

Host-to-host transmission of viruses is ideally studied in vivo in the natural host. Veterinary viruses such as Marek’s disease virus (MDV) are, therefore, models of choice to explore these aspects. The natural host of MDV, the chicken, is small, inexpensive, and economically important. MDV is a deadly and contagious herpesvirus that can kill infected animals in less than 4 weeks. The virus naturally infects epithelial cells of the feather follicle epithelium from where it is shed into the environment. In this study, we demonstrate that the viral protein pUL47 is an essential factor for bird-to-bird transmission of the virus. We provide some molecular basis to this function by showing that pUL47 enhances the splicing and the expression of another viral gene, UL44, which is essential for viral transmission. pUL47 may have a similar function in human herpesviruses such as varicella-zoster virus or herpes simplex viruses.
KEYWORDS: Marek’s disease virus, UL47, glycoproteins, Herpesviridae, tegument, transmission
ABSTRACT
Viral tropism and transmission of herpesviruses are best studied in their natural host for maximal biological relevance. In the case of alphaherpesviruses, few reports have focused on those aspects, primarily because of the few animal models available as natural hosts that are compatible with such studies. Here, using Marek’s disease virus (MDV), a highly contagious and deadly alphaherpesvirus of chickens, we analyze the role of tegument proteins pUL47 and pUL48 in the whole life cycle of the virus. We report that a virus lacking the UL48 gene (vΔUL48) is impaired in growth in cell culture and has diminished virulence in vivo. In contrast, a virus lacking UL47 (vΔUL47) is unaffected in its growth in vitro and is as virulent in vivo as the wild-type (WT) virus. Surprisingly, we observed that vΔUL47 was unable to be horizontally transmitted to naive chickens, in contrast to the WT virus. In addition, we show that pUL47 is important for the splicing of UL44 transcripts encoding glycoprotein gC, a protein known as being essential for horizontal transmission of MDV. Importantly, we observed that the levels of gC are lower in the absence of pUL47. Notably, this phenotype is similar to that of another transmission-incompetent mutant ΔUL54, which also affects the splicing of UL44 transcripts. This is the first study describing the role of pUL47 in both viral transmission and the splicing and expression of gC.
IMPORTANCE Host-to-host transmission of viruses is ideally studied in vivo in the natural host. Veterinary viruses such as Marek’s disease virus (MDV) are, therefore, models of choice to explore these aspects. The natural host of MDV, the chicken, is small, inexpensive, and economically important. MDV is a deadly and contagious herpesvirus that can kill infected animals in less than 4 weeks. The virus naturally infects epithelial cells of the feather follicle epithelium from where it is shed into the environment. In this study, we demonstrate that the viral protein pUL47 is an essential factor for bird-to-bird transmission of the virus. We provide some molecular basis to this function by showing that pUL47 enhances the splicing and the expression of another viral gene, UL44, which is essential for viral transmission. pUL47 may have a similar function in human herpesviruses such as varicella-zoster virus or herpes simplex viruses.
INTRODUCTION
Herpesviruses are complex viruses both in their structure and in their life cycle. They are strongly adapted to their natural host and the specific cell types that they naturally infect. Unraveling the molecular basis of this adaptation is made difficult by the numerous different proteins encoded by this family of viruses. To finely study the cellular tropism of a virus requires to use complex ex vivo methods such as organoids or in vivo methods. In the latter case, the most relevant information for herpesviruses is obtained when the natural host is used. In this regard, veterinary herpesviruses are models of choice. Gallid Herpesvirus 2 (GaHV2), commonly named Marek’s disease virus (MDV), is an avian alphaherpesvirus of chickens. It induces a highly contagious and deadly disease characterized by the appearance of tumors, mostly in the viscera of infected animals. These tumors are T-cell lymphomas originating from virally transformed T lymphocytes, which are the site of latency for MDV. The natural life cycle of MDV starts with the virus entering the host through the respiratory route from where it infects macrophages, B and T cells. In cells where the virus establishes latency (mostly CD4+ T cells), the genome is found integrated into cellular DNA (1, 2). MDV also displays a strong tropism for epithelial cells from the feather follicle epithelium (FFE). In this compartment, efficient viral lytic replication leads to the formation of infectious viral particles, which are shed as dander in the environment as early as 7 days postinfection (3). Unlike most of the other herpesviruses, the shed virus is stable and remains infectious for several weeks, complicating the disinfection of contaminated litter. Chickens are now protected from the disease thanks to the continuous use of vaccines since the late 1960s. Although these vaccines prevent the appearance of the tumors and the subsequent death of the animals (for reviews, see references 4 and 5), they do not prevent the reinfection. Therefore, they may contribute to the spread and evolution of wild-type (WT) strains of MDV toward increasing levels of virulence (6). Unlike most vaccines, the preparation of MDV vaccines consists essentially of infected primary cells rather than purified particles, because MDV is avidly cell associated. In this context, the development of a new generation, cell-free vaccine that can prevent MDV spread and evolution of WT strains is essential. Central to this challenge is the FFE. Unlike cell culture or lymphoid cells, which display a limited number of essentially nonenveloped cytosolic capsids, epithelial cells from the FFE can produce numerous infectious cell-free particles which are fully enveloped (7–11). Lack of envelopment in non-FFE cells may indicate that one or several viral components are missing or not functional in these cells. Tegument proteins are known to be important for the envelopment of herpesviruses.
The pUL47 (or VP13/14) protein is an abundant protein in the tegument of model alphaherpesviruses such as human herpes simplex virus 1 (HSV-1) or pseudorabies virus (PrV) (12, 13). It is a viral determinant of skin tropism for varicella-zoster virus (VZV), a human alphaherpesvirus biologically related to MDV (14). Interestingly, MDV pUL47 is strongly expressed in the FFE of infected chickens, whereas it is poorly expressed in other tissues and cultured cells (15–17). pUL48 (or VP16) is another tegument protein with similar properties. pUL48 is a known transcription factor of herpesviruses with many functions in regulating the lytic cycle or reactivation from latency (18–22). Here, we investigate the role of these two proteins in the tropism of MDV for the skin and the FFE and the subsequent horizontal transmission of the virus. To this aim, we obtained viruses unable to encode one or the other protein and characterized them in vitro and in vivo.
RESULTS
In vitro growth properties of MDV mutants lacking the UL47 or the UL48 genes.
It was previously shown that neither UL47 nor UL48 is essential for the in vitro growth of the cell-adapted strain Bac20 of MDV in primary chicken embryonic skin cells (CESCs) or quail muscle cells (QC7) (23). As we aimed at identifying a possible role of pUL47 and pUL48 in skin tropism, mutants with a complete deletion of either gene were engineered by en passant recombination of a bacterial artificial chromosomes (BAC) containing the complete genome of a very virulent strain (vvRB-1B) of MDV. The removal in the BAC of the entire UL47 gene or the UL48 gene yielded BACs pΔUL47 and pΔUL48, respectively. Each deletion was confirmed by RFLP, followed by pulse-field electrophoresis and by PCR (Fig. 1A). In addition, to ensure that no second site mutations occurred during the recombination process, the complete genomes of pΔUL47 and pΔUL48, as well as the parental pRB1B, were sequenced using the Illumina MiSeq platform (Fig. 1B and C). This confirmed that the UL47 and UL48 genes were wholly deleted and that no relevant additional mutations were present (see Materials and Methods for details). Following the transfection of purified BACs pΔUL47 and pΔUL48 into CESCs, infectious virus (vΔUL47 and vΔUL48, respectively) could be recovered, confirming that neither gene is essential for MDV replication. Measurement of plaques size on CESCs showed that plaques of vΔUL47 were 88% of the size of the plaques of the parental virus vRB1B, whereas plaques of vΔUL48 were only 30% the size of the parental virus (Fig. 2A and B). Similar results were observed with chicken keratinocytes, with a median plaque size of vΔUL47 of 94% of the size of the plaques of vRB1B and a median size of vΔUL48 plaques of 37% of the size of vRB1B plaques (Fig. 2A and C). As in CESCs, the mild difference of size between vRB1B and vUL47 plaques was not statistically significant. Altogether, these data indicate that vΔUL48 is impaired in its growth in CESCs and keratinocytes, while the growth of vΔUL47 is not significantly affected.
FIG 1.

Genomic characterization of vΔUL47 and vΔUL48. (A) Restriction fragment length polymorphism analysis of BACs pRB1B, pΔUL47, and pΔUL48. Purified BAC DNA was digested with XhoI and examined for the absence of an 1,818-bp band indicative of the deletion of the UL47 and UL48 genes (green arrows). The absence of the larger UL47 gene is also shown by the shift of a 3.8-kb band to 3.2 kb (red arrows). Note that this shift is absent in the vΔUL48 BAC. (B and C) List of all differences observed between the sequence of pRB1B and pΔUL47 (B) and pΔUL48 (C). Many of the putative mutations listed here are common artifacts linked to the multiple homopolymeric repeats present in the genome. The deletions in UL36 listed at positions 78879 in pΔUL48 and 78993 in pΔUL47 were absent from the corresponding BAC, as assessed by Sanger sequencing of these regions. Note that many mutations being present in inverted repeated regions, including insertions of telomeric sequences in a-like sequences, they are present twice. ND, not determined; Hyp protein, hypothetical protein; SNP, single nucleotide polymorphism. *, RB-1B reference genome accession number EF523390.
FIG 2.

Growth characteristics of vΔUL47 and vΔUL48 in comparison to vRB1B in CESCs and keratinocytes. The area of individual plaques of vRB1B, vΔUL47, and vΔUL48 viruses on CESCs or keratinocytes were measured 4 dpi. (A) Viral plaques were visualized using a cocktail of MAbs directed against ICP4, VP22, and gB. (B and C) A minimum of 50 plaques was measured for each virus. The results are shown as box plots for CESCs (B) or keratinocytes (C). Significant differences in the median of plaque areas were determined using a Wilcoxon-Mann-Whitney test. ns, not significant (P > 0.5); ****, P < 0.0001. Scale bars, 200 μm.
