

Abstract

Enzymatically degradable polymeric micelles have great potential as drug delivery systems, allowing the selective release of their active cargo at the site of disease. Furthermore, enzymatic degradation of the polymeric nanocarriers facilitates clearance of the delivery system after it has completed its task. While extensive research is dedicated toward the design and study of the enzymatically degradable hydrophobic block, there is limited understanding on how the hydrophilic shell of the micelle can affect the properties of such enzymatically degradable micelles. In this work, we report a systematic head-to-head comparison of well-defined polymeric micelles with different polymeric shells and two types of enzymatically degradable hydrophobic cores. To carry out this direct comparison, we developed a highly modular approach for preparing clickable, spectrally active enzyme-responsive dendrons with adjustable degree of hydrophobicity. The dendrons were linked with three different widely used hydrophilic polymers—poly(ethylene glycol), poly(2-ethyl-2-oxazoline), and poly(acrylic acid) using the CuAAC click reaction. The high modularity and molecular precision of the synthetic methodology enabled us to easily prepare well-defined amphiphiles that differ either in their hydrophilic block composition or in their hydrophobic dendron. The micelles of the different amphiphiles were thoroughly characterized and their sizes, critical micelle concentrations, drug loading, stability, and cell internalization were compared. We found that the micelle diameter was almost solely dependent on the hydrophobicity of the dendritic hydrophobic block, whereas the enzymatic degradation rate was strongly dependent on the composition of both blocks. Drug encapsulation capacity was very sensitive to the type of the hydrophilic block, indicating that, in addition to the hydrophobic core, the micellar shell also has a significant role in drug encapsulation. Incubation of the spectrally active micelles in the presence of cells showed that the hydrophilic shell significantly affects the micellar stability, localization, cell internalization kinetics, and the cargo release mechanism. Overall, the high molecular precision and the ability of these amphiphiles to report their disassembly, even in complex biological media, allowed us to directly compare the different types of micelles, providing striking insights into how the composition of the micelle shells and cores can affect their properties and potential to serve as nanocarriers.
Introduction
Polymeric nano-assemblies, amongst them polymeric micelles, have shown great potential as drug delivery systems (DDS) as well as in many other biomedical applications.1−3 This is due to the ability to dramatically increase the very low water solubility of lipophilic drug molecules by encapsulating them inside the hydrophobic cavities of the assemblies, simultaneously shielding them from the hostile biological environment. In addition, in many cases, these assemblies have sizes that allow passive accumulation in cancerous or inflamed tissues due to the enhanced permeability and retention (EPR) effect.4,5 Despite all advantages proven in numerous scientific reports, there are still challenges to overcome to increase the translation of polymeric DDS from academic research into the clinic.6 Hence, it is essential to further conduct fundamental research in this important area in order to gain deeper understanding of the parameters that govern the stability and functionality of such carriers and open the way for their broader application in biomedicine.
It is clear that DDS should be, on one hand, extremely stable to withstand the high dilution and interactions with blood components in order to allow their circulation in the body while maintaining their cargo of active drug molecules.7 On the other hand, the carriers should be able to release the drugs when the DDS has reached the target site.8−11 To address this need, over the last three decades, there has been a great interest in utilizing stimuli-responsive polymeric micelles as DDS to allow selective release of their therapeutic cargo.12,13 There are many reported examples of polymeric micelles that disassemble due to changes in pH,14−17 temperature,18−22 or redox potential,23−26 while there are significantly fewer examples of polymeric nanocarriers that can disassemble due to the presence of a designated enzyme.27−30 Enzymes are very appealing for triggering the disassembly of drug containing micelles since they are already present in the body, known for their high substrate specificity and in many cases specific enzymes are overexpressed in diseased tissues.31−33 Polymeric micelles are typically formed by the self-assembly of amphiphilic diblock copolymers so that the hydrophobic block forms the core and the hydrophilic block forms the micellar corona. It is clear that in the biological environment, most of the interactions between the micelle and its surroundings occur through the micelle’s corona.34,35 It is interesting to note that although most reported DDS are based on poly(ethylene glycol) (PEG),36,37 the use of additional types of promising hydrophilic polymers such as poly(2-oxazoline)s38−40 and polyacrylates41 has also been reported, inspired by the increasing human population that carries anti-PEG antibodies leading to an immune-response upon treatment with PEG-based therapeutics.42,43 To allow the rational design of DDS, it is critical to compare and study the behavior of different corona forming polymers in order to rationally select the most suited hydrophilic block.44 For a direct comparison between different hydrophilic polymers as micellar shells, it is essential that the hydrophobic core-forming block will be identical. Using dendrons as the hydrophobic core forming block provides many advantages compared to linear hydrophobic polymer blocks due to their well-defined structure and monodispersity45−48 and hence can be ideal for the purpose of the abovementioned comparison.
Here, we report the synthesis of clickable, fluorescently labeled enzyme-responsive dendrons, which can be reacted with a variety of azide-functionalized hydrophilic polymers. In this work, we studied and compared three types of commonly used hydrophilic polymers: PEG, poly(2-ethyl-2-oxazoline) (PEtOx), and poly(acrylic acid) (PAA). PEG and PEtOx, which are noncharged polymers, were selected as they are considered to be “stealth” polymers that minimize interactions with native proteins and other biomolecules and hence elongate circulation time of their conjugates in vivo.49−52 PAA is polyanionic at physiological pH and is much more hydrophilic than the other two polymers. Because of the comparative nature of this work, we decided to use polymers with similar molecular weights of approximately 5 kDa. Each type of polymer was clicked with two types of dendrons that differ in their hydrophobicity containing either four hexyl or four nonyl ester end-groups (Scheme 1). This allowed both head-to-head comparison between the three hydrophilic polymers and also to understand how altering the hydrophilic/hydrophobic ratio affects the micelles that are formed from each polymer.
Scheme 1. Synthesis of (A) Thiol Containing Enzyme-Cleavable Hydrophobic End-Group, (B) Clickable Fluorescently Labeled Enzyme-Responsive Hydrophobic Dendron, (C) Its Click Reaction with Terminal Azide-Functionalized Polymers to Yield Linear-Dendron Block Copolymer Amphiphiles, and (D) Types of Hydrophilic Polymers Compared in This Work.

Experimental Section
Instrumentation
HPLC: All measurements were recorded on a Waters Alliance e2695 separation module equipped with a Waters 2998 photodiode array detector. All solvents were purchased from Bio-Lab Chemicals and were used as received. All solvents are of high-performance liquid chromatography (HPLC) grade. 1H and13C NMR: spectra were recorded on Bruker AVANCE I and AVANCE III 400 MHz spectrometers, as indicated. Chemical shifts are reported in parts per million and referenced to the solvent. SEC: all measurements were recorded on Viscotek GPCmax by Malvern using the refractive index detector, and PEG standards (purchased from Sigma-Aldrich) were used for calibration. Absorbance and fluorescence spectra: measurements were recorded on a Tecan Infinite M200Pro device or Agilent Technologies Cary Eclipse Fluorescence Spectrometer. MALDI-TOF MS: analysis was conducted on a Bruker AutoFlex MALDI-TOF MS (Germany). An α-cyano-4-hydroxycinnamic acid matrix was used. High-resolution MS: analysis was conducted on Autospec HRMS (EI) Micromass (UK) or Synapt High Definition MS (ESI), Waters Inc. (USA). Dynamic light scattering (DLS): all measurements were recorded on a Corduran technology VASCOγ—particle size analyzer. Confocal microscopy: imaging was performed on a Zeiss LSM 800 confocal microscope, using 63× plan-apochromat oil immersion objective. Flow cytometry: analysis was performed using a spectral analyzer (Sony SA3800) flow cytometer, 96/384w.
Materials
Poly(ethylene glycol) methyl ether (5 kDa), allyl bromide (99%), 3-mercaptopropionic acid (98%), 2,2-dimethoxy-2-phenylacetophenone (99%), 4-(dimethylamino)pyridine (99%), ethyl α-bromoisobutyrate (98%), Fmoc-Lys(Boc)-OH (98%), N-hydroxysuccinimide (NHS, 99%), copper(I) bromide (CuBr, 98%), N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA, 99%), 3-bromopropionic acid (97%), bovine serum albumin (BSA), Triton X-100, porcine liver esterase (PLE), and Sephadex LH20 were purchased from Sigma-Aldrich. 1-Hexanol (98%) was purchased from Acros Organics. Propargyl bromide (80% in toluene), chlorotriphenylmethane (Trt–Cl, 98%), 4-nitrophenol (99%), triethylsilane (98%), N,N′-dicyclohexylcarbodiimide (99%), propargyl amine (98%), tert-butyl acrylate (tBA, 99%), and anhydrous K2CO3 (99%) were purchased from Alfa Aesar. 3,5-Dihydroxy benzoic acid was purchased from Apollo scientific. Cy5-NHS was purchased from Lumiprobe. Cystamine hydrochloride (98%), potassium hydroxide, N,N-diisopropylethylamine, 1-nonanol, and sodium azide (NaN3) were purchased from Merck. Silica gel (60 Å, 0.040–0.063 mm), sodium hydroxide, anhydrous Na2SO4 (granular, 10–60 mesh), piperidine (peptide synthesis), N,N-dimethylformamide (DMF, peptide synthesis), trifluoroacetic acid (HPLC grade), and all solvents were purchased from Bio-Lab and were used as received. Deuterated solvents for NMR were purchased from Cambridge Isotope Laboratories (CIL), Inc. PrestoBlue and Dulbecco’s modified Eagle medium (DMEM) with high glucose and pyruvate, fetal bovine serum, trypsin with 0.25% EDTA, and penicillin–streptomycin were purchased from Thermo Fisher. The dendrons and azide-functionalized hydrophilic polymers were synthesized, as detailed in the Supporting Information.
