

Abstract
Protein mass spectrometry (MS) is an enabling technology that is ideally suited for precision diagnostics. In contrast to immunoassays with indirect readouts, MS quantifications are multiplexed and include identification of proteoforms in a direct manner. Although widely used for routine measurements of drugs and metabolites, the number of clinical MS-based protein applications is limited. In this paper, we share our experience and aim to take away the concerns that have kept laboratory medicine from implementing quantitative protein MS. To ensure added value of new medical tests and guarantee accurate test results, five key elements of test evaluation have been established by a working group within the European Federation for Clinical Chemistry and Laboratory Medicine. Moreover, it is emphasized to identify clinical gaps in the contemporary clinical pathways before test development is started. We demonstrate that quantitative protein MS tests that provide an additional layer of clinical information have robust performance and meet long-term desirable analytical performance specifications as exemplified by our own experience. Yet, the adoption of quantitative protein MS tests into medical laboratories is seriously hampered due to its complexity, lack of robotization and high initial investment costs. Successful and widespread implementation in medical laboratories requires uptake and automation of this next generation protein technology by the In-Vitro Diagnostics industry. Also, training curricula of lab workers and lab specialists should include education on enabling technologies for transitioning to precision medicine by quantitative protein MS tests.
Introduction
Proteins have been measured in clinical laboratories as markers for several disease states since the early examples of urinary albumin as indicator of kidney disease in 1827 and the first tumor marker (Bence Jones protein) in 1845.1 Nowadays, proteins are routinely quantified using automated immunoassays, but it is widely acknowledged that this technique has several major drawbacks.2 In the past decades, mass spectrometry (MS) has demonstrated feasibility for quantitation of protein biomarkers with clear advantages compared to immunoassays.1,2 Such MS protein tests follow a bottom-up strategy, where peptide quantities are used to determine concentrations of protein targets and isoforms for specific clinical indications.
Hitherto, the adoption of MS-based protein tests in medical laboratories is rather limited, likely because the workflow of multiplexed (proteoform) testing is relatively complex, quality requirements with regard to allowable measurement uncertainty are stringent, and automation is not in place yet. It is of key importance to follow quality requirements that are common practice for all tests within medical laboratories. The earliest guidance for evaluation of liquid chromatography (LC) MS-based methods started from experience with small molecules3,4 and was based on recommendations of the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA). More specific LC–MS requirements have been available since the publication of guideline C50-A from the Clinical and Laboratory Standard Institute (CLSI) in 2007,5 which was later revised into a more systematic approach for development and validation of LC–MS methods in the CLSI C62-A guideline.6,7 At the same time, the MS-based proteomics community proposed a three tier system using a fit-for-purpose approach for the discovery of protein biomarkers and anticipated translation into a medical test.8 This latter guidance document provides a good starting point for analytical validation but it does not include clinical validation. For the development of tier 1 assays that are suitable for implementation as a clinical test, it does not address their adoption in medical laboratories. A CLSI document C64 dedicated to quantitation of proteins by MS techniques is currently being drafted.9 As of May 2022, in Europe, the development of MS-based tests will also have to be compliant with the new regulations for In-Vitro Diagnostics (IVDR) 2017/746.10 A working group within the European Federation for Clinical Chemistry and Laboratory Medicine (EFLM) has established a cyclical framework for test evaluation of both CE-IVDs and laboratory-developed tests (LDTs) (Figure 1).11,12 Importantly, development of an LC–MS method for quantitation of (multiple) biomarkers should only be commenced if there is an unmet clinical need in the clinical care pathway.11−13 Recently, a checklist was reported to aid in the discussions with clinicians to identify and verify current clinical needs.14 Once the clinical gaps are identified, and the decision is made to invest in solving the gap, a test can be developed. This process consists of a preparation phase, followed by the method development phase and subsequent analytical and clinical validation of the anticipated test (Table 1), which should be in compliance with the upcoming IVDR 2017/746.10,15 The total test process including implementation in a routine setting should be considered at an early stage in the development. This ensures high quality tests that meet predefined analytical and clinical performance specifications. From a technical point of view, proteotypic peptides are commonly measured using a triple quadrupole (QQQ)-instrument.8,16,17 The peptide quantities reflect protein concentrations, provided that digestion conditions allow equimolar conversions.18,19 Veracity of the protein quantity is inferred via agreement between multiple proteotypic peptides.20 Furthermore, it is assumed that the protein of interest is present in the intact form in the matrix and that digestion is not hampered by potential post-translational or chemical modifications.21 Ideally, these assumptions should be checked and corrected for during rigorous analytical method validation.
Figure 1.
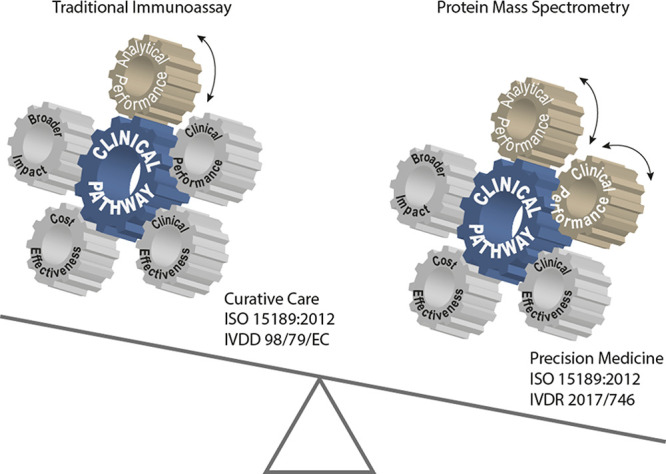
Protein MS as an alternative technology for immunoassay-based testing, potentially providing an additional layer of clinical information. The interplay between the key elements of the test evaluation process is summarized in a cyclical framework. The wheels dynamically link the elements that are driven by the clinical needs and intended use of the envisioned test in a clinical pathway aiming for improved patient outcome. In this paper, analytical and clinical performance of MS-based protein testing is discussed. Evaluation of the other three elements of the cyclical framework requires more data that will come available once MS-based protein testing is adopted in medical laboratories and used for longer time to evaluate thousands of specimens.
Table 1. Quality Criteria during Preparation and Implementation Phases of a Quantitative Protein MS Method to Be Used in Clinical Chemistry Routine Practice and Ahead of Clinical Evaluations.
| no. | phase | action | key ref |
|---|---|---|---|
| 0 | identifying and verifying an unmet clinical need | (11, 12) | |
| 1 | preparation phase including preanalytics | defining the measurand(s) | (22, 23) |
| setting clinical and analytical performance specifications; conceptualize total testing process | (30−33) | ||
| preanalysis; prevention of diagnostic errors at the stage of specimen collection | (36, 38, 39) | ||
| 2 | method development and analytical performance evaluation | selection of proteotypic peptides; quantitation and confirmation | (19, 46, 47) |
| sample preparation; digestion conditions and use of proteases | (50, 51, 54) | ||
| internal standard strategies; from SIL peptide to SIL protein and cost effectiveness | (20, 71) | ||
| peptide prepurification; sample clean up and solid phase extraction (SPE) | (76, 77) | ||
| LC–MRM–MS/MS analysis; data evaluation, separation of peptides, (dynamic) MRM detection, ion ratio selectivity | (7, 49, 78) | ||
| calibration and traceability to certified reference materials | (10, 79) | ||
| analytical validation | (6, 15, 81) | ||
| maintaining long-term accuracy; documented in quality management system (QMS) | (81) | ||
| 3 | implementation | education and training; theoretical and practical | (83, 84) |
| achieving consistent instrument performance | (81) | ||
| maintenance plans for LC–MS equipment | |||
| system suitability testing | (49,85) | ||
| application checks | |||
| internal quality control | (81, 89, 90) | ||
| external quality assessment | (93−95) | ||
| total testing checks | |||
| troubleshooting: manual/instrument mistakes | |||
In this paper, we focus on the potential of MS-based protein testing as an alternative technology for immunoassay-based testing. Stringent analytical performance and robustness have to be demonstrated before this enabling technology can be adopted into clinical practice. Different phases of test development will be highlighted as well as the rational selection of analytical performance specifications needed for ensuring the intended use of the test. The latter is based on the Milan hierarchy and goes beyond the specific LC–MS criteria for clinical adoption that we reported earlier.4
We conclude with presenting longitudinal, four-year analytical performance data of a multiplexed test for the quantitation of serum apolipoproteins.
Achieving Long-Term Robustness: The Preparation Phase
Test development should start with identification and verification of unmet clinical needs within the current clinical pathway (Table 1). Next, the candidate markers that address this need are determined11,12 followed by a preparation phase that includes defining the measurand(s), predefining analytical and clinical performance specifications, and the purpose and mode of the test in the clinical pathway. Preferably a business-case is also developed. Short-sightedness during the preparation phase will undoubtedly lead to difficulties during method development and possibly result in suboptimal test performance and/or research waste.
Defining the Measurand(s)
A measurand is defined as “a quantity intended to be measured”. Although it may seem trivial to identify the protein measurand, it is emphasized that products from a single gene can be presented as a plethora of so-called proteoforms.22 These proteoforms arise from genetic variations or alternative splicing, but importantly, they can also be the result of post-translational modifications (PTMs). Potential PTMs have been reviewed earlier23 and include, but are not limited to phosphorylation, methylation, acetylation and glycosylation. For these reasons a protein measurand should be defined at the molecular level. Depending on the clinical relevance of the proteoforms, the measurand could be defined as the sum of all proteoforms originating from a single gene24 (e.g., cardiac troponin25), a subgroup thereof (e.g., specific isoform such as IgG426), or even a single proteoform (e.g., carbohydrate deficient transferrin).27,28 The clear definition of the measurand is thus needed to ensure the intended analyte is indeed targeted and quantified. Besides the molecular definition of the analyte the matrix from which the analyte is intended to be measured also requires careful consideration.