In vivo replication of vΔUL47 and vΔUL48.
In order to assess the role of pUL47 and pUL48 in MDV’s tropism for the skin, 3,000 PFU of the respective null viruses, as well as the parental vRB1B virus, were inoculated by the intramuscular route to 10 White Leghorn chickens of 2 weeks of age. Five naive contact chickens were added to each group to test for the horizontal transmission of the virus. As shown in Fig. 3A, the mortality and the kinetics of disease were similar between animals inoculated with vRB1B or vΔUL47, with 100% mortality reached at 46 and 49 days postinoculation, respectively, and median survivals of 29 and 28.5 days. This shows that pUL47 is not a factor of virulence. In addition, at necropsy, all animals had tumors, indicating that pUL47 is not necessary for tumorigenesis. In contrast, only 65% of vΔUL48-inoculated chickens died by the end of the experiment (78 days postinoculation; median survival, 55 days), and all of these animals had tumors. The surviving animals showed no symptoms of the disease and had no tumors at the end of the experiment. However, viral genomes could be detected in their blood as early as 21 days postinoculation (Fig. 3B and Fig. 4A). Moreover, the first animals with symptoms of Marek’s disease appeared later when infected with vΔUL48 (36 days postinoculation) than with vΔUL47 (26 days) or vRB1B (24 days). Of note, animals that died of the vΔUL48 infection showed minimal symptoms before their death or none that could be noticed. Altogether, these results show that pUL48 is not necessary for tumorigenesis but contributes to the virulence of the virus.
FIG 3.
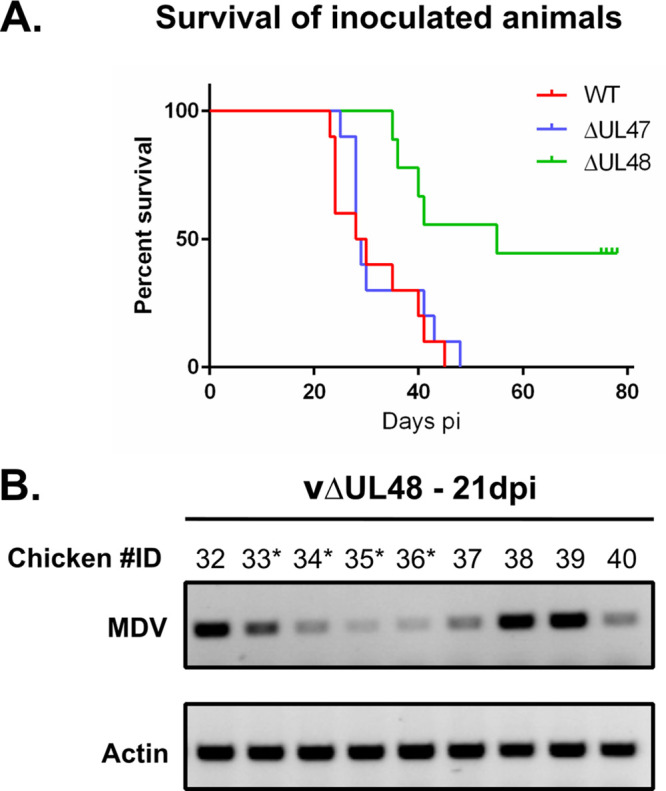
In vivo replication of vΔUL47 and vΔUL48 in comparison to vRB1B after muscular inoculation. (A) Ten 2-week-old white Leghorn chickens were inoculated with 3,000 PFU of vRB1B, vΔUL47, or vΔUL48. The percentages of surviving animals are shown for vRB1B (WT, red line), vΔUL47 (blue line), and vΔUL48 (green). (B) DNA was extracted from PBMCs of animals inoculated with vΔUL48 and was analyzed by PCR for the presence of the viral genome at 21 dpi. Actin was used as a loading control. Chicken identification numbers with an asterisk indicate animals that survived until the experiment terminated. Of note, one animal in the vΔUL48 group died 2 days postinoculation, possibly of a wound contracted at the foot.
FIG 4.

Viral genome loads in the blood and feathers of inoculated chickens. (A and B) DNA was extracted from PBMC (A) or feathers (B) of chickens inoculated with vRB1B (in red), vΔUL47 (in blue) or vΔUL48 (in green) at 7, 14, and 21 days postinoculation. Viral genomes were quantified by real-time quantitative PCR, and their numbers are indicated per million of cells. In addition, viral genome loads were quantified in PBMCs obtained at 44 days postinoculation from the four surviving ΔUL48-infected chickens. Significance of differences between the median viral loads in vΔUL47- or vΔUL48-infected birds and those in vRB1B-infected birds were determined using a Wilcoxon-Mann-Whitney test. ns, not significant (P > 0.5); **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
pUL47 and pUL48 are not necessary for the skin tropism of MDV.
During the first 4 weeks of the experiment, blood and growing feathers were collected from the animals every week, and the presence of viral genomes was assessed using PCR and qPCR. Figure 4 shows the results obtained during the first 3 weeks of the experiment (50% of animals inoculated with vRB1B and vΔUL47 were dead by week 4). The kinetics of viral replication and viral loads were globally similar between animals infected with vRB1B and vΔUL47. Viral loads in the blood of vΔUL47-infected animals were even 2.2 and 3.3 times higher than for vRB1B-infected animals at 7 and 14 days postinfection (dpi), respectively, indicating that UL47 is dispensable for initiating the infection. At 21 days postinoculation, the medians of viral loads were 5.7 × 105 and 1.9 × 106 copies/million cells, respectively, in the blood (Fig. 4A, red and blue boxes) and 1.6 × 107 and 6.4 × 106 copies/million cells in feathers (Fig. 4B, red and blue boxes). In comparison, the viral load of vΔUL48-infected animals at 21 days postinoculation in the blood and feathers were very low: 3 × 104 and 1.9 × 105 copies/million cells, respectively (Fig. 4, green boxes). There was no significant difference in the number of viral genomes found at 21 dpi in the blood of vΔUL48-infected chickens between animals that died and those which survived the infection. Surviving birds had no tumors and had viral genome numbers ranging from 1,380 to 5,130 copies/millions of cells at 45 days postinoculation (Fig. 4A, dark green box). These numbers were lower than those observed in the blood of all vΔUL48-infected birds in the course of infection, including as early as 7 dpi, suggesting that viral replication was under control in the surviving birds. These numbers confirm that viral replication in the blood and feather epithelia of vΔUL48-infected birds was impaired, whereas that of vΔUL47 is comparable to vRB1B. These numbers also confirm that both pUL47 and pUL48 are dispensable for the tropism of MDV for the skin.
Horizontal transmission of MDV is strongly impaired or absent in the absence of pUL48 or pUL47, respectively.
Out of the five naive chickens added to each group of 10 inoculated chickens, four of them showed symptoms and were euthanized in the vRB1B group between days 50 and 56 postinoculation (Fig. 5A). The lone surviving animal had multiple tumors at the end of the experiment, and it was probably destined to die. In contrast, none of the contact chickens from the vΔUL47 or the vΔUL48 group died, and none of them presented clinical symptoms or tumors by the end of the experiment. PCR and qPCR analysis failed to detect viral genomes in the blood of these animals at 28, 44, 58, and 71 dpi, except in one bird from the vΔUL48 group (positive from 44 dpi and quantified at 6,600 copies/million cells in the blood at 71 dpi) (animal 43 in Fig. 5B). In order to determine whether the absence of viral transmission was due to poor or absent viral excretion from the inoculated animals, dust samples were collected from all isolation units at 21 dpi and total DNA was extracted. qPCR analysis showed that viral loads were very high and similar in the dust from units containing birds shedding vRB1B (9.22 × 107 copies/million cells) and vΔUL47 (7.25 × 107 copies/million cells) units (Fig. 5C). In contrast, viral loads in the dust from units containing vΔUL48-infected birds were more than 60 times lower than that observed in units containing vRB1B shedding birds (1.46 × 106 copies/million cells). This is congruent with the small amounts of viral genomes found in vUL48-infected birds. In conclusion, the absence of transmission of vUL47 could not be directly linked to an absence of viral excretion from infected birds.
FIG 5.

Horizontal transmission of vRB1B, vΔUL47, and vΔUL48. (A) Five noninfected chickens per group were housed together with the inoculated chickens at the time of viral inoculation (D0). The percentages of surviving animals are shown for vRB1B (WT, red line), ΔUL47, and ΔUL48 animals (purple line). (B) The PBMCs of surviving ΔUL48-inoculated animals, as well as of all contact animals, were analyzed by PCR for the presence of the viral genome at 44 days postinoculation. Actin was used as a loading control. (C) The amount of viral genomes was determined by qPCR from DNA extracted from the dust accumulated during 1 week (between 21 and 28 dpi) and collected from the three different isolators.
Deletion of UL47 affects the splicing of UL44 transcripts.