General Procedure for CuAAC Click Reaction between Polymer-N3 and Dendron
CuBr (2 equiv with respect to polymer–N3) was loaded in a 4 mL glass vial, which was sealed with a rubber septum. The vial was deoxygenated by three vacuum-nitrogen cycles and backfilled with nitrogen. In a separate 4 mL vial polymer–N3 (1 equiv), dendron (1.3 equiv) and PMDETA (2 equiv) were dissolved in DMF (100–200 mg polymer/mL) and purged with nitrogen for 2 min. This mixture was added into the CuBr containing vial using the nitrogen flushed syringe and needle. The vial was thoroughly vortexed until clear green solution was obtained (approximately 30 s). The reaction was stirred at room temperature for 1 h, filtered through syringe filter [0.44 μm, hydrophilic poly(tetrafluoroethylene)], purified using LH20 (Sephadex) size exclusion column, and eluted with MeOH. Fractions that contained the product (identified by bright yellow color) were unified, MeOH was evaporated to dryness, and the product was dried on high vacuum. All polymers were obtained as bright yellow solids.
CMC
Preparation of Diluent
Nile Red stock solution (0.88 mg/mL in ethanol) was diluted into a phosphate buffer (PB) (100 mM, pH 7.4) to afford a final concentration of 1.25 μM.
Preparation and Measurement of Samples
The polymer–dendron amphiphile was directly dissolved in the diluent to give a final concentration of 250 μM. Solution was vortexed vigorously until the amphiphile completely dissolved and further sonicated for 15 min in an ultrasonic bath. This solution was consecutively diluted by a factor of 1.5 with the diluent to afford a series of 24 samples. 150 μL of each sample were loaded onto a 96 well plate, and a fluorescence emission scan was performed for each well. In order to determine the amphiphile’s critical micelles’ concentration (CMC)—the maximum emission of Nile Red (at about 630 nm) was plotted versus the amphiphile’s concentration. This procedure was repeated three times for each amphiphile, and mean value is reported as the CMC and the standard deviation as the measurement error.
Enzymatic Degradation Experiments
A micellar solution of the tested amphiphile was prepared by directly adding PB (pH 7.4) to solid polymer to a final concentration of 160 μM. The vial was vortexed until full solubility was obtained and then placed in an ultrasonic bath for 15 min. PLE stock solution or PB was added (30 μL into 1470 μL or 14 μL into 686 μL for HPLC or fluorescence experiments, respectively, to yield the final PLE concentration of 1.4 μM), and degradation was followed at 37 °C either by monitoring the area under the peak of the parent amphiphile by HPLC or the fluorescence emission at 540 nm. Each experiment was conducted thrice, and the reported values in each time point are the mean valued and the standard deviation is the error.
CPT and PTX Encapsulation Procedure
The tested amphiphile was dissolved in MeOH (1 mg/mL). 1 mL of amphiphile solution was mixed with 1 mL of drug solution (0.5 mg/mL in DCM). Solvents were removed in vacuum forming a thin film layer, which was further dried on high vacuum for 1 h. Then, 1 mL of PB was added, and the vial content was stirred vigorously and placed in an ultrasonic bath for 30 min. The undissolved drug was filtered off using a syringe filter (0.45 μm hydrophilic Nylon), and the clear solution was analyzed by HPLC. The drug concentration was calculated by calibration curve at 360 nm for camptothecin (CPT) and 224 nm for paclitaxel (PTX).
Blood Protein Interaction: Micelle Incubation with BSA-Cy5
The interaction between micelles and Cy5-labeled BSA was measured using Förster resonance energy transfer (FRET). Micelles solution in PBS (pH 7.4) at 145.5 μM was mixed with BSA (10% v/v labeled with cyanine 5) at the 5.5 μM final concentration or with the same volume of PBS. Each sample was 60 μL in final volume inside a 96-well plate (flat-bottom, transparent, NUNC). The samples were excited at the 420 nm wavelength, and a fluorescence spectra was collected between 450 and 750 nm, with a 5 nm step. A reading was performed for each well every 15 min for 5 h, with the initial time point being approximately 15 min after BSA addition (or PBS, respectively).
Cytotoxicity with PrestoBlue
The cellular toxicity was assessed using the PrestoBlue assay (ThermoFisher). HeLa cells were seeded at a density of 5000 cells/well in a 96-well plate (Nunc, transparent, flat-bottom plate). After 24 h, micelles were added to the final concentration of 160 μM in full DMEM. The same volume of PBS was added in the negative control, while Triton X-100 0.01% v/v was used as the positive control. The cells were incubated for 24 h at 37 °C 5% CO2, then PrestoBlue was added 10% v/v and incubated for 1 h at 37 °C 5% CO2. Fluorescence was measured in a multimode microplate reader (Infinite M200 Pro from Tecan) by sampling the emission from the bottom well at 600 nm, while exciting at 550 nm. Each sample was taken in three replicates, distributed randomly on each row (using randomizer.org). The signal was normalized using the negative and positive controls between 0 and 100%, respectively.
Micelle Incubation with HeLa Cells (Microscopy)
HeLa cells were seeded at 30000 cells/well density in 8-well LabTek, 200 μL/well, 24 h prior to the experiment. Medium was changed with fresh DMEM (10% FBS), and micelles were diluted 3× to the final concentration of 160 μM on the cells. Samples were imaged in a confocal microscope at 37 °C, 5% CO2. The fluorescence signal with 405 nm excitation 1% (diode laser, 5 mW) was acquired in two channels representing unimers (446–500 nm) and micelles (526–589 nm), with equal gain.
Data analysis was carried out on Fiji ImageJ. Total fluorescence images were obtained by summing unimer and micelle channels, then applying “Cyan hot” lookup table. For ratiometric images, background was removed using a mask obtained from the sum image that was multiplied with unimer and micelle images. Ratiometric images were obtained by dividing the background-removed unimer to micelle images.
Flow Cytometry on HeLa Cells Incubated with Micelles
HeLa cells were seeded in a 24-well plate at 120000 cells/well in 1 mL/well 24 h before the experiment. Micelles were added by diluting 3× to the 160 μM final concentration, for either 1 or 6 h of incubation. Cells were washed 2× with warm PBS, then trypsinized with 250 μL/well for 3–4 min at 37 °C, mixed with 750 μL/well full DMEM, centrifuged 3 min at 180 g, and then resuspended in 1 mL/well warm PBS.
The fluorescence spectra with 405 nm excitation was recorded for 8000–10000 cells per sample using a spectral analyzer (Sony SA3800) flow cytometer. For data analysis, the spectral signal was gated in two “channels” representing unimers (420–500 nm) and micelles (550–700 nm).
Encapsulation Stability in the Presence of BSA
Micellar solution of the tested amphiphile was prepared in PBS (176 μM). Hydrophobic Cy5 derivative was added directly (2 μL/mL from 2 mM Cy5 solution in EtOH), and solution was thoroughly vortexed. Then, 50 μL of either BSA solution (55 mg/mL in PBS) or PBS were added into 450 μL of the above solution, and solution was vortexed to obtain final concentrations of 160 and 4 μM for amphiphile and Cy5, respectively, and 5.5 mg/mL for BSA. Absorbance of all final solutions was measured at T0, and the emission spectra was recorded every 30 min for 2 h (λEx = 420 nm).
Imaging of Cy5 Release Experiments on HeLa Cells
Micelle solution in PBS (480 μM) was mixed with Cy5 solution in ethanol (2 mM) to the final Cy5 concentration 12 μM. Mixture was vortexed and filtered through nylon 0.45 μm syringe filters (PureTech). Then, each solution was diluted with fresh DMEM (10% FBS) by three fold to final micelle and Cy5 concentration of 160 and 4 μM, respectively. Absorbance of each solution was measured in order to verify similarity in concentrations. Imaging was performed similarly to the cell internalization experiment. One field of view was followed for 1 h for each sample. For 405 nm excitation (5 mW diode laser, 1%), the acquisition was split in three channels representing unimer (400–500 nm), micelle (500–617 nm), and possible FRET fluorescence (656–700 nm). For total Cy5 fluorescence, 640 nm excitation (5 mW diode laser, 0.2%) was used, with 656–700 nm acquisition. Also, an electronically switchable illumination and detection module (ESID,bright field-like) signal was acquired using a 488 nm (10 mW) diode laser.
For image analysis (on Fiji ImageJ), regions of interest were drawn manually either outside the cells or containing cell cytoplasm and membrane, without the nucleus. Mean fluorescence inside cells and median outside cells were plotted using GraphPad Prism.
Results and Discussion
Molecular Design and Synthesis
We chose 7-(diethylamino)coumarin-3-carboxylic acid (7-DEAC) as the fluorescent tag due to its excimer formation ability that allows to distinguish whether the amphiphiles disassembled into unimers or remained as micelles under various conditions.53,54 Once 7-DEAC dyes are forced to be in close proximity within the micelles, their emission maxima shifts from 480 to ∼540 nm, and once the micelles disassemble, the emission shifts back to 480 nm. Our synthetic methodology allows simple preparation of libraries of amphiphilic diblock copolymers that can be examined and compared in many aspects ranging from micellar stability and enzymatic degradability to more complex biological studies that are enabled due to the dendron’s unique fluorescent response.