Setting Clinical and Analytical Performance Specifications
To ensure that a developed test is fit for clinical purpose, the predefined clinical performance specifications (CPS) should determine the required analytical performance specifications (APS) prior to the initiation of the method development. A hierarchy for APS has initially been developed in the (Stockholm) consensus agreement formulated in 1999 and has been revisited, named the Milan hierarchy, in 2014 by the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM).29 To ensure that the APS of new tests are in agreement with the required clinical performance, analytical performance criteria should ideally be set based on clinical outcome studies. However, as such data is often not available,30 a second strategy is to use biological variation data from intraindividual and interindividual variations (CVw and CVg, respectively). In this approach, biological variation is the basis for establishing the minimal, desirable and optimal goals for imprecision (CVa) and bias (Ba) and calculation of total allowable error for the biomarker test.31,32 Recently a checklist was established to evaluate studies assessing biological variation33 and a database with biological variation data may be found at https://biologicalvariation.eu. If biological variation data is not available, APS should be set based on the state-of-the-art performance. For LC–MS based protein quantitation, analytical CVs of <10% should be possible based on our own experience.34,35 A final consideration in the preparation phase addresses the clinical pathway and role of the test in the pathway.
Preanalysis
The preanalytical phase occurs both outside and inside a medical laboratory. The former includes patient identification and specimen collection and transport, whereas the latter starts with specimen arrival and pre-examination, followed by possible centrifugation (of blood), decapping, and aliquoting. It should be recognized that the preanalysis is very critical for good biomarker research. Therefore, the provenance and quality of the clinical specimens should be documented and validated.36 These challenges are often underestimated. Earlier, the HUPO plasma proteome project identified that the sample processing (time on the bench, centrifugation speed) all affect the quality of the specimen and subsequent analyte recovery.37,38 Moreover, the efficiency of enzymatic digestion may be altered depending on the matrix,37,39,40 and stability of proteins or proteoforms may vary in certain matrices. Therefore, the preanalytical phase and specimen collection tubes require careful consideration. In addition, the absolute concentration of the measurand determines the sample preparation strategy, as preconcentration might be necessary, e.g., using MSIA or SISCAPA (Mass Spectrometric ImmunoAssay or Stable Isotope Standards and Capture by Anti-Peptide Antibodies) approaches.41,42 Historically, serum has been the matrix of choice for archiving patient materials in medical laboratories, but in general matrix choice is highly laboratory dependent.37,39,40 However, it should be emphasized that development and validation of an MS-based protein test in a specific matrix certainly does not guarantee accurate results in a different matrix. Therefore, all of these parameters have to be thoroughly investigated.
Achieving Long-Term Robustness: Method Development and Analytical Performance Evaluation
Quantitation of proteins by MS via the bottom-up proteomics approach involves protein digestion with subsequent MS detection of the proteotypic peptides measured against an internal standard, thus reflecting the original concentration of the target protein.16 A three tier system8 has been proposed for the different levels of confidence and rigor of the technique used, with only tier 1 type tests suitable for implementation in the medical laboratory. In our previous work, various quality criteria were summarized including clinical chemistry principles for method development of quantitative bottom-up proteomics from the preanalysis to postanalysis.4 Furthermore, a recently proposed guideline structures the minimum information on a developed method that is required for publication of quantitative bottom-up proteomics tests in a clinical setting.43 For absolute quantitation of target proteins, the establishment of a traceability chain according to ISO 17511:2020 is essential.
Peptide Selection
Quantitation of proteins via the bottom-up approach involves digestion of proteins by proteases (generally trypsin) into “signature” or proteotypic peptides that are detected using multiple reaction monitoring (MRM) in a triple quadrupole MS-system combined with LC. It is of great importance to benchmark genetic variations of a certain protein across populations, and to periodically keep track of reported mutations.44 To further ensure accurate quantitation of proteins, it is recommended to measure at least two peptides per protein. It may be worthwhile to follow up multiple peptides in the early stage of method development19,45 in order to have two remaining peptides per protein at the final stage. Some general rules apply for the best choice of peptides for optimal quantitation.46,47 Preferably, proteotypic peptides should not contain missed cleavage variations or unstable amino acids (cysteine, methionine, tryptophan, asparagine, or glutamine) that could undergo chemical modifications19,48 and they should not contain any PTMs or any known amino acid changes due to genetic variation. Moreover, transitions with interferences should be avoided.49 Finally, when (two) optimal peptides are selected for a protein the best performing peptide may then serve as the quantifying peptide while the other is used for (qualitative) confirmation.35
Proteolysis
The procedure of bottom-up sample preparation for protein quantitation should be tightly controlled and lead to equimolar conversion of proteins into quantifier peptides for the sake of accurate test results. Therefore, the experimental conditions should be optimized to ensure the stable production of peptides without missed cleavages.50,51 Standard procedure for protein digestion involves denaturation and reduction of the proteins and alkylation of SH-groups. The fully unfolded proteins can then be split by proteolytic digestion cutting at specific residues (lysine and arginine for trypsin).52,53 The efficiency of the tryptic digestion has been tested for various combinations of temperature, solvents, chaotropic agents and surfactants and optimal conditions can be demonstrated by time curves for the peptide generation.54 However, the time curves for peptide generation nicely demonstrate that optimal conditions for different peptides are not unambiguous and some peptides are produced faster and/or with higher yield using different surfactant or solvents.54 For quantitation of a large panel of protein biomarkers it will therefore be a major challenge to create uniform optimal conditions for absolute yields of all signature peptides. Next to optimal digestion conditions, activity and constant quality of the proteolytic enzyme for cleaving the protein should be guaranteed. Variability of proteolytic activity has been described for different types and sources of trypsin, which can influence digestion efficiency.55,56 Important for trypsin stability is preventing autoproteolysis.57−60 Digestion performance can be tested in various ways, e.g. by monitoring missed cleavages or other peptide variants19,61 or by using commercially available quality control samples.62
Internal Calibration Strategies
With regard to internal standards (IS), several strategies can be used from addition of the stable isotope labeled (SIL) peptide only, to addition of a full length SIL protein.63 The use of SIL peptide can correct for losses of the target peptide. It is expected that modifications to the endogenous peptide during digestion will also occur for the SIL peptide and the relative ratio between endogenous and SIL peptide thus remains constant.19,64 Addition of SIL peptides with different labels at different time points before and after the digestion period enables the comparison of endogenous peptides produced relative to the labeled peptides present during the digestion and thus may be used to demonstrate changes of the (SIL) peptides during the sample preparation workflow.61,65,66 Other solutions for internal standard strategies that have been explored are winged peptides (with sequences that still require tryptic cleavage).67,68 Also SIL peptides can be obtained from so-called Protein Epitope Signature Tags (PrESTs),69 containing a short and unique sequence of a protein of interest which can be produced in E. coli. Alternatively, artificial proteins can also be produced as concatamers of standard (Q) peptides (QCATs or QconCATs) which have the advantage of generating (SIL) peptides by endoproteolytic or chemical cleavage at once for multiple proteins.70 The use of full length SIL proteins may be considered the “gold standard” for the accurate quantitation of proteotypic peptides by LC-MRM-MS/MS.71−73 However, SIL versions of proteins are often not available, or at best as recombinant versions that are extremely costly. To this end, it is noted that digestion kinetics of a recombinant protein are not necessarily identical to those isolated from a native human matrix. The recombinant protein may differ with regard to secondary, tertiary or quaternary structure from the endogenous one, or differ in PTM content (or proteoform profile), thereby introducing a digestion bias. This difference between native and recombinant proteins should also be considered when selecting calibration materials.20 Since cost effectiveness is also a key component in the cyclical framework during evidence-based test evaluation30 the use of SIL proteins would make the test unaffordable, especially for highly multiplex protein assays.
Peptide Prepurification
For LC–MS analysis of crude serum samples after digestion cleanup of the sample reagent mixture is wanted in order to protect the analytical column and mass spectrometer from digest reagents and other serum or sample components. While it may be necessary to employ immunoaffinity based SISCAPA to enhance analytical sensitivity, peptide prepurification using either an in-line trapping column or cartridge or plate based solid phase extraction is often sufficient. A trapping column may be used online where peptides are bound and contaminating components are removed to waste before the start of the gradient elution. Next, the flow is switched in line with the analytical column for separation and elution of peptides and their subsequent MS measurement.35,45 However, thorough cleanup of the sample digests by offline solid phase extraction (SPE), may be preferred.74 Specific anion- or cation-exchange cartridges have been shown useful for peptide purification.75 SPE with hydrophilic lipophilic balanced (HLB) cartridges has been applied in various studies.76,77 In case that peptides elute directly from a trapping column onto the analytical column potential losses can become apparent from, for example blank runs between different samples. In case SPE is performed in an offline manner, clean up and recovery of the peptides will depend on both the pH and the percentage of organic solvent used for elution. Optimal conditions can be established experimentally for the set of peptides applicable.
The SPE cartridges efficiently remove matrix interferences, such as phospholipids and salts and thus potentially reduce ion suppression. The removal of matrix interferences by SPE is an important step in the long term protection of the LC column and stability of the MS triple quadrupoles as compared to the recurrent use of a trapping column. Furthermore, the 96-well format of SPE cartridges enables concentration of the sample in a (semi)-automated fashion.
LC–MRM–MS/MS Analysis
For the LC–MS measurement of multiple proteins a quantitation and confirmation peptide are measured for each protein. For accurate quantification of molecular proteoforms and/or specific proteins the metrological traceability concept has to be implemented according to ISO 17511:2020 calibration hierarchies. Key for accurate quantification is the unequivocal definition of the measurand. Clearly, when applying quantitative bottom up proteomics, the intact proteoform is not measured but rather a proteotypic peptide that is formed after equimolar digestion. It is emphasized that evaluation of peptide–protein equimolarity is crucial for each selected proteotypic peptide. For each peptide at least two, but preferably three MRM transitions are selected with one serving as the quantifying transition and others as qualifier transitions. The peptide MRM transitions should confirm ion ratios for both the endogenous and SIL-peptide. In order to minimize total analysis times, the high-throughput requirements determine limited LC-runtimes for routine applications in medical laboratories.