The UL47 gene product could be either directly involved in horizontal transmission, or its deletion could affect the expression of other genes known to be essential for transmission such as UL13, UL54, or UL44 (24–27), encoding a viral kinase, regulatory protein ICP27, and glycoprotein gC, respectively. To test this, RNAs were extracted from CESCs or keratinocytes infected with vRB1B, vΔUL47, or vΔUL48. Initially, RT-PCR was carried out with primers specific for UL47 neighboring genes to verify that the deletion had no impact on their expression. As shown in Fig. 6A, the expression of UL46 and UL48 was unaffected by the deletion of UL47 in both cell types. In subsequent RT-PCR experiments, the expression of other transmission-related genes, UL13 and UL54, remained unaffected in cells infected with vΔUL47. This was not the case for the expression of UL44 transcripts. Previously, it has been reported that three different transcripts exist due to the existence of two spliced forms (Fig. 6B): one transcript lacking 104 nucleotides after splicing (referred to as “UL44-104” here), one lacking 145 nucleotides (“UL44-145”), and the unspliced transcripts (“UL44-full”) (28). While UL44-full encodes full-length gC (“gC-full”), the splicing of UL44 transcripts lead to two different frameshifts, resulting in two truncated forms of gC (“gC-104” and “gC-145”) lacking the transmembrane domain. All three forms of gC are important for horizontal transmission of MDV since the expression of any one of them alone results in poor transmission (28). A pair of primers encompassing the splicing sites was designed to detect all three forms (Fig. 6B). In contrast to UL13 and UL54, the profile of UL44 transcripts differed in vΔUL47-infected cells compared to vRB1B or vΔUL48-infected cells. In vΔUL47-infected cells, the unspliced transcripts of UL44 (UL44-full) were more abundant than in the other infected cells (Fig. 6A). In order to quantify this observation made on both cell lines, primers specific for each of the three types of transcripts were designed, as well as primers specific for the total UL44 transcripts. These primers were used to quantify the relative abundance of all types of UL44 transcripts by RT-qPCR (Fig. 7). This confirmed that the unspliced transcripts of UL44 were more abundant in CESCs in the absence of UL47 (+84%), whereas the amount of UL44-104 and UL44-145 spliced transcripts decreased (−45% and −33%, respectively). Similarly, an increase of unspliced UL44 transcripts was observed in keratinocytes in the absence of UL47 (+63%), but no significant change was detected for the spliced forms. In cells infected with vΔUL48, changes were mild and nonsignificant. These results show that the splicing of UL44 is impaired in the absence of pUL47 but not in the absence of pUL48.
FIG 6.

Expression profiles of viral genes in cells infected with vRB1B, vΔUL47, or vΔUL48. (A) RNA was extracted from primary CESCs or chicken keratinocytes that were either mock infected or infected with vRB1B, vΔUL47, or vΔUL48. After cell lysis, RNA was extracted and used for RT-PCRs using primers to detect the UL46, UL47, UL48, UL44, UL13, and UL54 (ICP27). In addition, primers were used to detect cellular GADPH transcripts as a loading control and viral capsid protein gene UL35 as a control for infection levels. Reactions without reverse transcriptase [RT(–)] were also carried out to control for the absence of DNA contamination (here shown for UL44). (B) Splicing of the UL44 transcripts and associated predicted proteins. UL44 primers used for RT-PCR assays, as shown in panel A, are indicated in red. Two overlapping introns (104 and 145 bp) are present within the UL44 gene with a common acceptor site (A) and two donor sites (D1/D2). Splicing of these introns yields two transcripts of 1,402 and 1,361 bp, respectively, containing one frameshift each. The translation of these transcripts (UL44-104 and UL44-145) is expected to result in two truncated proteins of similar size but with differing C-terminal sequences (gC-104 and gC-145). The truncated proteins do not contain the transmembrane domain and are secreted.
FIG 7.

Quantification of the different UL44 transcripts in vΔUL47 and vΔUL48-infected CESCs or keratinocytes relative to vRB1B-infected cells. The same samples as in Fig. 6 were used to quantify the relative abundances of the three different forms of the UL44 transcripts by RT-qPCR using primers specific for each form. The abundance of each transcript form was normalized to the quantities of total UL44 transcripts. These normalized quantities in vΔUL47 and vΔUL48 were then quantified relative to the WT (red line) (no variation = 0%). The experiment was done three times in triplicates. Error bars indicate standard deviations. The significance of differences was determined using a paired two-tailed Student t test on ΔCq values. *, P < 0.5; ***, P < 0.001.
Splicing of UL44 transcripts is also regulated by ICP27, another viral gene involved in horizontal transmission.
ICP27 is a viral protein with a well-characterized role in the regulation of splicing of cellular pre-mRNAs. More specifically, ICP27 can inhibit splicing (29) or regulate alternative splicing of cellular (30) or viral (31) transcripts. Interestingly, it was recently demonstrated that ICP27 is necessary for horizontal transmission of MDV and the production of gC (27). However, the splicing of UL44 in the absence of ICP27 was not investigated. Since both pUL47 and ICP27 are involved in transmission and because pUL47 is involved in the splicing of UL44 transcripts, we compared the functions of ICP27 and pUL47 in the splicing of UL44 transcripts.
We engineered a new virus named vΔUL54, which lacks the complete open reading frame (ORF) of the ICP27-coding UL54 gene in the backbone of the RB-1B BAC. It has been shown previously that ICP27 is dispensable for MDV growth in vitro, although it forms plaques that are ∼50% smaller than the parental virus (27).
RNAs were purified from CESCs infected with vRB1B or vΔUL54 and RT-PCR was carried out using the primers encompassing the regions specific for the three forms of UL44. As shown in Fig. 8A, the expression of genes neighboring UL54 (i.e., UL53 and L-ORF9), as well as UL13 and UL47, were unaffected by the deletion of UL54. However, the distribution of the different UL44 transcripts was altered by the mutation as we observed an increase of UL44-145 transcripts (+30%) and a decrease of unspliced transcripts (−22%) in the absence of ICP27 compared to vRB1B-infected cells (Fig. 8B). UL44-104 transcripts levels were not affected. Since ICP27 is an efficient splicing regulator of cellular and viral genes, we checked whether ICP27 was sufficient for the modulation of the splicing of UL44 transcripts. To this aim, we used ESCDL-1 cells, which are derived from chicken embryonic stem cells and are fully permissive to MDV (32), and efficiently transfected by standard methods, as opposed to primary CESCs. Transfection was done using plasmids encoding gC, ICP27, and/or pUL47. Expression of UL44 from a plasmid resulted in all three transcripts, albeit with proportions differing from what was observed in infected CESCs: the levels of UL44-104 and UL44-145 were roughly similar and lower than those for the unspliced transcripts (Fig. 8C). Interestingly, UL44-145 transcripts could not be detected by RT-PCR in cells transfected with the ICP27-coding plasmid (lanes 2 and 4). In contrast, expression of UL47 did not alter the pattern of expression of all three transcripts. RT-qPCR assays showed that levels of UL44-145 were reduced by 58% and those of unspliced UL44 were increased by 22% in the presence of ICP27 compared to cells transfected with the UL44 plasmid alone (Fig. 8D). This result mirrors what was observed in infected CESCs and confirms that ICP27 inhibits the splicing of UL44 transcripts that results in the UL44-145 form.
FIG 8.

ICP27 is another viral gene involved in horizontal transmission that affects splicing of UL44 transcripts. (A) RNA was extracted from primary CESCs that were either mock infected or infected with vRB1B or vΔUL54 and used for RT-PCRs using primers to detect the UL53, UL54, L-ORF9, UL13, UL47, and UL44 transcripts. In addition, primers were used to detect cellular GADPH transcripts as a loading control and viral capsid gene UL18 as a control for infection levels. Reactions without reverse transcriptase [RT(–)] were also carried out to control for the absence of DNA contamination (here shown for UL44). (B) RT-qPCR quantification of the UL44 transcripts using primers specific for each form. The abundance of each transcript form was normalized to the quantities of total UL44 transcripts. These normalized quantities in vΔUL54 were then quantified relative to the vRB1B levels (no variation = 0%). The experiment was done three times in triplicates. Error bars indicate standard deviations. Significance of differences was determined using a paired two-tailed Student t test on ΔCq values. **, P < 0.01; ***, P < 0.001; ns, not significant. (C) RNA was extracted from ESCDL-1 cells that were transfected with plasmids encoding gC and ICP27 and/or pUL47. RT-PCR was carried out using primers encompassing all three forms of UL44 transcripts as described in Fig. 6B. (D) The cDNA obtained under two conditions (gC alone) (condition 1) and (gC+ICP27) (condition 2) were used to quantify the different UL44 transcripts using primers specific for each form. The abundance of each transcript form was normalized to the quantities of total UL44 transcripts. These normalized quantities under condition 2 (gC+ICP27) were then quantified relative to the levels under condition 1 (gC alone) (no variation = 0%). The experiment was done three times in triplicates. Error bars indicate standard deviation. Significance of differences was determined by using a paired two-tailed Student t test on ΔCq values. **, P < 0.01; ****, P < 0.0001; ns, not significant.
pUL47 and ICP27 are important for gC expression and/or stability.
Since gC is essential for transmission, we measured the impact of the absence of pUL47 on the production of gC. In infected CESCs, most of gC is secreted and is therefore almost undetectable from cell lysates (28). However, the treatment of cells with brefeldin A (BFA), a drug that blocks the transport of secreted proteins (33), allows for the accumulation of the secreted form of gC in cells and its subsequent detection. Cells were infected for 4 days with vRB1B, vΔUL47 or a virus unable to produce gC (vΔgC). At 6 h before harvesting, the cells were left untreated or treated with 10 μg/ml of BFA. Western blot analysis of cell lysates using the A6 monoclonal antibody (MAb) specific for gC showed the presence of a single band of apparent molecular weight of ∼55 kDa that was particularly visible in lysates from vRB1B-infected cells treated with BFA (Fig. 9A). The expected apparent molecular weight for secreted MDV gC in cell culture is 57 to 65 kDa (34), making it likely that secreted gC was detected in these Western blots. Importantly, the amount of gC blocked in cells by BFA in vΔUL47-infected cells represented only 26% of the amounts in vRB1B-infected cells. The same experiment was carried out using vΔUL54-infected cells and no secreted gC could be detected in those cells (Fig. 9B), which is in agreement to the observation by Ponnuraj et al. (27). These experiments show that in the absence of pUL47, the amount of secreted gC is sharply reduced in cell culture, and they confirm that in the absence of ICP27, secreted gC is undetectable.
FIG 9.