The synthetic route of the dendron is based on high yielding converging synthesis (Scheme 1). The synthesis started from the thiol–yne reaction55 between the AB2 branching unit and pre-made thiol-functionalized degradable hydrophobic end-groups, followed by coupling with a propargylated lysine-based taggable unit for fluorescent labeling. The dendron was then deprotected and labeled with the coumarin dye. In parallel, PEtOx-N3 (5 kDa) with narrow molecular weight distribution was synthesized by cationic ring opening polymerization of 2-ethyl-2-oxazoline using methyl tosylate as the initiator and terminated by sodium azide as previously reported.39,56 mPEG-N3 (5 kDa) was synthesized from commercially available mPEG-OH in three high yielding synthetic steps (Figure S21). PAA-based amphiphiles were obtained by atom transfer radical polymerization57 of tert-butyl acrylate using ethyl α-bromoisobutyrate as the initiator in the presence of CuBr and PMDETA.58 After polymerization, the terminal bromide was substituted with sodium azide. In the last step of the synthesis of the amphiphiles, a copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) between the hydrophilic polymers and the labeled dendrons was carried out using the CuBr/PMDETA catalyst as reported by Matyjaszewski.59 All polymers reacted neatly, and full conversion was confirmed by 1H NMR spectroscopy and size exclusion chromatography (SEC). Purification of the amphiphiles after the click reaction was performed using preparative SEC, and all polymers were obtained in high purity and yield. In the case of the PAA-based amphiphiles, an additional deprotection step of the t-butyl esters was performed. All final amphiphiles were characterized by 1H and 13C NMR, HPLC, SEC (PAA injected in its protected form), MALDI (except PAA), and UV–vis spectroscopy.
Micellization
Upon completing the synthesis of the six amphiphiles, we studied their self-assembly in aqueous media (PB, pH 7.4, at 37 °C). At first, the CMC of each polymer was determined using the Nile Red method.60 The CMC values of the amphiphiles with hexyl end-groups were determined to be 5 ± 1, 6 ± 1, and 10 ± 2 μM for PEtOx-Hex, PEG-Hex, and PAA-Hex, respectively (Figures S42–S47). This elucidates the tremendous effect of the hydrophobic block on the thermodynamic stability of polymeric micelles. The CMC values of the more hydrophobic nonyl amphiphiles were, as expected, slightly lower than those of the hexyl polymers and were determined to be 3 ± 1, 5 ± 1, and 9 ± 1 μM for PEtOx-Non, PEG-Non, and PAA-Non, respectively. Even though there is a substantial difference in the chemical composition of the micelle coronas, the CMCs of the three amphiphiles in each series are very similar with slightly higher CMC value for the PAA-based amphiphiles, most likely due to the greater hydrophilicity and repulsion of the charged PAA chains. Next, we used DLS to measure the diameters of the different micelles (Figure 1). The hexyl polymers self-assembled into micelles with diameters of 20 ± 2, 21 ± 2, and 20 ± 4 nm for PEtOx-Hex, PEG-Hex, and PAA-Hex, respectively, and the nonyl ones into 28 ± 5, 26 ± 5, and 32 ± 5 nm for PEtOx-Non, PEG-Non, and PAA-Non, respectively. It was fascinating to see the similar sizes of the micelles despite the different hydrophilic shells, demonstrating the key contribution of the hydrophobic block in directing the self-assembly of these polymeric amphiphiles into micelles.
Figure 1.
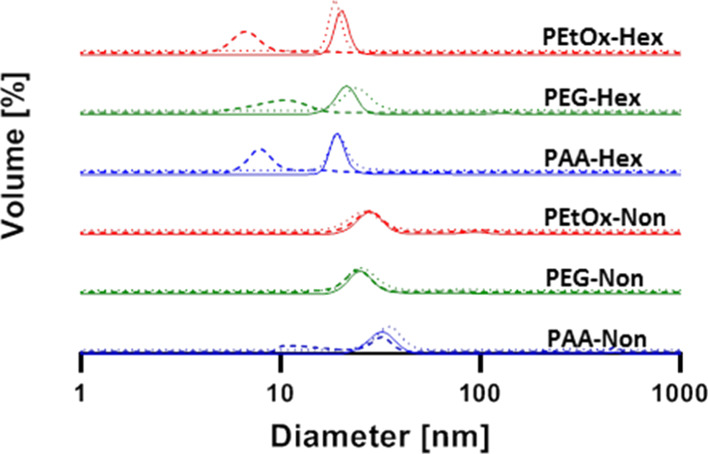
DLS measurements of the different micelles ([amphiphile] = 160 μM) before (solid lines) and after 24 h incubation in PB 7.4 with (dashed lines) and without (dotted lines) PLE (1.4 μM) at 37 °C.
Enzymatic Degradation Rate
After characterizing the self-assembly of the amphiphiles, we studied the enzymatic degradation rates of the six amphiphiles with PLE. PLE can selectively cleave the ester bonds between the dendron and the hydrophobic end-groups, exposing highly hydrophilic carboxylic acids on the dendron chain ends. This enzymatically induced modification will turn the polymer from amphiphilic into fully hydrophilic and therefore should cause the disassembly of the micelles (Figure 2A). Three methods were used to monitor the degradation of the amphiphiles and the disassembly of the micelles. Due to the high precision and purity of the polymers, we could directly monitor the degradation of the starting material and the appearance of the degraded polymer using HPLC (Figure 2D,E, solid lines and Figures S33 and S34). Simultaneously, we followed the enzymatically induced disassembly by monitoring 7-DEAC fluorescent response under the same conditions. Once an amphiphile’s end-groups are cleaved, it becomes fully hydrophilic and diffuses away from the micelle. This leads to a decrease in 7-DEAC excimer formation, and this spectral response can be quantified by measuring the decrease in fluorescence emission at 540 nm and the increase at 480 nm (Figure 2B–E, dashed lines). Finally, DLS was used to determine whether micelles are present in solution before and after 24 h incubation at 37 °C with or without PLE (Figure 1, dashed lines). Notably, the nonyl amphiphiles showed high stability in the presence of PLE even at concentration as high as 1.4 μM. PEtOx-Non and PEG-Non showed approximately 10% degradation over 24 h, and around 20% degradation was observed for PAA-Non (Figure 2E).
Figure 2.

(A) Schematic representation of the enzymatic degradation of the hydrophobic end-groups turning the polymeric amphiphile into fully hydrophilic polymers leading to micelle disassembly. Pictures of the fluorescence of PEG-Hex after 30 h incubation without (B) and with (C) PLE demonstrating the system’s spectral response. Enzymatic degradation profiles of hexyl- (D) and nonyl- (E) based micelles as monitored by HPLC (solid lines) and fluorescence spectroscopy (normalized decrease of emission intensity at 540 nm, dashed lines). [amphiphile] = 160 μM, [PLE] = 1.4 μM.
Strikingly, although the change in the hydrophilic to hydrophobic ratio is relatively small for the nonyl and hexyl series as reflected by the small differences in CMC values, the hexyl-based amphiphiles were significantly more susceptible to enzymatic degradation. While full degradation of PEG-Hex required longer time than PEtOx-Hex, their t1/2 was almost identical. PAA-Hex, in contrast, showed ultrafast response to PLE and was fully degraded after less than 30 min (Figure 2D, full lines). The faster degradation of the PAA amphiphile may be related to the higher hydrophilicity of the PAA compared to PEG and PEtOx, while the longer chain length of PEG-5k compared to PEtOx-5k and/or the slightly better anti-fouling behavior of PEG52 may be responsible for the slower degradation of the PEG amphiphile. Fluorescence measurements showed a decrease in the longer wavelength emission of the micelle and increase in unimer emission, indicating that the enzyme led to disassembly of the micelles. Excellent correlations were observed between the degradation kinetics obtained by HPLC and the decrease in fluorescence intensity at 540 nm (Figure 2D,E), dashed lines), which is indicative of the disassembly of the cleaved polymers into unimers. This clearly demonstrates that, indeed, it is the cleavage of the hydrophobic end-groups by the enzyme that caused the micelles to disassemble. Last, DLS measurements after incubation with PLE confirmed the disassembly of the hexyl-based micelles, while the nonyl-based series stayed intact (Figure 1, dashed lines). In the control experiments conducted in the absence of PLE, all amphiphiles showed micelles with similar sizes to the ones measured at t0 after 24 h incubation (Figure 1, dotted lines, and Figures S48 and S49). The similarities in CMCs and micelle sizes did not hint at the extreme difference in enzymatic degradation kinetics between the hexyl and nonyl polymers. The best example for this difference is seen for the two PAA-based amphiphiles, whereby PAA-Hex degraded in minutes while PAA-Non showed a very limited degree of degradation even after 24 h of incubation. This shows once more the tremendous effect of the hydrophobic block on the micellar dynamics and the importance of molecular precision when designing enzyme-responsive polymeric amphiphiles.61,62
Drug Encapsulation Capacity
Next, we investigated how the different hydrophilic shells affect the micelles encapsulation capacity for hydrophobic cargo. For this purpose, we chose the widely used hydrophobic anticancer drug CPT, which has very poor aqueous solubility. The CPT-loaded micelles were made by the thin-film hydration method, as previously reported.63 Unencapsulated CPT was removed by filtration through a 0.45 μm filter, and the filtrate was analyzed by HPLC to determine the drug concentration and the drug to polymer weight ratio. The CPT encapsulation results for the hexyl series were 5 ± 1, 18 ± 3, and 8 ± 1 weight percentage for PEtOx-Hex, PEG-Hex, and PAA-Hex, respectively (Figure 3A, black columns and Table 1). Surprisingly, the nonyl series showed lower encapsulation capacities of 1.0 ± 0.1, 14 ± 5, and 4.0 ± 0.3 weight percentages for PEtOx-Non, PEG-Non, and PAA-Non, respectively (Figure 3A, pink columns). These results are in good agreement with previous reports by Luxenhofer and co-workers, who observed a reduction in drug loading capacity upon increasing the hydrophobicity of the hydrophobic block in poly(2-oxazoline)-based micellar nanocarriers.64,65 In both, amphiphile series PEG micelles showed a higher encapsulation capacity than PAA micelles while PEtOx micelles showed the lowest encapsulation capacity. To determine whether the encapsulation ability of the different polymers is general or depends on the payload as previously suggested,66 we also prepared formulations of another, slightly less, hydrophobic anti-cancer drug, paclitaxel (PTX).67 Surprisingly, the trends with PTX were completely different and PEtOx micelles showed significantly higher encapsulation capacity than PEG micelles and PAA micelles (Figure 3B and Table 1). It is important to point out that the chemical composition of the micellar core in each of the hexyl and nonyl series is completely identical and, therefore, the observed differences in encapsulation capacities are solely dictated by the different micellar coronas. This may mean that the common way of thinking and illustrating micelles with their hydrophobic cargo trapped only inside the micelle core is not accurate and a significant amount of the cargo might be in the corona, solubilized also between the hydrophilic polymer chains.35,44,68 In addition, the hydrophilic corona may influence the core packing and hydration, which can also influence the drug encapsulation behavior for drugs with different hydrophobicity. After comparing the observed trends in encapsulation capacities, it is clear that the type of hydrophilic polymers that are chosen for DDS should be carefully selected to fit with the drug to be encapsulated.