Data Evaluation
Peptide retention times have to remain stable between runs and from batch to batch in order to ensure proper integration of peak areas. In general multiple product ions of a peptide precursor ion will be detected and the combination of both with the most stable (and often highest) signal is used as quantifying transition as opposed to the sum of multiple transitions also used in research grade assays. For the other product ions a certain transition ratio will be found compared to the quantifying transition which can be used for confirmation of identity of the peptide. In case of interfering peptides with the same combination of precursor and product ion eluting at (almost) the same retention time the transition ratios will be influenced.49,78 According to CLSI guidelines5,6 deviation of transition ratios should be <20% from the standard ratio and such interferences should not influence the calculated concentrations of the peptide (protein) based on the quantifying transition7 which would be the case with summation of all transitions measured. An interference could be visible by changes in peak shape of a quantifying or qualifying transition. Outlier set up for peak width, symmetry and full width at half-maximum (fwhm) will detect double peaks or obvious peak broadening for one of the transitions thus identifying interferences of different precursor/product combination. In case interferences are observed a different choice of transitions could solve the problem. Alternatively, specific SPE conditions for cleanup of the sample could be altered or separation of the peptides could be adjusted by changing gradient conditions for elution.
Calibration and Metrological Traceability: Selection of Calibrators
In laboratory medicine the concept of metrological traceability is defined by the Joint Committee for Guides in Metrology as “the property of a measurement result whereby the result can be related to a reference through a documented unbroken chain of calibrations, each contributing to the measurement uncertainty”.79 This concept is the basis for standardization and harmonization of medical tests; making test results comparable to allow universal exchange of patient laboratory data between institutions, nationally and at a global level.10 For quantitative proteomics tests aimed for clinical application a calibration strategy is required that takes into account metrological traceability of test results to the highest possible order if available. Therefore, calibrators should be used that guarantee traceability to secondary and finally primary reference materials and behave in a common way. An example of a traceability chain to SI is depicted in Figure 2 (source: ISO 17511:2020), recently discussed in more detail.80
Figure 2.
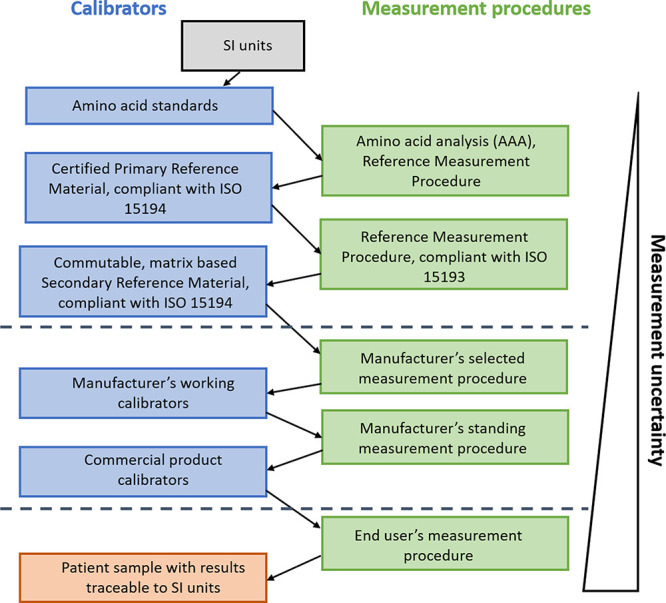
Full metrological traceability toward International System Units (SI). Modified from ISO 17511:2020.
Analytical Validation
Thorough analytical validation is a prerequisite for ISO 1518981 accreditation (https://www.iso.org/standard/56115.html) and will also be required under the new EU regulatory framework IVDR 2017/746.82 A structured framework and detailed guideline for validation of new MS methods that may also be valid for protein quantitation has been developed within the CLSI guideline C62-A.6 Analytical performance recommendations are defined for bias, imprecision, sensitivity, specificity, matrix effects, carry over, interference, and stability. Various CLSI EP protocols are mentioned that will provide further information on the exact procedures to be applied. Acceptance criteria for e.g. imprecision, trueness and carry over are deduced from biological variation (if known) and should be selected in such a way that the test can meet its intended use.6 In case a test is foreseen on dedicated LC–MS instrument, the criteria for cleanliness and compatibility of the samples are much less stringent than when the test is performed on an instrument already in use for other applications. In the latter case, care should be given not to introduce additional test-to-test interferences.
Achieving Long-Term Robustness: Implementation
To achieve long-term accuracy of quantitative proteomics MS tests both the analytical system (all equipment needed for the test), and the application should consistently meet the predefined analytical performance specifications as outlined in the preparation phase. Adequate implementation of the complex MS technology within the routine medical laboratory setting is therefore important. In the laboratory quality management system (QMS), special attention should be given to the education of personnel and staff. Moreover, checks should be established to ensure consistent performance of the total testing process, including, the instrumentation as well as the application.
Education of Medical Laboratory Technicians and Staff
To ensure that medical tests provide accurate clinical information, ISO guidelines require in general proper training of the work force on the procedures needed for the application.81 In the case of mass spectrometry, most technicians and lab staff do not have a background in analytical chemistry and are less familiar with mass spectrometry.83 Often, they have to be introduced to the complex analysis of quantitative bottom-up proteomics. MS-based quantitation of protein biomarkers is specialized work, which requires a thorough educational process that should be provided to employees at different levels from hands on technical staff to those dealing with management and directing biomarker quantitation for clinical utility.84 The training program should at least consist of a theoretical section, that could be attended by both technicians and staff, and hands-on training, preferably specifically on the intended application, for the technicians. In our setting we entrust the development of MS-based methods to analytical chemists whereas the stable operation of LDTs is done by clinically trained lab technicians. For safeguarding the long-term robustness of the LDTs supervision of lab technicians on quality assurance aspects such as monitoring SST and QC results are a prerequisite. To make the MS-based test development sustainable, the head of the clinical chemistry department has established a multidisciplinary collaboration and alliance with analytical chemists (mass spectrometrists) for initial development but also throughout to provide an entry for troubleshooting and test improvement. Bridging technological knowledge on one side and clinical quality—according to ISO 15189:2012—on the other side required continuous exchange, collaboration, and communication.
Theoretical Training
The theoretical training program should ideally cover all phases of method development (see previous sections), to ensure that all individuals working on the LC–MS-based protein applications are aware of the strengths and potential flaws of the technique. Therefore, the theoretical section should include an introduction to QQQ-MS to understand the potential for analytical sensitivity and specificity, the basics of bottom-up proteomics (including the essentials of sample preparation, the use of internal standards and the design of calibration strategies) and, importantly, also data analysis, with an emphasis on the use of retention time, peak shape and area, ion ratios and concordance between peptides to monitor quality of the results. Such theoretical training programs focused on proteomics applications are generally not available and hence, the training should be tailor-made. Alternatively, the knowledge may be acquired through several summer schools, short courses and workshops. Several courses on the basics of mass spectrometry are available, including the North American Mass Spectrometry Summer School (https://www.ncqbcs.com/resources/training/summer-school/) or the summer school in Mass Spectrometry in Biotechnology and Medicine (http://msbm.org/). Similarly, summer schools dedicated to proteomics are also available (summer school advanced proteomics, http://www.proteomic-basics.eu). An alternative option could be short courses offered in concordance with scientific meeting such as Mass Spectrometry: Applications to the Clinical Laboratory (MSACL, https://www.msacl.org/) or the American Society of Mass Spectrometry (ASMS, https://www.asms.org). More recently, the American Association for Clinical Chemistry (https://www.aacc.org) now also offers online LC–MS education for clinical applications.
Practical Training
When starting with LC–MS in a clinical laboratory, the technicians that will be running the applications may not be familiar with the concept of quantitative proteomics and practical training is necessary. Courses on proteomics and MS should cover theoretical aspects as well as contain hands-on practical sessions. It is noted that the exact training needed is highly dependent on the application, and therefore, it is advised to do practical training on-site on the specific application and instrumentation. The results of an educational program should be documented in the QMS. Moreover, roles and responsibilities for employees should be based on their level of training and expertise and should be identified within the QMS of the lab.
Achieving Consistent Instrument Performance
Besides proper training of personnel, precautionary measures should be in place to ensure that the performance of both the instrumentation as well as the application are robust. To ensure that the instrumentation performs accurately, it should be maintained properly. However, just proper maintenance is no guarantee for accurate performance. Therefore, system suitability checks should also be performed. To ensure accurate test results, matrix-based internal and external quality control materials are used, similarly to other (non LC–MS based) test applications (Figure 3).
Figure 3.
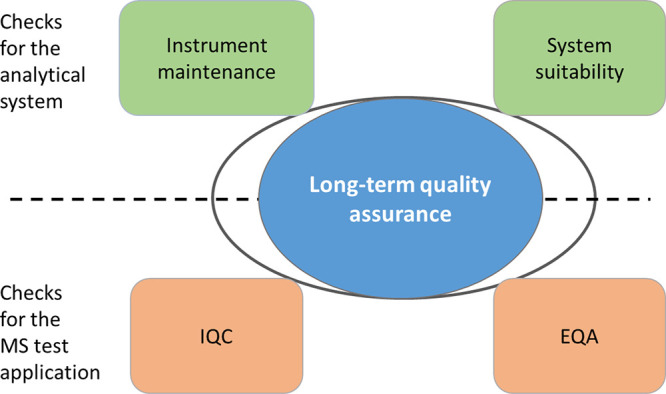
Long-term accuracy requires meeting instrument performance indicators and test application performance criteria.
