

Abstract
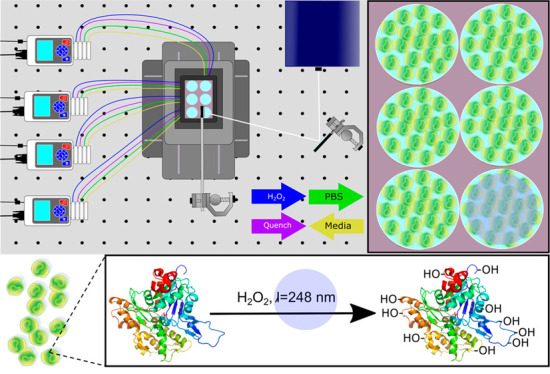
Fast photochemical oxidation of proteins (FPOP) is a protein footprinting technique that is being increasingly used in MS-based proteomics. FPOP is utilized to study protein–protein interactions, protein–ligand interactions, and protein conformational dynamics. This method has recently been extended to protein labeling in live cells (IC-FPOP), allowing the study of protein conformations in the complex cellular environment. Traditionally, IC-FPOP has been executed using a single cell flow system, in which hydrodynamic focusing drives cells along in a single file line, keeping the cells from clumping and thus ensuring equal exposure to the laser irradiation required for photochemical oxidation. Here, we introduce a novel platform that allows IC-FPOP to occur in a sterile incubation system complete with a mobile stage for XY movement, peristaltic pumps equipped with perfusion lines for chemical transport, and mirrors for laser beam guidance. This new system, called Platform Incubator with movable XY stage (PIXY), also utilizes software enabling automated communication between equipment and execution of the entire system. Further, comparison with a standard IC-FPOP flow system results reveal that this platform can successfully be used in lieu of the flow system while also decreasing the time to complete analysis of a single sample.
Cellular processes by nature are temporally dynamic, can respond to environmental perturbations, and occur based on the biological needs of the cell. The varied time scales of these processes ensure a cellular steady state.1 Some processes such as gene splicing occur relatively slowly on the seconds to mintues time scale,2 while from a protein perspective, time-dependent processes include protein folding and signaling, which occur on the millisecond to seconds time scale.3 Optimized methods for studying biomolecules in cells must be able to detect changes that occur over a wide range of time scales.
In recent years, mass spectrometry (MS)-based methods have been used to study protein interactions and conformations.4 In particular, protein footprinting methods have become integral constituents of the MS-based structural biology toolbox. A wide variety of chemical probes have been used to study protein–protein and protein–ligand interactions as well as conformational changes.5−9 These methods probe the solvent accessibility of proteins and hydrogen bonding in the case of hydrogen–deuterium exchange, which changes upon alterations in protein structure. Based on the chemistry of the covalent labeling technique, proteins are modified on various time scales from nanoseconds to minutes. Specific labeling methods such as glycine ethyl ester (GEE)10−12 and DEPC13 are relatively slow and modify proteins on the minutes time scale. In contrast, hydroxyl radical protein footprinting (HRPF) methods can modify proteins on the subsecond time scale. The specific time scale of labeling is dependent on the method used to generate hydroxyl (OH) radicals for labeling. Radical generation via synchrotron radiolysis of water leads to protein modification in milliseconds.14 However, laser-based HRPF methods modify proteins in the nanosecond to microsecond time regime.15,16 One of these methods, fast photochemical oxidation of proteins (FPOP), utilizes a KrF pulsed laser at 248 nm to oxidatively modify proteins on the microsecond time scale.17 FPOP has the ability to report protein transient dynamics and aid in epitope mapping.18−21 Owing to the rapid time scale of labeling, FPOP is especially suited to study fast biological processes such as protein folding. FPOP-based folding experiments include the oxidative modification of proteins at different time points after a temperature jump22,23 or pH-induced unfolding24,25 to determine protein folding intermediates. In both cases, FPOP is performed at various time points to investigate folding intermediates.