Expression of gC in CESCs infected with vΔUL47, vΔUL54, vΔgC, and vRB1B. Primary CESCs were infected with vRB1B, vΔUL47 (A), vΔUL54 (B), or a virus that did not express gC (vΔgC). At 6 h before harvesting, the cells were treated or not with 10 μg/ml brefeldin A (BFA) to block the secretion of gC and concentrate it within the cell. Cell lysates were analyzed by Western blot with MAb A6 against gC, MAb H18 against VP5, and an anti-tubulin antibody. Viral protein quantities were normalized to the amount of VP5, and the levels of gC in vΔUL47 and vΔUL54 samples were calculated relative to the levels in the vRB1B sample. Tubulin was used as a loading control.
DISCUSSION
Investigating the role of viral proteins in host-to-host transmission of herpesviruses is only possible in animal models where the virus infects its natural host. In this regard, MDV is ideal because its natural host, the chicken, is small and can be used in relatively large numbers. Moreover, MDV is highly infectious and contagious with a short life cycle (30 to 45 days for very virulent strains), thus facilitating experimental design requirements. Here, we unraveled the importance of two tegument proteins in horizontal transmission of the virus. Although transmission of vΔUL48 is strongly impaired (one contact bird out of five infected), the role of pUL48 in horizontal transmission cannot be directly compared to that of pUL47. Indeed, birds inoculated with vΔUL48 showed strong attenuation of viral replication, contrary to birds inoculated with vΔUL47. The median of survival was high (55 days) compared to the vRB1B or vΔUL47 groups (less than 30 days), and viral loads were constantly low in the blood of these animals. Importantly, surviving vΔUL48-inoculated animals showed low viral loads in the blood as late as 44 dpi. This strongly suggests that viral replication was attenuated in these birds. pUL48 (also called VP16) is a known factor of reactivation for alphaherpesviruses (21). It is therefore likely that its absence impairs with viral reactivation from latency and that the low viral loads observed in the blood of surviving birds represent latent genomes. In addition to triggering reactivation, VP16 is an important viral transactivator, which is either essential (HSV-1 [35]) or very important (PrV [36]) for replication of some other alphaherpesviruses. In its absence, viral replication is expected to be low as was observed in vitro and in birds inoculated with vΔUL48. Thus, it is possible that the poor horizontal transmission of vΔUL48 may be due to a low quantity of infectious viruses produced by inoculated birds or that the attenuation of the virus makes it unable to efficiently replicate in the respiratory tract of newly infected chickens to initiate a new viral cycle. The small amounts of viral genomes found in the dust of the incubator housing the vUL48-inoculated birds (1.46 × 106 copies per million cells as opposed to 7.25 × 107 copies/million cells for vUL47) supports the first postulate.
pUL47, on the other hand, has never been shown to our knowledge as a transmission factor or a transactivation factor of herpesviruses, although it is a virulence factor of BHV-1 in cattle (37). Its known functions are in the export of nuclear capsid (HSV-1 [38]), skin tropism (VZV [39]), and the regulation of the immune response (BHV [40]). Our study here excludes the role of MDV pUL47 in skin tropism since high viral loads in the skin were statistically similar between vΔUL47 and vRB1B-infected birds. Moreover, the growth of vΔUL47 was not impaired in vitro (Fig. 2) and was not attenuated in vivo (Fig. 3), in contrast to vΔUL48. This is surprising for a protein which has been described as a major tegument component in other alphaherpesviruses (12, 41–43). Interestingly, although viral loads in feathers were as high in vΔUL47-infected birds as in vRB1B-infected birds, we repeatedly failed to detect viral genomes in the blood of vΔUL47 contact chickens despite a high number of genomes detected in the dust of their isolation unit. This remarkable finding indicates that UL47 encodes a function that is either directly relevant for bird-to-bird viral transmission or affects the activity of host or viral proteins involved in this transmission. In testing the second hypothesis, we discovered that pUL47 plays an important role in the splicing of UL44 transcripts, a function that was not described until now. In the absence of pUL47, the amount of spliced products of UL44 decreased, while the amount of unspliced products increased, i.e., pUL47 enhances the splicing of UL44 transcripts. UL44 encodes glycoprotein gC, a protein essentially known as dispensable in cell culture for herpesviruses (44–46), which facilitates adsorption of viral particles onto cells by binding to heparan sulfates (47). Its other roles include immune modulation (48, 49) and, more importantly, it is necessary for horizontal transmission, as shown with MDV (28). Three differently spliced forms of the UL44 transcripts have been described. The unspliced form encodes the full-length protein, whereas the two spliced forms encode truncated versions that lack the transmembrane domain and are therefore secreted (28). It was demonstrated that the expression of any one of these three forms alone results in poor transmission, indicating that the balance of expression between each form of gC is important for efficient viral transmission (28).
In the course of this study, we were unable to detect full-length gC, a result compatible with previous reports showing that more than 94% of gC is secreted in cell culture (34, 50). Brefeldin A was used to disrupt the secretory pathway and to accumulate the secreted isoform of gC in cells. This showed that secreted gC migrated as an ∼55-kDa protein, in line with previous biochemical characterizations (50).
Since the absence of pUL47 leads to a defect in the splicing of UL44 transcripts, a decrease in the production of secreted gC, which is produced out of the translation of the spliced transcripts, was expected. We indeed observed a decrease of secreted gC in cells that amounted to up to 74% compared to vRB1B. Because gC is essential for viral transmission, it is possible that the small amounts of gC produced in the absence of pUL47 may be below the minimum threshold required for efficient viral transmission.
In an effort to understand whether horizontal transmission and splicing of UL44 are linked, we tested the importance of another viral gene, UL54, that encodes the “splicing” protein ICP27 for its effect on the splicing of UL44. We show that the absence of UL54 resulted in a modified splicing profile of UL44 transcripts where splicing is more efficient and leads to an increase in the UL44-145 form and a decrease in the unspliced transcript. Interestingly, this effect is mirrored in transfected cells where the sole expression of UL54 results in a strong decrease of the splicing of UL44 transcripts, leading to the UL44-145 form (Fig. 8). Although pUL47 and ICP27 have opposing effects on the splicing of UL44, it is significant that both proteins contribute to the correct splicing of UL44 transcripts and that their deletion results in poor or absent expression of the secreted forms of gC, ultimately resulting in the lack of horizontal transmission. It remains to be determined whether the altered splicing pattern of UL44 directly and/or solely causes the complete absence of transmission.
For VZV, it has been reported that the ectodomain of gC (which would correspond to the secreted forms of gC produced by the spliced transcripts of UL44) binds and potentiates chemokine activity, thus contributing to the enhancement of T-cell migration to the site of infection (51). This is particularly important in the first steps of infection when viral particles of MDV or VZV enter the body and need to find target cells to latently infect (B and T cells for VZV and MDV). One hypothesis linking the role of MDV pUL47 and ICP27 in the efficient splicing of UL44 and its role in horizontal transmission is that in the lung of newly infected animals, the splicing of UL44 in the absence of pUL47 or ICP27 is particularly inefficient, leading to the production of small amounts of the secreted forms of gC. The reduction in the levels of the chemokine-potentiating form of gC may result in an aborted infection due to the inability to locally recruit T cells.
Although never reported so far to our knowledge, a role for pUL47 in RNA splicing is in line with previous reports in various alphaherpesviruses describing its interaction with RNA (14, 39, 52–54), splicing-related ICP27 (55), and cellular protein p32, a component of the SF2 splicing complex (56). It would be of interest to test whether pUL47 of MDV have similar properties and how this could relate to the splicing of UL44 transcripts, the expression of gC, and the subsequent horizontal transmission of the virus.
MATERIALS AND METHODS
Plasmids.
All recombinant bacterial artificial chromosomes (BACs) used in this study were derived from the repaired pRB-1B BAC previously described (24, 25). The UL47, UL48, or UL54 open reading frame (ORF) was deleted using the two-step Red recombination technology (57), yielding BACs pRB1B-ΔUL47, pRB1B-ΔUL48, and pRB1B-ΔUL54, respectively, named pΔUL47, pΔUL48, and pΔUL54 here. The sequences of the primers used for mutagenesis, PCR, RT-PCR, and quantitative PCR are listed in Table 1.
TABLE 1.
