those detected in the adult pool. This was likely a reflection of multiple vaccinations and/or exposures of adults with A(H1N1)pdm09 viruses compared to children in the cohort tested. While vaccinated children may have lower titers of cross-protective antibodies compared to adults, population immunity to the A(H1N1)v and swine A(H1N1)pdm09 viruses is expected to be high.
The identifications of A/Iowa/33/2017 and A/Michigan/288/2019 mark the first occurrence in the United States of an A(H1N1)pdm09 virus infection in a human arising from a swine source. While the swine-origin of the 2009 A(H1N1) virus pandemic and the subsequent and reoccurring transmissions of human-origin A(H1N1)pdm09 viruses into swine are well documented, the genetic similarities among currently circulating swine and human viruses makes differentiation between the host of origin potentially difficult. The host-specific markers at nucleotide positions 954 and 966 of the HA gene coding region were identified at higher frequencies in circulating swine-associated A(H1N1)pdm09 viruses compared to human-associated viruses. Evaluation of these markers using only HA gene sequences could identify potential variant viruses in the future when epidemiological links and/or exposure history suggest swine-to-human transmission. As described here, routine seasonal influenza virus surveillance networks should include monitoring people throughout the year for signs of zoonotic influenza A virus infections and include whole-genome sequence analyses whenever possible for the detection of swine-origin viruses, which may otherwise be missed. The continual reintroduction and persistent circulation of A(H1N1)pdm09 viruses in U.S. swine populations will eventually lead to antigenically diverse viruses over time that may maintain their capacity for human-to-human transmission. While year-round surveillance for influenza viruses in the U.S. swine population indicated that during a 2-year period from April 2018 to March 2020 viruses with the A(H1N1) pdm09 virus HA and NA genes made up only 5.3% of all influenza viruses detected in swine, these data suggest even less frequently identified subtypes have the potential for spill-over into humans (13–14). Additional steps should be taken by health care providers and researchers to assess the potential for zoonotic influenza virus infections in persons with direct or indirect swine exposure.
MATERIALS AND METHODS
Specimen collection and preliminary diagnosis by real-time PCR and next-generation sequencing.
Case number 1. In October 2017, a 33-year-old female with no underlying medical conditions developed an influenza-like illness in Iowa. The person sought medical care at an emergency department and a nasopharyngeal swab specimen was collected for testing. A rapid influenza diagnostic test used at the hospital laboratory indicated an influenza A virus infection. The specimen was forwarded to the State Hygienic Laboratory at the University of Iowa as part of routine influenza surveillance activities, where real-time PCR (RT-PCR) testing using seasonal influenza typing and subtyping assays was conducted, as previously described (15). The specimen was then forwarded to the Wisconsin State Laboratory of Hygiene, National Influenza Reference Center, per national surveillance specimen submission guidelines. RNA from the sample was extracted and each gene segment amplified by multisegment RT-PCR (MRT-PCR) (16). Full-genome sequencing using Illumina MiSeq technology was performed, as previously described (17).
Case number 2. In May 2019, an elderly male residing in Michigan developed chills, cough, shortness of breath, and fatigue. The patient had multiple underlying medical conditions (i.e., chronic lung disease, multiple chronic heart/circulatory diseases, and diabetes mellitus) and sought medical care at an emergency department, where he was later admitted to the hospital, treated with oseltamivir, and fully recovered. A respiratory specimen was collected and forwarded to the Michigan State Public Health Laboratories, where the sample tested positive for A(H1N1)pdm09 virus prior to being forwarded to the Wisconsin State Laboratory of Hygiene. RNA from the sample was extracted and each gene segment amplified by multisegment RT-PCR (MRT-PCR) and full-genome sequencing using Illumina MiSeq technology was performed, as described above.
Swine and human influenza A(H1N1)pdm09 virus genomes analyzed.
