

Attachment factors and receptors are important determinants of dissemination and tropism during reovirus-induced disease. In a CRISPR cell survival screen, we discovered two genes, Cmas and Slc35a1, which encode proteins required for sialic acid expression on the cell surface and mediate reovirus infection of microglial cells. This work elucidates host genes that render microglial cells susceptible to reovirus infection and expands current understanding of the receptors on microglial cells that are engaged by reovirus. Such knowledge may lead to new strategies to selectively target microglial cells for oncolytic applications.
KEYWORDS: solute carrier family 35 member A1, sialic acid, viral attachment, reovirus, microglia, cytidine monophosphate N-acetylneuraminic acid synthetase
ABSTRACT
Engagement of cell surface receptors by viruses is a critical determinant of viral tropism and disease. The reovirus attachment protein σ1 binds sialylated glycans and proteinaceous receptors to mediate infection, but the specific requirements for different cell types are not entirely known. To identify host factors required for reovirus-induced cell death, we conducted a CRISPR-knockout screen targeting over 20,000 genes in murine microglial BV2 cells. Candidate genes required for reovirus to cause cell death were highly enriched for sialic acid synthesis and transport. Two of the top candidates identified, CMP N-acetylneuraminic acid synthetase (Cmas) and solute carrier family 35 member A1 (Slc35a1), promote sialic acid expression on the cell surface. Two reovirus strains that differ in the capacity to bind sialic acid, T3SA+ and T3SA−, were used to evaluate Cmas and Slc35a1 as potential host genes required for reovirus infection. Following CRISPR-Cas9 disruption of either gene, cell surface expression of sialic acid was diminished. These results correlated with decreased binding of strain T3SA+, which is capable of engaging sialic acid. Disruption of either gene did not alter the low-level binding of T3SA−, which does not engage sialic acid. Furthermore, infectivity of T3SA+ was diminished to levels similar to those of T3SA− in cells lacking Cmas and Slc35a1 by CRISPR ablation. However, exogenous expression of Cmas and Slc35a1 into the respective null cells restored sialic acid expression and T3SA+ binding and infectivity. These results demonstrate that Cmas and Slc35a1, which mediate cell surface expression of sialic acid, are required in murine microglial cells for efficient reovirus binding and infection.
IMPORTANCE Attachment factors and receptors are important determinants of dissemination and tropism during reovirus-induced disease. In a CRISPR cell survival screen, we discovered two genes, Cmas and Slc35a1, which encode proteins required for sialic acid expression on the cell surface and mediate reovirus infection of microglial cells. This work elucidates host genes that render microglial cells susceptible to reovirus infection and expands current understanding of the receptors on microglial cells that are engaged by reovirus. Such knowledge may lead to new strategies to selectively target microglial cells for oncolytic applications.
INTRODUCTION
The first interaction between a virus and a host cell occurs at the cell membrane, where the virus binds to attachment factors and entry receptors to initiate an infectious cycle. Identification of host factors that enable cell attachment of viruses is essential to understand this initial infection step. Genome-wide screens have been used to identify host molecules essential for viral attachment and infection. Selective disruption of target gene function using CRISPR-Cas9 offers a powerful approach to identify host genes and define cellular pathways required for early steps in virus infection. This method was used to identify CD300lf as a receptor for murine norovirus (1) and Mxra8 as an entry mediator for alphaviruses (2). This new knowledge expands our understanding of infectious diseases and may aid in the development of therapeutics and vaccines targeting these pathogenic microorganisms.
Mammalian orthoreoviruses (reoviruses) are nonenveloped viruses with a segmented double-stranded RNA (dsRNA) genome. Reovirus packages 10 dsRNA gene segments within two concentric protein shells. The reovirus S1 gene encodes the σ1 protein, a filamentous trimer that mediates viral attachment (3). Binding of σ1 to cell surface sialic acid (SA) initiates reovirus infection and is required for efficient infection of many cell types in vitro. Reoviruses have a broad host range, with the capacity to infect most mammalian species (4). While reovirus-induced diseases are similar in many mammals (4–6), most reovirus tropism studies employ mice. After establishing a primary infection in mice, reovirus disseminates systemically to several sites of secondary replication, including the central nervous system (CNS), where it causes hydrocephalus or lethal encephalitis, depending on the viral serotype (7, 8).
Reoviruses infect multiple cell types in the CNS, including neurons, ependymal cells, and microglia (7–9). Microglia are specialized phagocytic cells in the CNS that function to remove damaged neurons and maintain homeostasis (10). Following reovirus infection, microglia become activated as part of the innate immune response (11). As with many other cell types, reovirus infection of microglia induces apoptosis, a form of programmed cell death (12). Although microglia succumb to reovirus infection (9), the specific cellular determinants required for reovirus replication and subsequent cell killing are unknown. Expanding knowledge of the interactions between reovirus and microglia may help to further define mechanisms of reovirus tropism and neurovirulence.
Reovirus strains T3SA+ and T3SA− differ in the capacity to engage cell surface SA (13). T3SA+ was recovered following serial passage of strain T3C44, a type 3 (T3) reovirus field-isolate strain incapable of binding SA (14), in murine erythroleukemia (MEL) cells, which are poorly susceptible to infection by non-SA-binding reovirus strains (15). An SA-binding variant, termed T3C44-MA, was recovered and found to have a single point mutation in the σ1 protein (Leu204→Pro) (16). The S1 genes of strains T3C44-MA and T3C44 were introduced into the genetic background of strain type 1 Lang by genetic reassortment to produce T3SA+ and T3SA−, respectively (13).
Using strains T3SA+ and T3SA−, reoviruses were found to transiently bind SA with low affinity until a higher-affinity receptor, such as junctional adhesion molecule-A (JAM-A) or Nogo receptor 1 (NgR1), is encountered to initiate cell entry (13). In this way, SA strengthens cell adhesion. Interactions between reovirus and SA are required for efficient reovirus binding and infection of many types of cells in culture. In addition to MEL cells, only glycan-binding reovirus strains can infect murine embryonic fibroblasts (MEFs) (17, 18). Reovirus pathogenesis also is enhanced by SA engagement (19). Glycan-binding reovirus strains are more virulent in the CNS than their glycan-blind counterparts (20, 21). Additionally, cholestatic liver disease results from T3SA+ infection of the bile duct epithelium in mice but is not observed following infection with T3SA− (19). Viral dissemination from the intestine to sites of secondary replication is enhanced by the capacity of the virus to efficiently bind glycans (19). Defining host genes in the SA synthesis pathway required for reovirus infection will further our understanding of the function of SA engagement in reovirus attachment and infection.
SAs are a highly diverse family of acidic sugars that are most commonly appended to the terminal branches of glycan chains conjugated to proteins or lipids in which the entire molecule is referred to as a glycoprotein or glycolipid, respectively. SAs function to stabilize membranes, facilitate interactions with the environment, enhance cell-cell adhesion and signaling, and regulate ligand affinity for receptors (22, 23). Several cellular genes coordinate SA transport and conjugation. SA linkage to form the cytidine-5′-monophospho-SA (or “CMP-SA”) nucleotide donor occurs in the nucleus and is catalyzed by CMP-SA synthase, which is encoded by Cmas (24). Following this conversion, CMP-SA is transported to the cytosol, where it is delivered into the Golgi lumen by the CMP-SA transporter encoded by Slc35a1 (25). The final product, a glycoconjugate complex, is transported to the cell membrane (26). Ablation of either Cmas or Slc35a1 diminishes transport of SA to the cell surface.