Figure 3.
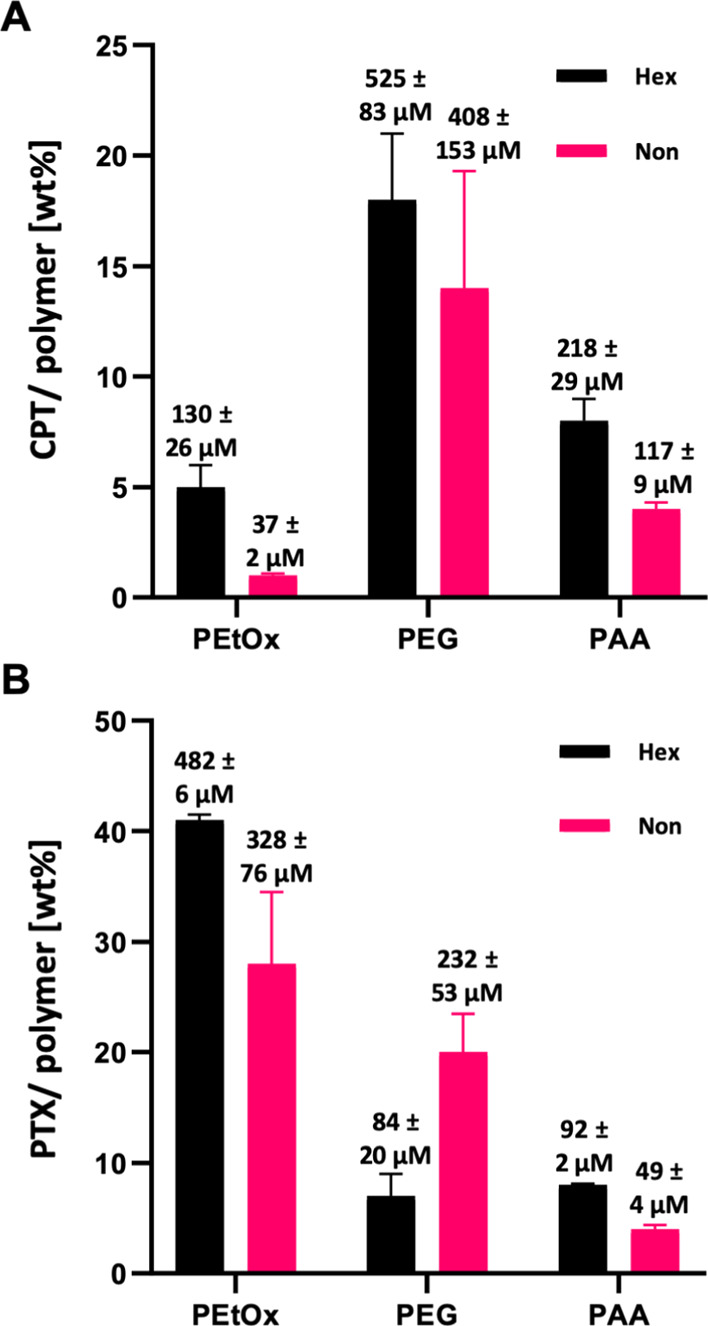
Encapsulation capacities of (A) CPT and (B) PTX in the different micelles (in μM and drug/polymer wt %). [amphiphile] = 160 μM in PB 7.4.
Table 1. Drug/Polymer Molar Ratio and Loading Capacity.
| molar
ratio ([drug]/[polymer]) |
loading capacity [%] |
|||
|---|---|---|---|---|
| CPT | PTX | CPT | PTX | |
| PEtOx-Hex | 0.84 ± 0.16 | 3.09 ± 0.04 | 4.3 ± 0.9 | 29.2 ± 0.3 |
| PEG-Hex | 3.36 ± 0.53 | 0.54 ± 0.13 | 15.5 ± 2.4 | 6.7 ± 1.6 |
| PAA-Hex | 1.40 ± 0.19 | 0.59 ± 0.01 | 7.1 ± 1.0 | 7.3 ± 0.1 |
| PEtOx-Non | 0.24 ± 0.01 | 2.16 ± 0.50 | 1.3 ± 0.1 | 21.9 ± 5.1 |
| PEG-Non | 2.69 ± 1.01 | 1.53 ± 0.35 | 12.5 ± 4.7 | 16.6 ± 3.7 |
| PAA-Non | 0.77 ± 0.06 | 0.32 ± 0.03 | 3.9 ± 0.3 | 4.0 ± 0.3 |
Interaction with Blood Proteins
After analyzing the in vitro properties of the six types of micelles, we moved on to study how these structural features influence their biological interactions. One of the first things that occur upon introduction of nanoparticles into the bloodstream, in addition to significant dilution, is the interactions with proteins and their adsorption resulting in the formation of a protein corona,69−72 which has been demonstrated to be key for shielding DDS from the immune system.73 Therefore, we investigated the interactions between the different micelles and albumin, which is the most abundant serum protein,74 as an initial evaluation of micelle–protein interactions. In order to understand the differences in protein adsorption, we incubated the micelles with BSA labeled with Cy5, which serves as a FRET acceptor for the micelle fluorescence (and much less with the shorter wavelength unimer’s fluorescence).54 FRET is highly dependent on spatial proximity (1–10 nm)75 and therefore the intensity of Cy5 emission qualitatively correlates with the interaction between the micelles and BSA. BSA is known to have hydrophobic regions that can interact with unimers and therefore significantly destabilize the micelles.76−78 This type of interaction translates into an increase in the unimer emission (480 nm) and a decrease in micelle emission (540 nm), as illustrated in Figure 4A and B. The increase in the hydrophobicity of the dendron changes the unimer–micelle equilibrium and decreases the interactions between unimers and BSA, indicating an increase in micellar stability (Figure 4C). PEtOx and PEG micelles showed overall similar behavior with slightly weaker interactions of PEtOx micelles with BSA indicated by a smaller peak of the FRET signal. For both PEtOx and PEG hexyl micelles, there was a moderate increase in the unimer emission, which is indicative of their interaction with BSA (Figure 4C). Almost no changes were observed for PEtOx and PEG nonyl micelles, which did not show an increase in the unimer peak upon incubation with BSA. On the other hand, PAA-Hex showed complete disassembly due to interactions with BSA as indicated by the total disappearance of the micelle fluorescence in addition to a significant increase in the unimer signal intensity (Figure 4C and D). The reason for this increase might be the change in the 7-DEAC microenvironment that can influence its quantum yield dramatically. PAA-Non also showed an increase in the unimer signal intensity, but the micelle signal still partially remains, and the Cy5 signal is the most intense amongst all micelles, suggesting the strongest micelle–BSA interactions among the different amphiphiles. To monitor the destabilization of the micelles over time, we calculated the unimer to the micelle fluorescence ratio (Figure S50). All ratios were nearly constant during 5 h, except for PAA-Non, which showed an increase in the unimer/micelle ratio. Overall, this suggests that for all micelles, the interactions with BSA happen almost immediately after addition, leading to various types and degrees of interactions. While the more hydrophobic nonyl micelles generally interact less with BSA, the PAA-Non showed significantly stronger interaction of both micelles and unimers with BSA.
Figure 4.

(A) Illustration of the two main possible interaction pathways with Cy5-labeled BSA: interaction of BSA either with unimer (left) or micelle (right). (B) Selected fluorescence emission spectrum with arrows highlighting the contribution of the different species to the spectrum. (C) Fluorescence spectra of the micelles with (red lines) and without (blue lines) BSA-Cy5 and (D) zoom out into PAA amphiphiles emission spectra. [amphiphile] = 160 μM, [BSA-Cy5] = 5.5 mg/mL, λEx = 420 nm.
Cell Internalization
After assessing the micellar stability in the presence of BSA and confirming the lack of cytotoxicity of the different micelles (Figure S51), the next step was to investigate the internalization of the micelles into HeLa cells. Therefore, the micelles were incubated with cells in full DMEM medium (with 10% fetal bovine serum) at a final micelle concentration of 160 μM and imaged with confocal microscopy.
Upon 405 nm excitation, we could differentiate the assembly states by separating the signal into two distinct channels: one for the unimer signal (400–500 nm) and one for the micelle signal (500–700 nm). Finally, we obtained total fluorescence and ratiometric images, by either combining the two channels or by dividing the unimer to micelle signal after background removal. Images with total fluorescence allowed a direct comparison of the internalization efficiency of the different amphiphiles as unimers and/or micelles, also showing the distribution inside different cellular compartments.
Ratiometric images allowed the visualization of the assembly state of the amphiphiles within any given pixel, enabling a deeper understanding of the behavior of the different micelles.
