

Abstract
The initiation and maintenance of cholinergic-induced status epilepticus (SE) are associated with decreased synaptic gamma-aminobutyric acid A receptors (GABAAR) and increased N-methyl-D-aspartate receptors (NMDAR) and amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR). We hypothesized that trafficking of synaptic GABAAR and glutamate receptors is maladaptive and contributes to the pharmacoresistance to anti-seizure drugs; targeting these components should ameliorate the pathophysiological consequences of refractory SE (RSE). We review studies of rodent models of cholinergic-induced SE, in which we used a benzodiazepine allosteric GABAAR modulator to correct loss of inhibition, concurrent with the NMDA antagonist ketamine to reduce excitation caused by increased synaptic localization of NMDAR and AMPAR, which are NMDAR-dependent. Models included lithium/pilocarpine-induced SE in rats and soman-induced SE in rats and in carboxylesterase knockout mice, which similar to humans lack plasma carboxylesterase, and may better model soman toxicity. These model human soman toxicity and are refractory to benzodiazepines administered at 40 min after seizure onset, when enough synaptic GABAAR may not be available to restore inhibition. Ketamine-midazolam combination reduces seizure severity, epileptogenesis, performance deficits and neuropathology following cholinergic-induced SE. Supplementing that treatment with valproate, which targets a non-benzodiazepine site, effectively terminates RSE, providing further benefit against cholinergic-induced SE. The therapeutic index of drug combinations is also reviewed and we show the improved efficacy of simultaneous administration of midazolam, ketamine and valproate compared to sequential drug administration. These data suggest that future clinical trials should treat both the lack of sufficient inhibition and the excess excitation that characterize RSE, and include early combination drug therapies.
Keywords: ketamine, seizure, pharmacoresistance, organophosphorus, soman, pilocarpine
1. Introduction
Chemical warfare nerve agents (CWNAs) are organophosphorus chemical agents that inhibit acetylcholinesterase and lead to uncontrollable status epilepticus (SE) and pose a risk to both military and civilian populations. Effective treatment of nerve agent poisoning is likely to be delayed in the event of mass civilian casualties, as in sarin exposures in Syria, or in cases where the chemical class is a contact hazard with delayed onset of action and/or not quickly identified, as in a recent assassination in Malaysia and assassination attempts in the United Kingdom. Currently, the standard treatment against a CWNA exposure consists of the anticholinergic compound atropine to reduce peripheral side effects, an oxime for reactivation of acetylcholinesterase (such as pralidoxime chloride; 2-PAM), and a benzodiazepine anticonvulsant (such as diazepam or midazolam; reviewed in Newmark, 2019). Benzodiazepines are commonly used as a first-line therapy to treat acute seizures and SE, including CWNA-induced seizures (Shih et al., 1999; Reddy and Reddy 2015). Although early administration of benzodiazepines is effective at reducing cholinergic-induced seizure (Lipp, 1972), the therapeutic window for terminating seizure is limited to 40 min or less after seizure onset (McDonough and Shih, 1993, 1995; Shih et al., 1999; Schultz et al., 2012), and treatment success with benzodiazepines is inversely correlated with seizure duration (McDonough et al., 1995; Mazarati et al., 1998; Goodkin et al., 2003). When left un-treated or treatment is delayed, soman-induced seizure progresses to SE, performance deficits and epileptogenesis and can lead to seizure-related brain damage (Shih et al., 2003; de Araujo Furtado et al., 2010; Moffett et al., 2011; Langston et al., 2012; Schultz et al., 2012; Aroniadou-Anderjaska et al., 2016).
Convulsive SE is considered a time-sensitive medical emergency with seizure duration associated with high mortality and morbidity, poor functional outcome and increased risk of epilepsy (Lowenstein, 2015; Hill et al., 2017; Sanchez-Fernandez et al., 2019; Prasad, 2014). Yet in the event of CWNA exposure that involves delayed onset of toxicity, territorial denial or mass casualties, early treatment may not be feasible. Time to treatment of overt generalized SE is at least as important as the specific anti-seizure drug being used (Treiman et al., 1998; Madzar et al., 2018). According to recent review of electronic databases, 17% to 64% of patients have a delay of over 30 min before treatment (Hill et al., 2017). The Rapid Anticonvulsant Medication Prior to Arrival Trial (RAMPART) showed clinical efficacy of intramuscular (IM) midazolam in the treatment of SE and highlighted the importance of early treatment (Silbergleit et al., 2012; 2013). Since midazolam is water soluble, is rapidly absorbed with fast onset of action, and can be administered IM, replacement of IM diazepam with IM midazolam for treatment of SE is recommended (Towne and DeLorenzo, 1999; Newmark, 2019). However, since the effectiveness of benzodiazepines against cholinergic-induced SE diminishes with time, adjunct and/or alternative therapies are needed.
1.1. Cholinergic-induced pharmacoresistant SE
While considerable progress has been made in treating CWNA-induced toxicity, treating CWNA-induced SE remains a therapeutic challenge. Chemical warfare nerve agent-induced SE shares many characteristics with other forms of SE (Chen and Wasterlain, 2006); it quickly becomes self-sustaining and is independent of its original cholinergic trigger, refractory to benzodiazepines (Shih et al., 1999), and partially responsive to NMDA blockers (Dorandeu et al., 2007). Such characteristics have been found in SE of a wide variety of etiologies, including SE induced by perforant path stimulation (Mazarati et al., 1998, Mazarati and Wasterlain, 1999), amygdala stimulation (Nissinen et al., 2000), or lithium and pilocarpine (Morrisett et al., 1987; Suchomelova et al., 2006). Insights from the studies reviewed herein (Lumley et al., 2019a; Niquet et al., 2017a, 2017b, 2019; Marrero-Rosado et al., 2018, 2020; Kundrick et al., 2020) may also have applications for the treatment of human SE, which affects an estimate of 102–152,000 individuals per year in the USA (DeLorenzo et al., 1996), as well as for organophosphate (OP) insecticide poisoning from agricultural exposures or self-poisoning, a worldwide clinical issue with approximately 200,000 deaths annually (Eddleston, 2000; Gunnell et al., 2007).