In this study, we identified, evaluated, and validated Cmas and Slc35a1 as host genes required for serotype 3 reovirus infection of microglial cells. These genes and their protein products serve important functions in early steps of reovirus infection. Disruption of these genes in microglial cells results in decreased levels of SA at the cell surface and decreased reovirus binding and infectivity. These studies advance knowledge of reovirus replication and identify two host genes required for infection of microglia by serotype 3 reoviruses. These results provide potential targets for the development of new therapeutics to limit viral neuropathogenesis.
RESULTS
A CRISPR screen identifies host factors required for reovirus replication.
We hypothesized that host genes required for reovirus replication could be identified using a CRISPR-Cas9 survival screen. To test this hypothesis, we selectively disrupted target gene function across the entire mouse genome to identify host genes that encode proteins required for reovirus-induced cell killing. Two reovirus strains, T3SA+ and T3SA−, were individually used to infect a BV2 mouse microglial CRISPR cell library containing gene disruptions targeting over 20,000 genes (27). BV2 cells were chosen for their ease of genetic manipulation (1), susceptibility to reovirus-induced cell killing, and potential to inform mechanisms of reovirus neurovirulence and innate immune sensing in the brain (28). T3SA+ and T3SA− prototype reovirus strains were chosen because of differences in their capacity to bind SA. These strains differ by a single amino acid polymorphism in the σ1 body domain (13) that allows T3SA+ to engage cell surface SA, while T3SA− does not. Nine days postinoculation (pi), genomic DNA (gDNA) was isolated from surviving cells and subjected to deep sequencing. An analysis performed with STARS software was conducted to identify enriched CRISPR single-guide RNAs (sgRNAs) within the surviving cell population.
We considered genes that showed a false-discovery rate (FDR) of less than 0.25 in either screen to score as a candidate (1, 29). We found 895 genes and 52 genes in BV2 cells inoculated with T3SA+ and T3SA−, respectively, that scored as candidates (see Table S1 in the supplemental material). Strikingly, the top four candidates in the T3SA+ screen encode proteins essential for SA expression on the cell surface (Fig. 1A). This enrichment was not observed following infection with T3SA− (Fig. 1B). Four genes involved in the synthesis or cellular transport of SA, Nans, St3gal4, Slc35a1, and Cmas, appear to be required for infection and cell death following infection with T3SA+. Nans encodes an enzyme member of the biosynthetic SA pathway, which operates in the phosphorylation of SA (30). St3gal4 encodes a member of the sialyltransferase 29 protein family, which functions in the glycosylation and production of α2,3-linked sialoglycoconjugates (22). Two other identified genes, Cmas and Slc35a1, are involved in critical steps in the synthesis pathway for all SAs, including α2,3-, α2,6-, and α2,8-linked sialoglycoconjugates (22, 31). Mutations introduced into Cmas disrupt the synthesis of all SAs and result in an intracellular accumulation of free SAs; mutations introduced into Slc35a1 disrupt the generation of sialo conjugates (32). The enrichment of genes required for SA synthesis, in addition to our prior understanding of SA as an attachment factor for reovirus, made these genes attractive candidates for further studies of their function in reovirus replication.
FIG 1.
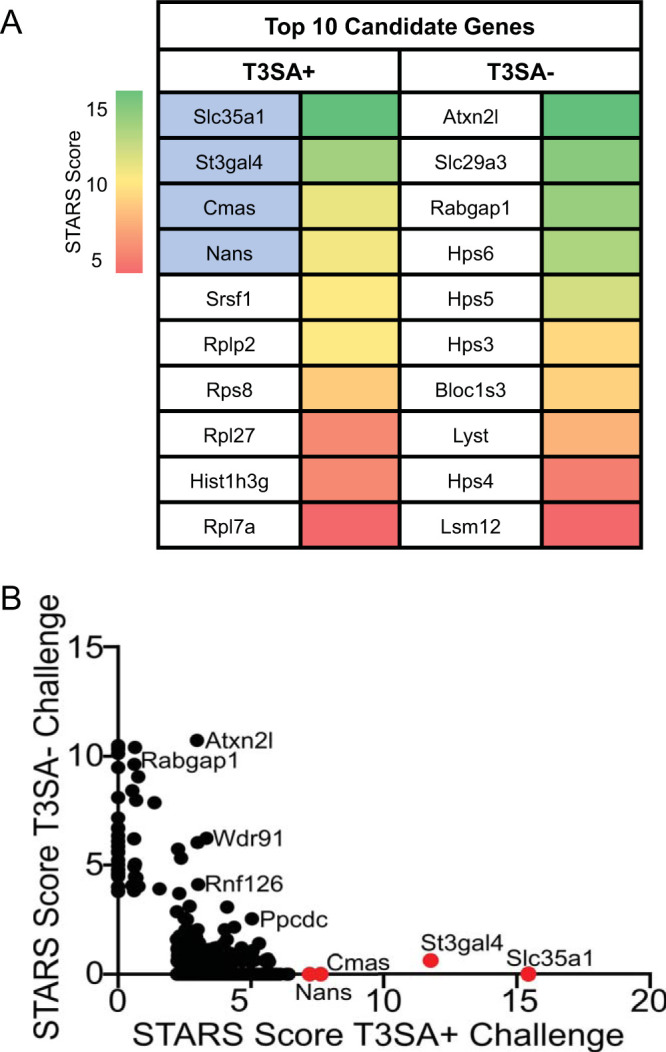
Whole-genome CRISPR screen identifies sialic acid (SA) synthesis genes as required for reovirus-induced death of BV2 cells. (A) The top 10 candidates from CRISPR screens using reovirus strains T3SA+ and T3SA− are ranked by their STARS scores. The heat map indicates STARS values. Genes encoding proteins involved in SA synthesis are indicated by blue shading. (B) Comparison of STARS scores from the candidates of the BV2 CRISPR screen between the T3SA+ (x axis) and T3SA− (y axis) conditions. Genes that did not meet the criteria to receive a STARS score are assigned a value of 0. Genes linked to SA metabolism are colored in red.
Complementation studies validate selective disruption of Cmas and Slc35a1 genes by CRISPR.
Considering the importance of Cmas and Slc35a1 in the SA synthesis pathway, we sought to evaluate the importance of these genes during reovirus infection. We engineered cell lines lacking either the Cmas (ΔCmas) or the Slc35a1 (ΔSlc35a1) gene using CRISPR-mediated gene ablation to test the hypothesis that these genes are required for reovirus infection.
To assess the expression of SA on the cell surface, we used a fluorescein-labeled lectin. Wheat germ agglutinin (WGA), derived from Triticum vulgaris, binds numerous sialoglycoconjugates that terminate in α2,3-, α2,6-, or α2,8-linked SA residues (33). The resulting ΔCmas or ΔSlc35a1 cell populations from CRISPR ablation were mixed populations (data not shown); approximately 80% of the cells contained gene ablations for Cmas or Slc35a1, and approximately 20% of the cells did not incorporate the sgRNA and, therefore, the targeted gene remained functional. To establish a uniform population of cells lacking SA, a bulk population of ΔCmas or ΔSlc35a1 cells was selected and propagated followed by single-cell sorting to establish clonal populations. Expression of SA on the cell surface was quantified based on fluorescent lectin binding using flow cytometry. The clonal population of cells binding the smallest amount of lectin was further propagated.