Swine and seasonal human virus sequences were downloaded from the Influenza Research Database using the following criteria: (i) full genome segments; (ii) virus type A; (iii) A(H1N1)pdm09 subtype; (iv) HA segment complete sequence; (v) geographic region of North America; (vi) date range between 2015 and 2019; and (vii) host identified as either swine or human. In total, 10,451 influenza virus genome sequences were downloaded from the Influenza Research Database (IRD) (https://www.fludb.org/brc/home.spg?decorator=influenza) with 3,381 human and 159 swine host sequences. After deduplication of sequences, there were 2,166 human and 149 swine strains used in the analysis. Analyses requiring down-sampling of human sequences was completed by randomly selecting subsets from the collections of gene segments.
Phylogenetic analysis of viral genomes.
A phylogenetic tree was built for each of the 8 influenza virus genome segments. The sequences for each strain included (as described above) were aligned using the ClustalW application within the MEGA7.0 software (18, 19). Neighbor joining phylogenetic trees were built using MEGA7.0 with 1,000 bootstraps and the Jukes-Cantor model of evolution with uniform rates. Secondary analysis was performed using MAFFT and IQ-tree, confirming the locations and fidelity of the represented NJ phylogenetic tree topology (20–23).
HA amino acid difference analysis.
The circulating swine and human host A(H1N1)pdm09 viruses were used as input sequences to generate a mature HA1 protein amino acid difference comparison table. The nearest vaccine virus was used as a reference sequence for comparison to identify differences in amino acids relative to circulating swine influenza viruses and the two A(H1N1) variant (A[H1N1]v) viruses identified (Table S1A and B). Ambiguous nucleotides had all potential amino acid outcomes reported. Amino acid differences were identified using software scripts written for the BioEdit platform (24). Amino acid residue locations were shown relative to the mature HA1 protein locations.
Identification of persistently circulating swine-associated A(H1N1)pdm09 viruses.
The identification of viruses belonging to persistently circulating A(H1N1)pdm09 swine virus populations in the United States were performed based on the qualifications by Nelson et al. (4). In brief, the 149 swine-associated viruses with multiple, spatiotemporally related swine viruses and within monophyletic branches were considered persistently circulating. Specifically, singletons and groups with long and well-separated branches, not closely related to the currently circulating human seasonal viruses, were selected subjectively and considered as circulating in swine as they were unlikely to be from a previously unsampled human circulating population of influenza viruses (4).
K-mer set difference analysis.
Identification of k-mer sequences unique to U.S. swine-associated HA gene segments was performed using the set difference functions in standard Python 3.7. In brief, the groups previously defined as human-associated and swine-associated were made based on phylogeny and host with 4 random viruses selected from either group. All swine-associated groups contained an additional gene segment, the HA gene segment from A/Iowa/33/2017. These groups were then split into a collection of all unique k-mers with a k-mer size of 11 and compared to identify k-mers unique to the swine-associated strains. The unique k-mers were then used to search for similar fragments across all inclusive (circulating swine-associated group) and exclusive (circulating human-associated group) data sets to determine the relative frequency and uniqueness of each identified k-mer. The highly unique fragments were then mapped to the reference virus, A/Iowa/33/2017, to determine their relative overlap and identify the length of any unique fragments.
Random forest analysis.
The equal populations of human-associated and circulating swine-associated viruses detected in the United States based on the previously defined groups (i.e., defined by both phylogeny and host of isolation) were used as input to determine sites that suggested association with a human versus swine host. Duplicate nucleotide sequences of HA within and between the host levels were removed from further analysis, reducing the total sequences from 3,188 to 2,166 sequences. The human-associated sample population was then randomly down-sampled to match the number of swine-associated viruses available. The sequences were then aligned using MAFFT, and the resulting alignment was converted to matrix format and used for further analysis (25). The matrix of aligned nucleotide sequences, along with the host-level metadata, were used as input for the random forest algorithm from the VSURF package for the R statistical language, R code available in Fig. S7 (26, 27). The R code, in brief, split the data into training and testing portions, 90% and 10%, respectively. The training data were then split using a 5-fold cross validation method to determine the frequency and error surrounding each site produced by the model. Error rates of less than 5% were considered acceptable, and sites identified below the error threshold were used in the analysis of the testing data. The entire process was performed three times, each time with a new sample of 149 strains from human-associated strains to match the number of available swine samples.