FPOP has recently been extended for modification of proteins in live cells.26 The development of in-cell FPOP (IC-FPOP) opens the door to studying signaling cascades, protein folding, and other fast biological processes in their native cellular environment. Imperative to the success of IC-FPOP was the development of a single cell flow system. This allowed equal exposure of individual cells to laser irradiation and led to a 13-fold increase in the number of oxidatively modified proteins.27 A disadvantage of this flow system is that it takes 10 min to oxidatively modify a single sample of cells and then another 10 min to wash the system before another sample can be analyzed. This 20 min time frame precludes using IC-FPOP for the study of transient species such as short-lived intermediates in protein folding or interaction network changes that occur in signaling. This shortcoming motivated the design of a new higher throughput IC-FPOP platform.
For IC-FPOP to be high throughput, it is essential that cell culture be performed directly at the location of laser irradiation. This requires an enclosed, sterile environment where temperature, humidity, and CO2 infusion can be controlled for cell growth and health. Here, we report a new platform for IC-FPOP that meets these requirements and is higher throughput than the flow system. This new platform, entitled Platform Incubator with XY movement (PIXY), utilizes a stage-top incubator so that cell culture can be performed directly on the optical bench. IC-FPOP is performed on adherent cells in six-well plates housed within the incubator. Laser irradiation is reflected downward onto an individual well, and a positioning stage moves the incubator so that each well is individually irradiated.
We demonstrate that IC-FPOP can be performed in this new system on a shorter time frame than the flow system. IC-FPOP in the PIXY system also leads to higher modification coverage per protein. The development of this new FPOP platform will increase both the applicability of the method and the information that can be obtained from cellular experiments.
Experimental Section
Platform Incubator with Movable XY Platform (PIXY)
The stage-top incubator was temporarily relocated to a standard cell culture hood to maintain sterile environment. A six-well plate seeded with human embryonic kidney (HEK293T) cells was placed in the stage-top incubator. In the culture hood, tygon tubing (1.59 mm ID) was fed through the custom ports around the incubator. Six 33 mm PLA filament rings were placed in each well to make the tubing flush with the walls of the well, keeping the tubing out of the path of the laser beam. The assembled incubator was then returned to the laser platform and secured to the positioning system. HEK cells were grown to 80% confluency in the stage-top incubator in 3 mL of DMEM supplemented with FBS and streptomycin–penicillin. Peristaltic pump tubing (3.18 mm ID) was connected via connectors to 1.59 mm ID tubing. Cell culture media was then removed via peristaltic pumps and replaced with 2 mL of sterile DPBS. Each well has four tubing connections to accommodate the following: withdrawal of media, infusion of DPBS, infusion of H2O2, and infusion of quench solutions. The flow rate used for experimentation is 35 mL/min, which ensures sufficient dispersion and mixing of reagents. Using the LabVIEW software (National Instruments), a command script was designed that enabled the pumps to infuse reagents in series. The laser pulse and stage movement were performed manually.
Results and Discussion
Design, Assembly, and Optimization
The complete PIXY system consists of a stage-top incubator, movable stage, and peristaltic pumps (Figure 1). The custom designed stage-top incubator from Okolabs allows for cell culture to be performed directly at the laser platform. The completely enclosed system has temperature and humidity control and the ability to perfuse carbon dioxide for proper cell growth. The top of the incubator was designed with a quartz top instead of the standard glass to make it compatible with the 248 nm excimer laser used for FPOP. The system has a total of 36 ports available to accommodate lines for perfusion of H2O2 and quench reagents as well as other reagents required for specific experiments such as drug treatment for target engagement studies. The incubator is mounted onto a custom piezo stage from Mad City Labs that moves in both the X and Y dimensions. Four peristaltic pumps with four individual channels that both infuse and withdraw solutions are included in the PIXY platform. In order to reflect the laser irradiation downward onto the incubator, a two-mirror configuration similar to a previously published study by Riaz et al.28 was utilized. The first mirror was set at a 90° angle from the laser aperture to capture the laser beam (19 × 7 mm), and the second 50 mm mirror was placed at approximately 45° to the first mirror angle downward in the direction of the stage-top incubator. To irradiate an entire 35 mm diameter well with a single laser pulse, the mirrors were used to increase the laser beam diameter to approximately 30 mm, so an entire well can be irradiated with a single shot of the laser pulse. A minimal energy loss, <2%, is observed with this two-mirror configuration. The two-mirror configuration provides the ability to irradiate an entire well in one shot while not compromising the efficacy of IC-FPOP.