To determine whether the lectin- and virus-binding phenotypes observed in studies of ΔCmas and ΔSlc35a1 cells were due to the targeted gene disruptions, we complemented the CRISPR-ablated clones with wild-type (WT) alleles. Complementation was achieved by stable transfection of plasmid harboring Cmas or Slc35a1 into ΔCmas or ΔSlc35a1 cells, respectively. Cells expressing Cmas or Slc35a1 were preferentially selected using resistance markers conferred by the plasmids. In the same manner as with the CRISPR-ablated cells, clonal populations of complemented cell lines (ΔCmas+Cmas and ΔSlc35a1+Slc35a1 cells) were established by bulk cell sorting followed by single-cell sorting, using lectin binding as a surrogate for cell surface SA expression.
To interrogate SA expression on the cell surface of WT, ΔCmas, ΔSlc35a1, ΔCmas+Cmas, and ΔSlc35a1+Slc35a1 cells, we quantified lectin binding using flow cytometry after several passages of the cells in culture (Fig. 2A). The level of lectin binding to the WT, ΔCmas+Cmas, and ΔSlc35a1+Slc35a1 cells was significantly greater than that to ΔCmas and ΔSlc35a1 cells (Fig. 2B). These data demonstrate that disruption of Cmas or Slc35a1, two genes involved in the SA synthesis pathway, efficiently impairs SA expression on the cell surface.
FIG 2.

SA expression is diminished following CRISPR-mediated gene disruption and restored by stable complementation of Cmas and Slc35a1 genes. Cells were incubated with fluorescein-labeled WGA to assess cell surface expression of SA. WGA binding to cells was detected by flow cytometry. (A) Representative flow cytometric profiles of fluoresceinated WGA binding to WT, CRISPR-ablated, and complemented cells. (B) The mean fluorescence intensity (MFI) of WGA binding was quantified. The data represent results from three independent experiments each with duplicate samples. Error bars indicate standard error of the mean (SEM). *, P < 0.05; **, P < 0.01; ns, not significant, as determined by Welch’s analysis of variance (ANOVA) with Dunnett’s multiple-comparison test.
Cells lacking functional Cmas or Slc35a1 display increased viability during reovirus infection.
Cell viability, as assessed by the metabolic activity of cells in a population, was used to determine whether the Cmas and Slc35a1 host genes identified in the CRISPR screen are required for reovirus infection. To determine the effect of Cmas or Slc35a1 gene disruption on reovirus-induced cell death, WT, ΔCmas, ΔSlc35a1, ΔCmas+Cmas, and ΔSlc35a1+Slc35a1 BV2 cells were inoculated with reovirus strain T3SA+ or strain T3SA−. Using these viruses, which differ in SA-binding capacity, phenotypic changes caused by a disruption in Cmas and Slc35a1 could be directly correlated with SA engagement. At 24 and 48 h postinoculation (hpi) with T3SA− (Fig. 3A), the levels of cell viability were comparable in all five cell lines. At 24 and 48 hpi with T3SA+ (Fig. 3B), the ΔCmas and ΔSlc35a1 cell lines displayed significantly enhanced viability compared with WT, ΔCmas+Cmas, and ΔSlc35a1+Slc35a1 cells. The viability of ΔCmas and ΔSlc35a1 cells after inoculation with T3SA+ was comparable to the viability observed after inoculation with T3SA− (Fig. 3). These data are consistent with the results of the CRISPR screen and indicate that genetic disruption of Cmas or Slc35a1 protects murine microglial cells from reovirus-induced cell death.
FIG 3.
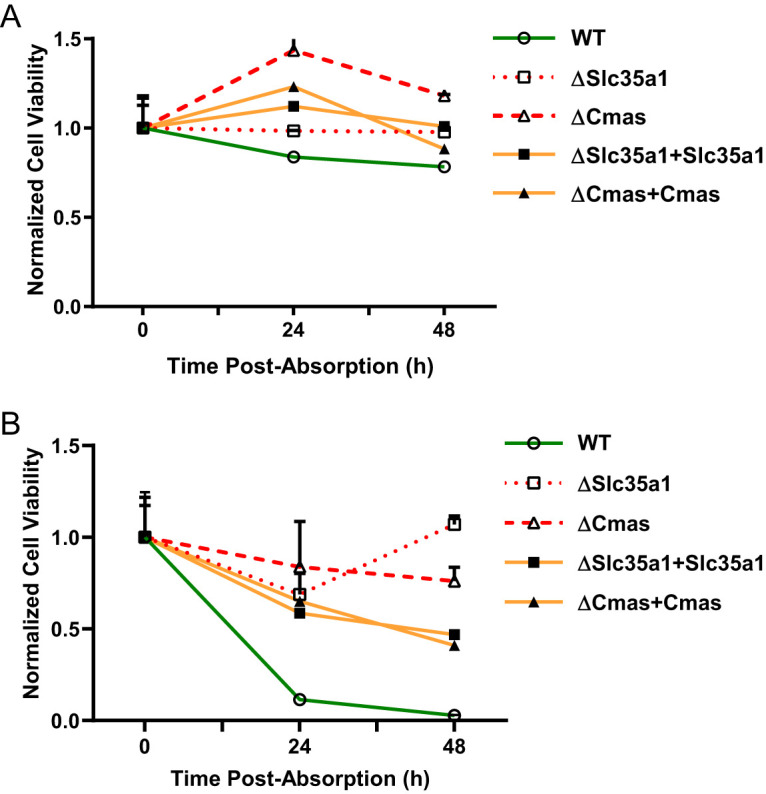
BV2 cells lacking Cmas or Slc35a1 expression are protected from reovirus-induced cell death, while complemented cells are not. The cell lines shown were adsorbed with (A) T3SA− or (B) T3SA+ at an MOI of 100 PFU/cell, and cell viability was quantified using a PrestoBlue assay at the intervals shown. Data are normalized to a mock-infected condition (not shown) and the 0 h average for each respective cell line. Data are representative of results from nine technical replicates from three independent experiments. Error bars indicate SEM.
The efficiency of reovirus infection is diminished by disruptions in Cmas or Slc35a1.
We hypothesized that the differences in the viability of the cells that differed in expression of Cmas or Slc35a1 following infection by reovirus T3SA+ indicate that Cmas and Slc35a1 encode host factors required for productive infection of SA-binding reovirus strains. To test this hypothesis, we determined the capacity of T3SA+ and T3SA− to infect cells with Cmas or Slc35a1 gene disruptions. WT, ΔCmas, ΔSlc35a1, ΔCmas+Cmas, and ΔSlc35a1+Slc35a1 cells were adsorbed with T3SA+ or T3SA− at a multiplicity of infection (MOI) of 100 PFU/cell, as determined by plaque assay using highly susceptible L929 cells. BV2 infectivity was quantified at 24 hpi by indirect immunofluorescence. T3SA+ infected WT, ΔCmas+Cmas, and ΔSlc35a1+Slc35a1 cells significantly more efficiently than ΔCmas and ΔSlc35a1 cells (Fig. 4), consistent with results obtained from the viability studies. The level of T3SA+ infectivity following inoculation of ΔCmas and ΔSlc35a1 cells was comparable to that observed following inoculation of T3SA−, which is incapable of binding SA. These data indicate that reovirus is incapable of efficiently infecting murine microglial cells lacking Cmas and Slc35a1.