1.2. The receptor trafficking hypothesis of status epilepticus
An understanding of the molecular changes that are triggered by prolonged seizure activity is paramount for the identification of novel pharmacological approaches to control the severity, duration, and long-term effects of seizures. We and others found that cholinergic SE causes internalization/inactivation of synaptic γ-aminobutyric acid type A receptors (GABAAR) (Naylor et al., 2005a; Goodkin et al., 2008) as well as the trafficking of N-methyl-D-aspartate receptors (NMDAR) (Naylor et al., 2013) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) (Rajasekaran et al., 2012) in the opposite direction so that “spare” subunits assemble into functional receptors that move to the perisynaptic area. The result is a reduction of GABAergic inhibition (Kapur et al., 1989; Rice and DeLorenzo, 1999; Naylor and Wasterlain, 2005b) and an increase of glutamatergic excitation (Kapur and Lothman, 1990; Wasterlain et al., 2000; Naylor et al., 2013). Both the decrease in synaptic GABAAR and the increase in glutamate receptors are maladaptive and tilt the balance between excitation and inhibition toward greater seizure activity and SE. We hypothesized that polytherapy aimed at correcting the consequences of receptor trafficking can reduce SE severity. Muscarinic antagonists can effectively block SE when given in the early stage of nerve agent intoxication, but cannot quickly stop SE once it has become established (McDonough and Shih, 1993; McDonough et al., 2000; Schultz et al., 2012). Blocking muscarinic receptors would not reverse the reduction of synaptic GABAAR or the increase in synaptic NMDAR and AMPAR. This also suggests that blocking one pathway (e.g., benzodiazepine monotherapy) is unlikely to stop established SE. Polytherapy is needed to simultaneously activate the remaining GABAAR (effectiveness depends on number of GABAAR that are still available) and block NMDAR activation. When treatment is late, there may not be enough synaptic benzodiazepine receptors left to restore inhibition with benzodiazepines and it may be useful to also enhance inhibition by a non-GABA, non-NMDA mechanism.
1.3. Ketamine as neuroprotectant against cholinergic-induced status epilepticus
A potential candidate for the pharmacological treatment of CWNA-induced SE is ketamine, a well-known non-competitive antagonist of NMDAR. Ketamine has numerous mechanisms of actions, including the inhibition of current through the NMDAR in rat hippocampal neurons (Zeilhofer et al., 1992) and the inhibition of M1 muscarinic receptor signaling (inhibition of acetyl-β-methylcholine-induced Ca2+ release in xenopus oocytes transfected with rat M1 receptors) (Durieux, 1995). Ketamine also inhibits currents through nicotinic acetylcholine receptors (Friederich et al., 2000) and has been shown to positively modulate α6β2δ α6β3δ GABAAR subtypes at low concentrations and to directly activate these receptor subtypes at high concentrations (Hevers et al., 2008). Given its multiple molecular targets, the anticonvulsive and neuroprotective properties of ketamine should come as no surprise. The anticonvulsive properties of ketamine, marketed as an anesthetic in human and veterinary uses, are apparent in preclinical animal models of cholinergic-induced SE (reviewed in Dorandeu et al., 2013). Preclinical studies of CWNA-induced SE have also demonstrated the benefits of including ketamine as treatment. Racemic ketamine and atropine sulfate administered to guinea pigs at repeated time points starting 30 min following soman exposure protect against lethality and arrest seizure activity (Dorandeu et al., 2005). The S(+) isomer of ketamine, which has approximately 4-fold greater affinity than racemic ketamine for the NMDA receptor, provided similar protection against lethality and seizure-related brain damage; guinea pigs treated with repeated (3 x, 30-minute interval) 15–20 mg/kg S(+) ketamine received similar protection from lethality and neuropathology to that of 40–60 mg/kg of R-ketamine, suggesting that the therapeutic efficacy is linked to NMDA antagonism (Dorandeu et al., 2007). Studies have also demonstrated the safe and effective use of ketamine combined with benzodiazepines for the treatment of cholinergic-induced SE. Treatment of cholinergic-induced SE with combinations of a GABAAR agonist and an NMDAR antagonist, such as diazepam and ketamine (Ballough et al., 2008; Martin and Kapur, 2008) or midazolam and ketamine (Niquet et al., 2016; Lumley et al., 2019a) is effective in reducing the severity of seizures and brain pathology. Table 1 summarizes some of the preclinical studies evaluating ketamine against cholinergic-induced toxicity when used in combination with benzodiazepine and/or atropine (for additional review beyond what is shown in the table, see Dorandeu et al., 2013).
Table 1.
Efficacy of ketamine as treatment of cholinergic-induced status epilepticus.
| Toxicant | Species | Dose of Toxicant | Route of Toxicant Exposure | Treatment | Outcomes | Reference |
|---|---|---|---|---|---|---|
| Sarin | Rats | 105 μg/kg | Subcutaneous | ● 1 min after exposure: 2-PAM and AMN ● 50 min after seizure onset: ○ MDZ (0.66 mg/kg, once; IM) ○ KET (5 injections of 7.5 mg/kg, 90 min apart; IM) ○ MDZ (0.66 mg/kg, once; IM) + low dose KET (5 injections of 2.5 mg/kg, 90 min apart; IM) ○ MDZ (0.66 mg/kg, once; IM) + high dose KET (5 injections of 7.5 mg/kg, 90 min apart; IM) |
MDZ + high dose KET: ● Reduction in electroencephalographic (EEG) abnormalities. ● Improved Morris water maze performance ● Decreased neuronal necrosis |
Lewine et al. 2018 |
| Soman | Rats | 60 μg/kg | Subcutaneous (at 31 °C) | ● 75 min after exposure: KET (75 mg/kg; IP) and AS (5 mg/kg; IP) | ● Reduction of convulsion-induced hyperthermia ● No treatment effect on convulsion-induced changes in mRNA levels in brain. |
Barbier et al. 2015 |