Figure 1.
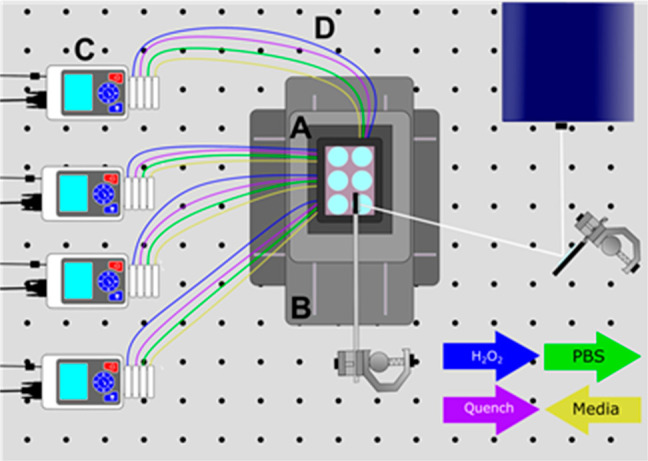
Schematic of the PIXY system. The system consists of a stage-top incubator (A), XY movable stage (B), and four peristaltic pumps (C) equipped with perfusion (D) lines for chemical transfer. Cell culture media is withdrawn from each well. DPBS, hydrogen peroxide, and quench solutions are infused into each well as indicated by the colored arrows. White lines indicate the laser path for irradiation.
The configuration of the new static platform required a systematic approach for testing. As a first step in evaluating the system, we tested cell culture conditions in the stage-top incubator on the optical bench. For IC-FPOP, it is important that cells are incubated at optimal conditions so that normal cellular functions are maintained. To ensure the stage-top incubator conditions were amenable for cell culture, we used transient transfection in order to express the fluorescent protein GCaMP2. GCaMP2, a chimera of green fluorescent protein (GFP) and calmodulin, is used as an intracellular calcium indicator. HEK cells were transfected with a pET28-GFP plasmid containing the GCaMP2 gene, and the HEK cells were incubated in our standard cell culture conditions both in standard CO2 incubators and in PIXY. Fluorescent imaging of the cells was used to inform GCaMP2 expression (Figure S1). Imaging of GCaMP2 fluorescence 2 days post-transfection indicates higher fluorescence intensity in the cells incubated in PIXY (Figure 2A). For a more quantitative comparison, a luciferase assay was performed to measure transfection efficiency. Results show PIXY outperformed the standard incubator with a higher level of transfected protein expression levels (Figure 2B). These results demonstrate that cell function is preserved in the PIXY system, and cell culture can be performed directly on the optical bench.
Figure 2.
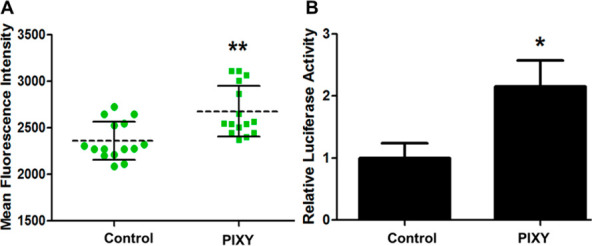
HEK cell transfection efficiency. (A) Mean fluorescent intensity of GCaMP2 transfected cells grown in the standard incubator (control) and stage-top incubator (PIXY). Each dot and square (green) represents a single measurement acquired in a well. (B) Quantitation and validation of the transfection efficiency using a luciferase assay. HEK cells were transfected with pRL-TK plasmid. P-value < 0.005.