FIG 4.
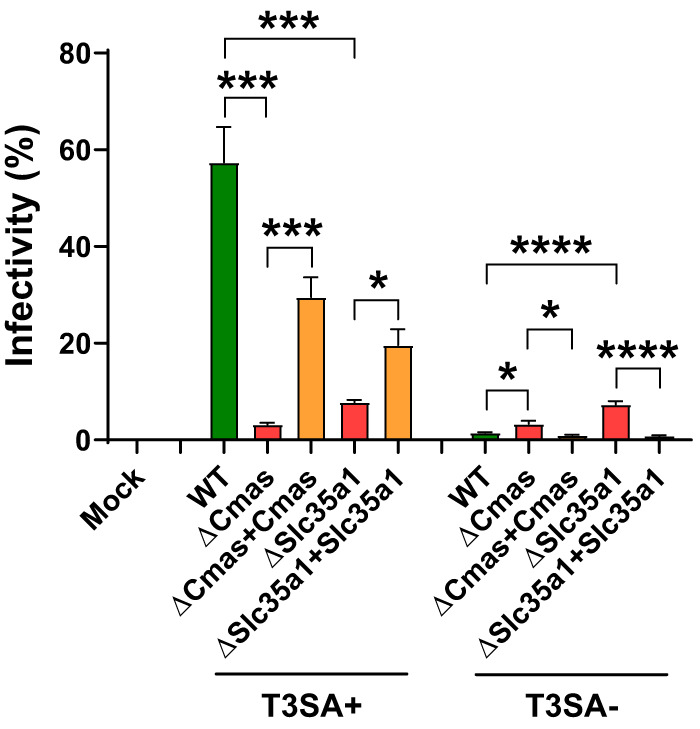
Expression of Cmas and Slc35a1 is required for reovirus infection of BV2 cells. The cell lines shown were adsorbed with T3SA+ or T3SA− at an MOI of 100 PFU/cell. The percentage of infected cells was determined by enumeration of reovirus-positive cells at 24 h postadsorption from immunofluorescence images. The data are representative of results from nine technical replicates from three independent experiments. Error bars indicate SEM. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001, as determined by Welch’s ANOVA with Dunnett’s multiple-comparison test.
Reovirus does not bind to cells lacking Cmas or Slc35a1.
To explore the initial interactions between reovirus and cells with Cmas or Slc35a1 gene disruptions, we studied reovirus binding to gene-altered cells. We hypothesized that T3SA+, a virus capable of engaging SA, would bind to WT, ΔCmas+Cmas, and ΔSlc35a1+Slc35a1 cells more efficiently than to ΔCmas and ΔSlc35a1 cells and that T3SA−, a virus incapable of engaging SA, would not bind efficiently to any of these cell types. To test this hypothesis, we incubated cells with fluorescently labeled T3SA+ (Fig. 5A) or T3SA− (Fig. 5B) at 4°C and quantified bound virus using flow cytometry. The level of T3SA+ bound to the surface of WT, ΔCmas+Cmas, and ΔSlc35a1+Slc35a1 cells was significantly greater than that bound to ΔCmas and ΔSlc35a1 cells (Fig. 5C). Moreover, the binding of T3SA+ to ΔCmas and ΔSlc35a1 cells was comparable to the binding of T3SA− to all cells tested. Collectively, these results demonstrate that expression of Cmas and Slc35a1 in murine microglial cells is required for efficient reovirus binding and infection.
FIG 5.

Expression of Cmas and Slc35a1 confers reovirus binding to BV2 cells. The cell lines shown were adsorbed with 105 particles/cell of Alexa Fluor 647-labeled T3SA+ (A) or T3SA− (B). Virus binding was quantified using flow cytometry. Representative flow cytometric profiles are shown. (C) The MFI of Alexa Fluor 647 was quantified. The data are representative of results from six technical replicates from three independent experiments. Error bars represent SEM. **, P < 0.01; ***, P < 0.001; ns, not significant, as determined by Welch’s ANOVA with Dunnett’s multiple-comparison test.
DISCUSSION
At the cell surface, reovirus interactions with SA and proteinaceous receptors are essential for cell entry. However, a complete understanding of the host genes required for efficient reovirus cell binding is lacking. In this study, we used a CRISPR cell survival screen to identify host genes required for reovirus infection. We found two host genes that govern early entry steps in reovirus infection of murine microglial cells, Cmas and Slc35a1, which are required for synthesis of sialylated cell surface glycoproteins and glycolipids. Disruption of these genes in microglial cells resulted in a decreased abundance of SA at the cell surface (Fig. 2) and diminished attachment of an SA-binding reovirus strain (Fig. 5). These data further our understanding of host genes that regulate reovirus infection of microglial cells, an important component of the innate immune response in the CNS, illuminate potential targets to ameliorate reovirus disease, and guide development of highly selective reovirus oncolytic therapeutics.
Analysis of disrupted host genes in microglial cells that survived infection by reovirus strain T3SA+ or strain T3SA− revealed little overlap (Fig. 1B). We hypothesize that differences in the genes identified in the screens using these strains are mediated by the efficiency (Fig. 3) or mechanism (19) of cell killing induced by the two strains over the 9-day interval of infection. Differences in the genes identified following T3SA+ or T3SA− infection also could be due to differences in the cell entry mechanisms used by the viruses such as has been observed for SA-binding mutants of influenza virus (34). The screen using the SA-binding T3SA+ reovirus strain revealed a specific enrichment of genes involved in the SA synthesis pathway (Fig. 1). The top four candidates identified in the CRISPR screen following T3SA+ infection were Nans, St3gal4, Slc35a1, and Cmas. Nans is required for the phosphorylation of all types of SAs (35). St3gal4 is required for the sialylation of α2,3-linked glycoconjugates (36). Slc35a1 and Cmas are required for early steps in the SA synthesis pathway and thus are essential to produce all types of SAs. Interestingly, Slc35a1 also is required for influenza virus cell entry (37). We showed that T3SA+ does not efficiently infect CRISPR-ablated cells that lack Cmas or Slc35a1 expression (Fig. 4), suggesting that both host genes contribute to reovirus infection of microglial cells. This reduction in infectivity also correlates with reduced T3SA+ binding to Cmas or Slc35a1 CRISPR-ablated cells relative to WT cells (Fig. 5), indicating that these host genes have important functions in the initial interactions between reovirus and microglial cells.