Antigenic analysis.
Postinfection ferret immune sera were raised to a panel of A(H1N1) and A(H1N2) viruses, including select A(H1N1)pdm09 viruses and recent A(H1N1)v and A(H1N2)v viruses. Antisera were harvested from ferrets 14 days postinfection following an intranasal infectious dose of 106 50% egg infectious doses. The panel of antisera was used to test two-way hemagglutination inhibition (HI) titers of each virus to assess cross-reactivity between various genetic lineages of A(H1N1) and A(H1N2) swine and human viruses. Each serum was treated at a 1:4 dilution with receptor-destroying enzyme (RDE) for 18 h at 37°C, with a total dilution of 1:10 using physiological saline, followed by adsorption with turkey red blood cells. All antigens were standardized to 8 HAU/50 μl prior to 30 min incubation at room temperature with 2-fold serial dilutions of antiserum. Turkey red blood cells (0.5%) were used to evaluate hemagglutination inhibition.
Supplementary Material
ACKNOWLEDGMENTS
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention or the Agency for Toxic Substances and Disease Registry.
P.W.C. was supported by the Association of Public Health Laboratories. N.Z. was supported by Chickasaw Nation Industries.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Nelson MI, Stratton J, Killian ML, Janas-Martindale A, Vincent AL. 2015. Continual reintroduction of human pandemic H1N1 influenza A viruses into swine in the United States, 2009 to 2014. J Virol 89:6218–6226. doi: 10.1128/JVI.00459-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simon G, Larsen LE, Dürrwald R, Foni E, Harder T, Van Reeth K, Markowska-Daniel I, Reid SM, Dan A, Maldonado J, Huovilainen A, Billinis C, Davidson I, Agüero M, Vila T, Hervé S, Breum SØ, Chiapponi C, Urbaniak K, Kyriakis CS, Brown IH, Loeffen W, ESNIP3 consortium. 2014. European surveillance network for influenza in pigs: surveillance programs, diagnostic tools and swine influenza virus subtypes identified in 14 European countries from 2010 to 2013. PLoS One 9:e115815-21. doi: 10.1371/journal.pone.0115815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chastagner A, Hervé S, Bonin E, Quéguiner S, Hirchaud E, Henritzi D, Béven V, Gorin S, Barbier N, Blanchard Y, Simon G. 2018. Spatiotemporal distribution and evolution of the A/H1N1 2009 pandemic influenza virus in pigs in France from 2009 to 2017: identification of a potential swine-specific lineage. J Virol 92:440–442. doi: 10.1128/JVI.00988-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson MI, Wentworth DE, Culhane MR, Vincent AL, Viboud C, LaPointe MP, Lin X, Holmes EC, Detmer SE. 2014. Introductions and evolution of human-origin seasonal influenza a viruses in multinational swine populations. J Virol 88:10110–10119. doi: 10.1128/JVI.01080-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Q, Ma J, Liu H, Qi W, Anderson J, Henry SC, Hesse RA, Richt JA, Ma W. 2012. Emergence of novel reassortant H3N2 swine influenza viruses with the 2009 pandemic H1N1 genes in the United States. Arch Virol 157:555–562. doi: 10.1007/s00705-011-1203-9. [DOI] [PubMed] [Google Scholar]
- 6.Belser JA, Gustin KM, Maines TR, Blau DM, Zaki SR, Katz JM, Tumpey TM. 2011. Pathogenesis and transmission of triple-reassortant swine H1N1 influenza viruses isolated before the 2009 H1N1 pandemic. J Virol 85:1563–1572. doi: 10.1128/JVI.02231-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao S, Anderson TK, Walia RR, Dorman KS, Janas-Martindale A, Vincent AL. 2017. The genomic evolution of H1 influenza A viruses from swine detected in the United States between 2009 and 2016. J Gen Virol 98:2001–2010. doi: 10.1099/jgv.0.000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chastagner A, Enouf V, Peroz D, Hervé S, Lucas P, Quéguiner S, Gorin S, Béven V, Behillil S, Leneveu P, Garin E, Blanchard Y, van der Werf S, Simon G. 2019. Bidirectional human-swine transmission of seasonal influenza A(H1N1)pdm09 virus in pig herd, France, 2018. Emerg Infect Dis 25:1940–1943. doi: 10.3201/eid2510.190068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. 1982. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31:417–427. doi: 10.1016/0092-8674(82)90135-0. [DOI] [PubMed] [Google Scholar]