FPOP in the PIXY Platform
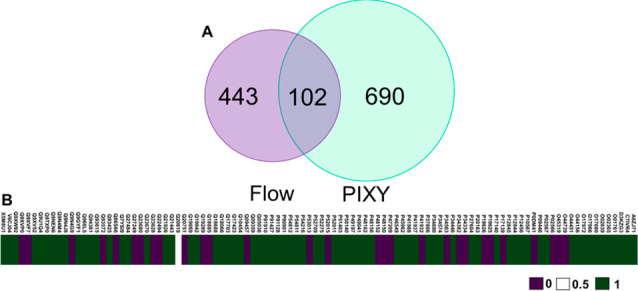
The next step in testing the system was to evaluate its suitability for IC-FPOP. For IC-FPOP in PIXY, HEK cells were seeded in a six-well plate and placed in the stage-top incubator all within a sterile cell culture hood. Cells were grown at the optical bench to ∼80% confluency (24 h growth) prior to IC-FPOP. Because cell culture media contain several components that can scavenge OH radicals, the media is perfused out of an individual well and replaced with 2 mL of DPBS prior to H2O2 infusion. Using a LabVIEW custom designed automated command script builder, H2O2 and quench solutions are infused using individual channels (four channels per pump) on one of the peristaltic pumps at specific time points before (H2O2) and after (quench) a manually initiated laser pulse. The HEK cells were incubated in the H2O2 for 10 s followed by a single laser pulse (50 Hz, 160 mJ) and then the addition of quench containing solution. The total time for IC-FPOP of a single sample in PIXY is 20 s, which is in stark contrast to the 20 min per samples required for the flow system. In total, 728 proteins were oxidatively modified in PIXY across two biological replicates, demonstrating IC-FPOP can be performed in this system (Figure 3A and Tables S3 and S4). A comparison of IC-FPOP in PIXY and in the flow system reveals a 1.5-fold increase in the number of oxidatively modified proteins in PIXY in comparison to the flow system in which 486 proteins were modified (Figure 3A and Tables S3 and S4). There is little overlap in the modified proteins that are identified using the two methods. These discrepancies are most likely attributed to protein expression abundance differences between the cells in the two experiments. Previous studies have demonstrated that expression abundance differences play a role in variations in modified proteins.29 The GCaMP2 transfection results establish that culturing cells in PIXY does lead to differences in protein expression compared to the standard CO2 incubator.
Figure 3.
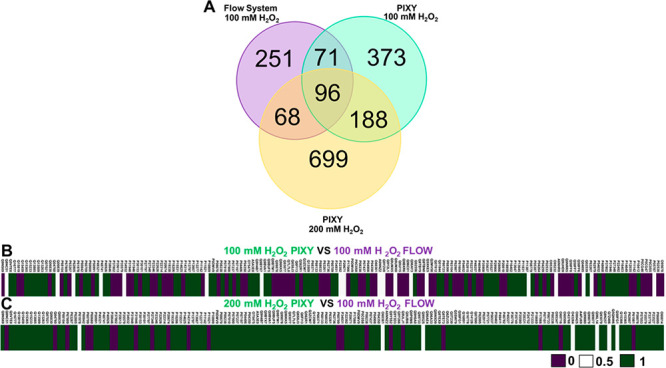
Comparison of proteins modified in the single cell flow system and PIXY. (A) Venn diagram of proteins modified using 100 mM H2O2 in the flow system (purple), 100 mM H2O2 in PIXY(green), and 200 mM H2O2 in PIXY (yellow). (B) Comparison of the percent FPOP residue-level coverage for 100 mM H2O2 PIXY vs 100 mM H2O2 flow and (C) 200 mM H2O2 PIXY vs 100 mM H2O2 flow. For percent FPOP coverage that is larger in PIXY, the corresponding heat map values were set to 1 (green). For percent FPOP coverage that is smaller in PIXY, the corresponding heat map values were set to 0 (purple). If PIXY and flow have the same FPOP coverage, the value was set to 0.5 (white). Proteins are indicated using UniProt IDs.