To verify that the observed reduction in reovirus binding and infectivity of BV2 cells was specific to the Cmas and Slc35a1 gene disruptions, we reintroduced WT Cmas and Slc35a1 alleles into the knockout cells. We demonstrated that T3SA+ binding (Fig. 5) and infection (Fig. 4) of complemented Cmas and Slc35a1 CRISPR knockout cells were significantly increased compared with the results seen with their CRISPR-ablated counterparts. However, T3SA+ binding and infection were not restored in complemented knockout cells to levels equivalent to those in WT BV2 cells. We think it possible that the introduction of Cmas or Slc35a1 cDNA does not fully restore functionality of the SA synthesis pathway. Therefore, the SAs expressed on the surface of complemented cells may not be structurally identical to those expressed on WT cells. Further experiments are required to clarify this possibility.
Receptor binding often triggers signaling cascades that mediate viral entry and subsequent infection of cells. Reovirus infection induces apoptosis of many types of cells, a process triggered by recognition of the virus by cellular pattern recognition receptors (38). Cmas and Slc35a1 CRISPR-ablated cells displayed increased cell viability compared with WT BV2 cells following infection with T3SA+ (Fig. 3). This finding is consistent with previous studies demonstrating that SA-binding reovirus strains induce apoptosis more efficiently than non-SA-binding strains (17). Engagement of SA may contribute to apoptosis by enhancing reovirus entry and activation of pattern recognition receptors or by stimulating signaling pathways initiated by SA binding. Regardless of the precise mechanism, disruption of Cmas or Slc35a1 protects murine microglial cells from reovirus-induced cell death.
Microglial cells are an essential component of the innate immune response in the CNS. These cells comprise the resident phagocytic cell population of the myeloid linage and function to remove damaged neurons and maintain CNS homeostasis (39). Serotype 3 reovirus strains infect microglial cells during infection of mice (40), and reovirus strain serotype 3 Abney can infect cultured amoeboid microglial cells, a subpopulation of activated microglia (9). However, expression of reovirus receptors JAM-A and NgR1 in BV2 microglial cells is not detectable by immunoblotting (data not shown). Thus, it is not apparent how reovirus is internalized into these cells. Interestingly, WT BV2 cells displayed generally poor susceptibility to reovirus infection, with less than 60% of cells scoring positive for infection despite the high MOI used in our experiments. This percentage of infectivity is low compared with other cell lines commonly used for reovirus infection studies, such as HeLa cells, L929 cells, and MEL cells (19). It is possible that reovirus uses SA on microglial cells as an attachment factor and an unidentified proteinaceous receptor to induce viral entry, bypassing a requirement for JAM-A or NgR1. The capacity of reovirus to infect microglial cells could allow propagation within the CNS and evasion of innate immune detection.
Virus-receptor interactions often regulate tissue tropism and pathogenesis. The host determinants of reovirus tropism in the mammalian CNS are not fully understood. Reovirus transmission occurs primarily through fecal-oral pathways (41). Following primary replication in the intestine, serotype 3 reoviruses spread by neural and hematogenous routes to infect neurons of the CNS, where these viruses cause apoptosis and lethal encephalitis (7). Following direct intracranial inoculation, T3SA+ is more neurovirulent, produces higher titers, and causes apoptosis more efficiently than T3SA− (19, 42). However, T3SA+ and T3SA− display comparable degrees of tropism (42). It is possible that the neurovirulence of serotype 3 reovirus is influenced by the capacity of the virus to infect microglial cells, a process regulated by SA binding. If so, therapeutic interruption of reovirus-SA engagement may attenuate reovirus-induced CNS injury.
Work presented here used a genetic screen to identify Cmas and Slc35a1 as host genes required for reovirus infection of microglial cells. Protein products of these genes function to promote early steps of reovirus infection. It is important to understand interactions between reovirus and cell surface moieties, such as SA, to expand current understanding of the receptors on microglial cells that allow reovirus infection and provide potential therapeutic targets to limit reovirus neuropathogenesis or guide the targeting of oncolytic reoviruses to tumors of microglial origin.
MATERIALS AND METHODS
Cells and viruses.
BV2 cells were cultivated in Dulbecco’s modified Eagle medium (DMEM; Gibco) supplemented to contain 10% fetal bovine serum (FBS; VWR), 1% HEPES (Gibco), 100 units/ml of penicillin, and 100 μg/ml of streptomycin (Gibco) (referred to as BV2 maintenance medium). Puromycin (Sigma-Aldrich) (2.5 μg/ml) and blasticidin (Thermo Fisher Scientific) (4 μg/ml) or Geneticin (300 μg/ml; Gibco) were added to the medium as appropriate (see below). The medium with both puromycin and blasticidin added is referred to as BV2 selection medium. The medium with Geneticin added is referred to as BV2 transfection medium.
Parental (WT), CRISPR-edited parental, CRISPR-edited bulk-sorted, and CRISPR-edited single-cell sorted (ΔCmas and ΔSlc35a1) BV2 cells (where the “Δ” symbol signifies disruption of either the Slc35a1 gene or the Cmas gene) were cultivated in BV2 selection medium unless otherwise noted. Complemented ΔCmas and ΔSlc35a1 BV2 (ΔCmas+Cmas and ΔSlc35a1+Slc35a1) cells, which were stably transfected with plasmids expressing Cmas and Slc35a1, respectively, were cultivated in BV2 maintenance medium or BV2 transfection medium by alternating the medium used with each passage.
Reovirus strains T3SA+ and T3SA− were recovered using plasmid-based reverse genetics as previously described (21). T3SA− differs from strain T3SA+ by a single point mutation in the S1 gene (encoding Leu204 in T3SA− σ1 and Pro204 in T3SA+ σ1). Virus was purified from infected L929 cells by cesium chloride gradient centrifugation (43), and viral titers were determined by plaque assay using L929 cells (44).
Reovirus particle concentration was estimated by spectral absorbance at 260 nm (optical density at 260 nm [OD260] of 1 = 2.1 × 1012 particles/ml). Reovirus virions were labeled with succinimidyl-ester Alexa Fluor 647 (Thermo Fisher Scientific) to produce fluorescently labeled particles (45).
CRISPR screen.
The screen was conducted and transduction validated as previously described (1). BV2 cells were transduced with pXPR_101 lentivirus encoding Cas9 (Addgene; 52962) and propagated for 11 days in BV2 maintenance medium supplemented to contain blasticidin. These parental BV2 or BV2-Cas9 cells were transduced for 2 days with pXPR_011 lentivirus expressing enhanced green fluorescent protein (eGFP) (Addgene; 59702) and an sgRNA targeting eGFP at a multiplicity of infection (MOI) of less than 1 PFU/cell. Cells were selected for 5 days with BV2 selection medium.
Four pools of the murine Asiago sgRNA CRISPR library, which contains six independent genome-wide pools with unique sgRNAs targeting 20,077 mouse genes represented 500 times, were transduced into 5 × 107 BV2 cells at an MOI of 0.2 PFU/cell to establish four BV2 libraries (27). Two days postransduction, cells were transferred to BV2 selection medium and propagated for 5 days. Under each set of experimental conditions, 107 BV2 library cells expressing Cas9 and sgRNAs were seeded in duplicate into T175 tissue culture flasks (Greiner Bio-One). Cells were inoculated with Dulbecco’s phosphate-buffered saline without calcium, magnesium, and phenol red (PBS−/−; mock) or with reovirus strain T3SA+ or strain T3SA− at an MOI of 100 PFU/cell diluted in Opti-MEM (Gibco). Cells were incubated at room temperature (RT) for 1 h, followed by the addition of 20 ml of DMEM supplemented to contain 10% FBS, 100 units/ml of penicillin, 100 μg/ml of streptomycin, 1% sodium pyruvate, and 1% sodium bicarbonate. After 2 days (mock condition) or 9 days (T3SA+ or T3SA− condition) postinoculation, surviving cells were harvested, and genomic DNA (gDNA) was isolated from surviving cells using a QIAamp DNA minikit (Qiagen) according to the manufacturer’s instructions. At 9 days postinoculation, a cell death rate of ∼95% or more was apparent under both virus conditions.