- 10.Sharma A, Zeller MA, Li G, Harmon KM, Zhang J, Hoang H, Anderson TK, Vincent AL, Gauger PC. 2020. Detection of live attenuated influenza vaccine virus and evidence of reassortment in the U.S. swine population. J Vet Diagn Invest 32:301–311. doi: 10.1177/1040638720907918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Squires RB, Noronha J, Hunt V, García-Sastre A, Macken C, Baumgarth N, Suarez D, Pickett BE, Zhang Y, Larsen CN, Ramsey A, Zhou L, Zaremba S, Kumar S, Deitrich J, Klem E, Scheuermann RH. 2012. Influenza research database: an integrated bioinformatics resource for influenza research and surveillance. Influenza Other Respir Viruses 6:404–416. doi: 10.1111/j.1750-2659.2011.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rajao DS, Gauger PC, Anderson TK, Lewis NS, Abente EJ, Killian ML, Perez DR, Sutton TC, Zhang J, Vincent AL. 2015. Novel reassortant human-like H3N2 and H3N1 influenza A viruses detected in pigs are virulent and antigenically distinct from swine viruses endemic to the United States. J Virol 89:11213–11222. doi: 10.1128/JVI.01675-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeller MA, Anderson TK, Walia RR, Vincent AL, Gauger PC. 2018. ISU FLUture: a veterinary diagnostic laboratory web-based platform to monitor the temporal genetic patterns of Influenza A virus in swine. BMC Bioinformatics 19:397. doi: 10.1186/s12859-018-2408-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Animal Plant Health Inspection Service Veterinary Services. 2020. Animal disease information. https://www.aphis.usda.gov/animal_health/animal_dis_spec/swine/downloads/fy2020quarter2swinereport.pdf. (Data accessed 21 September 2020)
- 15.Centers for Disease Control and Prevention. 2013. CDC laboratory support for influenza surveillance (CLSIS). https://www.cdc.gov/flu/clsis/.
- 16.Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J Virol 83:10309–10313. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shepard SS, Meno S, Bahl J, Wilson MM, Barnes J, Neuhaus E. 2016. Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genomics 17:708–718. doi: 10.1186/s12864-016-3030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 19.Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katoh K, Misawa K, Kuma K-I, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalyaanamoorthy S, Minh BQ, Wong TKF, Haeseler von A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen L-T, Schmidt HA, Haeseler von A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trifinopoulos J, Nguyen L-T, Haeseler von A, Minh BQ. 2016. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res 44:W232–5. doi: 10.1093/nar/gkw256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser (Oxf) 41:95–98. [Google Scholar]
- 25.Katoh K, Toh H. 2008. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 26.R Core Team. 2017. R: a language and environment for statistical computing. R Core Team, Vienna, Austria. [Google Scholar]
- 27.Genuer R, Poggi J-M, Tuleau-Malot C. 2015. VSURF: an R package for variable selection using random forests. https://journal.r-project.org/archive/2015/RJ-2015-018/RJ-2015-018.pdf.
- 28.Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 29.Holmes S. 2003. Bootstrapping phylogenetic trees: theory and methods. Statist Sci 18:241–255. doi: 10.1214/ss/1063994979. [DOI] [Google Scholar]
- 30.Jukes TH, Cantor CR. 1969. Evolution of protein molecules, p 21–132. In Munro HN (ed), Mammalian protein metabolism. Academic Press Elsevier, Inc., New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.