| Mice | 172 μg/kg | Subcutaneous | ● 5 min prior to exposure: HI-6 (50 mg/kg, IP) ● Group 1, 30 min post-exposure: KET (25 mg/kg; IP) + AS (10 mg/kg, IP); 6 injections every 30 min ● Group 2, 60 min post-exposure: KET (100 mg/kg; IP) + AS (10 mg/kg, IP); 2 injections, 60 min apart |
● In all KET groups, convulsions stopped within 1–2 min after first treatment injection ● No convulsions observed after the 6th injection in soman-poisoned mice given 25 mg/kg KET, and after recovery of anesthesia in mice treated with 100 mg/kg KET. ● In 25 mg/kg KET group, loss of motor activity within 1–2 min of injection. Improved motor activity after recovery from anesthesia. ● Decreased brain damage in various regions at 48 h and 7d post-exposure. ● Reduction in GFAP immunostaining, but not complete. |
Dhote et al. 2012 | |
| Guinea pigs | 62 μg/kg | Subcutaneous | ● 30 min prior to exposure: PYR (26 μg/kg; IM) ● 1 min after exposure: AMN (4 mg/kg; IM) ● When multiple KET injections were given, time between injections was 30 min. ● 1–6x injections of KET (10, 40, or 60 mg/kg) + AS (2 or 10 mg/kg) at various time points after exposure (30, 60, 90, or 120 min). ● Two groups included MDZ: ○ KET (40 mg/kg) + AS (10 mg/kg) + MDZ (1 mg/kg); 3 injections at 60 min after exposure. ○ KET (60 mg/kg) + AS (10 mg/kg) + MDZ (1 mg/kg); 3 injections at 120 min after exposure |
● Sedation and disappearance of tremors and clonic movements within 1–2 min. ● Reduction of amplitude in electrocorticographic activity; change to high frequency, low amplitude signal. ● When KET/AS/MDZ combination is administered at 120 min after exposure, it significantly decreased the time to seizure suppression compared with KET monotherapy. ● An overall reduction of soman-induced lethality in KET/AS-treated animals was observed. ● Reduction in brain damage. ● Longer delays in treatment require higher doses of KET to be effective at seizure cessation, but spontaneous recurrent seizures were still observed. |
Dorandeu et al. 2005 | |
| Rats | Rats 132 μg/kg | Subcutaneous | ● 1 min after exposure: AS (2 mg/kg; IM) + HI-6 (118 mg/kg; IM) ● 40 min exposure: MDZ (3 mg/kg; IP); KET (10–90 mg/kg; IP), or MDZ+KET |
● 60 mg/kg KET monotherapy increased survival. ● When combined with MDZ, a dose of 30 mg/kg KET was able to increase survival and reduce EEG power integral ● One month after exposure, MDZ+KET group showed improved spatial memory acquisition performance |
Lumley et al. 2019 | |
| Rats | 180 μg/kg | Subcutaneous | ● 30 min prior to exposure: HI-6 (125 mg/kg, IP) ● 1 min after exposure: AMN (2 mg/kg, IM) ● 20 min after seizure onset: KET (30 or 45 mg/kg, IP), among other neuroprotectants. |
● Increased survival to 56% compared to 33% in soman-exposed animals without neuroprotectant. ● KET did not turn off seizures with 100% of animals showing continuous seizure activity for 24 h after exposure. ● At 24 h after exposure, lower overall neuropathology score was observed, but little to no effect on neuropathology score in specific brain regions (cerebral cortex, piriform cortex, amygdala, hippocampus, thalamus, and striatum). |
Acon-Chen et al. 2016 | |
| Dichlorvos | Rats | 20 mg/kg | Subcutaneous | ● 5 minutes prior to exposure: glycopyrrolate (3 mg/kg, IP), varying doses of test antidotes, including KET (10, 18, 31, and 55 mg/kg, SC) | ● Up-and-down method was used to estimate median effective dose. ● Primary outcome was survival to 10 min ● Poor survival when KET was used as pre-treatment. |
Sivilotti et al. 2006 |
| DFP | Mice | 7.6 mg/kg | Subcutaneous | ● Immediately after exposure: KET (6.25, 12.5, 25, or 50 mg/kg, IP) + AS (4 mg/kg, IP) | ● ncrease in survival | Klemm et al., 1985 |
| LiCl-pilocarpine | Rats (postnatal day 15) | 3 mEq/kg of lithium followed 12–18 h later by 60 mg/kg of pilocarpine | Intraperitoneal | ● Group 1, 15 min after pilocarpine exposure: KET (22.5 mg/kg, IP) ● Group 2, 60 min after pilocarpine exposure: KET (22.5 mg/kg, IP) |
● Both treatment groups showed reduction of some toxic signs within 5 min of treatment. ● SE-induced motor activity was halted by 70 min following treatment. ● Reduced FluoroJade C staining in various brain regions. ● KET treatments decreased long-term effects on elevated plus maze (time spent in open arms). |
Loss et al. 2012 |
| LiCl-pilocarpine | Rats | 5 mEq/kg of lithium followed 16 h later by 320 mg/kg of pilocarpine and 1 mg/kg of scopolamine methyl bromide | Subcutaneous (lithium) and intraperitoneal (pilocarpine and scopolamine) | ● Group 1, 40 min after seizure onset: MDZ (9 mg/kg; IP) ● Group 2, 40 min after seizure onset: KET (90 mg/kg, IP) ● Group 3, 40 min after seizure onset: MDZ (4.5 mg/kg; IP) + KET (45 mg/kg, IP) |
● MDZ+KET was only treatment that decreased EEG power to below baseline levels. ● MDZ+KET was the fastest treatment to decline EEG signal amplitude. ● KET monotherapy and combined with MDZ increased survival. ● Neuronal cell injury in various brain regions was significantly reduced with KET and MDZ+KET treatment. ● MDZ+KET treatment prevented epileptogenesis. ● MDZ+KET decreased Morris water maze deficits. |
Niquet et al., 2016 |
| Pilocarpine | Mice | 200 mg/kg initially, then 100 mg/kg every 30 min until SE onset | Intraperitoneal | ● 30 min prior to exposure: scopolamine methylnitrate (1 mg/kg, IP) ● Group 1, 60 min after seizure onset: DZP (10 mg/kg; SC), followed 60 min after by KET (25 mg/kg; SC) ● `Group 2, 60 min after seizure onset: KET (25 mg/kg; SC), followed 60 min after by DZP (10 mg/kg; SC) |
● Faster behavioral recovery period when DZP administered prior to KET. ● Both injection protocols reduced power in gamma EEG frequency range. ● Epileptiform activity was still observed up to 3 h after either of the injection protocols were administered. This is followed by a pattern of interictal spiking. |
Wang et al., 2019 |
2-PAM = pralidoxime; AMN = atropine methyl nitrate; AS = atropine sulfate; DZP = diazepam; KET = ketamine; MDZ = midazolam; PYR = pyridostigmine; DFP = di-isopropylfluorophosphate. For additional review beyond what is shown in this table, see Dorandeu et al., 2013.