CRISPR screen sequencing and analysis.
Illumina sequencing and STARS analyses were conducted as previously described (27). The gDNA was aliquoted into a 96-well plate (Greiner Bio-One) with up to 10 μg gDNA in a total volume of 50 μl per well. A PCR master mix containing Ex Taq DNA polymerase (Clontech), Ex Taq buffer (Clontech), deoxynucleoside triphosphates (dNTPs), P5 stagger primer, and water was prepared. PCR master mix (40 μl) and 10 μl of a barcoded primer were added to each well containing gDNA. Samples were amplified using the following protocol: 95°C for 1 min, followed by 28 cycles of 94°C for 50 s, 52.5°C for 30 s, and 72°C for 30 s and ending with a final 72°C extension for 10 min. The PCR product was purified using Agencourt AMPure XP solid-phase reversible-immobilization (SPRI) beads (Beckman Coulter) according to the manufacturer’s instructions. Samples were sequenced using a HiSeq 2000 sequencer (Illumina).
Following deconvolution of the barcodes in the P5 primer, sgRNA sequences were mapped to a reference file of sgRNAs from the Asiago library. To account for the differing numbers of reads under the various conditions, read counts per sgRNA were normalized to 107 total reads per sample. Normalized values were then subjected to log-2 transformation. The sgRNAs that were not detected were arbitrarily assigned a read count value of 1. Frequencies of sgRNAs were analyzed using STARS software to produce a rank-ordered score for each gene. This score correlated with the sgRNA candidates that represented above 10% of the total sequenced sgRNAs. Gene targets scoring above this threshold in either of the two independent subpools and in at least two of the four independent genome-wide pools were assigned a STAR score. In addition to the STAR score, screen results were compared using FDR analysis to distinguish gene-specific signal from background noise. Statistical values of independent replicates were averaged.
Production of Cmas and Slc35a1 knockout cells.
To establish ΔCmas and ΔSlc35a1 BV2 cell lines, WT BV2 cells expressing Cas9 were transduced with lentiviruses expressing different sgRNAs and a puromycin resistance gene as follows: for Cmas sgRNA, 5′-CACCGGCAACTTTCTGGAGGTCAGT-3′; for Slc35a1 sgRNA, 5′-CACCGTATCACTTCTGTGATACACA-3′.
Cells with disrupted Cmas or Slc35a1 genes were preferentially selected (see clonal cell selection information below).
cDNA transfection of ΔCmas and ΔSlc35a1 cells.
Plasmids containing the mouse Cmas (GenBank accession no. NM_009908.2) and Slc35a1 (accession no. NM_011895.3) cDNAs in pcDNA3.1+/C-(K)-DYK and pcDNA3.1(+)-N-DYK vectors, respectively, were obtained from GenScript.
One day prior to transfection, ΔCmas and ΔSlc35a1 cells were cultivated in BV2 maintenance medium at a density of 5 × 105 cells per well in 6-well tissue culture plates. On the day of transfection, BV2 maintenance medium was removed and replaced with Opti-MEM (Gibco). Cells were transfected with either Cmas or Slc35a1 DNA using FuGene 6 (Promega) according to the manufacturer’s instructions. At 24 h posttransfection, the medium was removed and replaced with BV2 transfection medium, which was replaced every 3 days. Cells were selected for 10 days before further experiments. Stably transfected ΔCmas and ΔSlc35a1 cells are denoted as ΔCmas+Cmas and ΔSlc35a1+Slc35a1 cells, respectively.
Selection of clonal cell populations using flow cytometry.
ΔCmas, ΔSlc35a1, ΔCmas+Cmas, or ΔSlc35a1+Slc35a1 BV2 cells were detached from tissue culture plates using CellStripper dissociation reagent (Corning) and quenched with double the volume of BV2 selection medium. Cells were centrifuged at 1,500 rpm after quenching to form a pellet, washed twice with PBS−/−, and maintained at 4°C. Cells were adsorbed with fluorescein-labeled wheat germ agglutinin (WGA; Vector Laboratories) (0.005 mg/ml) at 4°C for 60 min, and unbound lection was removed by washing twice with PBS−/−. Cell populations binding the smallest amount (ΔCmas and ΔSlc35a1 cells) or largest amount (ΔCmas+Cmas and ΔSlc35a1+Slc35a1 cells) of lectin were isolated using a FACSAria flow cytometer (BD Biosciences). A total of 10,000 ΔCmasBulk, ΔSlc35a1Bulk, ΔCmas+CmasBulk, or ΔSlc35a1+Slc35a1Bulk BV2 cells were selected and seeded into wells of a 6-well tissue culture plate containing 2 ml of BV2 maintenance medium. Following propagation of the bulk-sorted populations, lectin binding was reassessed. Single cells from ΔCmasBulk, ΔSlc35a1Bulk, ΔCmas+CmasBulk, or ΔSlc35a1+Slc35a1Bulk cell populations were sorted into wells of a 96-well tissue culture plate containing 100 μl of BV2 maintenance medium and propagated to establish clonal populations. The clones binding lectin the least (ΔCmas and ΔSlc35a1 clones) or most (ΔCmas+Cmas and ΔSlc35a1+Slc35a1 clones) were used for all subsequent experiments.
Quantification of reovirus binding using flow cytometry.
WT, ΔCmas, ΔSlc35a1, ΔCmas+Cmas, and ΔSlc35a1+Slc35a1 BV2 cells were detached from tissue culture plates using CellStripper dissociation reagent, quenched with BV2 selection medium, and washed twice with PBS−/−. Cells were resuspended in PBS−/− supplemented to contain 105 particles/cell of fluorescently labeled T3SA+ or T3SA− or vehicle control and incubated on a rotor at 4°C for 1 h. Cells were washed twice with PBS−/− to remove unbound virus and fixed in PBS−/− supplemented to contain 1% paraformaldehyde. Propidium iodide (1 μl/sample) was added to all samples except the unstained control. Cells were analyzed using a LSRII flow cytometer (BD Biosciences). Results were analyzed using FlowJo V10 software.
Cell viability assay.
WT, ΔCmas, ΔSlc35a1, ΔCmas+Cmas, and ΔSlc35a1+Slc35a1 BV2 cells were plated at a density of 104 cells/well in 96-well tissue culture plates and incubated at 37°C overnight. Cells were adsorbed with reovirus at an MOI of 100 PFU/cell and incubated at RT for 1 h. The virus inoculum was removed and replaced with 200 μl of BV2 selection medium (for the WT, ΔCmas, and ΔSlc35a1 cells) or BV2 maintenance medium (for the ΔCmas+Cmas and ΔSlc35a1+Slc35a1 cells). At various intervals postinoculation (0, 24, and 48 h), 20 μl of PrestoBlue cell viability reagent (Thermo Fisher Scientific) was added to wells, plates were incubated at 37°C for 10 min, and total well fluorescence at 570 nm was quantified using a Synergy H1 microplate reader (BioTek).