Oligonucleotides used in this study
| Primer | Role | Sequence (5′–3′) |
|---|---|---|
| dUL47_for | Deletion of the UL47 ORF | AAACGGCGTAGTTTTGATAACAGTATGCTGGTAGCACATTCCACCGAAGAAGGATGACGACGATAAGTAGGG |
| dUL47_rev | Deletion of the UL47 ORF | AAATGCCATTATTCTACATCCGGAGTAAAAGTCCCGCCCTCTTCCCTACGTCTTCGGTGGAATGTGCTACCAGCATACTGTTATCAAAACTACGCCGTTTACAACCAATTAACCAATTCTGATTAG |
| dUL48_for | Deletion of the UL48 ORF | TTAAAAACTAGTACATATATATCTTATCTACTCATTATTGTATAGTGTGAAGGATGACGACGATAAGTAGGG |
| dUL48_rev | Deletion of the UL48 ORF | TATAGTTTCGTTCTGCCAACTCACCATCATACTAATAGACAAATACCTCCTCACACTATACAATAATGAGTAGATAAGATATATATGTACTAGTTTTTAAACAACCAATTAACCAATTCTGATTAG |
| dUL54_for | Deletion of the UL54 ORF | CTCAAGTGTACCTAATGGACTATTATCTATATCAAGATTAAACAAAAAAAAGGATGACGACGATAAGTAGGG |
| dUL54_rev | Deletion of the UL54 ORF | AGAAGAAATTGTGTAGAATACTAAATATAGGATGTCTTTTAGATAACTATTTTTTTTGTTTAATCTTGATATAGATAATAGTCCATTAGGTACACTTGAGACAACCAATTAACCAATTCTGATTAG |
| UL51_checkF | Detection of viral genomes | CTGTACGTTCTAGCCAATGATTG |
| UL51_checkR | Detection of viral genomes | GCTTGGATCCCAATAGCCGATAAC |
| 332-bactinTLVI_S | Detection of chicken beta-actin gene (cellular genomes) | CATCACCATTGGCAATGAGAGG |
| 333-bactinTLVI_R | Detection of chicken beta-actin gene (cellular genomes) | GCATACAGATCCTTACGGATATCC |
| UL46_RTf | Detection of UL46 transcripts | GTTGATCTCCCGGGAACTCG |
| UL46_RTr | Detection of UL46 transcripts | GCCATTGACAACATGTCCAC |
| UL47_RTf | Detection of UL47 transcripts | GGAGAACACTTGTAGAAGCG |
| UL47_RTr | Detection of UL47 transcripts | GAATGTCATAATTAATCGGCG |
| UL13_RTf | Detection of UL13 transcripts | CTGAGTTACTTATGACATTG |
| UL13_RTr | Detection of UL13 transcripts | CACATCGAACCTGGATTTCAGG |
| gE_RTf | Detection of Us8 transcripts | GTTGGAGATACTAAATGCTAGCC |
| gE_RTr | Detection of Us8 transcripts | CATAAATCGTGACGAACTTGC |
| Us1_RTf | Detection of Us1 transcripts | CCGAGGACGCAGCGATTGGGTG |
| Us1_RTr | Detection of Us1 transcripts | GTCAGATAATGAGAGCGACGACG |
| ICP27_RTf | Detection of UL54 transcripts | GGTCCTCAGATAACATGGGAG |
| ICP27_RTr | Detection of UL54 transcripts | CATTCTCCAGGTACATAGTC |
| UL48RT_checkF | Detection of UL48 transcripts | GAAGTCAGAACCCAATCTATTC |
| UL48RT_checkR | Detection of UL48 transcripts | GTCGCTGTGATACATCCCGCG |
| gC-RTPCRf2 | Detection of all forms of UL44 transcripts | GAGCTACGTTGGTTTCTACAATAAC |
| gC-RTPCRrev2 | Detection of all forms of UL44 transcripts | CATAGGGCAGTCATGATTATCC |
| ggGAPDH_f | Detection of chicken GAPDH transcripts | GTGAAAGTCGGAGTCAACGGATTTG |
| ggGAPDHr2 | Detection of chicken GAPDH transcripts | GGCCAAGGGTGCCAGGCAGTTGG |
| UL35_RT_f | Detection of UL35 transcripts | CTCGTGCATCATCCCAACAGGC |
| UL35_RT_r | Detection of UL35 transcripts | CGTCGATATATCATCATCTGAC |
| gC145RTPCR_4ntF | Detection of 145-bp spliced UL44 transcripts only (qPCR) | CTATCCCGACGGTCTCT |
| gC-RTPCRrev3 | Detection of UL44 transcripts (qPCR) | CTTCATCGAAGGGGTAGCC |
| gC-RTPCRr104+ | Detection of 104-bp spliced UL44 transcripts only (qPCR) | GGTCCGGCAGAGACCCATC |
| gC-RTPCR-FLf | Detection of unspliced UL44 transcripts only (qPCR) | GCAATATGTACTATAGAATGTGTCC |
| gC-RTPCR-FLr | Detection of unspliced UL44 transcripts only (qPCR) | CCTGTAACCACAGTATTATAAGTTG |
| UL44GFP_ktrlF | Detection of all forms of UL44 transcripts (qPCR) | GCCGCATTCCAGTATGGGAC |
| UL36mut2_checkF | Detection of the putative UL36 deletion + SNP | GGAAGCAGAAGGAGACTTG |
| UL36mut2_checkR | Detection of the putative UL36 deletion + SNP | GTGATCCGATTGATGAAACC |
| Rep_chck150Kf | Sanger control of homopolymeric repetitions in the 150k region | CTCCACAGCATTGTTATACG |
| Rep_chck150Kr | Sanger control of homopolymeric repetitions in the 150k region | CGCGGTTAATGTTTGCTGCTC |
| UL53_RTr | Detection of UL53 transcripts | CGCCCAAACAATGCAATTTG |
| UL53_RTf | Detection of UL53 transcripts | GGCTCAGTGTCTTTGTCTAGCG |
| RLORF9_RTr | Detection of R-LORF9 transcripts | GATGGCCCTGAAATTGCACGAAC |
| RLORF9_RTf | Detection of R-LORF9 transcripts | CTGTGTTCCAGGAATAAAACC |
| UL18_RTf | Detection of UL18 transcripts | CAAATGCCCCCTCCTACCAG |
| UL18qPCR_r | Detection of UL18 transcripts | CGCTTTTATATTGGCAGGGC |
| Telo_check2F | Detection of telomeric repeats in a-like sequence | GGGATGTGGTCTACGAACCGG |
| Telo_check2r | Detection of telomeric repeats in a-like sequence | GGAGCTGCCGCCAAACTTGTGCC |
| Seq_3045f | Sanger control for mutation in position 3045/136487 | GCGCATGCGCAGATACACTCC |
| Seq_3045r | Sanger control for mutation in position 3045/136488 | GAACTGTCGGTGCCGGTACG |
| Seq_4025f | Sanger control for mutation in position 4025/135499 | GAGCTGGCCACAGGAGAGGGAC |
| Seq_4025r | Sanger control for mutation in position 4025/135500 | CATCTCTCGGCCCCAGACTGC |
| Seq_5764f | Sanger control for mutation in position 5764/133769 | GCCTAGGAATCAGTGTGCGG |
| Seq_5764r | Sanger control for mutation in position 5764/133770 | GGATTTGGAATAACCGAATTC |
| Seq_9003f | Sanger control for mutation in position 9003/130529 | CGCCATGTGTACTGCTTGTATG |
| Seq_9003r | Sanger control for mutation in position 9003/130530 | CGTCTGTATATACGCACGAAC |
| ICP27L_XhoIr | Cloning of UL54 into pCDNA3-V5 | GATCTCGAGTTACATACCAAACAGAGTATTGCAG |
| ICP27L_EcoRI0f | Cloning of UL54 into pcDNA3-V5 | CAAGAATTCTGTCTGTAGATGCATTCTCTCG |
| UL47_EcoRI1noATf | Cloning of UL47 into pCDNA3-V5 | CGAGAATTCCAAATGCCTTCTATGCATCGGTATG |
| UL47_NotIr | Cloning of UL47 into pCDNA3-V5 | TCTGCGGCCGCTCAATTCGCCCGTTGTGGCTCACG |
Plasmids used for testing the specificity of the primers used in RT-qPCR to detect the unspliced form of UL44 only were constructed by cloning the three different RT-PCR products obtained using the UL44 screening primers on primary skin cells cDNA (as shown in Fig. 6B) into the pGEM-T-Easy vector (Promega). These plasmids were subsequently sequenced to determine what form of the UL44 transcripts was cloned. The plasmid thus obtained with unspliced UL44 was termed “gCfull plasmid,” and the plasmid obtained with UL44 missing the 145-bp intron was termed “gC145 plasmid.”
The UL47 and UL54 ORFs were amplified from pRB1B using specific primers and cloned into pcDNA3-V5 using restriction sites EcoRI/NotI and EcoRI/XhoI, respectively. The resulting plasmids were named pV5-UL47 and pV5-UL54 and encoded their respective proteins with the V5 tag at the N terminus of the viral protein. The UL44 ORF was cloned into pEGFP-N1 between the EcoRI and BamHI sites, yielding plasmid pgC-GFP. Protein expression upon transfection was controlled by Western blotting (data not shown).
Cells and viruses.
Primary chicken embryonic skin cells (CESCs) were obtained from 12-day-old, specific-pathogen-free White Leghorn embryos as described previously (58) and maintained in William’s modified E medium containing 1.5% chicken serum and 1% fetal calf serum. These cultures contain mainly dermal fibroblasts and some myoblasts. Chicken keratinocytes derived from chicken embryonic stem cells were used, maintained, and infected as previously described (59, 60). ESCDL-1 cells are derived from chicken embryonic stem cells and have been characterized and previously validated for MDV replication (32). These cells, which are fully permissive to MDV, can be efficiently transfected by standard methods such as lipofection, as opposed to primary skin cells. A total of 106 cells were transfected using 5 μl of Lipofectamine 2000 (Thermo) and 0.5 μg of each plasmid. The cells were lysed for RNA extraction 24 h later.
Recombinant viruses were all derived from the cloned genome of the very virulent strain RB-1B. They were obtained after transfection of 5 × 106 CESCs with 4 μg of recombinant infectious genomes cloned as BACs by electroporation using the Amaxa Nucleofector apparatus (program 024) with the basic Nucleofector kit for primary mammalian fibroblasts (Lonza). After 4 days of incubation, viral plaques were observed. Viruses obtained from the repaired pRB1B BAC, the pΔUL47, pΔUL48, and the pΔUL54 BACs were named vRB1B, vΔUL47, vΔUL48, and vΔUL54, respectively.
The vΔgC virus was derived from the original unrepaired RB-1B BAC in which mutations are present in genes RLORF1, UL13, UL44 (gC), and Us6 (gD) (24, 25, 61, 62) that result in frameshifting (RLORF1) and the usage of alternative start codons (UL13, UL44, and Us6). These mutations do not alter viral replication in cell culture, as described earlier (61), and the virus was therefore used as a gC-negative control.
The viruses used in this study were all derived from recombineered BACs and were not passaged more than three times and only in CESCs.
Antibodies.
The following primary MAbs were used: anti-gC MDV (clone A6) (63), anti-VP5 MDV (clone F19 and clone H18) (23), anti-gB MDV (clone K11) (24), anti-VP22 MDV (clone B17) (64), anti-ICP4 MDV (clone E21) (24), and anti-alpha tubulin (clone DM1A, Sigma).
Western blotting.
For Western blot assays, cellular extracts were obtained by resuspending the cell pellet (approximatively 3 × 106 cells) in 90 μl of phosphate-buffered saline and adding of 30 μl of 4× Laemmli buffer. Then, 12 μl was loaded on a 10% SDS-PAGE gel. Proteins were transferred onto Porablot nitrocellulose membranes (Macherey-Nagel), which were probed with MAbs against MDV VP5, gC, or alpha-tubulin. Horseradish peroxidase-conjugated goat anti-mouse antibody (Sigma) was used for secondary detection in combination with the Immobilon Western HRP substrate (Millipore). Quantification on Western blots was done using the plotting function of ImageJ software.