Just as important as the overall number of proteins modified by IC-FPOP is the number of modifications per protein. More structural information can be gained if higher FPOP coverage across a protein is achieved. We compared FPOP modification coverage of the 167 proteins that were modified using both the flow system and PIXY. Modification coverage heatmaps indicate that proteins labeled with the flow system had a higher number of modified amino acid residues compared with PIXY (Figure 3B). Of the 167 proteins, 95 of them had a higher number of modifications with the flow system, while 72 proteins were more highly modified with PIXY. Interestingly, the average numbers of modifications for both methods were similar, with 10.9 and 11.1 modifications per protein for the flow system and PIXY, respectively (Table S1). Since viability studies indicated that >90% of HEK cells are viable in peroxide concentrations ranging from 20 to 200 mM in the time frame of a PIXY experiment (Figure S2), we tested higher concentrations of H2O2 to increase the modification coverage. IC-FPOP with 200 mM H2O2 with PIXY resulted in the oxidative modification of 1051 proteins 1.4- and 2.2-fold increases compared to 100 mM H2O2 with PIXY and the flow system, respectively. A comparison between the 164 proteins modified with 100 mM H2O2 in the flow system and with 200 mM H2O2 in PIXY indicates 133 proteins have a higher number of oxidatively modified residues per protein in PIXY (Tables S3–S5). The average number of modifications per protein in PIXY with 200 mM H2O2 is 41 (Table S1). Owing to a large variation in the number of amino acids per protein, the standard deviation for these values is greater than the average (Table S7). However, this does not negate the validity of the results. Since the goal is to optimize the conditions for the PIXY system, we did not test 200 mM H2O2 with the flow system, which has already been benchmarked with 100 mM H2O2.
Localization of IC-FPOP Modifications
To further demonstrate the advantage of higher modification coverage across a protein, we performed peptide-level quantitation of the extent of FPOP modification for the protein actin. Actin, the free monomer in the family of actin proteins, contains 375 amino acids. In the case of 100 mM H2O2 with the flow system, only two peptides were modified, providing limited structural information (Figure 4A). In contrast, five peptides spanning the actin sequence were modified in the 200 mM H2O2 PIXY samples. There was a single peptide, 316–326, that was modified in both systems. Tandem MS spectra indicate that a single amino acid (Pro322) was modified for both experiments (Figure 4B). Since the same residue is modified, the extent of modification can be compared without normalizing for changes in OH radical reactivity. There was not a statistically significant difference in the extent of oxidation on this peptide (P-value = 0.060). This demonstrates the additional H2O2 does not alter the levels of oxidation but rather modifies additional residues. The 5 peptides modified in the PIXY samples contain 12 modified amino acids, while only 4 amino acids were modified with the flow system (Figure 4C). The increase in modification coverage is further demonstrated with the protein tubulin, where 67 residues were modified with PIXY, while only 15 were modified using the standard IC-FPOP conditions with the flow system (Figure S3).
Figure 4.

Localization of IC-FPOP modifications. (A) Oxidatively modified peptides within G-actin from the flow system (purple) vs PIXY (green). (B) Tandem MS spectra of modified and unmodified G-actin peptide 316–326 showing b- and y-ions and a +16 FPOP modification on proline in both systems. The MS spectrum of the unmodified peptide is shown at the bottom. (C) FPOP modified residues of G-actin (PDB: 6ZXJ, chain A) are represented in stick representation: 11 modified residues in PIXY (green), 3 modified residues in flow (purple), 1 overlapping modified residue (yellow).
Owing to actin having both a monomeric and polymeric form, we cannot rule out the possibility that the low number of modifications with the flow system are due to the polymerized, more protected, form (F-actin) being the dominant species in these samples. We calculated the solvent accessible surface area (SASA) (Table S2) of the F-actin polymerized structure and found residues M190, P322, M305, and K326 to all be solvent accessible. While all four of these residues are modified with PIXY, only P322 is modified with the flow system. Interestingly, the highly reactive methionine residues are not modified with the flow system, even though they are solvent accessible. In both cases, we do see residues with low accessibility modified, which could be a result of the difference in solution and static crystal structure conditions. It also could be due to the effects of molecular crowding within the cell that are not considered in the crystal structure. There are two peptides 119–147 and 148–177 that are not solvent accessible in the F-actin structure. This raises the question as to whether the increased peroxide leads to overoxidation and protein unfolding. These residues could be accessible in monomeric actin, but this cannot be verified, because there is not a structure available for the monomer. However, unfolding is unlikely owing to the large number of macromolecules present in the cell that scavenge the radical.