Quantification of reovirus infectivity.
WT, ΔCmas, ΔSlc35a1, ΔCmas+Cmas, and ΔSlc35a1+Slc35a1 BV2 cells were plated at a density of 104 cells/well in 96-well tissue culture plates and incubated at 37°C overnight. Cells were adsorbed with reovirus at an MOI of 100 PFU/cell and incubated at RT for 1 h. The virus inoculum was removed, and 100 μl of BV2 selection medium (WT, ΔCmas, and ΔSlc35a1 cells) or BV2 maintenance medium (ΔCmas+Cmas and ΔSlc35a1+Slc35a1 cells) was added to the cells. Cells were incubated at 37°C for 24 h, washed once with PBS−/−, and fixed with 100 μl of ice-cold methanol at −20°C for at least 30 min. Fixed cells were washed twice with PBS−/−, blocked with 1% bovine serum albumin (BSA) for 30 min, and incubated with reovirus-specific rabbit antiserum diluted 1:1,000 in PBS−/− containing 0.5% Triton X-100 at RT for 1 h. Cells were washed twice with PBS−/− and incubated with Alexa Fluor 488-conjugated anti-rabbit antibody (Thermo Fisher) at a dilution of 1:1,000 at RT for 1 h. Cells were washed two times with PBS−/−, and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific) at 1:2,000 dilution at RT for 5 min. Cells were imaged for reovirus antigen and DAPI using a Lionheart FX automated imager (BioTek) equipped with a 20× air objective. The percentage of cells infected with reovirus was determined using Gen5+ software (BioTek).
Statistical analysis.
All statistical tests were conducted using Prism 8 (GraphPad Software). P values of less than 0.05 were considered to be statistically significant. Descriptions of the specific tests used are provided in the figure legends.
Supplementary Material
ACKNOWLEDGMENTS
We thank Adaeze Izuogu and Nicole McAllister for critically reviewing the manuscript. We are grateful to members of the Dermody and Virgin laboratories for useful discussions throughout this study. We thank Joshua Michel and Alexis Styche for technical assistance with flow cytometry and data analysis.
This work was supported by U.S. Public Health Service awards R01 AI038296 (K.U., D.M.S., J.J.K., P.A., G.M.T., and T.S.D.), R00 DK116666 (R.C.O.), K08 AI128043 (C.B.W.), and U19 AI109725 (H.W.V.) and a Burroughs Welcome Fund Career Award for Medical Scientists (C.B.W.). Additional funding was provided by the Heinz Endowments.
We declare that no conflicts of interest exist. H.W.V. is currently an employee of VIR Biotechnology. The work reported here was conducted at the Washington University School of Medicine and was not supported by VIR.
K.U. wrote the manuscript. K.U., D.M.S., R.C.O., and C.B.W. designed and conducted experiments and analyzed results. J.J.K. and P.A. provided crucial reagents and reviewed the manuscript. G.M.T. designed experiments and reviewed the manuscript. H.W.V. and T.S.D. designed experiments, analyzed results, and wrote the manuscript.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Orchard RC, Wilen CB, Doench JG, Baldridge MT, McCune BT, Lee YC, Lee S, Pruett-Miller SM, Nelson CA, Fremont DH, Virgin HW. 2016. Discovery of a proteinaceous cellular receptor for a norovirus. Science 353:933–936. doi: 10.1126/science.aaf1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang R, Kim AS, Fox JM, Nair S, Basore K, Klimstra WB, Rimkunas R, Fong RH, Lin H, Poddar S, Crowe JE, Jr, Doranz BJ, Fremont DH, Diamond MS. 2018. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 557:570–574. doi: 10.1038/s41586-018-0121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chappell JD, Prota A, Dermody TS, Stehle T. 2002. Crystal structure of reovirus attachment protein sigma1 reveals evolutionary relationship to adenovirus fiber. EMBO J 21:1–11. doi: 10.1093/emboj/21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tyler KL. 2001. Mammalian reoviruses, p 1729–1745. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 5.Raine CS, Fields BN. 1973. Reovirus type 3 encephalitis-a virologic and ultrastructural study. J Neuropathol Exp Neurol 32:19–33. doi: 10.1097/00005072-197301000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Wu AG, Pruijssers AJ, Brown JJ, Stencel-Baerenwald JE, Sutherland DM, Iskarpatyoti JA, Dermody TS. 2018. Age-dependent susceptibility to reovirus encephalitis in mice is influenced by maturation of the type-I interferon response. Pediatr Res 83:1057–1066. doi: 10.1038/pr.2018.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oberhaus SM, Smith RL, Clayton GH, Dermody TS, Tyler KL. 1997. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J Virol 71:2100–2106. doi: 10.1128/JVI.71.3.2100-2106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pruijssers AJ, Hengel H, Abel TW, Dermody TS. 2013. Apoptosis induction influences reovirus replication and virulence in newborn mice. J Virol 87:12980–12989. doi: 10.1128/JVI.01931-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goody RJ, Hoyt CC, Tyler KL. 2005. Reovirus infection of the CNS enhances iNOS expression in areas of virus-induced injury. Exp Neurol 195:379–390. doi: 10.1016/j.expneurol.2005.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan WY, Kohsaka S, Rezaie P. 2007. The origin and cell lineage of microglia: new concepts. Brain Res Rev 53:344–354. doi: 10.1016/j.brainresrev.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 11.Schittone SA, Dionne KR, Tyler KL, Clarke P. 2012. Activation of innate immune responses in the central nervous system during reovirus myelitis. J Virol 86:8107–8118. doi: 10.1128/JVI.00171-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goody RJ, Beckham JD, Rubtsova K, Tyler KL. 2007. JAK-STAT signaling pathways are activated in the brain following reovirus infection. J Neurovirol 13:373–383. doi: 10.1080/13550280701344983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem 276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 14.Dermody TS, Nibert ML, Bassel-Duby R, Fields BN. 1990. A sigma 1 region important for hemagglutination by serotype 3 reovirus strains. J Virol 64:5173–5176. doi: 10.1128/JVI.64.10.5173-5176.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubin DH, Wetzel JD, Williams WV, Cohen JA, Dworkin C, Dermody TS. 1992. Binding of type 3 reovirus by a domain of the sigma 1 protein important for hemagglutination leads to infection of murine erythroleukemia cells. J Clin Invest 90:2536–2542. doi: 10.1172/JCI116147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chappell JD, Gunn VL, Wetzel JD, Baer GS, Dermody TS. 1997. Mutations in type 3 reovirus that determine binding to sialic acid are contained in the fibrous tail domain of viral attachment protein sigma 1. J Virol 71:1834–1841. doi: 10.1128/JVI.71.3.1834-1841.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Connolly JL, Barton ES, Dermody TS. 2001. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J Virol 75:4029–4039. doi: 10.1128/JVI.75.9.4029-4039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reiss K, Stencel JE, Liu Y, Blaum BS, Reiter DM, Feizi T, Dermody TS, Stehle T. 2012. The GM2 glycan serves as a functional co-receptor for serotype 1 reovirus. PLoS Pathog 8:e1003078. doi: 10.1371/journal.ppat.1003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barton ES, Youree BE, Ebert DH, Forrest JC, Connolly JL, Valyi-Nagy T, Washington K, Wetzel JD, Dermody TS. 2003. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J Clin Invest 111:1823–1833. doi: 10.1172/JCI16303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stencel-Baerenwald J, Reiss K, Blaum BS, Colvin D, Li XN, Abel T, Boyd K, Stehle T, Dermody TS. 2015. Glycan engagement dictates hydrocephalus induction by serotype 1 reovirus. mBio 6:e02356. doi: 10.1128/mBio.02356-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sutherland DM, Aravamudhan P, Dietrich MH, Stehle T, Dermody TS. 2018. Reovirus neurotropism and virulence are dictated by sequences in the head domain of the viral attachment protein. J Virol 92:e00974-18. doi: 10.1128/JVI.00974-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varki A, Schauer R. 2009. Sialic acids. In Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME (ed), Essentials of glycobiology, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed] [Google Scholar]
- 23.Schauer R, Kamerling JP. 2018. Exploration of the sialic acid world. Adv Carbohydr Chem Biochem 75:1–213. doi: 10.1016/bs.accb.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Münster AK, Eckhardt M, Potvin B, Mühlenhoff M, Stanley P, Gerardy-Schahn R. 1998. Mammalian cytidine 5'-monophosphate N-acetylneuraminic acid synthetase: a nuclear protein with evolutionarily conserved structural motifs. Proc Natl Acad Sci U S A 95:9140–9145. doi: 10.1073/pnas.95.16.9140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hadley B, Litfin T, Day CJ, Haselhorst T, Zhou Y, Tiralongo J. 2019. Nucleotide sugar transporter SLC35 family structure and function. Comput Struct Biotechnol J 17:1123–1134. doi: 10.1016/j.csbj.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitajima K, Varki N, Sato C. 2015. Advanced technologies in sialic acid and sialoglycoconjugate analysis. Top Curr Chem 367:75–103. doi: 10.1007/128_2013_458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE. 2016. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bocchini V, Mazzolla R, Barluzzi R, Blasi E, Sick P, Kettenmann H. 1992. An immortalized cell line expresses properties of activated microglial cells. J Neurosci Res 31:616–621. doi: 10.1002/jnr.490310405. [DOI] [PubMed] [Google Scholar]
- 29.Orchard RC, Sullender ME, Dunlap BF, Balce DR, Doench JG, Virgin HW. 2018. Identification of antinorovirus genes in human cells using genome-wide CRISPR activation screening. J Virol 93:e01324-18. doi: 10.1128/JVI.01324-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawrence SM, Huddleston KA, Pitts LR, Nguyen N, Lee YC, Vann WF, Coleman TA, Betenbaugh MJ. 2000. Cloning and expression of the human N-acetylneuraminic acid phosphate synthase gene with 2-keto-3-deoxy-D-glycero- D-galacto-nononic acid biosynthetic ability. J Biol Chem 275:17869–17877. doi: 10.1074/jbc.M000217200. [DOI] [PubMed] [Google Scholar]
- 31.Kauskot A, Pascreau T, Adam F, Bruneel A, Reperant C, Lourenco-Rodrigues MD, Rosa JP, Petermann R, Maurey H, Auditeau C, Lasne D, Denis CV, Bryckaert M, de Lonlay P, Lavenu-Bombled C, Melki J, Borgel D. 2018. A mutation in the gene coding for the sialic acid transporter SLC35A1 is required for platelet life span but not proplatelet formation. Haematologica 103:e613–e617. doi: 10.3324/haematol.2018.198028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abeln M, Borst KM, Cajic S, Thiesler H, Kats E, Albers I, Kuhn M, Kaever V, Rapp E, Munster-Kuhnel A, Weinhold B. 2017. Sialylation is dispensable for early murine embryonic development in vitro. Chembiochem 18:1305–1316. doi: 10.1002/cbic.201700083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwarz RE, Wojciechowicz DC, Picon AI, Schwarz MA, Paty PB. 1999. Wheatgerm agglutinin-mediated toxicity in pancreatic cancer cells. Br J Cancer 80:1754–1762. doi: 10.1038/sj.bjc.6690593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leung HS, Li OT, Chan RW, Chan MC, Nicholls JM, Poon LL. 2012. Entry of influenza A virus with a α2,6-linked sialic acid binding preference requires host fibronectin. J Virol 86:10704–10713. doi: 10.1128/JVI.01166-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willems AP, Sun L, Schulz MA, Tian W, Ashikov A, van Scherpenzeel M, Hermans E, Clausen H, Yang Z, Lefeber DJ. 2019. Activity of N-acylneuraminate-9-phosphatase (NANP) is not essential for de novo sialic acid biosynthesis. Biochim Biophys Acta Gen Subj 1863:1471–1479. doi: 10.1016/j.bbagen.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 36.Marx N, Grunwald-Gruber C, Bydlinski N, Dhiman H, Ngoc Nguyen L, Klanert G, Borth N. 2018. CRISPR-based targeted epigenetic editing enables gene expression modulation of the silenced beta-galactoside alpha-2,6-sialyltransferase 1 in CHO cells. Biotechnol J 13:e1700217. doi: 10.1002/biot.201700217. [DOI] [PubMed] [Google Scholar]
- 37.Han J, Perez JT, Chen C, Li Y, Benitez A, Kandasamy M, Lee Y, Andrade J, tenOever B, Manicassamy B. 2018. Genome-wide CRISPR/Cas9 screen identifies host factors essential for influenza virus replication. Cell Rep 23:596–607. doi: 10.1016/j.celrep.2018.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Connolly JL, Rodgers SE, Clarke P, Ballard DW, Kerr LD, Tyler KL, Dermody TS. 2000. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. J Virol 74:2981–2989. doi: 10.1128/jvi.74.7.2981-2989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, Peterson PK. 2004. Role of microglia in central nervous system infections. Clin Microbiol Rev 17:942–964. doi: 10.1128/CMR.17.4.942-964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richardson-Burns SM, Tyler KL. 2005. Minocycline delays disease onset and mortality in reovirus encephalitis. Exp Neurol 192:331–339. doi: 10.1016/j.expneurol.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 41.Dermody TS, Parker JS, Sherry B. 2013. Orthoreoviruses, p 1304–1346. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 42.Frierson JM, Pruijssers AJ, Konopka JL, Reiter DM, Abel TW, Stehle T, Dermod TS. 2012. Utilization of sialylated glycans as coreceptors enhances the neurovirulence of serotype 3 reovirus. J Virol 86:13164–13173. doi: 10.1128/JVI.01822-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol 62:246–256. doi: 10.1128/JVI.62.1.246-256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Virgin HW, Bassel-Duby R, Fields BN, Tyler KL. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J Virol 62:4594–4604. doi: 10.1128/JVI.62.12.4594-4604.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mainou BA, Dermody TS. 2012. Transport to late endosomes is required for efficient reovirus infection. J Virol 86:8346–8358. doi: 10.1128/JVI.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.