1.4. Animal model of CWNA-induced toxicity in support of the FDA Animal Rule
Since exposing human subjects to CWNA for the purpose of evaluating the efficacy of medical countermeasures is unethical, the Food & Drug Administration (FDA) developed a set of regulations, commonly known as the Animal Rule, for the approval of drugs without human efficacy studies. Importantly, the predictive efficacy of novel therapies against CWNA-induced SE highly benefits from the development and characterization of animal models that accurately mimic nerve agent toxicity in humans. Studies of the effectiveness of prophylaxis and treatment of nerve agent-induced SE have frequently made use of rodent animal models, albeit with the major caveat that mice and rats contain plasma carboxylesterase (CaE) activity. Carboxylesterase has a role in the detoxification of certain organophosphorus compounds, particularly the nerve agent soman, and serves as an endogenous bioscavenger of such toxicants, thus increasing the amount of toxicant required to produce a response in the animal. In contrast, humans have undetectable plasma CaE activity. The importance of CaE on soman toxicity has been previously demonstrated. Treatment of mice, rats, guinea pigs and rabbits with cresylbenzodioxaphosphorin oxide (CBDP), a preferential inhibitor of CaE, prior to soman exposure results in a significant reduction of the species-specific median lethal doses (Maxwell et al., 1987). However, CBDP at high doses may confound soman toxicity observations due to its inhibiting effect on acetylcholinesterase and its ability to inhibit activity of CaE in the liver and kidney (Maxwell et al., 1987). Es1 gene knockout (Es1−/−) mice were developed to specifically lack plasma CaE while containing normal CaE activity levels in organs (Duysen et al., 2011). As expected, Es1−/− mice exhibit lower median lethal doses (LD50) of parathion, chlorpyrifos, and soman when compared to wild‐type mice (Duysen et al., 2012; Marrero-Rosado et al., 2018). Our laboratory has characterized the seizurogenic, ictogenic, epileptogenic, and neuropathological consequences of soman exposure in Es1−/− mice (Marrero-Rosado et al., 2018, 2020; Kundrick et al., 2020). In agreement with findings in our rat model of soman-induced toxicity (de Araujo Furtado et al., 2010; Moffett et al., 2011; Langston et al., 2012; Schultz et al., 2012, 2014; Lumley et al., 2019a), the subcutaneous exposure of Es1−/− mice to a seizure-inducing dose of soman (approximately 4 LD50) results in severe seizures that become refractory to benzodiazepine treatment when treatment is administered at 15–40 min after seizure onset. In the days following soman-induced seizure, the development of spontaneous recurrent seizures (SRS) occurs, as well assevere neuropathology (loss of neurons and robust neuroinflammation) (Marrero-Rosado et al., 2018, 2020; Kundrick et al., 2020). In line with several models of cholinergic-induced SE demonstrating the benefits of using ketamine as a treatment to effectively reduce the duration and/or seizure severity (see Table 1), soman-induced SE in the Es1−/− mouse decreases in severity of seizures when ketamine is combined with midazolam, which further validates the use of the Es1−/− mouse as a reliable model for the identification of novel therapies against SE induced by acute CWNA exposure.
The following sections review findings from preclinical studies conducted in our laboratories that demonstrate the benefits of simultaneous administration of anti-seizure drugs as adjunct to midazolam in ameliorating the long-term effects of cholinergic-induced SE. These studies highlight the need to target both the reduced inhibition and excess excitation that characterizes RSE. We discuss the rationale for these studies as well as the clinical implications of these findings.
2. Methods
The detailed methods have been published: Schultz et al., 2014, Schultz, 2015; Niquet et al., 2016, 2017 a, b; Lumley et al., 2019a; and Marrero-Rosado et al., 2020. A brief description also follows.
2.1. Animals
Adult male Sprague-Dawley rats (Charles River) were individually housed with food and water ad libitum. Rats were 350–400 g for soman and 200–300 g for the lithium/pilocarpine studies. Groups used for behavioral assessments in soman studies were maintained on a reverse 12 h:12 h light-dark cycle. For soman studies in Es1−/− mice, male (24–28 g) and female (17–22 g) mice were obtained at 8–9 weeks of age from a breeding colony established at the United States Army Medical Research Institute of Chemical Defense (USAMRICD). Mice were single-housed, with food and water available ad libitum, on a 12 h:12 h light:dark cycle with lights on at 6:00 AM. The experimental protocols were approved by the Animal Care and Use Committee at the United States Army Medical Research Institute of Chemical Defense, and all procedures were conducted in accordance with the principles stated in the Guide for the Care and Use of Laboratory Animals (National Research Council, 2011), the Public Health Service Policy on Humane Care and Use of Laboratory Animals and the Animal Welfare Act of 1966 (P.L. 89e544), as amended.
2.2. Implantation of Electrodes
Under isoflurane anesthesia, the animals were implanted with stainless steel screws for use as recording electrodes. For the soman studies, telemetry transmitters (Data Sciences International; DSI™; F40-EET for rats; ETA-F10 for mice) were implanted subcutaneously (SC) to continuously record electroencephalographic (EEG) activity. In rats, four cortical stainless steel screw electrodes were implanted on the skull at 2 mm bilaterally to the midline and 1.6 mm anterior and 4 mm posterior to bregma. In mice, two cortical stainless steel screw electrodes were placed at 1.5 mm right of the midline, 1.5 mm anterior, and 3.0 mm posterior to bregma. Stainless steel wires from the transmitter were implanted SC, wrapped around the electrodes, and secured in place using dental acrylic.
For lithium/pilocarpine studies, two electrodes were used for bipolar recording and were located 3 mm anterior to lambda and 4 mm left and right of the medial suture. A third electrode served as reference and was located 1 mm anterior to bregma and 1 mm to the right of the mid-line defined by the medial suture. The electrodes were connected to a tri-polar connector (Plastics One), and dental cement covered the electrodes so that only the connector was exposed. In both soman and pilocarpine studies, animals received analgesia (buprenorphine or Buprenex® SR) after surgery followed by 1–2 weeks of surgical recovery.
2.3. Induction of SE and administration of medical countermeasures
Soman (GD, pinacolyl methylphosphonofluoridate) was obtained from the U.S. Army Combat Capabilities Development Command Chemical Biological Center (Aberdeen Proving Ground, MD, USA). Rats were SC administered 1.2 x median lethal dose (LD50) of soman based on Shih et al, 1991 (132 µg/kg;) or Wright et al., 2016, (118 µg/kg) and 1 min later administered an IM injection with an admix of atropine sulfate (2 mg/kg) and the oxime HI-6 dimethanesulfonate salt (DMS; 118.5 mg/kg). Forty minutes after seizure onset, midazolam (3 mg/kg), ketamine (30 mg/kg), and/or valproate (90 mg/kg) was administered intraperitoneally (IP).
In Es1−/− mice, the LD50 of soman was determined via stage-wise adaptive dose design (Feder et al., 1991) in males and in females across estrus. Es1−/− mice, which similar to humans lack plasma CaE, have an LD50 of soman (Marrero-Rosado et al., 2018; Kundrick et al., 2020) that is approximately one-fourth of the LD50 in C57Bl/6 wildtype mice, which is 83 ug/kg (Clement et al., 1981). In mice treated with atropine sulfate and HI-6, a dose approximately four times the lethal dose is required to consistently induce SE with approximately 50% survival (Marrero-Rosado et al., 2018; Kundrick et al., 2019). Male and female Es1−/− mice were exposed SC to either saline (No GD; control group) or 80–82 μg/kg of soman. Soman-exposed mice were treated IP with an admix of atropine sulfate (4 mg/kg) and HI-6 (50 mg/kg) at 1 min after exposure. Midazolam (3 mg/kg; IP), ketamine (30 mg/kg; IP) or a combination of midazolam and ketamine (IP) was administered at 40 min after seizure onset. Saline-exposed mice received midazolam (3 mg/kg; IP) at 50 min after saline administration.
Seizure onset was on average 5 minutes in rats and 4 min in mice, so treatment was approximately 45 and 44 min after exposure, respectively. Following exposures, EEG activity was monitored in real-time, and seizure onset was defined as the appearance of rhythmic high-amplitude spikes (>2 × baseline) that lasted at least 10 s (based on Nissinen et al., 2001). EEG data were continuously recorded at least 24 h before soman exposure up to study endpoint and digitized at 250 Hz to evaluate seizure onset, acute seizure, and the development of SRS.