IV-FPOP in the PIXY Platform
Recently, we demonstrated that FPOP can be performed in C. elegans, an animal model for human disease.29 This in vivo FPOP (IV-FPOP) method required the design of a new flow system to accommodate the size of the worms and their ability to curl their tails during locomotion. These properties led to reduced recovery of worms even at higher inner diameters of the flow tube, where only 63–89% of worms were recovered after FPOP.29 The PIXY system may be better suited for IV-FPOP, because the size of an individual well in a six-well plate can better accommodate the worms. For IV-FPOP in PIXY, worms were incubated in the stage-top incubator at 20 °C, an optimal temperature for worm growth. Approximately 10 000 worms were placed in each well and used for each flow system sample. LC–MS/MS analysis reveals that 792 proteins were modified by IV-FPOP in PIXY, a 1.5-fold increase over the 545 proteins modified with the flow system (Figure 5A and Table S6). In addition, 72 of the 102 common modified proteins showed higher modification coverage in PIXY (Figure 5B). IV-FPOP in PIXY had an average of 14.5 modifications per protein, while the flow system had 13.1 modifications per protein (Table S1). These results demonstrate the potential of the PIXY platform for in vivo experiments.
Figure 5.
(A) Comparison of FPOP modified proteins in C. elegans by flow vs PIXY. There is a .45-fold increase in oxidatively modified proteins using PIXY when compared to the flow system. (B) Comparison of the percent FPOP residue-level coverage between PIXY and flow. For percent FPOP coverage that is larger in PIXY, the corresponding heat map values were set to 1 (green). For percent FPOP coverage that is smaller in PIXY, the corresponding heat map values were set to 0 (purple). If PIXY and flow have the same FPOP coverage, the value was set to 0.5 (white). Flow IV-FPOP data was adapted from ref (24).
Conclusions
We describe a new platform for in-cell FPOP that eliminates the need for flow. The static PIXY platform permits the growth of cells directly on the optical bench. The higher transfection efficiency of GCaMP2 in PIXY could be related to the small footprint of the stage-top incubator leading to a better distribution of CO2. PIXY also provides the ability to perform FPOP on a high throughput scale. The single pulse of irradiation for an entire sample well reduces the time frame of FPOP by 60-fold with only 20 s of analysis time. This time scale puts IC-FPOP in the regime required to study cellular processes such as protein folding and signaling cascades in a time-dependent manner. With complete automation of the system, including laser pulse initiation and movement of the well plate, this time could be further decreased. PIXY is also versatile with the ability to be used for both in-cell and in vivo FPOP. The use of well plates overcomes the difficulties with flowing C. elegans and allows for complete sample recovery. This well plate format may also increase the applicability of FPOP to study other nontraditional systems such as spheroids, tissues, and organoids.
Acknowledgments
This work was supported by a grant from the NIH R01 GM128983-01.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b04933.
Additional experimental details and supporting figures showcasing HEK cell viability, residue modifications, and tables of protein modifications obtained in each system (PDF)
Author Contributions
L.M.J. and A.G. devised the project, D.T.J., B.P.-S., and J.A.P performed the experiments, and L.M.J. and D.T.J. wrote the manuscript.
The authors declare no competing financial interest.
Notes
The research presented herein is associated with the below referenced patent application that was published on October 3, 2019 under publication number: WO 2019/191499; PCT Patent Application Number: PCT/US2019/024691; Title: Device and Method for Determining In-Cell Protein Folding; UMB Docket Number: LJ-2018-104; UMass Amherst ref. No.: UMA 18-059.