Instrument Maintenance
To ensure that the instruments used for quantitative proteomics by LC–MS are in good order, maintenance should be performed at three levels: regularly, annually and incidental in the case of malfunction. A laboratory technician should be trained to perform regular maintenance. This includes at least cleaning of the ion source in a weekly or biweekly interval, as well as cleaning of the inlet capillary every 3–6 months, depending on the occupancy rate of the instrument. Annual maintenance mostly pertains to cleaning of the LC, with an emphasis on the pump heads and changing of the pump oil of the MS rough pumps. Annual maintenance may be performed using a well-trained technical specialist, or could be outsourced to the instrument manufacturer or dedicated maintenance companies, for instance through service contracts. If a laboratory chooses not to use a service contract, it should ensure to have a well-trained LC–MS technical specialist who is capable of annual maintenance and incidental repairs.
System Suitability Testing
System suitability testing (SST) is a highly valuable procedure that is often overlooked during method development and implementation. Often, laboratories believe that the use of internal quality control (IQC) is sufficient to detect method errors and incorrect results. However, underlying instrument errors are sometimes masked by accurate IQC results.85 Moreover, SST aids in detection of instrument errors and convincing of instrument manufacturers of malfunctioning instrumentation.
System suitability testing should consist of two phases: one is the accurate and consistent use of logbooks. It is important to monitor the vacuum, column temperature, flow rate, column pressure and precision of calibration, as well as any maintenance procedure.86 Besides logbooks, system suitability testing should comprise repeated measurement of a well-defined sample, that reflects the biological analytes. For peptide-based quantitation, this can easily be accomplished using a mixture of synthetic peptides reflecting both endogenous and stable isotope labeled peptides. Briscoe et al.85 described a design for an SST used in biopharmaceutical analysis. In the design, samples are tested at the start, middle, and end of a batch of samples. Using this strategy, the SST can detect changes in analytical sensitivity, analytical specificity, precision and carry over for relevant compounds of a test. Abbatiello et al.49 described a system suitability test making use of a predigested sample of six proteins that was used for evaluation of different LC–MRM–MS/MS platforms in 11 laboratories. Here, several chromatographic and MS performance metrics were compared such as retention time drifts, MS detector responses, chromatographic resolution, peak capacity and tailing and carry over, demonstrating abnormalities in LC and MS performance among the participating laboratories. The aim of SST was to define the criteria to optimize replication of results within the LC–MRM–MS/MS platform. For comparison of SST at multiple sites longitudinal monitoring has been proposed based on Statistical Process Control (SPC) using the software package MSstatsQC in accordance with that for quality control samples using Levey-Jenning plots.87,88 Automatic flagging for unwanted deviations can help to detect failures of the system regarding response, signal-to-noise, retention times and peak shape.88 Only when the system fulfills the criteria of SST it is allowed to proceed with measurement of the actual samples, calibrators and IQCs.
Application Checks
Besides ensuring that the instrument is performing according to the specifications, it is also necessary to be convinced of the accurate performance of the full application. This may be done according to typical laboratory medicine procedures of IQC and external quality assessment (EQA). The procedures may be very similar to the ones already in use for batch processes, although in a multimarker quantitative protein MS test the selection of samples with appropriate levels for all proteins may be difficult.
Internal Quality Control (IQC)
Quality control samples for any diagnostic test are compulsory for ISO 1518981 accreditation, and often at least bilevel IQC samples are used. IQC should automatically detect batches in which errors occur, without rejecting accurate batches.4 Ideally, this is performed through a standard procedure, often based on Westgard rules,89 and results are monitored in the laboratory information system (LIS). The selection of IQC samples should be done carefully. Ideally, IQC-samples are native, matrix-based materials just like the clinical samples to ensure similar behavior during sample preparation and analysis. Moreover, their stability should be assessed to ensure that the IQC materials are appropriate for longer-term monitoring of test performance. A general recommendation is to select IQC concentrations at or near medical decision points, but this is often not feasible for multiplexed tests. Behavior and trends of the IQC results around their respective targets can be followed in the so-called Levey-Jennings plots87 and violation of the rules will be checked and should lead to corrective measures. For uniplex tests the IQC is typically performed at two levels, a physiological and pathological concentration, but it should be noted that this is often not feasible for multimarker panels. Here, a pragmatic choice may be taken by selecting available samples as long as there is spread between the samples for all important analytes. For both IQC levels concentrations are established with appropriate CLSI protocols with 1, 2, and 3 SD boundaries.89 Importantly, these levels of imprecision should be in line with the allowable imprecision and bias according to the predefined APS.90 In case of multimarker panels, each analyte should be monitored individually, and boundaries should be set individually. It has to be noted that correction for multiple comparisons may be necessary to avoid a high false rejection rate.91 In recent years, the six sigma model has been introduced for the evaluation of IQC samples,92 but so far, it has not yet been applied to LC–MS proteomics tests.
External Quality Assessment (EQA)
EQA or proficiency testing (PT) programs are meant to investigate comparability of test results between laboratories. For new LC–MS protein biomarkers EQA is often not available and in that case prolonged follow-up is required according to CLSI GP29.93 EQA materials should be targeted either by a gold standard method or assigned by a peer group means. The importance of using commutable materials for correct value assignment has been clearly stated by Miller et al.94 who described that the use of noncommutable materials will cause a matrix related bias, and inaccuracy of test results. Furthermore, when commutable samples have been used, agreement of results will reflect what would be seen for patient samples. As an example, in the Dutch EQA program the organizers make use of commutable, value-assigned materials and JCTLM listed reference measurement procedures (RMPs). This approach enables systematic monitoring of standardization efforts for years and demonstrates improvement of interlaboratory CVs for various analytes.95
Total Testing Process Checks
Besides measures to ensure accurate analytical performance of a test, the embedding of the new test in the clinical care pathway should also be in place to enable future clinical validation and implementation of a novel MS-based quantitative protein test. Therefore, a concise implementation plan is required that contains multiple checks before final approval for clinical use may be obtained. This plan should at least contain information on the embedding of the test in the clinical care pathways, routing of samples in the laboratory, implementation of the test in the laboratory information management system (LIMS), decisions on turn-around times, reference intervals or decision limits, the confirmation and authorization of patient results, and communication to the relevant medical doctors.
Robust Performance of Quantitative Bottom up Proteomics: The Example of Serum Apolipoproteins
Within our laboratory, we have recently introduced quantitation of apolipoproteins for patient diagnostics. It is well-known that current cardiovascular disease (CVD) risk assessment based on serum lipid profiling overlooks a large group of CV risk patients (including women96 and individuals with familiar dyslipidemias such as remnant disease and hyperlipoproteinemia (a)97). Moreover, a reduction in low-density lipoprotein (LDL) cholesterol through therapeutic intervention is often not sufficient to avoid further cardiovascular complications.98 There is now a substantial body of evidence for the quantitation of the functional proteins on lipoprotein particles as markers for CVD risk assessment,99−102 and it is believed that apolipoprotein quantitation could therefore fulfill the currently unmet clinical need for cardiovascular patients.103
An MS-based method was developed in accordance with CLSI guideline C62A and its intended use for precision diagnostics. First, the apolipoproteins of interest were selected: apoA-I, apoB, apoC-I, apoC-II, apoC-III, apoE, apo(a), apoA-II, and apoA-IV. For apoE the phenotype originating from its genetic polymorphism was also clinically relevant. Therefore, proteotypic peptides for each of these proteins, as well as the peptides specific for the apoE phenotypes were targeted in our LC–MS method. It was not possible to define analytical performance specifications based on outcome studies, therefore, biological variation derived analytical performance data were assessed. For apoA-I, apoB and apo(a) this resulted in TEa of 9.1%, 11.6%, and 24.1%, respectively. For the other apolipoproteins, a TEa of 20% was predefined based on the state of the art. The method development and provisional analytical validation has been described elsewhere.35 Moreover, the apoE phenotyping was in good concordance with a genotyping method.104 Analytical performance of the apolipoprotein multiplex test for precision diagnostics is optimized in such a way that the required (predefined) clinical performances for detecting and/or monitoring residual cardiovascular disease can be met. To prove the clinical value of the apo multiplex panel in addition to the conventional lipid panel, clinical outcome studies are underway that evaluate the predictive value of, e.g., the baseline apolipoprotein panel for predicting recurrent cardiovascular events.
A system suitability sample (SSS) was developed consisting of all synthetic versions of both the endogenous and stable isotope labeled peptides at a concentration of 0.15 μmol/L. Five replicate injections of (two μL of) this sample are followed by injection of a blank (the same solution without peptides) prior to a run, as well as immediately following a run.85 To ensure sufficient analytical sensitivity, a threshold was specified for the area counts of all internal standard signals (all signals >20.000 counts). For the analytical specificity, the CVs of the ion ratios (ratio between the different transitions of a single peptide6) have to be within 15%; similarly, to ensure sufficient precision, the CVs over the relative responses (endogenous/SIL) had to be <10% within five replicates, and <15% difference between the SSS before and after the run. To ensure that the full peak is being detected, the optimum of the peak should deviate no more than 12 s from the retention time specified in the method, and carry-over is monitored for all peptides as the SIL peptide area (blank)/SIL peptide area (last SSS). The maximum allowable carry-over was set at 1%. If the system suitability test failed for any of the criteria, the instrument should be checked prior to the analysis of the clinical samples.
To ensure long-term stability of the method, an external calibration procedure was developed using native human serum based calibrators, that are traceable to WHO-IFCC international reference materials SP1-01 and SP3-07 for apoA-I and apoB, respectively. Traceability of apo(a) was secondary through immunoassay measurement to SRM 2B.105 Currently, there are no reference materials available for apoC-I, C-II, C-III and E, but a working group of the International Federation for Clinical Chemistry (IFCC WG APO-MS, https://www.ifcc.org/ifcc-scientific-division/sd-working-groups/wg-apo-ms/) has developed a complete reference measurement system for global standardization of seven serum apolipoproteins.80 Long-term stability in our laboratory is exemplified for three apolipoproteins in two QC samples (Figure 4, with Levey-Jennings plots of apoA-I; (1.32 g/L) ± 0.07 (1SD 5.3%), apoB; (1.02 g/L) ± 0.04 (1SD 4.2%) and apo(a); (102.0 nmol/L) ± 7.4 (1SD 7.3%)106). We have been able to run the multiplexed apolipoprotein test stably for more than four years, even though a number of amendments were made (multiple instruments, introduction of SPE cleanup and adding additional apolipoproteins, several trypsin lots and several laboratory technicians performed the application). Given the experience with the multiplex apolipoprotein test we stress that stringent quality requirements during test development are essential for validation and implementation to achieve robust test performance.