Looking at the total intensity inside the cells, we could see a remarkable difference in the degree of internalization of the different micelles (Figure 5A top row and 5B). The micelles of PEtOx-Non and PEG-Non that were shown to be more stable showed a very weak signal within the cells. While PEtOx-Hex and PEG-Hex had similar distributions in intracellular vesicles, PAA-Hex and PAA-Non bound mostly to the cell membrane and had the most intense fluorescence emissions, similar to the trend observed for the incubation with BSA. Thus, we can assume that the hydrophilic block directs the cellular fate of the micelles toward the endo-lysosomal compartments for PEtOx and PEG and membrane-bound for PAA. Interestingly, the relatively small change in hydrophobicity causes a notable decrease in internalization efficiency for the nonyl micelles. To assess whether this difference is due to disassembly of the micelles outside or inside the cells, we analyzed the ratiometric images (Figure 5A bottom row). For all amphiphiles except PAA-Hex, the ratiometric analysis indicated the presence of micelles outside the cells, while inside cells or on the cell membrane, all amphiphiles were mostly in their unimer form (Figure 5C). However, slightly more micelles were observed inside the cells for the more stable PEtOx-Non and PEG-Non (Figure 5B,C). We can assume that the less stable PAA micelles disassembled outside the cells more readily into unimers that could then intercalate into the plasma membrane. For the polymers that were localized in endosomal vesicles, it may be that they internalized as micelles and very rapidly disassembled inside the endosomal vesicles into unimers. To achieve higher degree of internalization and obtain more intense fluorescence signal inside the cells, the experiment was repeated with a longer incubation time of 6 h (Figure S52). The longer incubation time indeed led to a stronger signal within the cells, which was mostly observed in the unimer channel. Although the ratiometric images indicated the presence of unimers inside the cells, it is most likely that the increased internalization cannot be attributed to the disassembly of the micelles outside of the cells over time. This assumption is based on the high micellar stability for PEtOx-Non and PEG-Non, which remained highly stable when incubated with BSA, PLE, and outside of the cells. The degree of internalization was further validated by spectral flow cytometry after incubation times of 1 and 6 h using both unimer and micelle channels (Figure S53).
Figure 5.

Internalization of micelles into HeLa cells after 1 h of incubation in DMEM with 10% FBS. Images show total fluorescence signal with 405 nm excitation (A, top row) or ratiometric images of unimer/micelle pixel ratio after background removal (A, bottom row). The green color indicates the micellar form, and magenta indicates the unimer form. The scale bar is 10 μm. The median fluorescence for control areas (inside nucleus as a negative control and in solution outside cells as a positive control) were plotted along with the mean fluorescence inside the cytoplasm or in the membrane area (n = 8–10 cells) for either total fluorescence (B) or unimer/micelle ratio (C).
Encapsulation Stability and Cargo Release in the Presence of BSA and Cell Culture
In addition to the crucial effect of micellar stability on the ability of the micelles to retain their molecular cargo, one of the major drawbacks in physical encapsulation of hydrophobic compounds is the possibility of premature leakage, that is, before reaching the tumor through passive targeting via the EPR effect, due to migration of the cargo into native hydrophobic regions in the surroundings of the carrier such as proteins or membranes.35,79 Hence, after studying the stability of the micelles, we evaluated their ability to retain their cargo of hydrophobic molecules in a biological environment. Therefore, another FRET-based experimental setup was designed in order to study how encapsulated hydrophobic cargo behaves in the presence of BSA. A hydrophobic Cy5 derivative (Figure S17), which served as a model for an encapsulated lipophilic drug, was physically encapsulated within the different micelles, and non-labeled BSA was added to the micellar solution. The encapsulated Cy5-derivative undergoes significant FRET with the fluorescence of the micelles while its migration from the micelles to BSA or its precipitation due to micelle disassembly into unimers should translate into a reduction in FRET efficiency (Figure 6A).
Figure 6.

(A) Illustration of the two main possible interaction pathways between micelles with encapsulated Cy5 and BSA. (B) Selected fluorescence emission spectrum with arrows highlighting the contribution of the different species to the spectrum. (C) Fluorescence spectra of the micelles with encapsulated Cy5 with (red lines) and without (blue lines) BSA and (D) zoom out into PAA-Hex emission spectra. [amphiphile] = 160 μM, [Cy5] = 4 μM, [BSA] = 5.5 mg/mL, λEx = 420 nm.
Prior to the fluorescence measurements, the absorbance spectra of all tested solutions containing micelles and BSA, as well as the controls without BSA, were measured to verify that the concentrations of the polymers and Cy5 were similar for all solutions (Figure S54). Next, fluorescence spectra were measured every 30 min over 2 h. Upon addition of BSA, both PEtOx and PEG micelles showed a decrease of ∼30% in FRET-related emission, while a slight increase in both the micelle and unimer emission was observed (Figure 6B,C). These results indicate that the majority of encapsulated Cy5 molecules remained entrapped inside the micelles as complete Cy5 release would lead to a complete disappearance of the FRET signal and a substantial increase in micelle fluorescence would be expected. A slight increase in unimer emission is attributed to the interaction of the micelles with the BSA, as described above (Figure 4). Interestingly, the least stable PAA-Hex micelles showed a significantly lower FRET signal in the absence of BSA, which was sustainably reduced by 50% upon the addition of BSA. In addition, a strong increase in the unimer emission was also observed, indicating the low stability of the PAA-Hex micelles and their tendency to disassemble due to interaction with BSA. Unlike the PAA-Hex, the PAA-Non-based micelles showed only a moderate decrease of ∼30% of the FRET signal in the presence of BSA, similar to the PEG and PEtOx micelles. The notable increase in the unimer emission of the PAA-Non can again be attributed to the stronger interaction with BSA (Figure 6A). The response to BSA was extremely fast in this experiment as noted before in the Cy5-labeled BSA assay, and no major changes were observed over time, which is indicative of the high encapsulation stability of the PEG, PEtOx, and PAA-Non micelles (Figure S55).
Intrigued by the encapsulation stability of the micelles in the presence of BSA, we decided to study the release of the Cy5 dyes in HeLa cell culture. Two possible mechanisms for the release of physically encapsulated cargo from polymeric micelles can be envisioned: (i) spontaneous leakage or (ii) disassembly and release. Spontaneous leakage would leave the micelles intact, while the Cy5 molecules would exit and accumulate inside cells. On the other hand, disassembly based release could occur outside the cells followed by internalization of the unimers together with the released Cy5 dyes. Alternatively, the Cy5 containing micelles could be internalized followed by disassembly and cargo releasing in the endosomal vesicles. Based on the inferior stability noted in all previous experiments for the PAA micelles, we expected that the release of encapsulated cargo would be significantly faster in the presence of cells as a large fraction of PAA micelles disassembled in the presence of HeLa cell culture. To study the effect of micellar corona composition on the release kinetics of hydrophobic cargo, we decided to prepare micelles only from the more stable nonyl amphiphiles. The three types of micelles were loaded with Cy5 and incubated with HeLa cells. The same cells were followed for 1 h, measuring the fluorescence of the unimer and micelle (excitation at 405 nm) as well as directly following the Cy5 (excited at 640 nm, Figures 7A and S57). For every time point, the fluorescence was measured inside the cytoplasm of 10 cells and the mean values were compared to the signal outside the cells (Figure 7B). This unique setup provided important new insights regarding the behavior of the different micelles. By monitoring the ratio of unimer/micelle fluorescence, the assembly state of the amphiphiles in each pixel is revealed (Figure 7C). In addition, the Cy5 channel enabled direct monitoring of the cell internalization kinetics of the encapsulated cargo (Figure 7B). We hypothesized that the combination of the ratiometric data together with the direct tracking of Cy5 release would shed light on the release mechanism of the different micelles. First, we examined the assembly state of the micelles. As seen in the previous internalization experiments (Figure 5), PAA-Non micelles showed significant disassembly both outside and inside the cells, which increased over time (Figure 7C).
Figure 7.

Release of encapsulated Cy5 onto HeLa cells over time. (A) Fluorescence with 640 nm excitation is shown inside HeLa cells at different time points for hydrophobic Cy5 encapsulated in micelles or free, in full DMEM (10% FBS), scale bar is 20 μm. The quantification of fluorescence from confocal images with (B) as mean fluorescence intensity in the Cy5 channel, with 640 nm excitation or (C) 405 nm excitation is shown as ratio of unimer/micelle signal. Regions of interest were manually drawn around cell cytoplasm, including the cell membrane and excluding the nucleus; n = 10 cells. [amphiphile] = 160 μM, [Cy5] = 4 μM.
As expected, the PEtOx-Non and PEG-Non micelles were much more stable than the PAA-Non micelles and almost no disassembly was observed, as indicated by the nearly constant unimer/micelle fluorescence ratio (Figure S57). Therefore, we were rather surprised to discover that the release and internalization of Cy5 was significantly slower from PAA-Non micelles compared with PEG-Non and PEtOx-Non. While both PEtOx-Non and PEG-Non samples showed dramatic increase in the Cy5 signal inside the cells, PAA-Non showed delayed and more gradual increase, even though its tendency to undergo micellar disassembly is much higher. The results of these experiments demonstrate that lower micellar stability does not always correlate with faster release kinetics, and that disassembly of the carrier is not essential for cargo release (Figure 8). Furthermore, these findings clearly emphasize the importance of tracking both the carrier and the cargo when studying the internalization mechanism and kinetics of DDS.
Figure 8.