For pilocarpine-induced SE, rats were administered lithium chloride (5 mEq/kg) SC, and 16 h later, SE was induced with pilocarpine hydrochloride (320 mg/kg; IP). Only lithium/pilocarpine-treated rats displaying behavioral /EEG seizures were used. All rats received scopolamine methyl bromide (1 mg/kg; IP), a muscarinic antagonist that does not cross the blood-brain barrier, at the same time as they received pilocarpine, to decrease peripheral cholinergic effects such as pulmonary secretions. Seizures occurred 7.6 ± 2.7 min. after pilocarpine injection so that the time from pilocarpine injection to mono or dual therapy was approximately 48 min. All animals subsequently received scopolamine (10 mg/kg; IP) to remove the original seizure trigger without stopping SE, and sham injection (control SE group), one drug (monotherapy), a combination of two drugs (dual therapy) or a combination of three drugs (triple therapy) IP at 40 min after EEG seizure onset, a time at which benzodiazepine pharmacoresistance and self-sustaining seizures were well established. Drugs for monotherapy groups included midazolam (9 mg/kg), ketamine (90 mg/kg), and sodium valproate (270 mg/kg). Dual therapy groups included the combination of 4.5 mg/kg midazolam with 45 mg/kg ketamine. Triple therapy groups included the combination of 3 mg/kg midazolam with 30 mg/kg ketamine and 90 mg/kg valproate.
2.4. Seizure recording and analysis of EEG power integral and spontaneous recurrent seizure
For EEG activity recording in soman studies, an RPC-1 physiotel receiver from DSI was placed under each animal’s home cage for continuous data collection (24 h/day) using Dataquest ART Acquisition software (DSI, Inc.). The signal was filtered using a Butterworth filter (pass band of 0.1–125 Hz; notch filter of 60 Hz) (de Araujo Furtado et al., 2009). Epileptiform activity was identified using Dataquest ART 4.1 (analysis software), and a customized MATLAB (MATLAB; Mathworks, 2008a) algorithm (de Araujo Furtado et al., 2009) and confirmed by visual screening. Data files were analyzed to identify the time spent in SE and the development of SRS. For the rat study, the EEG ratio power integral was calculated taking the average of power spectra of each hour period through a customized MATLAB algorithm and applying a formula, [decibels ¼ 10*(Log(V^2sample/V^2normal))]*60 min, resulting in decibels/h. The range of frequency analyzed was 0.1–100 Hz, and the data represent the full spectrum and the ratio of power of EEG signal during SE at specific time points after treatment. For the mouse study, power spectrum density (μV2/Hz; 0.1–125 Hz) was determined by integrating the power spectra calculated through fast Fourier transform in 60-second epochs. In addition, full EEG power spectrum data were further reduced by extracting the median power (10 min bins) in 60-minute intervals to obtain EEG power spectral density values at SE (20 min before treatment), and 1, 3, 6, and 12 h after treatment. For more details on the methods of EEG analysis following soman exposure see de Araujo Furtado et al., 2010, Marrero-Rosado et al., 2018, and Lumley et al., 2019a.
For the lithium-pilocarpine study, the BioPac Systems MP150 was used to record digital EEG using a BioPac UM100A preamplifier. Sampling rate was 200Hz. Recording was started before pilocarpine injection and was continuous for 24 h, which included an initial pre-pilocarpine segment of EEG, the development of SE, drug treatment, and the overnight recovery period. Outcome measure was the ratio of EEG power at T time (e.g. 1 hour after treatment) divided by the average baseline EEG power before pilocarpine injection. These results have been previously published (Niquet et al., 2016; Niquet et al., 2017a; Niquet et al, 2017b).
2.5. Morris water maze (MWM)
Spatial learning and memory were assessed in a 170 cm diameter pool filled with paint-blackened water in which a hidden platform was submerged in a fixed position 1–2 cm below the water surface. Soman-exposed rats were evaluated one month after exposure for spatial memory acquisition with water temperature at 26 ± 1 °C. Briefly, rats received four 60 s trials per session, 2 training sessions per day and a 30 min rest period between sessions for a total of 8 trials/day for three days. After three training days, the platform was removed, and two 60 sec probe trials were conducted. A tracking program (HVS Watermaze 2100, HVS Image) was used to measure latency to escape (time from the start of the trial for the rat to reach the platform and end the trial), target quad time (the percent of trial time that the rat spent in the target quadrant), thigmotaxis (the percent of trial time the rat spent in the perimeter of the water maze), distance (length of the path that the rat took from the starting location to its location at the end of the trial), speed, and Gallagher score (distance between rat and platform at every second of the trial). Similarly, pilocarpine-exposed rats were evaluated 6 weeks after exposure in eight 60 sec trials per day for 5 consecutive days with water temperature at 20 oC and using Ethovision tracking system (Noldus, Inc.) to assess spatial memory acquisition. Ten days later, long-term memory retention was evaluated. For detailed methods on MWM performance following soman exposure see Schultz et al., 2014, and Lumley et al., 2019a. See Niquet et al., 2017b, for detailed methods on pilocarpine-induced MWM impairment.
2.6. Tissue preparation for detection of neuronal injury
Rats and mice were deeply anesthetized with sodium pentobarbital (75 mg/kg, IP) and perfused with heparinized 0.9% saline in 0.1 M phosphate buffer (PB) followed by 4% paraformaldehyde in PB; brains were post‐fixed for 6 hours at 48 °C, cryoprotected in 20% sucrose in PB, and rapidly frozen and stored at −75 °C. Sectioning and staining were completed by FD Neurotechnologies, Inc., to include FD NeuroSilver™ stain to identify degenerating neuronal fibers in coronal sections (50 um) with damage scored on a scale of 0‐4: 0, normal tissue; 1, minimal (up to 10% damage); 2, mild (above 10% up to 25% damage); 3, moderate (above 25% up to 45% damage); and 4, severe (above 45% damage). For more details, see Schultz et al. (2012, 2014). Immunocytochemistry was performed on coronal sections (30 um) using antibodies against the neuronal nuclear protein (NeuN) and the ionized calcium-binding adaptor molecule 1 (Iba1), with cresyl violet used as a counterstain to visualize anatomical landmarks. NeuN profile density was quantified in various regions with Image Pro, as well as by using unbiased stereology with the optical dissector method in the CA1. For Iba1, expressed both in resting and activated microglia, density cell-body-to-cell-size ratio (an indicator of microglial activation) of Iba1-positive cells was determined using Image J software. For details, see Kundrick et al., 2019, and Marrero-Rosado et al., 2020.