Figure 4.
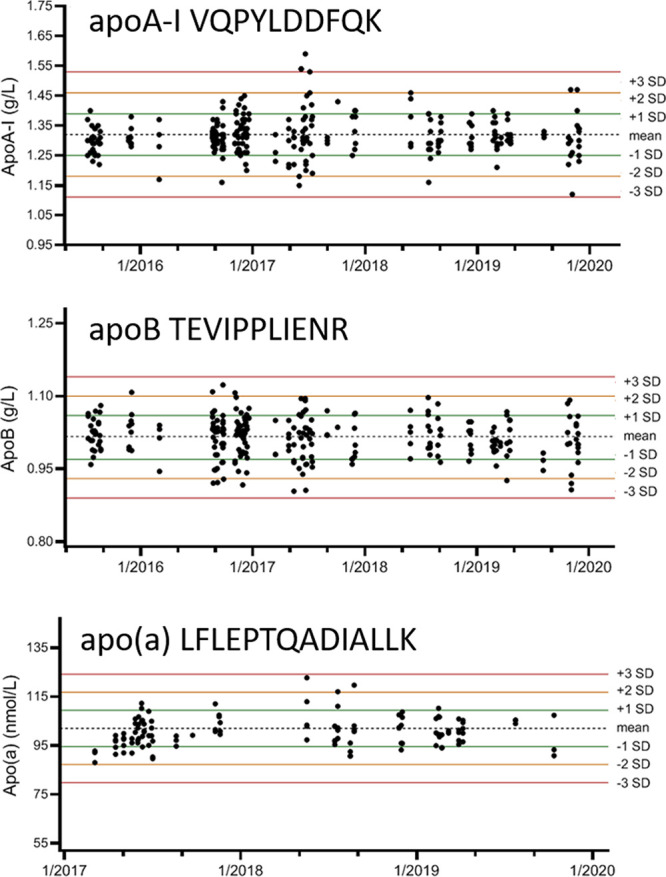
Levey–Jennings plots with concentrations of apolipoproteins A-I, B and (a) based on peptides VQPYLDDFQK, TEVIPPLIENR, and LFLEPTQADIALLK, respectively. Results from apoA-I and apoB were obtained from a single QC sample (n = 237) measured in 77 runs over four years (top and middle plot). Results from apo(a) were obtained from a second QC sample measured in 33 runs (n = 71) over three years (bottom plot). Recent measurements (2019-present) on a second Agilent 6495 QQQ-MS system in our laboratory have demonstrated similar long-term performance (data not shown).
Conclusions and Future Perspectives
In this era of steadily increasing needs for precision medicine, it is important to focus on test development and enabling technology that address clinical gaps in care pathways, in order to improve patient management and outcomes. Therefore, clinical needs should be properly identified and verified. Subsequent biomarker selection and test development and evaluation should be done in a precise manner, encompassing all key elements of the Test Evaluation cyclical framework established by the EFLM working group on Test Evaluation.11 In this paper, we present our practice for developing quantitative MS-based protein tests. Once candidate markers of interest are selected, method development should be done after considering the measurand(s) and predefining analytical and clinical performance specifications for the intended use of the test. Actual method development and analytical performance evaluation experiments should be designed according to CLSI recommendations and in agreement with ISO15189:2012. It is imperative to have proper instrument maintenance, system suitability testing, internal/external quality control procedures in place, as well as training programs for developing respectively applicating personnel. With the precautions mentioned, we have demonstrated that LC–MS based protein quantitation can be performed in a robust manner in medical laboratories for years. Yet, for worldwide uptake of these next generation MS-based protein tests we call upon the In-Vitro Diagnostics industry for automating and integrating the workflow of both the sample preparation and LC–MS technology into a convenient and robotized MS-system. Moreover, a concerted effort is needed between IVD-industry, laboratory specialists, clinicians and researchers to implement high-quality MS-based protein tests with proven clinical performance and clinical utility at affordable costs.
Acknowledgments
This work was partially funded by Roche Diagnostics NL, Leiden University Medical Centre, Leiden, NL, the Dutch Foundation for Clinical Chemistry and Laboratory Medicine to C.M.C., and an EU H2020 MSCA individual fellowship (No. 843615) to L.R.R.
The authors declare no competing financial interest.
References
- Hortin G. L.; Carr S. A.; Anderson N. L. Introduction: Advances in protein analysis for the clinical laboratory. Clin. Chem. 2010, 56, 149–151. 10.1373/clinchem.2009.132803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoofnagle A. N.; Wener M. H. The fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry. J. Immunol. Methods 2009, 347, 3–11. 10.1016/j.jim.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honour J. W. Development and validation of a quantitative assay based on tandem mass spectrometry. Ann. Clin. Biochem. 2011, 48, 97–111. 10.1258/acb.2010.010176. [DOI] [PubMed] [Google Scholar]
- Smit N.; van den Broek I.; Romijn F. P.; Haex M.; Deelder A. M.; van der Burgt Y.; van der Laarse A.; Cobbaert C. Quality requirements for quantitative clinical chemistry proteomics. Transl. Proteomics 2013, 2, 1–13. [Google Scholar]
- Chace D. H.; Barr J. R.; Duncan M. W.; Matern D.; Morris M. R.D.E.P.T.; Rockwood A. L.; Siuzdak G.; Urbani A.; Yergey A. L.; Chan Y. M. Mass Spectrometry in the Clinical Laboratory: General Principles and Guidance. Approved Guidelines, Clinical and Laboratory Standards Institute Guidelines 2007, 27, 94. [Google Scholar]
- Clarke W.; Molinaro R. J.; Bachmann L. M.; Botelho J. C.; Cao Z.; French D.; Garg S.; Gawoski J. M.; Grant R. P.; Hoofnagle A. N.; Iyer B.; Khulasingam V.; Mason D. S.; Rappold B.; Tacker D. H.; Truscott S. M.; Yu C.; Zhu Y. Liquid Chromatography-Mass Spectrometry Methods;. Approved Guidelines, Clinical and Laboratory Standards Institute Guidelines 2014, 34, 71. [Google Scholar]
- Lynch K. L. CLSI C62-A: A New Standard for Clinical Mass Spectrometry. Clin. Chem. 2016, 62, 24–29. 10.1373/clinchem.2015.238626. [DOI] [PubMed] [Google Scholar]
- Carr S. A.; Abbatiello S. E.; Ackermann B. L.; Borchers C.; Domon B.; Deutsch E. W.; Grant R. P.; Hoofnagle A. N.; Huttenhain R.; Koomen J. M.; Liebler D. C.; Liu T.; MacLean B.; Mani D. R.; Mansfield E.; Neubert H.; Paulovich A. G.; Reiter L.; Vitek O.; Aebersold R.; Anderson L.; Bethem R.; Blonder J.; Boja E.; Botelho J.; Boyne M.; Bradshaw R. A.; Burlingame A. L.; Chan D.; Keshishian H.; Kuhn E.; Kinsinger C.; Lee J. S.; Lee S. W.; Moritz R.; Oses-Prieto J.; Rifai N.; Ritchie J.; Rodriguez H.; Srinivas P. R.; Townsend R. R.; Van Eyk J.; Whiteley G.; Wiita A.; Weintraub S. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell Proteomics 2014, 13, 907–917. 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seger C.; Salzmann L. After another decade: LC–MS/MS became routine in clinical diagnostics. Clin. Biochem. 2020, 82, 2–11. 10.1016/j.clinbiochem.2020.03.004. [DOI] [PubMed] [Google Scholar]
- Cobbaert C.; Smit N.; Gillery P. Metrological traceability and harmonization of medical tests: a quantum leap forward is needed to keep pace with globalization and stringent IVD-regulations in the 21st century!. Clin. Chem. Lab. Med. 2018, 56, 1598–1602. 10.1515/cclm-2018-0343. [DOI] [PubMed] [Google Scholar]
- Horvath A. R.; Lord S. J.; StJohn A.; Sandberg S.; Cobbaert C. M.; Lorenz S.; Monaghan P. J.; Verhagen-Kamerbeek W. D.; Ebert C.; Bossuyt P. M. Test Evaluation Working Group of the European Federation of Clinical Chemistry Laboratory Medicine From biomarkers to medical tests: the changing landscape of test evaluation. Clin. Chim. Acta 2014, 427, 49–57. 10.1016/j.cca.2013.09.018. [DOI] [PubMed] [Google Scholar]
- Monaghan P. J.; Lord S. J.; St John A.; Sandberg S.; Cobbaert C. M.; Lennartz L.; Verhagen-Kamerbeek W. D.; Ebert C.; Bossuyt P. M.; Horvath A. R. Test Evaluation Working Group of the European Federation of Clinical Chemistry and Laboratory Medicine Biomarker development targeting unmet clinical needs. Clin. Chim. Acta 2016, 460, 211–219. 10.1016/j.cca.2016.06.037. [DOI] [PubMed] [Google Scholar]
- St John A. The Pursuit of Value in Laboratory Medicine - Progress and Challenges. Clin Biochem Rev. 2020, 41, 3–11. 10.33176/AACB-19-00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan P. J.; Robinson S.; Rajdl D.; Bossuyt P. M. M.; Sandberg S.; St John A.; O’Kane M.; Lennartz L.; Roddiger R.; Lord S. J.; Cobbaert C. M.; Horvath A. R. Practical guide for identifying unmet clinical needs for biomarkers. EJIFCC 2018, 29, 129–137. [PMC free article] [PubMed] [Google Scholar]
- EU . Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU Official Journal of the European Union, https://eur-lex.europa.eu/eli/reg/2017/746/oj.