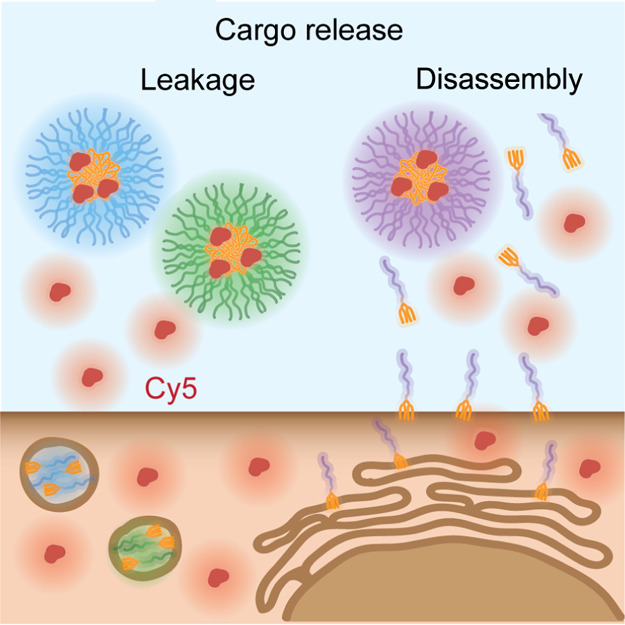
Schematic illustration of two mechanisms of cargo release by either leakage from stable micelles or disassembly of the polymeric carrier.
Conclusions
In summary, we designed a highly modular approach for preparing clickable spectrally active enzyme-responsive dendrons with adjustable degree of hydrophobicity. The dendrons were synthesized using a thiol–yne reaction and then conjugated by the CuAAC click reaction with three different hydrophilic polymers, namely PEG, PEtOx, and PAA, with similar molecular weights, enabling us to prepare six amphiphiles that differ either in the type of hydrophilic blocks or the lipophilicity of the hydrophobic dendrons. The high similarity of the hydrophobic blocks, which rises from the well-defined structure of the dendrons, allowed a head-to-head comparison of the effects of the hydrophilic blocks on the supramolecular behavior of the amphiphiles. The CMCs of the two series of amphiphiles were all below 10 μM and the nonyl containing amphiphiles, which had a higher degree of hydrophobicity, showed slightly lower values than the hexyl-based ones. Interestingly, the micelles in each of two series showed similar diameters, with the three amphiphiles bearing the more hydrophobic nonyl-based dendron having larger diameters than the hexyl-based ones. Unlike the CMCs and mean micelle diameters, which were affected mostly by the hydrophobicity of the dendrons, the enzymatic degradation rates of the micelles were found to be strongly dependent on both the hydrophilic and hydrophobic blocks. Drug encapsulation capacities of the six micelles were very sensitive to the type of the hydrophilic block, and the two drugs tested, CPT and PTX, had substantially different degrees of loadings. The drug loading results clearly indicate the importance of the nature of the hydrophilic shell for the encapsulation of hydrophobic drugs. In addition, the composition of the hydrophilic block had a strong effect on the interactions of both unimers and micelles with BSA, which in the case of PAA-Hex led to the complete disassembly of the micelles. Cell internalization experiments revealed a substantial difference in the membrane binding and internalization rate between PAA amphiphiles and the PEtOx and PEG amphiphiles. PAA-based amphiphiles localized more on the cell membrane and internalized to a greater extent than the PEG- and PEtOx-based amphiphiles. In all cases, the more hydrophobic nonyl amphiphiles internalized significantly more slowly than the hexyl ones. Using a hydrophobically modified Cy5 dye as a model for encapsulated drug molecules, it was interesting to see its slower release and cell internalization in the case of the less stable PAA-Non-based micelles, while much faster Cy5 cell internalization was observed for the PEG-Non and PEtOx-Non micelles, which seemed to self-assemble into significantly more stable micelles. These encapsulation and release experiments in cell culture revealed the complexity of studying release mechanisms and the importance of directly tracking both the carrier and the cargo. Overall, the ability to directly compare micelles with different shells and the resulting comparative results provide important fundamental insights into how the composition of the shell and core of such polymeric micelles can affect their properties and potential to serve as nanocarriers for DDS.
Acknowledgments
G.S. thanks the Marian Gertner Institute for Medical Nanosystems in Tel Aviv University for their financial support. R.H. and V.R.D.L.R are grateful to Ghent University and FWO for continuous financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.0c01708.
Synthetic procedures, amphiphiles and micelles characterization data, detailed experimental protocols, control experiments, cytotoxicity data, and additional confocal imaging (PDF)
Author Contributions
⧫ G.S. and A.R.O. authors contributed equally.
R.J.A. thanks the Israel Science Foundation (grant no. 1553/18) for the support of this research. This project has received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie [grant agreement no. 713673 (THERACAT)].
The authors declare the following competing financial interest(s): R.H. and V.R.D.L.R. are co-founders of Avroxa BVBA that commercializes poly(2-oxazoline)s as Ultroxa. The other authors have no conflicts of interest to declare.
Supplementary Material
References
- Langer R.; Tirrell D. A. Designing Materials for Biology and Medicine. Nature 2004, 428, 487–492. 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- Cabral H.; Kataoka K. Progress of Drug-Loaded Polymeric Micelles into Clinical Studies. J. Controlled Release 2014, 190, 465–476. 10.1016/j.jconrel.2014.06.042. [DOI] [PubMed] [Google Scholar]
- Torchilin V. P. Micellar Nanocarriers: Pharmaceutical Perspectives. Pharm. Res. 2007, 24, 1–16. 10.1007/s11095-007-9463-5. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Wu J.; Sawa T.; Matsumura Y.; Hori K. Tumor Vascular Permeability and the EPR Effect in Macromolecular Therapeutics: A Review. J. Controlled Release 2000, 65, 271–284. 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- Satchi-Fainaro R.; Puder M.; Davies J. W.; Tran H. T.; Sampson D. A.; Greene A. K.; Corfas G.; Folkman J. Targeting Angiogenesis with a Conjugate of HPMA Copolymer and TNP-470. Nat. Med. 2004, 10, 255–261. 10.1038/nm1002. [DOI] [PubMed] [Google Scholar]
- van der Meel R.; Lammers T.; Hennink W. E. Cancer Nanomedicines: Oversold or Underappreciated?. Expet Opin. Drug Deliv. 2017, 14, 1–5. 10.1080/17425247.2017.1262346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen S. C.; Chan D. P. Y.; Shoichet M. S. Polymeric Micelle Stability. Nano Today 2012, 7, 53–65. 10.1016/j.nantod.2012.01.002. [DOI] [Google Scholar]
- Karimi M.; Ghasemi A.; Sahandi Zangabad P.; Rahighi R.; Moosavi Basri S. M.; Mirshekari H.; Amiri M.; Shafaei Pishabad Z.; Aslani A.; Bozorgomid M.; Ghosh D.; Beyzavi A.; Vaseghi A.; Aref A. R.; Haghani L.; Bahrami S.; Hamblin M. R. Smart Micro/Nanoparticles in Stimulus-Responsive Drug/Gene Delivery Systems. Chem. Soc. Rev. 2016, 45, 1457–1501. 10.1039/c5cs00798d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S.; Shang Q.; Wang N.; Li Q.; Song A.; Luan Y. Rational Design of a Minimalist Nanoplatform to Maximize Immunotherapeutic Efficacy: Four Birds with One Stone. J. Controlled Release 2020, 328, 617–630. 10.1016/j.jconrel.2020.09.035. [DOI] [PubMed] [Google Scholar]
- Shang Q.; Zhou S.; Jiang Y.; Wang D.; Wang J.; Song A.; Luan Y. Rational Design of a Robust Antibody-like Small-Molecule Inhibitor Nanoplatform for Enhanced Photoimmunotherapy. ACS Appl. Mater. Interfaces 2020, 12, 40085–40093. 10.1021/acsami.0c11156. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Huang C.; Luan Y. Lactosylated Ir820/Dox Co-Assembled Nanodrug for Synergetic Antitumour Therapy. Int. J. Nanomed. 2020, Volume 15, 4431–4440. 10.2147/ijn.s247617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart M. A. C.; Huck W. T. S.; Genzer J.; Müller M.; Ober C.; Stamm M.; Sukhorukov G. B.; Szleifer I.; Tsukruk V. V.; Urban M.; Winnik F.; Zauscher S.; Luzinov I.; Minko S. Emerging Applications of Stimuli-Responsive Polymer Materials. Nat. Mater. 2010, 9, 101–113. 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- Movassaghian S.; Merkel O. M.; Torchilin V. P. Applications of Polymer Micelles for Imaging and Drug Delivery. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2015, 7, 691–707. 10.1002/wnan.1332. [DOI] [PubMed] [Google Scholar]
- Brooks W. L. A.; Vancoillie G.; Kabb C. P.; Hoogenboom R.; Sumerlin B. S. Triple Responsive Block Copolymers Combining pH-Responsive, Thermoresponsive, and Glucose-Responsive Behaviors. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 2309–2317. 10.1002/pola.28615. [DOI] [Google Scholar]
- Gillies E. R.; Fréchet J. M. J. pH-Responsive Copolymer Assemblies for Controlled Release of Doxorubicin. Bioconjugate Chem. 2005, 16, 361–368. 10.1021/bc049851c. [DOI] [PubMed] [Google Scholar]
- Müller S. S.; Fritz T.; Gimnich M.; Worm M.; Helm M.; Frey H. Biodegradable Hyperbranched Polyether-Lipids with in-Chain PH-Sensitive Linkages. Polym. Chem. 2016, 7, 6257–6268. 10.1039/c6py01308b. [DOI] [Google Scholar]