Plaque assays.
CESCs or keratinocytes seeded in 6-well plates were infected with 100 or 15,000 PFU, respectively, of vRB1B, vΔUL47, or vΔUL48. Cells were fixed 4 days later with 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100. The cells were next blocked using 1% of bovine serum albumin (Fraction V; GE Healthcare) for 20 min at room temperature. Plaques were labeled using a mix of monoclonal antibodies directed against gB, ICP4, and VP22 and Alexa Fluor 488-conjugated goat anti-mouse secondary antibodies (Molecular Probes). Images of at least 50 plaques per condition were obtained on an Axiovert 200M inverted microscope (Zeiss) using a 5× Fluar long-distance objective (NA = 0.25). The size of plaques was determined manually using Axiovision LE64 software (release 4.9.1; Zeiss).
Deep sequencing of BACs and control of putative deletions.
(i) Sequencing. The nucleotide sequences of the repaired pRB1B BAC and the pΔUL47 and the pΔUL48 BACs were determined using the Illumina MiSeq platform. Paired-end library preparation was performed by standard Illumina methods. The BAC DNA was sheared and size-fractionated to 500 bp in length and ligated to Illumina adapters with a unique barcode per sample. Each library was generated using the Illumina TruSeq Nano LT sample preparation kit, according to the manufacturer’s instructions. The quality and quantity of the DNA were assessed using an Agilent DNA high-sensitivity series ChIP assay and the Qubit BR dsDNA assay (Invitrogen). Libraries were standardized to 2 nM, and paired-end sequencing was performed on an Illumina MiSeq using version 2 kits to generate ca. 1 to 3 million 150-nucleotide paired-end reads per sample. Residual adapter-containing reads were removed, and reads were trimmed from the 3′ to a median Phred score of 30 and a minimum length of 50 nucleotides.
(ii) Sequence assembly. Read preprocessing and de novo assembly were performed using workflows run with Nextflow v18.10.1.5003 (65). Briefly, raw reads were quality-checked using FastQC v0.11.7 (66) and trimmed using Trim Galore v0.4.5 (67) (minimum quality = 8, minimum trimmed length = 40, stringency = 4). Host sequence removal was performed using the BBDuk tool of the BBTools package v.38.06 (68) (k = 35, hdist = 0). Digital normalization was performed using the BBNorm tool from BBTools (tgtcov = 75, minabund = 4, fixspikes=t). De novo assembly was performed using MIRA v4.9.6 (69) (the parameter file is available upon request). Circularization was performed using a modified Minimus2 script (70) from the AMOS package. Manual finishing was performed using Staden Gap5 v2.0.0b11 (71). Annotation was performed using an in-house avian herpesvirus-specific pipeline.
(iii) Control of mutations. In order to determine whether the putative mutations observed by deep-sequencing were genuine, primers were designed to encompass the area of interest (Table 1). A PCR was carried out using pRB1B, pΔUL47, and pΔUL48 as the templates, and the PCR products were cloned into the vector pGEM-T-Easy (Promega). Six individual Escherichia coli colonies per condition were analyzed by colony PCR. One or two products per construct were sent for Sanger sequencing. The results are listed in Fig. 1B and C. For the UL36 deletions, different inserts were found. The inserts with the highest molecular weight were indeed the expected full-length sequence, while the shorter products represented various deletions of the repeated motif of the region (AAGCCGGAGAGGGCTTGG). These deletions did not correspond to those found in the deep sequencing files. In addition, the A/T and A/G single nucleotide polymorphisms (SNPs) observed at positions 78898 and 78924 in pΔUL47 were absent from our Sanger analysis. We concluded that the repeated region of UL36 is a source of errors either for polymerases or for deep sequencing and that the deletions and SNPs in UL36 observed by deep sequencing for pΔUL47 and pΔUL48 are unlikely to be genuinely present in the BACs. Most of the other additional mutations were localized in homopolymeric stretches, regions that are not accurately sequenced using next-generation sequencers. Some were confirmed by Sanger sequencing, but they were localized in regions of the genome that do not encode known proteins. Insertions in the a-like sequence represent additional telomeric repeats which are naturally variable in number in MDV genomes (72) and are thus unlikely to contribute to the phenotypes of vΔUL47 and vΔUL48.
DNA isolation and quantification of viral genomes by TaqMan qPCR.
Venous occipital sinus blood samples were collected into tubes containing 3% sodium citrate. Peripheral blood mononuclear cells (PBMCs) were isolated using density gradient centrifugation on MSL (Eurobio, France). The DNA extractions were performed using the DNA purification “blood or body fluids spin” protocol of the QIAamp DNA minikit (Qiagen, Germany). Incubation time with proteinase K at 56°C was extended from 10 min to 2 h to increase DNA yield.
The feather tips of approximatively three to four growing feathers were manually collected, and DNA was extracted using the “tissues protocol” of the QIAamp DNA minikit (Qiagen). Samples were incubated overnight at 56°C with proteinase K to ensure efficient lysis.
DNA concentrations were measured with a NanoDrop spectrophotometer.
Real-time PCR was performed using TaqMan technology, as previously described (73, 74). Both iNos and the ICP4 probes were tagged with FAM-BHQ1. All qPCR analyses were performed independently with 250 ng of DNA, 10 pmol of each gene-specific primer, 5 pmol of the gene-specific probe in a 20-μl volume on a CFX96 Real-Time C1000 Touch thermal cycler (Bio-Rad, Marnes-la-Coquette, France). The results were analyzed using the CFX Manager software (version 3.1; Bio-Rad). For each sample, viral DNA (based on ICP4 gene) and cellular DNA (based on iNos) were quantified independently in triplicates. The positive cutoff points corresponded to 568 copies for ICP4 and 57 copies for iNos. For each sample, the number of MDV genome copies was calculated per 106 cells.
RNA isolation and RT-(q)PCR.
RNA from cultured cells was purified using a NucleoSpin RNA Plus kit (Macherey-Nagel) according to the manufacturer’s instructions.
Reverse transcription was carried out using 2 μg of DNase-treated RNA, 0.5 μg of oligo(dT) primer, and 200 U of MMLV-Reverse Transcriptase (Promega) according to the manufacturer’s protocol. After 1 h at 42°C, reverse transcriptase was inactivated at 70°C for 15 min before proceeding to the PCRs.
PCRs were done on 0.5 μl of cDNA in a volume of 25 μl using the GoTaq Flexi G2 Taq DNA polymerase (Promega) according to the manufacturer’s instructions. All PCRs were performed using the following program: 3 min of denaturation at 94°C and 30 to 35 cycles of denaturation at 94°C for 30 s, annealing at 52°C for 30 s, and extension at 72°C for 40 s. A final extension of 5 min was performed at the end of the program. For cDNA obtained from infected cells, 30 cycles of amplification were used, whereas for cDNA obtained from tissue, 35 cycles of amplification were used.
The relative abundance of the different forms of UL44 transcripts was determined by SYBR-green-based quantitative PCR assays (IQ SYBR green Supermix; Bio-Rad) using four pairs of primers specific for spliced transcripts gC104 (gC-RTPCRr104+ and gC-RTPCRf2), gC145 (gC145RTPCR_4ntF and gC-RTPCRrev3), unspliced UL44 (gC-RTPCR-FLf and gC-RTPCR-FLr), and total UL44 (UL44GFPktrlF and gC-RTPCR-rev3) (Table 1). Quantification was performed on a Bio-Rad CFX96 Real Time System-C1000 Touch cycler, and the cycling conditions were as follows: an initial step of 95°C for 5 min, followed by 39 cycles at 95°C for 15 s and 60°C for 30 s. A final step consisted of incubation from 55 to 95°C, followed by a final reading of the plate.
Animal experiments.
Specific-pathogen-free and MDV maternal antibody-free White Leghorn chicks (B13/B13 haplotype) of 2 weeks of age were obtained from INRA animal facilities. Animals were separated into three groups of 15 animals housed in three independent isolation units. Ten animals were inoculated with the virus, while five other animals were left uninfected (“contacts”) to measure viral transmission. Animals were inoculated by intramuscular injection of 3,000 PFU of vRB1B, vΔUL47, or vΔUL48 in 200 μl of William’s E medium. One animal in the vΔUL48 group died 2 days later in the isolation unit, possibly of a wound contracted at the foot.
Statistics.
A two-tailed Wilcoxon-Mann-Whitney test was used for qPCR experiments with small samples (<30) or experiments where the sample distribution was not Gaussian as determined by a Shapiro-Wilk test for P > 0.05. A paired two-tailed Student t test was used for RT-qPCR experiments using the ΔCq obtained between the Cq values of the total UL44 and the Cq values of the transcript (full, 104, or 145) to be quantified. All statistical analyses were performed using Prism6 software (GraphPad).
Ethical statement.
In vivo experiments were carried out according to the guidance and regulation of the French Ministry of Higher Education and Research (MESR) and the experimental protocol was approved by the regional ethics committee, CREEA VdL (Comité d’Éthique pour l’Expérimentation Animale Val de Loire), under number 8723-2017013011386459.
Data availability.
The full sequences of pRB-1B, pΔUL47, and pΔUL48 are accessible in GenBank under numbers MT272733, MT272734, and MT272735, respectively.
ACKNOWLEDGMENTS
All animal experiments were carried out at the Experimental Infectiology Facilities (PFIE) of INRA (Nouzilly). We are grateful to Patrice Cousin, Sébastien Lavillate, Arnaud Faurie, and Olivier Dubes for their invaluable help with animal care and handling. We also thank Evelyne Manet for precious advice regarding the design of primers specific to the different spliced forms of UL44 for the RT-qPCR assays.