- Domon B.; Aebersold R. Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 2010, 28, 710–721. 10.1038/nbt.1661. [DOI] [PubMed] [Google Scholar]
- Grant R. P.; Hoofnagle A. N. From lost in translation to paradise found: enabling protein biomarker method transfer by mass spectrometry. Clin. Chem. 2014, 60, 941–944. 10.1373/clinchem.2014.224840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenthal M. S.; Liang Y.; Phinney K. W.; Stein S. E. Quantitative bottom-up proteomics depends on digestion conditions. Anal. Chem. 2014, 86, 551–558. 10.1021/ac4027274. [DOI] [PubMed] [Google Scholar]
- van den Broek I.; Smit N. P.; Romijn F. P.; van der Laarse A.; Deelder A. M.; van der Burgt Y. E.; Cobbaert C. M. Evaluation of interspecimen trypsin digestion efficiency prior to multiple reaction monitoring-based absolute protein quantification with native protein calibrators. J. Proteome Res. 2013, 12, 5760–5774. 10.1021/pr400763d. [DOI] [PubMed] [Google Scholar]
- Shuford C. M.; Walters J. J.; Holland P. M.; Sreenivasan U.; Askari N.; Ray K.; Grant R. P. Absolute Protein Quantification by Mass Spectrometry: Not as Simple as Advertised. Anal. Chem. 2017, 89, 7406–7415. 10.1021/acs.analchem.7b00858. [DOI] [PubMed] [Google Scholar]
- van den Broek I.; Mastali M.; Mouapi K.; Bystrom C.; Bairey Merz C. N.; Van Eyk J. E. Quality control and outlier detection of targeted mass spectrometry data from multiplex protein panels. J. Proteome Res. 2020, 19, 2278–2293. 10.1021/acs.jproteome.9b00854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L. M.; Kelleher N. L. Consortium for Top Down Proteomics Proteoform: a single term describing protein complexity. Nat. Methods 2013, 10, 186–187. 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G. A.; Baliban R. C.; Floudas C. A. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci. Rep 2011, 1. 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R.; Agar J. N.; Amster I. J.; Baker M. S.; Bertozzi C. R.; Boja E. S.; Costello C. E.; Cravatt B. F.; Fenselau C.; Garcia B. A.; Ge Y.; Gunawardena J.; Hendrickson R. C.; Hergenrother P. J.; Huber C. G.; Ivanov A. R.; Jensen O. N.; Jewett M. C.; Kelleher N. L.; Kiessling L. L.; Krogan N. J.; Larsen M. R.; Loo J. A.; Ogorzalek Loo R. R.; Lundberg E.; MacCoss M. J.; Mallick P.; Mootha V. K.; Mrksich M.; Muir T. W.; Patrie S. M.; Pesavento J. J.; Pitteri S. J.; Rodriguez H.; Saghatelian A.; Sandoval W.; Schluter H.; Sechi S.; Slavoff S. A.; Smith L. M.; Snyder M. P.; Thomas P. M.; Uhlen M.; Van Eyk J. E.; Vidal M.; Walt D. R.; White F. M.; Williams E. R.; Wohlschlager T.; Wysocki V. H.; Yates N. A.; Young N. L.; Zhang B. How many human proteoforms are there?. Nat. Chem. Biol. 2018, 14, 206–214. 10.1038/nchembio.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soetkamp D.; Raedschelders K.; Mastali M.; Sobhani K.; Bairey Merz C. N.; Van Eyk J. The continuing evolution of cardiac troponin I biomarker analysis: from protein to proteoform. Expert Rev. Proteomics 2017, 14, 973–986. 10.1080/14789450.2017.1387054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisawa T.; Zen Y.; Pillai S.; Stone J. H. IgG4-related disease. Lancet 2015, 385, 1460–1471. 10.1016/S0140-6736(14)60720-0. [DOI] [PubMed] [Google Scholar]
- Trenchevska O.; Nelson R. W.; Nedelkov D. Mass Spectrometric Immunoassays in Characterization of Clinically Significant Proteoforms. Proteomes 2016, 4. 10.3390/proteomes4010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helander A.; Wielders J.; Anton R.; Arndt T.; Bianchi V.; Deenmamode J.; Jeppsson J. O.; Whitfield J. B.; Weykamp C.; Schellenberg F., on behalf of the International Federation of Clinical Chemistry and Laboratory Medicine Working Group on Standardisation of Carbohydrate-Deficient Transferrin (IFCC WG-CDT).Standardisation and use of the alcohol biomarker carbohydrate-deficient transferrin (CDT). Clin. Chim. Acta 2016, 459, 19−24, 10.1016/j.cca.2016.05.016. [DOI] [PubMed] [Google Scholar]
- Kenny D.; Fraser C. G.; Petersen P. H.; Kallner A. Consensus agreement. Scand. J. Clin. Lab Invest 1999, 59, 585–585. 10.1080/00365519950185409. [DOI] [PubMed] [Google Scholar]
- Horvath A. R.; Bossuyt P. M.; Sandberg S.; John A. S.; Monaghan P. J.; Verhagen-Kamerbeek W. D.; Lennartz L.; Cobbaert C. M.; Ebert C.; Lord S. J. Setting analytical performance specifications based on outcome studies - is it possible?. Clin Chem. Lab Med. 2015, 53, 841–848. 10.1515/cclm-2015-0214. [DOI] [PubMed] [Google Scholar]
- Fraser C. G. General strategies to set quality specifications for reliability performance characteristics. Scand. J. Clin. Lab. Invest. 1999, 59, 487–490. 10.1080/00365519950185210. [DOI] [PubMed] [Google Scholar]
- Biswas S. S.; Bindra M.; Jain V.; Gokhale P. Evaluation of imprecision, bias and total error of clinical chemistry analysers. Indian J. Clin. Biochem. 2015, 30, 104–108. 10.1007/s12291-014-0448-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aarsand A. K.; Roraas T.; Fernandez-Calle P.; Ricos C.; Diaz-Garzon J.; Jonker N.; Perich C.; Gonzalez-Lao E.; Carobene A.; Minchinela J.; Coskun A.; Simon M.; Alvarez V.; Bartlett W. A.; Fernandez-Fernandez P.; Boned B.; Braga F.; Corte Z.; Aslan B.; Sandberg S., on behalf of the European Federation of Clinical Chemistry and Laboratory Medicine Working Group on Biological Variation and Task and Finish Group for the Biological Variation Database.The Biological Variation Data Critical Appraisal Checklist: A Standard for Evaluating Studies on Biological Variation. Clin. Chem. 2018, 64, 501−514, 10.1373/clinchem.2017.281808. [DOI] [PubMed] [Google Scholar]
- Ruhaak L. R.; Romijn F.; Smit N. P. M.; van der Laarse A.; Pieterse M. M.; de Maat M. P. M.; Haas F.; Kluft C.; Amiral J.; Meijer P.; Cobbaert C. M. Detecting molecular forms of antithrombin by LC-MRM-MS: defining the measurands. Clin. Chem. Lab. Med. 2018, 56, 1704–1714. 10.1515/cclm-2017-1111. [DOI] [PubMed] [Google Scholar]
- van den Broek I.; Romijn F. P.; Nouta J.; van der Laarse A.; Drijfhout J. W.; Smit N. P.; van der Burgt Y. E.; Cobbaert C. M. Automated Multiplex LC–MS/MS Assay for Quantifying Serum Apolipoproteins A-I, B, C-I, C-II, C-III, and E with Qualitative Apolipoprotein E Phenotyping. Clin. Chem. 2016, 62, 188–197. 10.1373/clinchem.2015.246702. [DOI] [PubMed] [Google Scholar]
- Plebani M. Quality indicators to detect pre-analytical errors in laboratory testing. Clin Biochem Rev. 2012, 33, 85–88. [PMC free article] [PubMed] [Google Scholar]
- Rai A. J.; Gelfand C. A.; Haywood B. C.; Warunek D. J.; Yi J.; Schuchard M. D.; Mehigh R. J.; Cockrill S. L.; Scott G. B.; Tammen H.; Schulz-Knappe P.; Speicher D. W.; Vitzthum F.; Haab B. B.; Siest G.; Chan D. W. HUPO Plasma Proteome Project specimen collection and handling: towards the standardization of parameters for plasma proteome samples. Proteomics 2005, 5, 3262–3277. 10.1002/pmic.200401245. [DOI] [PubMed] [Google Scholar]
- Tammen H.; Schulte L.; Hess R.; Menzel C.; Kellmann M.; Mohring T.; Schulz-Knappe P. Peptidomic analysis of human blood specimens: Comparison between plasma specimens and serum by differential peptide display. Proteomics 2005, 5, 3414–3422. 10.1002/pmic.200401219. [DOI] [PubMed] [Google Scholar]
- Lundblad R. L. Considerations for the Use of Blood Plasma and Serum for Proteomic Analysis. Internet Journal of Genomics and Proteomics 2005, 1. 10.5580/5526e. [DOI] [Google Scholar]
- O’Keane M. P.; Cunningham S. K. Evaluation of three different specimen types (serum, plasma lithium heparin and serum gel separator) for analysis of certain analytes: clinical significance of differences in results and efficiency in use. Clin Chem. Lab Med. 2006, 44, 662–668. 10.1515/CCLM.2006.099. [DOI] [PubMed] [Google Scholar]
- Anderson N. L.; Anderson N. G.; Haines L. R.; Hardie D. B.; Olafson R. W.; Pearson T. W. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J. Proteome Res. 2004, 3, 235–244. 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- Krastins B.; Prakash A.; Sarracino D. A.; Nedelkov D.; Niederkofler E. E.; Kiernan U. A.; Nelson R.; Vogelsang M. S.; Vadali G.; Garces A.; Sutton J. N.; Peterman S.; Byram G.; Darbouret B.; Perusse J. R.; Seidah N. G.; Coulombe B.; Gobom J.; Portelius E.; Pannee J.; Blennow K.; Kulasingam V.; Couchman L.; Moniz C.; Lopez M. F. Rapid development of sensitive, high-throughput, quantitative and highly selective mass spectrometric targeted immunoassays for clinically important proteins in human plasma and serum. Clin. Biochem. 2013, 46, 399–410. 10.1016/j.clinbiochem.2012.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogeser M.; Schuster C.; Rockwood A. L. A proposal to standardize the description of LC–MS-based measurement methods in laboratory medicine. Clin Mass Spectrom 2019, 13, 36–38. 10.1016/j.clinms.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smigielski E. M.; Sirotkin K.; Ward M.; Sherry S. T. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 2000, 28, 352–355. 10.1093/nar/28.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit N. P.; Romijn F. P.; van den Broek I.; Drijfhout J. W.; Haex M.; van der Laarse A.; van der Burgt Y. E.; Cobbaert C. M. Metrological traceability in mass spectrometry-based targeted protein quantitation: a proof-of-principle study for serum apolipoproteins A-I and B100. J. Proteomics 2014, 109, 143–161. 10.1016/j.jprot.2014.06.015. [DOI] [PubMed] [Google Scholar]