- Ko J.; Park K.; Kim Y.-S.; Kim M. S.; Han J. K.; Kim K.; Park R.-W.; Kim I.-S.; Song H. K.; Lee D. S.; Kwon I. C. Tumoral acidic extracellular pH targeting of pH-responsive MPEG-poly(β-amino ester) block copolymer micelles for cancer therapy. J. Controlled Release 2007, 123, 109–115. 10.1016/j.jconrel.2007.07.012. [DOI] [PubMed] [Google Scholar]
- Boerman M. A.; Van der Laan H. L.; Bender J. C. M. E.; Hoogenboom R.; Jansen J. A.; Leeuwenburgh S. C.; Van Hest J. C. M. Synthesis of pH- and thermoresponsive poly(2-n -propyl-2-oxazoline) based copolymers. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 1573–1582. 10.1002/pola.28011. [DOI] [Google Scholar]
- André X.; Zhang M.; Müller A. H. E. Thermo- and pH-Responsive Micelles of Poly(acrylic acid)-block-Poly(N,N-diethylacrylamide). Macromol. Rapid Commun. 2005, 26, 558–563. 10.1002/marc.200400510. [DOI] [Google Scholar]
- Wang C.; Zhang G.; Liu G.; Hu J.; Liu S. Photo- and Thermo-Responsive Multicompartment Hydrogels for Synergistic Delivery of Gemcitabine and Doxorubicin. J. Controlled Release 2017, 259, 149–159. 10.1016/j.jconrel.2016.11.007. [DOI] [PubMed] [Google Scholar]
- Qiao J.; Qi L.; Shen Y.; Zhao L.; Qi C.; Shangguan D.; Mao L.; Chen Y. Thermal Responsive Fluorescent Block Copolymer for Intracellular Temperature Sensing. J. Mater. Chem. 2012, 22, 11543–11549. 10.1039/c2jm31093g. [DOI] [Google Scholar]
- Vanparijs N.; Nuhn L.; De Geest B. G. Transiently Thermoresponsive Polymers and Their Applications in Biomedicine. Chem. Soc. Rev. 2017, 46, 1193–1239. 10.1039/c6cs00748a. [DOI] [PubMed] [Google Scholar]
- Khatun Z.; Choi Y. S.; Kim Y. G.; Yoon K.; Nurunnabi M.; Li L.; Lee E.; Kang H. C.; Huh K. M. Bioreducible Poly(Ethylene Glycol)-Triphenylphosphonium Conjugate as a Bioactivable Mitochondria-Targeting Nanocarrier. Biomacromolecules 2017, 18, 1074–1085. 10.1021/acs.biomac.6b01324. [DOI] [PubMed] [Google Scholar]
- Sun B.; Luo C.; Yu H.; Zhang X.; Chen Q.; Yang W.; Wang M.; Kan Q.; Zhang H.; Wang Y.; He Z.; Sun J. Disulfide Bond-Driven Oxidation- and Reduction-Responsive Prodrug Nanoassemblies for Cancer Therapy. Nano Lett. 2018, 18, 3643–3650. 10.1021/acs.nanolett.8b00737. [DOI] [PubMed] [Google Scholar]
- Porsch C.; Zhang Y.; Montañez M. I.; Malho J.-M.; Kostiainen M. A.; Nyström A. M.; Malmström E. Disulfide-Functionalized Unimolecular Micelles as Selective Redox-Responsive Nanocarriers. Biomacromolecules 2015, 16, 2872–2883. 10.1021/acs.biomac.5b00809. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Zhang H.; He W.; Zhao D.; Song A.; Luan Y. Disulfide-Linked Amphiphilic Polymer-Docetaxel Conjugates Assembled Redox-Sensitive Micelles for Efficient Antitumor Drug Delivery. Biomacromolecules 2016, 17, 1621–1632. 10.1021/acs.biomac.5b01758. [DOI] [PubMed] [Google Scholar]
- Roy D.; Cambre J. N.; Sumerlin B. S. Future Perspectives and Recent Advances in Stimuli-Responsive Materials. Prog. Polym. Sci. 2010, 35, 278–301. 10.1016/j.progpolymsci.2009.10.008. [DOI] [Google Scholar]
- Mu J.; Lin J.; Huang P.; Chen X. Development of Endogenous Enzyme-Responsive Nanomaterials for Theranostics. Chem. Soc. Rev. 2018, 47, 5554–5573. 10.1039/c7cs00663b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Munkhbat O.; Secinti H.; Thayumanavan S. Disassembly of polymeric nanoparticles with enzyme-triggered polymer unzipping: polyelectrolyte complexes vs. amphiphilic nanoassemblies. Chem. Commun. 2020, 56, 8456–8459. 10.1039/d0cc03257c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarajeewa S.; Zentay R. P.; Jhurry N. D.; Li A.; Seetho K.; Zou J.; Wooley K. L. Programmed Hydrolysis of Nanoassemblies by Electrostatic Interaction-Mediated Enzymatic-Degradation. Chem. Commun. 2014, 50, 968–970. 10.1039/c3cc46013d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K.; Plaks V.; Werb Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q.; Katti P. S.; Gu Z. Enzyme-Responsive Nanomaterials for Controlled Drug Delivery. Nanoscale 2014, 6, 12273–12286. 10.1039/c4nr04249b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rica R.; Aili D.; Stevens M. M. Enzyme-Responsive Nanoparticles for Drug Release and Diagnostics. Adv. Drug Deliv. Rev. 2012, 64, 967–978. 10.1016/j.addr.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Kataoka K.; Harada A.; Nagasaki Y. Block Copolymer Micelles for Drug Delivery: Design, Characterization and Biological Significance. Adv. Drug Deliv. Rev. 2012, 64, 37–48. 10.1016/j.addr.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Lammers T.; Storm G.; Hennink W. E. Physico-Chemical Strategies to Enhance Stability and Drug Retention of Polymeric Micelles for Tumor-Targeted Drug Delivery. Macromol. Biosci. 2017, 17, 1600160. 10.1002/mabi.201600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gref R.; Domb A.; Quellec P.; Blunk T.; Müller R. H.; Verbavatz J. M.; Langer R. The Controlled Intravenous Delivery of Drugs Using PEG-Coated Sterically Stabilized Nanospheres. Adv. Drug Deliv. Rev. 2012, 64, 316–326. 10.1016/j.addr.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. Molecular Simulations of PEGylated Biomolecules, Liposomes, and Nanoparticles for Drug Delivery Applications. Pharmaceutics 2020, 12, 533. 10.3390/pharmaceutics12060533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caponi P.-F.; Winnik F. M.; Ulijn R. V. Charge Complementary Enzymatic Reconfigurable Polymeric Nanostructures. Soft Matter 2012, 8, 5127. 10.1039/c2sm07460e. [DOI] [Google Scholar]
- Sedlacek O.; Hoogenboom R. Drug Delivery Systems Based on Poly(2-Oxazoline)s and Poly(2-Oxazine)s. Adv. Ther. 2019, 3, 1900168. 10.1002/adtp.201900168. [DOI] [Google Scholar]
- Luxenhofer R.; Schulz A.; Roques C.; Li S.; Bronich T. K.; Batrakova E. V.; Jordan R.; Kabanov A. V. Doubly Amphiphilic Poly(2-Oxazoline)s as High-Capacity Delivery Systems for Hydrophobic Drugs. Biomaterials 2010, 31, 4972–4979. 10.1016/j.biomaterials.2010.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Munyaradzi O.; Xia X.; Green D. S.; Bong D. High-Capacity Drug Carriers from Common Polymer Amphiphiles. Biomacromolecules 2016, 17, 3060–3066. 10.1021/acs.biomac.6b00960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Sun F.; Liu S.; Jiang S. Anti-PEG Antibodies in the Clinic: Current Issues and beyond PEGylation. J. Controlled Release 2016, 244, 184–193. 10.1016/j.jconrel.2016.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma G. T.; Shimizu T.; Ishida T.; Szebeni J. Anti-PEG Antibodies: Properties, Formation, Testing and Role in Adverse Immune Reactions to PEGylated Nano-Biopharmaceuticals. Adv. Drug Deliv. Rev. 2020, 154–155, 163–175. 10.1016/j.addr.2020.07.024. [DOI] [PubMed] [Google Scholar]
- Haider M. S.; Lübtow M. M.; Endres S.; Forster S.; Flegler V. J.; Böttcher B.; Aseyev V.; Pöppler A.-C.; Luxenhofer R.; Luxenhofer R. Think beyond the Core: Impact of the Hydrophilic Corona on Drug Solubilization Using Polymer Micelles. ACS Appl. Mater. Interfaces 2020, 12, 24531–24543. 10.1021/acsami.9b22495. [DOI] [PubMed] [Google Scholar]
- Rajasekhar Reddy R.; Raghupathi K. R.; Torres D. A.; Thayumanavan S. Stimuli Sensitive Amphiphilic Dendrimers. New J. Chem. 2012, 36, 340. 10.1039/C2NJ20879B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado C. A.; Smith I. R.; Savin D. A. Self-Assembly of Oligo- and Polypeptide-Based Amphiphiles: Recent Advances and Future Possibilities. Macromolecules 2019, 52, 1899–1911. 10.1021/acs.macromol.8b02043. [DOI] [Google Scholar]
- Choi J.; Moquin A.; Bomal E.; Na L.; Maysinger D.; Kakkar A. Telodendrimers for Physical Encapsulation and Covalent Linking of Individual or Combined Therapeutics. Mol. Pharm. 2017, 14, 2607–2615. 10.1021/acs.molpharmaceut.7b00019. [DOI] [PubMed] [Google Scholar]
- Bolu B.; Sanyal R.; Sanyal A. Drug Delivery Systems from Self-Assembly of Dendron-Polymer Conjugates †. Molecules 2018, 23, 1570. 10.3390/molecules23071570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Photos P. J.; Bacakova L.; Discher B.; Bates F. S.; Discher D. E. Polymer Vesicles in Vivo: Correlations with PEG Molecular Weight. J. Controlled Release 2003, 90, 323–334. 10.1016/s0168-3659(03)00201-3. [DOI] [PubMed] [Google Scholar]