Supplementary Material
References
- Periyasamy S.; Gray A.; Kille P. The bottom-up approach to defining life: deciphering the functional organization of biological cells via multi-objective representation of biological complexity from molecules to cells. Front. Physiol. 2013, 4, 369. 10.3389/fphys.2013.00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmo-Fonseca M.; Kirchhausen T. The timing of pre-mRNA splicing visualized in real-time. Nucleus 2014, 5 (1), 11–4. 10.4161/nucl.28056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamir M.; Bar-On Y.; Phillips R.; Milo R. SnapShot: Timescales in Cell Biology. Cell 2016, 164 (6), 1302e1. 10.1016/j.cell.2016.02.058. [DOI] [PubMed] [Google Scholar]
- Kaur U.; Johnson D. T.; Chea E. E.; Deredge D. J.; Espino J. A.; Jones L. M. Evolution of Structural Biology through the Lens of Mass Spectrometry. Anal. Chem. 2019, 91 (1), 142–155. 10.1021/acs.analchem.8b05014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limpikirati P.; Liu T.; Vachet R. W. Covalent labeling-mass spectrometry with non-specific reagents for studying protein structure and interactions. Methods 2018, 144, 79–93. 10.1016/j.ymeth.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Serra A.; Guo T.; Park J. E.; Zhong Q.; Sze S. K. Application of Nanosecond Laser Photolysis Protein Footprinting to Study EGFR Activation by EGF in Cells. J. Proteome Res. 2017, 16 (6), 2282–2293. 10.1021/acs.jproteome.7b00154. [DOI] [PubMed] [Google Scholar]
- Li J.; Wei H.; Krystek S. R. Jr.; Bond D.; Brender T. M.; Cohen D.; Feiner J.; Hamacher N.; Harshman J.; Huang R. Y.; Julien S. H.; Lin Z.; Moore K.; Mueller L.; Noriega C.; Sejwal P.; Sheppard P.; Stevens B.; Chen G.; Tymiak A. A.; Gross M. L.; Schneeweis L. A. Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Anal. Chem. 2017, 89 (4), 2250–2258. 10.1021/acs.analchem.6b03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Bavro V. N.; D’Mello R.; Tucker S. J.; Venien-Bryan C.; Chance M. R. Conformational changes during the gating of a potassium channel revealed by structural mass spectrometry. Structure 2010, 18 (7), 839–46. 10.1016/j.str.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehmood S.; Domene C.; Forest E.; Jault J. M. Dynamics of a bacterial multidrug ABC transporter in the inward- and outward-facing conformations. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (27), 10832–6. 10.1073/pnas.1204067109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J.; Zhang H.; Gross M. L.; Blankenship R. E. Membrane orientation of the FMO antenna protein from Chlorobaculum tepidum as determined by mass spectrometry-based footprinting. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (15), 6134–9. 10.1073/pnas.0901691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Wen J.; Huang R. Y.; Blankenship R. E.; Gross M. L. Mass spectrometry-based carboxyl footprinting of proteins: method evaluation. Int. J. Mass Spectrom. 2012, 312, 78–86. 10.1016/j.ijms.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L. Y.; Salas-Solano O.; Valliere-Douglass J. F. Localized conformational interrogation of antibody and antibody-drug conjugates by site-specific carboxyl group footprinting. MAbs 2017, 9 (2), 307–318. 10.1080/19420862.2016.1268306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limpikirati P.; Pan X.; Vachet R. W. Covalent Labeling with Diethylpyrocarbonate: Sensitive to the Residue Microenvironment, Providing Improved Analysis of Protein Higher Order Structure by Mass Spectrometry. Anal. Chem. 2019, 91 (13), 8516–8523. 10.1021/acs.analchem.9b01732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselar J. G.; Maleknia S. D.; Sullivan M.; Downard K. M.; Chance M. R. Hydroxyl radical probe of protein surfaces using synchrotron X-ray radiolysis and mass spectrometry. Int. J. Radiat. Biol. 2002, 78 (2), 101–14. 10.1080/09553000110094805. [DOI] [PubMed] [Google Scholar]