2.7. Construction of 3-dimensional isobolograms for toxicity and efficacy
Isobolograms were prepared as described in Niquet et al., 2017a. We defined the median effective dose (ED50) as the dose that reduced a particular measure of seizure severity (i.e., EEG power) by 50%, and the median toxic dose (TD50) as the dose that induced half-maximal toxicity (i.e. severity of toxic signs). The severity of drug toxic signs was scored using the following scale: 0, normal gait; 1, ataxic; 2, unable to walk but able to crawl, righting reflex preserved; 3, righting reflex partially impaired; 4, complete loss of righting reflex; 5, no response to tail pinch; and 6, loss of corneal reflex. Dose-response curves were fit by the method of Chou (2006). The variances of the TD50s were calculated by the method of Tallarida (2000) (p 29, equation 2.9). The theoretical dosage sum was calculated from Tallarida (2000) (p 59, equation 4.1, and its variance from equation 4.2, p 60). For each isobologram, we calculated the ED50 and TD50 for each drug and for combinations, and built a plane connecting the ED50s and TD50s of the 3 individual drugs using SciDavis (http://scidavis.sourceforge.net). The observed toxicity or efficacy of the triple drug combination was then placed in the same 3-dimensional plot with the plane connecting TD50s or ED50s for individual drugs. When the toxicity or efficacy doses of the combination lies below the additivity plane in the isobolograms, a positive cooperativity or drug synergism is assumed. When the effects are simply additive, the values fall in the additivity plane. Negative cooperativity would result in values above that plane. Significance was determined by a modified t-test (Tallarida, 2000, Section 2.3, p 60–62).
2.8. Statistical analysis
For soman studies, median survival time and median SRS time were estimated and compared using a Kaplan-Meier analysis, followed by a log-rank test to determine treatment effect on the distributions. EEG power integral and measures in the MWM were analyzed using a repeated measures ANOVA. If there were significant interactions between group and repeat (time or trial), a one-way ANOVA was used to compare groups at each repeat and to compare times within each group. A silver stain neuronal damage score was analyzed using a Mann-Whitney U test. NeuN immunoreactivity was analyzed using a one-way ANOVA, followed by group comparisons using a Tukey’s test. For lithium-pilocarpine studies, when data showed a non-Gaussian distribution, they were presented as median values with the interquartile range, which is the difference between the 75th and 25th percentile, and analyzed with nonparametric statistical methods: Kruskal-Wallis test followed by Dunn’s multiple comparison test. MWM data was analyzed by 2-way ANOVA. Differences were considered statistically significant when p < 0.05.
3. Results and Discussion
3.1. Synergistic effects of the GABAA receptor agonist midazolam in combination with the NMDA receptor antagonist ketamine in treating benzodiazepine-refractory SE
The receptor trafficking hypothesis posits that seizure-induced receptor trafficking, consisting of a loss of synaptic GABAA receptor and an increase in synaptic ionotropic glutamate receptors, is a major contributor to the development of pharmacoresistance following cholinergic-induced SE. This does not discount a role for other physiological changes that occur during SE, such as changes in transmitter or neuropeptide release, extracellular potassium, and intracellular chloride, among others. However, it suggests that blocking receptor trafficking and/or its consequences may overcome pharmacoresistance and seizures may respond to drug treatment even when treatment is delayed.
Although current medical countermeasures when delayed will not prevent SE-associated receptor trafficking, receptor-targeted treatment can block its consequences. Drugs such as benzodiazepines, neurosteroids or other GABAergic drugs that stimulate remaining synaptic and extra-synaptic GABAAR can be used to increase inhibition. If treatment is late and too few GABAAR remain in synapses to fully restore inhibition by that mechanism, another drug that increases inhibition by a non-GABAergic mechanism (or reduces excitation) should be beneficial. In addition, antagonists of NMDAR and AMPAR would reduce excitation. Combining GABAAR agonist and NMDAR antagonist drugs in the initial treatment of SE is not recommended by current therapeutic guidelines (Glauser et al., 2016), but is effective in preclinical models of cholinergic-induced SE, including against high-dose lithium/pilocarpine exposure in rats (Niquet et al., 2016, 2017a, b; 2019), soman exposure in rats (Lumley et al., 2019a), and in Es1−/− mice (Marrero-Rosado et al., 2020), as shown below.
3.1.1. Soman-induced morbidity and SE in rats: Combination drug efficacy
Although early treatment with benzodiazepines (within 5–20 min of exposure) is effective at terminating seizure and seizure-associated brain damage following soman-induced SE, when treatment is delayed to 40 min after seizure onset, benzodiazepines are less effective at terminating SE (McDonough and Shih, 1993; Schultz et al., 2012). Midazolam or midazolam-ketamine combination improves survival over that in untreated rats or in those treated with ketamine monotherapy, but seizure severity is lessened in rats treated with midazolam-ketamine compared to untreated or to monotherapy. Rats treated with midazolam (3 mg/kg; 77.8% survival) or with midazolam and ketamine combination therapy (72.7% survival) 40 min after SE had greater 14-day survival compared to rats treated with saline (vehicle; 40% survival) or ketamine monotherapy (50% survival; Figure 1A). The majority of lethality occurred within the first day of exposure. The values of EEG power integral of treatment groups were compared in specific time periods (during pre-treatment SE, and during the first hour or 6 h after treatment). EEG power integral (a measure of EEG seizure severity) increased in all soman-exposed rats during SE with no differences between groups before treatment (Figure 1B). Rats treated with 30 mg/kg ketamine + 3 mg/kg midazolam after 40 min of soman-induced SE had reduced EEG power integral (reduced seizure severity) during the 1 h after treatment compared to vehicle-, midazolam- or ketamine-treated rats. In addition, during the 1 h and 6 h time periods after treatment, rats treated with midazolam-ketamine combination had a greater decrease in percent EEG power integral (70%) compared to those receiving midazolam monotherapy (14%) (p < 0.01 at 1 h, p < 0.05 at 6 h). The first hour is the critical time to reduce neuronal injury (which may appear within 20 min of SE onset; McDonough et al., 1995; Schultz et al., 2012) and pharmacoresistance, which increases with seizure burden (Niquet et al., 2016; 2019). Representative tracings of EEG signal are shown in Figure 1C for baseline (24 h prior to soman exposure), SE, and 1 h and 12 h after SE onset (20 min and 11 h 20 min after treatment, respectively).
Figure 1. Addition of ketamine to delayed midazolam treatment after soman (GD) exposure increased survival and reduced seizure severity.