This study was funded by a grant from the Institut Carnot Santé Animale/France Futur Elevage (ICSA-FFE) (Mardished, 75000073). A.C. was supported by an INRA/Région Centre Val-de-Loire fellowship.
REFERENCES
- 1.Delecluse HJ, Hammerschmidt W. 1993. Status of Marek’s disease virus in established lymphoma cell lines: herpesvirus integration is common. J Virol 67:82–92. doi: 10.1128/JVI.67.1.82-92.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaschka-Dierich C, Nazerian K, Thomssen R. 1979. Intracellular state of Marek’s disease virus DNA in two tumour-derived chicken cell lines. J Gen Virol 44:271–280. doi: 10.1099/0022-1317-44-2-271. [DOI] [PubMed] [Google Scholar]
- 3.Calnek BW, Adldinger HK, Kahn DE. 1970. Feather follicle epithelium: a source of enveloped and infectious cell-free herpesvirus from Marek’s disease. Avian Dis 14:219–233. doi: 10.2307/1588466. [DOI] [PubMed] [Google Scholar]
- 4.Reddy SM, Izumiya Y, Lupiani B. 2017. Marek’s disease vaccines: current status, and strategies for improvement and development of vector vaccines. Vet Microbiol 206:113–120. doi: 10.1016/j.vetmic.2016.11.024. [DOI] [PubMed] [Google Scholar]
- 5.Schat KA. 2016. History of the first-generation Marek’s disease vaccines: the science and little-known facts. Avian Dis 60:715–724. doi: 10.1637/11429-050216-Hist. [DOI] [PubMed] [Google Scholar]
- 6.Read AF, Baigent SJ, Powers C, Kgosana LB, Blackwell L, Smith LP, Kennedy DA, Walkden-Brown SW, Nair VK. 2015. Imperfect vaccination can enhance the transmission of highly virulent pathogens. PLoS Biol 13:e1002198. doi: 10.1371/journal.pbio.1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denesvre C, Blondeau C, Lemesle M, Le Vern Y, Vautherot D, Roingeard P, Vautherot JF. 2007. Morphogenesis of a highly replicative EGFPVP22 recombinant Marek’s disease virus in cell culture. J Virol 81:12348–12359. doi: 10.1128/JVI.01177-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denesvre C, Remy S, Trapp-Fragnet L, Smith LP, Georgeault S, Vautherot JF, Nair V. 2016. Marek’s disease virus undergoes complete morphogenesis after reactivation in a T-lymphoblastoid cell line transformed by recombinant fluorescent marker virus. J Gen Virol 97:480–486. doi: 10.1099/jgv.0.000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson EA, Burke CN, Fredrickson TN, DiCapua RA. 1975. Morphogenesis of Marek’s disease virus in feather follicle epithelium. J Natl Cancer Inst 55:89–99. doi: 10.1093/jnci/55.1.89. [DOI] [PubMed] [Google Scholar]
- 10.Nazerian K, Solomon JJ, Witter RL, Burmester BR. 1968. Studies on the etiology of Marek’s disease. II. Finding of a herpesvirus in cell culture. Proc Soc Exp Biol Med 127:177–182. doi: 10.3181/00379727-127-32650. [DOI] [PubMed] [Google Scholar]
- 11.Nazerian K, Witter RL. 1970. Cell-free transmission and in vivo replication of Marek’s disease virus. J Virol 5:388–397. doi: 10.1128/JVI.5.3.388-397.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michael K, Klupp BG, Mettenleiter TC, Karger A. 2006. Composition of pseudorabies virus particles lacking tegument protein US3, UL47, or UL49 or envelope glycoprotein E. J Virol 80:1332–1339. doi: 10.1128/JVI.80.3.1332-1339.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Che X, Oliver SL, Sommer MH, Rajamani J, Reichelt M, Arvin AM. 2011. Identification and functional characterization of the varicella-zoster virus ORF11 gene product. Virology 412:156–166. doi: 10.1016/j.virol.2010.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jarosinski KW. 2012. Marek’s disease virus late protein expression in feather follicle epithelial cells as early as 8 days postinfection. Avian Dis 56:725–731. doi: 10.1637/10252-052212-Reg.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jarosinski KW, Arndt S, Kaufer BB, Osterrieder N. 2012. Fluorescently tagged pUL47 of Marek’s disease virus reveals differential tissue expression of the tegument protein in vivo. J Virol 86:2428–2436. doi: 10.1128/JVI.06719-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jarosinski KW, Vautherot JF. 2015. Differential expression of Marek’s disease virus (MDV) late proteins during in vitro and in situ replication: role for pUL47 in regulation of the MDV UL46-UL49 gene locus. Virology 484:213–226. doi: 10.1016/j.virol.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 18.Kristie TM, Roizman B. 1987. Host cell proteins bind to the cis-acting site required for virion-mediated induction of herpes simplex virus 1α genes. Proc Natl Acad Sci U S A 84:71–75. doi: 10.1073/pnas.84.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Preston CM, Frame MC, Campbell ME. 1988. A complex formed between cell components and an HSV structural polypeptide binds to a viral immediate early gene regulatory DNA sequence. Cell 52:425–434. doi: 10.1016/S0092-8674(88)80035-7. [DOI] [PubMed] [Google Scholar]
- 20.Stern S, Tanaka M, Herr W. 1989. The Oct-1 homoeodomain directs formation of a multiprotein-DNA complex with the HSV transactivator VP16. Nature 341:624–630. doi: 10.1038/341624a0. [DOI] [PubMed] [Google Scholar]
- 21.Thompson RL, Preston CM, Sawtell NM. 2009. De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog 5:e1000352. doi: 10.1371/journal.ppat.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weir JP. 2001. Regulation of herpes simplex virus gene expression. Gene 271:117–130. doi: 10.1016/s0378-1119(01)00512-1. [DOI] [PubMed] [Google Scholar]
- 23.Dorange F, Tischer BK, Vautherot JF, Osterrieder N. 2002. Characterization of Marek’s disease virus serotype 1 (MDV-1) deletion mutants that lack UL46 to UL49 genes: MDV-1 UL49, encoding VP22, is indispensable for virus growth. J Virol 76:1959–1970. doi: 10.1128/jvi.76.4.1959-1970.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blondeau C, Chbab N, Beaumont C, Courvoisier K, Osterrieder N, Vautherot JF, Denesvre C. 2007. A full UL13 open reading frame in Marek’s disease virus (MDV) is dispensable for tumor formation and feather follicle tropism and cannot restore horizontal virus transmission of rRB-1B in vivo. Vet Res 38:419–433. doi: 10.1051/vetres:2007009. [DOI] [PubMed] [Google Scholar]
- 25.Jarosinski KW, Margulis NG, Kamil JP, Spatz SJ, Nair VK, Osterrieder N. 2007. Horizontal transmission of Marek’s disease virus requires US2, the UL13 protein kinase, and gC. J Virol 81:10575–10587. doi: 10.1128/JVI.01065-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jarosinski KW, Osterrieder N. 2010. Further analysis of Marek’s disease virus horizontal transmission confirms that U(L)44 (gC) and U(L)13 protein kinase activity are essential, while U(S)2 is nonessential. J Virol 84:7911–7916. doi: 10.1128/JVI.00433-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ponnuraj N, Tien YT, Vega-Rodriguez W, Krieter A, Jarosinski KW. 2018. The Herpesviridae conserved multifunctional infected-cell protein 27 (ICP27) is important but not required for replication and oncogenicity of Marek’s disease alphaherpesvirus. J Virol 93:e01903-18. doi: 10.1128/JVI.01903-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jarosinski KW, Osterrieder N. 2012. Marek’s disease virus expresses multiple UL44 (gC) variants through mRNA splicing that are all required for efficient horizontal transmission. J Virol 86:7896–7906. doi: 10.1128/JVI.00908-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardy WR, Sandri-Goldin RM. 1994. Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J Virol 68:7790–7799. doi: 10.1128/JVI.68.12.7790-7799.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang S, Patel A, Krause PR. 2016. Herpes simplex virus ICP27 regulates alternative pre-mRNA polyadenylation and splicing in a sequence-dependent manner. Proc Natl Acad Sci U S A 113:12256–12261. doi: 10.1073/pnas.1609695113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang S, Patel A, Krause PR. 2019. Hidden regulation of herpes simplex virus 1 pre-mRNA splicing and polyadenylation by virally encoded immediate early gene ICP27. PLoS Pathog 15:e1007884. doi: 10.1371/journal.ppat.1007884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vautherot JF, Jean C, Fragnet-Trapp L, Remy S, Chabanne-Vautherot D, Montillet G, Fuet A, Denesvre C, Pain B. 2017. ESCDL-1, a new cell line derived from chicken embryonic stem cells, supports efficient replication of mardiviruses. PLoS One 12:e0175259. doi: 10.1371/journal.pone.0175259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Misumi Y, Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y. 1986. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J Biol Chem 261:11398–11403. [PubMed] [Google Scholar]