- Ke P. C.; Lin S.; Parak W. J.; Davis T. P.; Caruso F. A Decade of the Protein Corona. ACS Nano 2017, 11, 11773–11776. 10.1021/acsnano.7b08008. [DOI] [PubMed] [Google Scholar]
- Nyström A. M.; Wooley K. L. The Importance of Chemistry in Creating Well-Defined Nanoscopic Embedded Therapeutics: Devices Capable of the Dual Functions of Imaging and Therapy. Acc. Chem. Res. 2011, 44, 969–978. 10.1021/ar200097k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgese G.; Verbraeken B.; Ramakrishna S. N.; Gombert Y.; Cavalli E.; Rosenboom J. G.; Zenobi-Wong M.; Spencer N. D.; Hoogenboom R.; Benetti E. M. Chemical Design of Non-Ionic Polymer Brushes as Biointerfaces: Poly(2-oxazine)s Outperform Both Poly(2-oxazoline)s and PEG. Angew. Chem. Int. Ed. 2018, 57, 11667–11672. 10.1002/anie.201805620. [DOI] [PubMed] [Google Scholar]
- Buzhor M.; Harnoy A. J.; Tirosh E.; Barak A.; Schwartz T.; Amir R. J. Supramolecular Translation of Enzymatically Triggered Disassembly of Micelles into Tunable Fluorescent Responses. Chem. - Eur. J. 2015, 21, 15633–15638. 10.1002/chem.201502988. [DOI] [PubMed] [Google Scholar]
- Feiner-Gracia N.; Buzhor M.; Fuentes E.; Pujals S.; Amir R. J.; Albertazzi L. Micellar Stability in Biological Media Dictates Internalization in Living Cells. J. Am. Chem. Soc. 2017, 139, 16677–16687. 10.1021/jacs.7b08351. [DOI] [PubMed] [Google Scholar]
- Lowe A. B. Thiol-yne ’click’/coupling chemistry and recent applications in polymer and materials synthesis and modification. Polymer 2014, 55, 5517–5549. 10.1016/j.polymer.2014.08.015. [DOI] [Google Scholar]
- Kempe K.; Hoogenboom R.; Jaeger M.; Schubert U. S. Three-Fold Metal-Free Efficient (″Click″) Reactions onto a Multifunctional Poly(2-oxazoline) Designer Scaffold. Macromolecules 2011, 44, 6424–6432. 10.1021/ma201385k. [DOI] [Google Scholar]
- Braunecker W. a.; Matyjaszewski K. Controlled/Living Radical Polymerization: Features, Developments, and Perspectives. Prog. Polym. Sci. 2007, 32, 93–146. 10.1016/j.progpolymsci.2006.11.002. [DOI] [Google Scholar]
- Davis K. A.; Matyjaszewski K. Atom Transfer Radical Polymerization oftert-Butyl Acrylate and Preparation of Block Copolymers. Macromolecules 2000, 33, 4039–4047. 10.1021/ma991826s. [DOI] [Google Scholar]
- Golas P. L.; Tsarevsky N. V.; Sumerlin B. S.; Matyjaszewski K. Catalyst Performance in “Click” Coupling Reactions of Polymers Prepared by ATRP: Ligand and Metal Effects. Macromolecules 2006, 39, 6451–6457. 10.1021/ma061592u. [DOI] [Google Scholar]
- Gillies E. R.; Jonsson T. B.; Fréchet J. M. J. Stimuli-Responsive Supramolecular Assemblies of Linear-Dendritic Copolymers. J. Am. Chem. Soc. 2004, 126, 11936–11943. 10.1021/ja0463738. [DOI] [PubMed] [Google Scholar]
- Amir R. Enzyme-Responsive PEG-Dendron Hybrids as a Platform for Smart Nanocarriers. Synlett 2015, 26, 2617–2622. 10.1055/s-0035-1560522. [DOI] [Google Scholar]
- Segal M.; Avinery R.; Buzhor M.; Shaharabani R.; Harnoy A. J.; Tirosh E.; Beck R.; Amir R. J. Molecular Precision and Enzymatic Degradation: From Readily to Undegradable Polymeric Micelles by Minor Structural Changes. J. Am. Chem. Soc. 2017, 139, 803–810. 10.1021/jacs.6b10624. [DOI] [PubMed] [Google Scholar]
- Slor G.; Papo N.; Hananel U.; Amir R. J. Tuning the Molecular Weight of Polymeric Amphiphiles as a Tool to Access Micelles with a Wide Range of Enzymatic Degradation Rates. Chem. Commun. 2018, 54, 6875–6878. 10.1039/c8cc02415d. [DOI] [PubMed] [Google Scholar]
- Lübtow M. M.; Keßler L.; Appelt-Menzel A.; Lorson T.; Gangloff N.; Kirsch M.; Dahms S.; Luxenhofer R. More Is Sometimes Less: Curcumin and Paclitaxel Formulations Using Poly(2-oxazoline) and Poly(2-oxazine)-Based Amphiphiles Bearing Linear and Branched C9 Side Chains. Macromol. Biosci. 2018, 18, 1800155. 10.1002/mabi.201800155. [DOI] [PubMed] [Google Scholar]
- Seo Y.; Schulz A.; Han Y.; He Z.; Bludau H.; Wan X.; Tong J.; Bronich T. K.; Sokolsky M.; Luxenhofer R.; Jordan R.; Kabanov A. V. Poly(2-Oxazoline) Block Copolymer Based Formulations of Taxanes: Effect of Copolymer and Drug Structure, Concentration, and Environmental Factors. Polym. Adv. Technol. 2015, 26, 837–850. 10.1002/pat.3556. [DOI] [Google Scholar]
- Hahn L.; Lübtow M. M.; Lorson T.; Schmitt F.; Appelt-Menzel A.; Schobert R.; Luxenhofer R. Investigating the Influence of Aromatic Moieties on the Formulation of Hydrophobic Natural Products and Drugs in Poly(2-Oxazoline)-Based Amphiphiles. Biomacromolecules 2018, 19, 3119–3128. 10.1021/acs.biomac.8b00708. [DOI] [PubMed] [Google Scholar]
- Singla A. K.; Garg A.; Aggarwal D. Paclitaxel and Its Formulations. Int. J. Pharm. 2002, 235, 179–192. 10.1016/s0378-5173(01)00986-3. [DOI] [PubMed] [Google Scholar]
- Cao C.; Zhao J.; Chen F.; Lu M.; Khine Y. Y.; Macmillan A.; Garvey C. J.; Stenzel M. H. Drug-Induced Morphology Transition of Self-Assembled Glycopolymers: Insight into the Drug-Polymer Interaction. Chem. Mater. 2018, 30, 5227–5236. 10.1021/acs.chemmater.8b01882. [DOI] [Google Scholar]
- Vilanova O.; Mittag J. J.; Kelly P. M.; Milani S.; Dawson K. A.; Rädler J. O.; Franzese G. Understanding the Kinetics of Protein-Nanoparticle Corona Formation. ACS Nano 2016, 10, 10842–10850. 10.1021/acsnano.6b04858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J.; Lee S. H.; Onogi S.; Shea K. J. Engineering the Protein Corona of a Synthetic Polymer Nanoparticle for Broad-Spectrum Sequestration and Neutralization of Venomous Biomacromolecules. J. Am. Chem. Soc. 2016, 138, 16604–16607. 10.1021/jacs.6b10950. [DOI] [PubMed] [Google Scholar]
- Obst K.; Yealland G.; Balzus B.; Miceli E.; Dimde M.; Weise C.; Eravci M.; Bodmeier R.; Haag R.; Calderón M.; Charbaji N.; Hedtrich S. Protein Corona Formation on Colloidal Polymeric Nanoparticles and Polymeric Nanogels: Impact on Cellular Uptake, Toxicity, Immunogenicity, and Drug Release Properties. Biomacromolecules 2017, 18, 1762–1771. 10.1021/acs.biomac.7b00158. [DOI] [PubMed] [Google Scholar]
- Settanni G.; Zhou J.; Suo T.; Schöttler S.; Landfester K.; Schmid F.; Mailänder V. Protein corona composition of poly(ethylene glycol)- and poly(phosphoester)-coated nanoparticles correlates strongly with the amino acid composition of the protein surface. Nanoscale 2017, 9, 2138–2144. 10.1039/c6nr07022a. [DOI] [PubMed] [Google Scholar]
- Schöttler S.; Becker G.; Winzen S.; Steinbach T.; Mohr K.; Landfester K.; Mailänder V.; Wurm F. R. Protein Adsorption Is Required for Stealth Effect of Poly(Ethylene Glycol)- and Poly(Phosphoester)-Coated Nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. 10.1038/nnano.2015.330. [DOI] [PubMed] [Google Scholar]
- Liu B.; Thayumanavan S. Importance of Evaluating Dynamic Encapsulation Stability of Amphiphilic Assemblies in Serum. Biomacromolecules 2017, 18, 4163–4170. 10.1021/acs.biomac.7b01220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teunissen A. J. P.; Pérez-Medina C.; Meijerink A.; Mulder W. J. M. Investigating supramolecular systems using Förster resonance energy transfer. Chem. Soc. Rev. 2018, 47, 7027–7044. 10.1039/c8cs00278a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.; Zhuang J.; Wang F.; Raghupathi K. R.; Thayumanavan S. Protein and Enzyme Gated Supramolecular Disassembly. J. Am. Chem. Soc. 2014, 136, 2220–2223. 10.1021/ja4108676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molla M. R.; Prasad P.; Thayumanavan S. Protein-Induced Supramolecular Disassembly of Amphiphilic Polypeptide Nanoassemblies. J. Am. Chem. Soc. 2015, 137, 7286–7289. 10.1021/jacs.5b04285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amado Torres D.; Garzoni M.; Subrahmanyam A. V.; Pavan G. M.; Thayumanavan S. Protein-Triggered Supramolecular Disassembly: Insights Based on Variations in Ligand Location in Amphiphilic Dendrons. J. Am. Chem. Soc. 2014, 136, 5385–5399. 10.1021/ja500634u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rösler A.; Vandermeulen G. W. M.; Klok H.-A. Advanced Drug Delivery Devices via Self-Assembly of Amphiphilic Block Copolymers. Adv. Drug Deliv. Rev. 2012, 64, 270–279. 10.1016/j.addr.2012.09.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.