Ketamine (KET; 30 mg/kg), midazolam (MDZ; 3 mg/kg) or KET/MDZ combination was administered IP at 40 min after seizure onset induced by SC exposure to GD (132 µg/kg). A) Soman-exposed rats treated with vehicle (GD/VEH; 40%; n=6/15) or with KET monotherapy (GD/KET; 50%; n= 4/8) had poor survival compared to rats administered MDZ (GD/MDZ; 77.8%; n=21/27) or MDZ/KET (GD/MDZ/KET; 72.7%; n=8/11) combination therapy. B) The EEG ratio power integral in rats treated with MDZ and KET was reduced 1 h after treatment compared to GD/MDZ, GD/VEH or to GD/KET. Data shown are mean ± SEM. C) Representative images of EEG tracings are shown at baseline, SE (60 s after SE onset), 1 h after SE onset, and 12 h after SE onset for GD-exposed animals treated with midazolam, ketamine, or midazolam-ketamine. *p < 0.05. Data modified from Lumley et al., 2019, and Niquet et al., 2019.
3.1.2. Soman-induced morbidity and SE in plasma CaE knockout (Es1−/−) mice: Combination drug efficacy
Similar to findings in rats, administration of ketamine as an adjunct to midazolam in Es1−/− mice improves outcome following soman-induced SE compared to midazolam treatment. In particular, midazolam-ketamine combination increases survival and reduces seizure severity compared to midazolam monotherapy. Male and female mice were exposed to a seizure-inducing dose of soman and administered delayed anticonvulsant therapy comprised of midazolam monotherapy or a midazolam-ketamine combination; their survival was monitored over the course of 14 days following exposure (Figure 2A). Logistic regression analysis detected a main effect of treatment without an effect of sex on survival by the end of the study. A chi-square analysis followed by Fisher’s exact test revealed that the percentage of animals surviving at the study endpoint in the midazolam monotherapy group (46.2%) was significantly reduced compared with the control (No soman) group (100%). Percent survival in the midazolam-ketamine combination therapy group (77.3%) was significantly higher compared with the midazolam monotherapy group. This mouse model is a severe model of soman-induced SE with high lethality that is reduced by treatment with the midazolam-ketamine combination.
Figure 2. Effect of delayed midazolam treatment combined with ketamine on survival and seizure severity following soman (GD)-induced status epilepticus in Es1−/− mice.

A) Mice exposed SC to 80 μg/kg of GD and treated with midazolam monotherapy (GD + MDZ) had a significantly lower percentage of survival compared with the No GD group. The animal group treated with midazolam-ketamine combination therapy (GD + MDZ/KET) had a percentage of survival that was higher than the GD + MDZ group and not significantly different from the No GD group. *p < 0.05, compared with No GD. ^p < 0.05, compared with GD + MDZ. B) Effect of ketamine as an adjunct to delayed midazolam on EEG power spectral density and frequency bands following GD exposure. Soman exposure increased EEG power spectral density during SE (−20 min relative to treatment) compared with the No GD group. At 1, 3, and 6 h after treatment, the power density of the group of mice that received midazolam monotherapy after GD exposure (GD + MDZ) continued to increase, while the midazolam-ketamine group (GD + MDZ/KET) had reduced EEG power spectral density compared with the No GD group; the effect of midazolam-ketamine treatment seems to wear off, indicated by the significant increased EEG power density at 12 h compared with the No GD group. *p < 0.05, ***p < 0.001, compared with the No GD group. ^p < 0.05, ^^^p < 0.001, compared with GD + MDZ group. C) Representative EEG tracings of 24-hour compressed signal and 10-second recordings at baseline (24 h prior to GD exposure), status epilepticus, 20 min after treatment, and 12 h after treatment. Modified from Marrero-Rosado et al., 2020.
Exposure to soman induced behavioral seizure with a mean (± SD) latency of 2.1 ± 1.3 min. Treatment with midazolam-ketamine therapy significantly reduced behavioral seizure score compared with midazolam therapy by 20 min after treatment and was lower than midazolam during most of the remaining 4 h of observations (Marrero-Rosado et al., 2020). Mice treated with midazolam or midazolam-ketamine combination therapy had initial seizure activity that lasted an average (± SD) of 620 ± 365 min and 529 ± 418 min, respectively, in the first 24 h following exposure with no sex or treatment differences. Soman-induced SE caused an increase in the EEG power density in Es1−/− mice that was estimated to be an average (± SD) of 615 ± 181.4% in the midazolam group and 557 ± 121.1% in the midazolam-ketamine group (Figure 2B). The midazolam monotherapy group showed increased EEG power density for up to at least 3 h compared with the control (No soman) group. In contrast, at 1, 3, and 6 h after treatment, the EEG power density in the midazolam-ketamine combination therapy was significantly lower than in the midazolam monotherapy group and was not significantly different from the control group. Representative images of EEG tracings at various time points are shown in Figure 2C for soman-exposed animals treated with midazolam or midazolam-ketamine combination. An increase in the power of EEG activity in the delta frequency (0.1–4.0 Hz) occurs in soman-exposed Es1−/− mice (Marrero-Rosado et al., 2018), and in rats this increase correlates with neuropathological damage (McDonough et al., 1998; Philippens et al., 1992; Schultz et al., 2015). Following administration, the midazolam-ketamine therapy caused a brief 40-minute decrease in delta power such that it was not significantly different from the No soman group, whereas midazolam therapy did not reduce the soman-induced increase in delta (Marrero-Rosado et al., 2020). However, neither midazolam nor midazolam-ketamine treatment was able to prevent the prolonged increase in the power of delta, suggesting that combination therapy may benefit from targeting an additional mechanism to better prevent the neuropathological sequelae that results from soman-induced seizure. We also observe that in soman-exposed rats treated with midazolam-ketamine combination, the increase in the power of delta caused by the occurrence of SE returns to levels similar to control much more rapidly than in animals treated with midazolam or ketamine monotherapy (Schultz, 2015).
3.1.3. Cholinergic-induced SE in rats: Efficacy of triple drug combination therapy
In the lithium/pilocarpine studies, EEG activity was recorded for 24 h following pilocarpine injection, and the midazolam-ketamine combination powerfully reduced EEG power and other measures of seizure severity, compared to higher-dose monotherapy (Niquet et al., 2016). Since we suspected that when treating at 40 min not enough synaptic GABAAR are left to fully restore inhibition with midazolam, we also studied the effect of supplementing the midazolam-ketamine combination with an anti-seizure drug acting at a non-benzodiazepine site and observed even better results (Figure 3). Valproate is a well-known anticonvulsant whose mechanism of action is still not well understood but may involve, among other mechanisms, the inhibition of rapidly inactivating sodium channels and the regulation of GABA metabolism (reviewed in Sills and Rogawski, 2020). In randomized double-blind studies in adults (Kapur et al., 2019) and children (Chamberlain et al., 2020), valproate showed similar anticonvulsant efficacy to two drugs commonly used to treat RSE, levetiracetam and fosphenytoin. We examined the effect of triple therapy and triple-dose monotherapies on the EEG power integral over the first hour posttreatment (Figure 3C). Kruskal-Wallis analysis showed that triple therapy significantly decreased EEG power over the first hour post-treatment compared to midazolam, ketamine and valproate monotherapy. Among all groups, the combination of midazolam–ketamine-valproate was the only treatment that decreased EEG power below pre–pilocarpine baseline. In addition, triple therapy significantly reduced the EEG power integral over the six hours posttreatment when compared to midazolam, ketamine and valproate monotherapy (Figure 3D), demonstrating that seizures did not recur. We also compared the effect of triple therapy with higher-dose midazolam 4.5 mg/kg-ketamine 45 mg/kg dual therapy. We found that midazolam-ketamine-valproate triple therapy (median = 5.95 min) was more efficient in reducing the duration of SE than was higher-dose midazolam 4.5 mg/kg-ketamine 45 mg/kg dual therapy (median = 8.45 min; p < 0.05).