- Bronsema K. J.; Bischoff R.; van de Merbel N. C. Internal standards in the quantitative determination of protein biopharmaceuticals using liquid chromatography coupled to mass spectrometry. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2012, 893, 1–14. 10.1016/j.jchromb.2012.02.021. [DOI] [PubMed] [Google Scholar]
- Picotti P.; Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Methods 2012, 9, 555–566. 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- Arsene C. G.; Ohlendorf R.; Burkitt W.; Pritchard C.; Henrion A.; O’Connor G.; Bunk D. M.; Guttler B. Protein quantification by isotope dilution mass spectrometry of proteolytic fragments: cleavage rate and accuracy. Anal. Chem. 2008, 80, 4154–4160. 10.1021/ac7024738. [DOI] [PubMed] [Google Scholar]
- Abbatiello S. E.; Mani D. R.; Keshishian H.; Carr S. A. Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin. Chem. 2010, 56, 291–305. 10.1373/clinchem.2009.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownridge P.; Beynon R. J. The importance of the digest: proteolysis and absolute quantification in proteomics. Methods 2011, 54, 351–360. 10.1016/j.ymeth.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Lawless C.; Hubbard S. J. Prediction of missed proteolytic cleavages for the selection of surrogate peptides for quantitative proteomics. OMICS 2012, 16, 449–456. 10.1089/omi.2011.0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switzar L.; Giera M.; Niessen W. M. Protein digestion: an overview of the available techniques and recent developments. J. Proteome Res. 2013, 12, 1067–1077. 10.1021/pr301201x. [DOI] [PubMed] [Google Scholar]
- Tsiatsiani L.; Heck A. J. Proteomics beyond trypsin. FEBS J. 2015, 282, 2612–2626. 10.1111/febs.13287. [DOI] [PubMed] [Google Scholar]
- Proc J. L.; Kuzyk M. A.; Hardie D. B.; Yang J.; Smith D. S.; Jackson A. M.; Parker C. E.; Borchers C. H. A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. J. Proteome Res. 2010, 9, 5422–5437. 10.1021/pr100656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart J. M.; Schumbrutzki C.; Wortelkamp S.; Sickmann A.; Zahedi R. P. Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics. J. Proteomics 2012, 75, 1454–1462. 10.1016/j.jprot.2011.11.016. [DOI] [PubMed] [Google Scholar]
- Vandermarliere E.; Mueller M.; Martens L. Getting intimate with trypsin, the leading protease in proteomics. Mass Spectrom. Rev. 2013, 32, 453–465. 10.1002/mas.21376. [DOI] [PubMed] [Google Scholar]
- Freije J. R.; Mulder P. P.; Werkman W.; Rieux L.; Niederlander H. A.; Verpoorte E.; Bischoff R. Chemically modified, immobilized trypsin reactor with improved digestion efficiency. J. Proteome Res. 2005, 4, 1805–1813. 10.1021/pr050142y. [DOI] [PubMed] [Google Scholar]
- van den Broek I.; Sobhani K.; Van Eyk J. E. Advances in quantifying apolipoproteins using LC–MS/MS technology: implications for the clinic. Expert Rev. Proteomics 2017, 1–12. 10.1080/14789450.2017.1374859. [DOI] [PubMed] [Google Scholar]
- Shah V.; Lassman M. E.; Chen Y.; Zhou H.; Laterza O. F. Achieving efficient digestion faster with Flash Digest: potential alternative to multi-step detergent assisted in-solution digestion in quantitative proteomics experiments. Rapid Commun. Mass Spectrom. 2017, 31, 193–199. 10.1002/rcm.7778. [DOI] [PubMed] [Google Scholar]
- Toth C. A.; Kuklenyik Z.; Jones J. I.; Parks B. A.; Gardner M. S.; Schieltz D. M.; Rees J. C.; Andrews M. L.; McWilliams L. G.; Pirkle J. L.; Barr J. R. On-column trypsin digestion coupled with LC–MS/MS for quantification of apolipoproteins. J. Proteomics 2017, 150, 258–267. 10.1016/j.jprot.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittremieux W.; Tabb D. L.; Impens F.; Staes A.; Timmerman E.; Martens L.; Laukens K. Quality control in mass spectrometry-based proteomics. Mass Spectrom. Rev. 2018, 37, 697–711. 10.1002/mas.21544. [DOI] [PubMed] [Google Scholar]
- Lebert D.; Louwagie M.; Goetze S.; Picard G.; Ossola R.; Duquesne C.; Basler K.; Ferro M.; Rinner O.; Aebersold R.; Garin J.; Mouz N.; Brunner E.; Brun V. DIGESTIF: a universal quality standard for the control of bottom-up proteomics experiments. J. Proteome Res. 2015, 14, 787–803. 10.1021/pr500834z. [DOI] [PubMed] [Google Scholar]
- van den Broek I.; Romijn F. P.; Smit N. P.; van der Laarse A.; Drijfhout J. W.; van der Burgt Y. E.; Cobbaert C. M. Quantifying Protein Measurands by Peptide Measurements: Where Do Errors Arise?. J. Proteome Res. 2015, 14, 928–942. 10.1021/pr5011179. [DOI] [PubMed] [Google Scholar]
- Shuford C. M.; Sederoff R. R.; Chiang V. L.; Muddiman D. C. Peptide production and decay rates affect the quantitative accuracy of protein cleavage isotope dilution mass spectrometry (PC-IDMS). Mol. Cell Proteomics 2012, 11, 814–823. 10.1074/mcp.O112.017145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourmaud A.; Gallien S.; Domon B. A quality control of proteomic experiments based on multiple isotopologous internal standards. EuPa Open Proteomics 2015, 8, 16–21. 10.1016/j.euprot.2015.07.010. [DOI] [Google Scholar]
- Gallien S.; Bourmaud A.; Domon B. A simple protocol to routinely assess the uniformity of proteomics analyses. J. Proteome Res. 2014, 13, 2688–2695. 10.1021/pr4011712. [DOI] [PubMed] [Google Scholar]
- Barnidge D. R.; Hall G. D.; Stocker J. L.; Muddiman D. C. Evaluation of a cleavable stable isotope labeled synthetic peptide for absolute protein quantification using LC–MS/MS. J. Proteome Res. 2004, 3, 658–661. 10.1021/pr034124x. [DOI] [PubMed] [Google Scholar]
- Kushnir M. M.; Rockwood A. L.; Roberts W. L.; Abraham D.; Hoofnagle A. N.; Meikle A. W. Measurement of thyroglobulin by liquid chromatography-tandem mass spectrometry in serum and plasma in the presence of antithyroglobulin autoantibodies. Clin. Chem. 2013, 59, 982–990. 10.1373/clinchem.2012.195594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiler M.; Straube W. L.; Lundberg E.; Uhlen M.; Mann M. A Protein Epitope Signature Tag (PrEST) library allows SILAC-based absolute quantification and multiplexed determination of protein copy numbers in cell lines. Mol. Cell Proteomics 2012, 11i, O111009613. 10.1074/mcp.O111.009613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beynon R. J.; Doherty M. K.; Pratt J. M.; Gaskell S. J. Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat. Methods 2005, 2, 587–589. 10.1038/nmeth774. [DOI] [PubMed] [Google Scholar]
- Brun V.; Dupuis A.; Adrait A.; Marcellin M.; Thomas D.; Court M.; Vandenesch F.; Garin J. Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol. Cell. Proteomics 2007, 6, 2139–2149. 10.1074/mcp.M700163-MCP200. [DOI] [PubMed] [Google Scholar]
- Kuhn E.; Whiteaker J. R.; Mani D. R.; Jackson A. M.; Zhao L.; Pope M. E.; Smith D.; Rivera K. D.; Anderson N. L.; Skates S. J.; Pearson T. W.; Paulovich A. G.; Carr S. A. Interlaboratory evaluation of automated, multiplexed peptide immunoaffinity enrichment coupled to multiple reaction monitoring mass spectrometry for quantifying proteins in plasma. Mol. Cell Proteomics 2012, 11, M111013854. 10.1074/mcp.M111.013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouri-Nigjeh E.; Zhang M.; Ji T.; Yu H.; An B.; Duan X.; Balthasar J.; Johnson R. W.; Qu J. Effects of calibration approaches on the accuracy for LC–MS targeted quantification of therapeutic protein. Anal. Chem. 2014, 86, 3575–3584. 10.1021/ac5001477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells D. A. (2018) Sample preparation for mass spectrometry applications. In Tietz Textbook of Clinical Chemistry and Molecular Diagnostics, 6th ed.; Rifai N., Ed.; Saunders, 2017. [Google Scholar]
- Kulak N. A.; Pichler G.; Paron I.; Nagaraj N.; Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- Keshishian H.; Addona T.; Burgess M.; Kuhn E.; Carr S. A. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 2007, 6, 2212–2229. 10.1074/mcp.M700354-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassine H. N.; Jackson A. M.; Reaven P. D.; Nedelkov D.; Nelson R. W.; Lau S. S.; Borchers C. H. The application of multiple reaction monitoring to assess ApoA-I methionine oxidations in diabetes and cardiovascular disease. Transl. Proteomics 2014, 4–5, 18–24. 10.1016/j.trprot.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnir M. M.; Rockwood A. L.; Nelson G. J.; Yue B. F.; Urry F. M. Assessing analytical specificity in quantitative analysis using tandem mass spectrometry. Clin. Biochem. 2005, 38, 319–327. 10.1016/j.clinbiochem.2004.12.003. [DOI] [PubMed] [Google Scholar]
- JCGM . ISO/IEC Guide 99; 2007. International vocabulary of metrology. Basic and general concepts and associated terms (VIM); 2007 [DOI] [PubMed] [Google Scholar]
- Cobbaert C. A. H.; Begcevic Brkovic I.; Ceglarek U.; Coassin S.; Delatour V.; Deprez L.; Dikaios I.; Dittrich J.; Hoofnagle A. N.; Kostner G.; Kronenberg F.; Kuklenyyik Z.; Prinzing U.; Vesper H. W.; Zegers I.; Ruhaak L. R. Towards an SI-traceable reference measurement system for seven serum apolipoproteins using bottom-up quantitative proteomics: Conceptual approach enabled by cross-disciplinary/cross-sector collaboration. Clin Chem. 2020, 66. 10.1093/clinchem/hvaa239. [DOI] [PubMed] [Google Scholar]
- ISO . ISO 15189. Medical laboratories — Requirements for quality and competence, 2012. [Google Scholar]
- EU. Regulation (EU) 2017/746 of the European Parliament and of the Council of April 5, 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU Official Journal of the European Union.