- 34.Isfort RJ, Stringer RA, Kung HJ, Velicer LF. 1986. Synthesis, processing, and secretion of the Marek’s disease herpesvirus A antigen glycoprotein. J Virol 57:464–474. doi: 10.1128/JVI.57.2.464-474.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weinheimer SP, Boyd BA, Durham SK, Resnick JL, O’Boyle DR, II. 1992. Deletion of the VP16 open reading frame of herpes simplex virus type 1. J Virol 66:258–269. doi: 10.1128/JVI.66.1.258-269.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuchs W, Granzow H, Klupp BG, Kopp M, Mettenleiter TC. 2002. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J Virol 76:6729–6742. doi: 10.1128/jvi.76.13.6729-6742.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lobanov VA, Maher-Sturgess SL, Snider MG, Lawman Z, Babiuk LA, van Drunen Littel-van den Hurk S. 2010. A UL47 gene deletion mutant of bovine herpesvirus type 1 exhibits impaired growth in cell culture and lack of virulence in cattle. J Virol 84:445–458. doi: 10.1128/JVI.01544-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y. 2014. Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J Virol 88:4657–4667. doi: 10.1128/JVI.00137-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Che X, Reichelt M, Sommer MH, Rajamani J, Zerboni L, Arvin AM. 2008. Functions of the ORF9-to-ORF12 gene cluster in varicella-zoster virus replication and in the pathogenesis of skin infection. J Virol 82:5825–5834. doi: 10.1128/JVI.00303-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afroz S, Brownlie R, Fodje M, van Drunen Littel-van den Hurk S. 2016. VP8, the major tegument protein of bovine herpesvirus 1, interacts with cellular STAT1 and inhibits interferon beta signaling. J Virol 90:4889–4904. doi: 10.1128/JVI.00017-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barber KA, Daugherty HC, Ander SE, Jefferson VA, Shack LA, Pechan T, Nanduri B, Meyer F. 2017. Protein composition of the bovine herpesvirus 1.1 virion. Vet Sci 4:11. doi: 10.3390/vetsci4010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carpenter DE, Misra V. 1991. The most abundant protein in bovine herpes 1 virions is a homologue of herpes simplex virus type 1 UL47. J Gen Virol 72:3077–3084. doi: 10.1099/0022-1317-72-12-3077. [DOI] [PubMed] [Google Scholar]
- 43.Kramer T, Greco TM, Enquist LW, Cristea IM. 2011. Proteomic characterization of pseudorabies virus extracellular virions. J Virol 85:6427–6441. doi: 10.1128/JVI.02253-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holland TC, Marlin SD, Levine M, Glorioso J. 1983. Antigenic variants of herpes simplex virus selected with glycoprotein-specific monoclonal antibodies. J Virol 45:672–682. doi: 10.1128/JVI.45.2.672-682.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruyechan WT, Morse LS, Knipe DM, Roizman B. 1979. Molecular genetics of herpes simplex virus. II. Mapping of the major viral glycoproteins and of the genetic loci specifying the social behavior of infected cells. J Virol 29:677–697. doi: 10.1128/JVI.29.2.677-697.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wathen MW, Wathen LM. 1986. Characterization and mapping of a nonessential pseudorabies virus glycoprotein. J Virol 58:173–178. doi: 10.1128/JVI.58.1.173-178.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herold BC, WuDunn D, Soltys N, Spear PG. 1991. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J Virol 65:1090–1098. doi: 10.1128/JVI.65.3.1090-1098.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Friedman HM, Cohen GH, Eisenberg RJ, Seidel CA, Cines DB. 1984. Glycoprotein C of herpes simplex virus 1 acts as a receptor for the C3b complement component on infected cells. Nature 309:633–635. doi: 10.1038/309633a0. [DOI] [PubMed] [Google Scholar]
- 49.Fries LF, Friedman HM, Cohen GH, Eisenberg RJ, Hammer CH, Frank MM. 1986. Glycoprotein C of herpes simplex virus 1 is an inhibitor of the complement cascade. J Immunol 137:1636–1641. [PubMed] [Google Scholar]
- 50.Long PA, Kaveh-Yamini P, Velicer LF. 1975. Marek’s disease herpesviruses. I. Production and preliminary characterization of Marek’s disease herpesvirus A antigen. J Virol 15:1182–1191. doi: 10.1128/JVI.15.5.1182-1191.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gonzalez-Motos V, Jurgens C, Ritter B, Kropp KA, Duran V, Larsen O, Binz A, Ouwendijk WJD, Lenac Rovis T, Jonjic S, Verjans G, Sodeik B, Krey T, Bauerfeind R, Schulz TF, Kaufer BB, Kalinke U, Proudfoot AEI, Rosenkilde MM, Viejo-Borbolla A. 2017. Varicella-zoster virus glycoprotein C increases chemokine-mediated leukocyte migration. PLoS Pathog 13:e1006346. doi: 10.1371/journal.ppat.1006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Donnelly M, Verhagen J, Elliott G. 2007. RNA binding by the herpes simplex virus type 1 nucleocytoplasmic shuttling protein UL47 is mediated by an N-terminal arginine-rich domain that also functions as its nuclear localization signal. J Virol 81:2283–2296. doi: 10.1128/JVI.01677-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Islam A, Schulz S, Afroz S, Babiuk LA, van Drunen Littel-van den Hurk S. 2015. Interaction of VP8 with mRNAs of bovine herpesvirus-1. Virus Res 197:116–126. doi: 10.1016/j.virusres.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 54.Sciortino MT, Taddeo B, Poon AP, Mastino A, Roizman B. 2002. Of the three tegument proteins that package mRNA in herpes simplex virions, one (VP22) transports the mRNA to uninfected cells for expression prior to viral infection. Proc Natl Acad Sci U S A 99:8318–8323. doi: 10.1073/pnas.122231699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dobrikova E, Shveygert M, Walters R, Gromeier M. 2010. Herpes simplex virus proteins ICP27 and UL47 associate with polyadenylate-binding protein and control its subcellular distribution. J Virol 84:270–279. doi: 10.1128/JVI.01740-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Z, Kato A, Oyama M, Kozuka-Hata H, Arii J, Kawaguchi Y. 2015. Role of host cell p32 in herpes simplex virus 1 de-envelopment during viral nuclear egress. J Virol 89:8982–8998. doi: 10.1128/JVI.01220-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 58.Silim A, El Azhary MA, Roy RS. 1982. A simple technique for preparation of chicken-embryo-skin cell cultures. Avian Dis 26:182–185. doi: 10.2307/1590039. [DOI] [PubMed] [Google Scholar]
- 59.Couteaudier M, Courvoisier K, Trapp-Fragnet L, Denesvre C, Vautherot JF. 2016. Keratinocytes derived from chicken embryonic stem cells support Marek’s disease virus infection: a highly differentiated cell model to study viral replication and morphogenesis. Virol J 13:7. doi: 10.1186/s12985-015-0458-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Couteaudier M, Trapp-Fragnet L, Auger N, Courvoisier K, Pain B, Denesvre C, Vautherot JF. 2015. Derivation of keratinocytes from chicken embryonic stem cells: establishment and characterization of differentiated proliferative cell populations. Stem Cell Res 14:224–237. doi: 10.1016/j.scr.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 61.Petherbridge L, Brown AC, Baigent SJ, Howes K, Sacco MA, Osterrieder N, Nair VK. 2004. Oncogenicity of virulent Marek’s disease virus cloned as bacterial artificial chromosomes. J Virol 78:13376–13380. doi: 10.1128/JVI.78.23.13376-13380.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spatz SJ, Zhao Y, Petherbridge L, Smith LP, Baigent SJ, Nair V. 2007. Comparative sequence analysis of a highly oncogenic but horizontal spread-defective clone of Marek’s disease virus. Virus Genes 35:753–766. doi: 10.1007/s11262-007-0157-1. [DOI] [PubMed] [Google Scholar]
- 63.Tischer BK, Schumacher D, Chabanne-Vautherot D, Zelnik V, Vautherot JF, Osterrieder N. 2005. High-level expression of Marek’s disease virus glycoprotein C is detrimental to virus growth in vitro. J Virol 79:5889–5899. doi: 10.1128/JVI.79.10.5889-5899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dorange F, El Mehdaoui S, Pichon C, Coursaget P, Vautherot JF. 2000. Marek’s disease virus (MDV) homologues of herpes simplex virus type 1 UL49 (VP22) and UL48 (VP16) genes: high-level expression and characterization of MDV-1 VP22 and VP16. J Gen Virol 81:2219–2230. doi: 10.1099/0022-1317-81-9-2219. [DOI] [PubMed] [Google Scholar]
- 65.Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C. 2017. Nextflow enables reproducible computational workflows. Nat Biotechnol 35:316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- 66.Andrew S. 2010. FastQC: a quality control tool for high throughput sequence data. Babraham Institute, Cambridge, UK. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. [Google Scholar]
- 67.Kruege F. 2019. A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files, with some extra functionality for MspI-digested RRBS-type (Reduced Representation Bisufite-Seq) libraries. Babraham Institute, Cambridge, UK. http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/. [Google Scholar]
- 68.Anonymous. . 2019. BBDuk: filters, trims, or masks reads with kmer matches to an artifact/contaminant file on Sourceforge. Slashdot Media, San Diego, CA. https://sourceforge.net/projects/bbmap/. [Google Scholar]
- 69.Chevreux BW, Suhai S. 1999. Genome sequence assembly using trace signal and additional sequence information. Comput Sci Biol GCB 1999:45–56. [Google Scholar]
- 70.Sommer DD, Delcher AL, Salzberg SL, Pop M. 2007. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics 8:64. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Staden RJ, Bonfield JK. 2003. Managing sequencing projects in the GAP4 environment. In Krawetz SA, Wombie DD (ed), Introduction to bioinformatics: a theoretical and practical approach. Springer, New York, NY. [Google Scholar]
- 72.Volkening JD, Spatz SJ. 2013. Identification and characterization of the genomic termini and cleavage/packaging signals of gallid herpesvirus type 2. Avian Dis 57:401–408. doi: 10.1637/10410-100312-Reg.1. [DOI] [PubMed] [Google Scholar]
- 73.Jarosinski KW, Yunis R, O’Connell PH, Markowski-Grimsrud CJ, Schat KA. 2002. Influence of genetic resistance of the chicken and virulence of Marek’s disease virus (MDV) on nitric oxide responses after MDV infection. Avian Dis 46:636–649. doi: 10.1637/0005-2086(2002)046[0636:IOGROT]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 74.Remy S, Blondeau C, Le Vern Y, Lemesle M, Vautherot JF, Denesvre C. 2013. Fluorescent tagging of VP22 in N terminus reveals that VP22 favors Marek’s disease virus (MDV) virulence in chickens and allows morphogenesis study in MD tumor cells. Vet Res 44:125. doi: 10.1186/1297-9716-44-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The full sequences of pRB-1B, pΔUL47, and pΔUL48 are accessible in GenBank under numbers MT272733, MT272734, and MT272735, respectively.