- Aye T. T.; Low T. Y.; Sze S. K. Nanosecond laser-induced photochemical oxidation method for protein surface mapping with mass spectrometry. Anal. Chem. 2005, 77 (18), 5814–22. 10.1021/ac050353m. [DOI] [PubMed] [Google Scholar]
- Gau B. C.; Sharp J. S.; Rempel D. L.; Gross M. L. Fast Photochemical Oxidation of Protein Footprints Faster than Protein Unfolding. Anal. Chem. 2009, 81 (16), 6563–6571. 10.1021/ac901054w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambly D. M.; Gross M. L. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 2005, 16 (12), 2057–63. 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Li J.; Chen G. The use of fast photochemical oxidation of proteins coupled with mass spectrometry in protein therapeutics discovery and development. Drug Discovery Today 2019, 24 (3), 829–834. 10.1016/j.drudis.2018.12.008. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Chen G.; Wei H.; Huang R. Y.; Mo J.; Rempel D. L.; Tymiak A. A.; Gross M. L. Fast photochemical oxidation of proteins (FPOP) maps the epitope of EGFR binding to adnectin. J. Am. Soc. Mass Spectrom. 2014, 25 (12), 2084–92. 10.1007/s13361-014-0993-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K. S.; Chen G.; Mo J.; Huang R. Y.; Deyanova E. G.; Beno B. R.; O’Neil S. R.; Tymiak A. A.; Gross M. L. Orthogonal Mass Spectrometry-Based Footprinting for Epitope Mapping and Structural Characterization: The IL-6 Receptor upon Binding of Protein Therapeutics. Anal. Chem. 2017, 89 (14), 7742–7749. 10.1021/acs.analchem.7b01748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D. T.; Di Stefano L. H.; Jones L. M. Fast photochemical oxidation of proteins(FPOP): A powerful mass spectrometry based structural proteomics tool. J. Biol. Chem. 2019, 294, 11969. 10.1074/jbc.REV119.006218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Rempel D. L.; Gross M. L. Temperature jump and fast photochemical oxidation probe submillisecond protein folding. J. Am. Chem. Soc. 2010, 132 (44), 15502–4. 10.1021/ja106518d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Rempel D. L.; Gau B. C.; Gross M. L. Fast photochemical oxidation of proteins and mass spectrometry follow submillisecond protein folding at the amino-acid level. J. Am. Chem. Soc. 2012, 134 (45), 18724–31. 10.1021/ja307606f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocks B. B.; Konermann L. Time-dependent changes in side-chain solvent accessibility during cytochrome c folding probed by pulsed oxidative labeling and mass spectrometry. J. Mol. Biol. 2010, 398 (2), 362–73. 10.1016/j.jmb.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Stocks B. B.; Sarkar A.; Wintrode P. L.; Konermann L. Early hydrophobic collapse of alpha(1)-antitrypsin facilitates formation of a metastable state: insights from oxidative labeling and mass spectrometry. J. Mol. Biol. 2012, 423 (5), 789–99. 10.1016/j.jmb.2012.08.019. [DOI] [PubMed] [Google Scholar]
- Espino J. A.; Mali V. S.; Jones L. M. In Cell Footprinting Coupled with Mass Spectrometry for the Structural Analysis of Proteins in Live Cells. Anal. Chem. 2015, 87 (15), 7971–8. 10.1021/acs.analchem.5b01888. [DOI] [PubMed] [Google Scholar]
- Rinas A.; Mali V. S.; Espino J. A.; Jones L. M. Development of a Microflow System for In-Cell Footprinting Coupled with Mass Spectrometry. Anal. Chem. 2016, 88 (20), 10052–10058. 10.1021/acs.analchem.6b02357. [DOI] [PubMed] [Google Scholar]
- Riaz M.; Misra S. K.; Sharp J. S. Towards high-throughput fast photochemical oxidation of proteins: Quantifying exposure in high fluence microtiter plate photolysis. Anal. Biochem. 2018, 561–562, 32–36. 10.1016/j.ab.2018.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espino J. A.; Jones L. M. Illuminating Biological Interactions with in Vivo Protein Footprinting. Anal. Chem. 2019, 91 (10), 6577–6584. 10.1021/acs.analchem.9b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.