- Jayathirtha M.; Whitham D.; Stradtman S.; Darie C. C. Recent Applications of Mass Spectrometry at Clarkson University. Adv. Exp. Med. Biol. 2019, 1140, 771–785. 10.1007/978-3-030-15950-4_46. [DOI] [PubMed] [Google Scholar]
- Stone J. A.; Fitzgerald R. L. Liquid Chromatography-Mass Spectrometry Education for Clinical Laboratory Scientists. Clin Lab Med. 2018, 38, 527–537. 10.1016/j.cll.2018.04.002. [DOI] [PubMed] [Google Scholar]
- Briscoe C. J.; Stiles M. R.; Hage D. S. System suitability in bioanalytical LC/MS/MS. J. Pharm. Biomed. Anal. 2007, 44, 484–491. 10.1016/j.jpba.2007.03.003. [DOI] [PubMed] [Google Scholar]
- McDowall E. The humble instrument logbook?. Spectroscopy 2017, 32, 8–12. [Google Scholar]
- Levey S.; Jennings E. R. The use of control charts in the clinical laboratory. Am. J. Clin. Pathol. 1950, 20, 1059–1066. 10.1093/ajcp/20.11_ts.1059. [DOI] [PubMed] [Google Scholar]
- Dogu E.; Mohammad-Taheri S.; Abbatiello S. E.; Bereman M. S.; MacLean B.; Schilling B.; Vitek O. MSstatsQC: Longitudinal System Suitability Monitoring and Quality Control for Targeted Proteomic Experiments. Mol. Cell Proteomics 2017, 16, 1335–1347. 10.1074/mcp.M116.064774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westgard J. O. Internal quality control: planning and implementation strategies. Ann. Clin. Biochem. 2003, 40, 593–611. 10.1258/000456303770367199. [DOI] [PubMed] [Google Scholar]
- Sandberg S.; Fraser C. G.; Horvath A. R.; Jansen R.; Jones G.; Oosterhuis W.; Petersen P. H.; Schimmel H.; Sikaris K.; Panteghini M. Defining analytical performance specifications: Consensus Statement from the 1st Strategic Conference of the European Federation of Clinical Chemistry and Laboratory Medicine. Clin Chem. Lab Med. 2015, 53, 833–835. 10.1515/cclm-2015-0067. [DOI] [PubMed] [Google Scholar]
- Bland J. M.; Altman D. G. Multiple significance tests: the Bonferroni method. BMJ. 1995, 310, 170. 10.1136/bmj.310.6973.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar B. V.; Mohan T. Sigma metrics as a tool for evaluating the performance of internal quality control in a clinical chemistry laboratory. J. Lab. Physicians 2018, 10, 194–199. 10.4103/JLP.JLP_102_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLSI . Assessment of Laboratory Tests When Proficiency Testing is Not Available; Approved Guideline GP29, 2008. [Google Scholar]
- Miller W. G.; Jones G. R.; Horowitz G. L.; Weykamp C. Proficiency testing/external quality assessment: current challenges and future directions. Clin. Chem. 2011, 57, 1670–1680. 10.1373/clinchem.2011.168641. [DOI] [PubMed] [Google Scholar]
- Cobbaert C.; Weykamp C.; Franck P.; de Jonge R.; Kuypers A.; Steigstra H.; Gunnewiek J. K.; van Loon D.; Jansen R. Systematic monitoring of standardization and harmonization status with commutable EQA-samples-Five year experience from the Netherlands. Clin. Chim. Acta 2012, 414, 234–240. 10.1016/j.cca.2012.09.027. [DOI] [PubMed] [Google Scholar]
- Mehta L. S.; Beckie T. M.; DeVon H. A.; Grines C. L.; Krumholz H. M.; Johnson M. N.; Lindley K. J.; Vaccarino V.; Wang T. Y.; Watson K. E.; Wenger N. K., on behalf of the American Heart Association Cardiovascular Disease in Women and Special Populations Committee of the Council on Clinical Cardiology, Council on Epidemiology and Prevention, Council on Cardiovascular and Stroke Nursing, and Council on Quality of Care and Outcomes Research.Acute Myocardial Infarction in Women: A Scientific Statement From the American Heart Association. Circulation 2016, 133, 916−947, 10.1161/CIR.0000000000000351. [DOI] [PubMed] [Google Scholar]
- Varbo A.; Benn M.; Nordestgaard B. G. Remnant cholesterol as a cause of ischemic heart disease: evidence, definition, measurement, atherogenicity, high risk patients, and present and future treatment. Pharmacol. Ther. 2014, 141, 358–367. 10.1016/j.pharmthera.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Libby P. The forgotten majority: unfinished business in cardiovascular risk reduction. J. Am. Coll. Cardiol. 2005, 46, 1225–1228. 10.1016/j.jacc.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Bodde M. C.; Hermans M. P. J.; Jukema J. W.; Schalij M. J.; Lijfering W. M.; Rosendaal F. R.; Romijn F.; Ruhaak L. R.; van der Laarse A.; Cobbaert C. M. Apolipoproteins A1, B, and apoB/apoA1 ratio are associated with first ST-segment elevation myocardial infarction but not with recurrent events during long-term follow-up. Clin. Res. Cardiol. 2019, 108, 520–538. 10.1007/s00392-018-1381-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechlaner R.; Tsimikas S.; Yin X.; Willeit P.; Baig F.; Santer P.; Oberhollenzer F.; Egger G.; Witztum J. L.; Alexander V. J.; Willeit J.; Kiechl S.; Mayr M. Very-Low-Density Lipoprotein-Associated Apolipoproteins Predict Cardiovascular Events and Are Lowered by Inhibition of APOC-III. J. Am. Coll. Cardiol. 2017, 69, 789–800. 10.1016/j.jacc.2016.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniderman A. D.; Williams K.; Contois J. H.; Monroe H. M.; McQueen M. J.; de Graaf J.; Furberg C. D. A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ Cardiovasc Qual Outcomes 2011, 4, 337–345. 10.1161/CIRCOUTCOMES.110.959247. [DOI] [PubMed] [Google Scholar]
- van Capelleveen J. C.; Bernelot Moens S. J.; Yang X.; Kastelein J. J. P.; Wareham N. J.; Zwinderman A. H.; Stroes E. S. G.; Witztum J. L.; Hovingh G. K.; Khaw K. T.; Boekholdt S. M.; Tsimikas S. Apolipoprotein C-III Levels and Incident Coronary Artery Disease Risk: The EPIC-Norfolk Prospective Population Study. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1206–1212. 10.1161/ATVBAHA.117.309007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhaak L. R.; van der Laarse A.; Cobbaert C. M. Apolipoprotein profiling as a personalized approach to the diagnosis and treatment of dyslipidaemia. Ann. Clin. Biochem. 2019, 56, 338–356. 10.1177/0004563219827620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhaak L. R.; Smit N. P. M.; Suchiman H. E. D.; Pieterse M. M.; Romijn F.; Beekman M.; Cobbaert C. M. MS-based proteomics: a metrological sound and robust alternative for apolipoprotein E phenotyping in a multiplexed test. Clin Chem. Lab Med. 2019, 57, e102-e104 10.1515/cclm-2018-0782. [DOI] [PubMed] [Google Scholar]
- Dati F.; Tate J. Reference Materials for the Standardization of the Apolipoproteins A-I and B, and Lipoprotein(a). EJIFCC 2001, 13, 73–79. [PMC free article] [PubMed] [Google Scholar]
- Ruhaak L. R.; Smit N. P. M.; Romijn F. P. H. T. M.; Pieterse M. M.; van der Laarse A.; van der Burgt Y. E. M.; Cobbaert C. M. Robust and Accurate 2-Year Performance of a Quantitative Mass Spectrometry-Based Apolipoprotein Test in a Clinical Chemistry Laboratory. Clin. Chem. 2018, 64, 747–749. 10.1373/clinchem.2017.285098. [DOI] [PubMed] [Google Scholar]