Figure 3. The midazolam-ketamine-valproate combination was more effective than triple-dose midazolam, ketamine or valproate monotherapy in reducing the severity of SE in both the lithium/pilocarpine model of SE and soman model of SE.
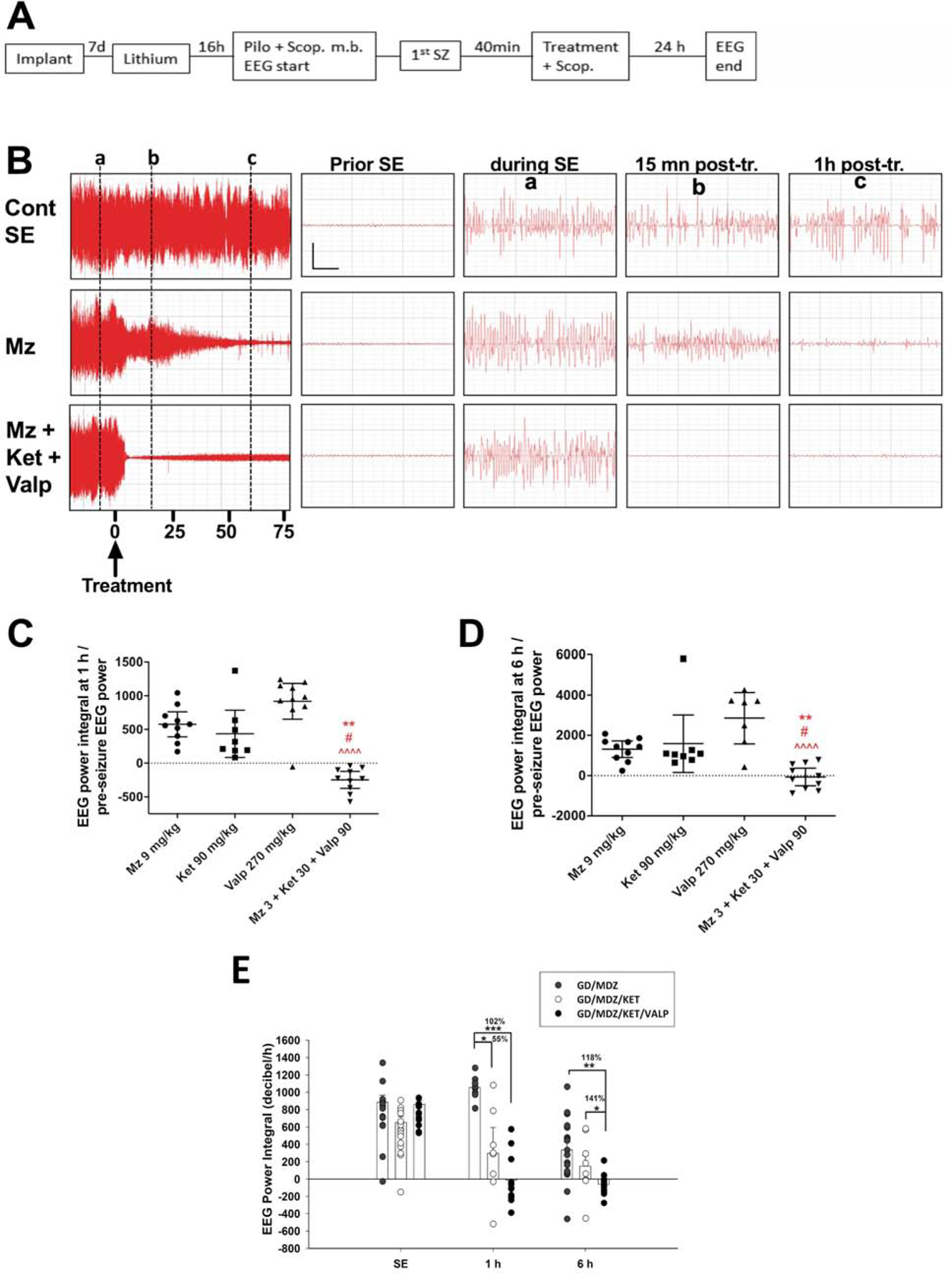
A) Experimental flow: A severe form of SE was induced by administration of a high dose of lithium + scopolamine methyl bromide (scop. m. b), followed by an injection of a high dose of pilocarpine. Drug(s) or vehicle and scopolamine (scop. m. b) were injected 40 min after seizure onset. Animals were implanted 1 week prior to SE induction and euthanized 48 h after SE onset. B) The left panels show the compressed EEG from SE control, midazolam, or midazolam-ketamine-valproate animals up to 75 min following treatment. The right panels show magnified 6 sec EEG tracings prior to SE or following SE (marked by vertical lines a‐c). Vertical bar = 0.5 mV; horizontal bar = 1 sec. C) This graph shows the ratio of EEG power integral over the first hour to initial EEG power at baseline, before pilocarpine injection. The midazolam-ketamine-valproate group (n=10), which displayed an EEG power that fell below the pre-pilocarpine baseline, is significantly different from the midazolam (n=10; ** p < 0.01), ketamine (n=8, # p < 0.05) and valproate (n=10, ^^^^ p < 0.0001) groups. D) This graph shows the ratio of EEG power integral over the first 6 h following treatment to initial EEG power at baseline, before pilocarpine injection. The midazolam-ketamine valproate group (n=10), which lowered the EEG power below pre-pilocarpine baseline, is significantly different from midazolam (n=10; ** p < 0.01), ketamine (n=8, # p < 0.05) and valproate (n=7, ^^^^ p < 0.0001) by Kruskal-Wallis, followed by Dunn’s test. E) Triple therapy with midazolam-ketamine-valproate was efficacious in reducing EEG seizure severity against soman (GD; 118.1 µg/kg, SC)-induced SE. Rats were treated 40 min after seizure onset with midazolam, midazolam-ketamine or midazolam-ketamine-valproate. The triple therapy reduced EEG power integral 1 and 6 h after SE compared to midazolam. The dual therapy of midazolam-ketamine reduced EEG power integral 1 h, but not 6 h after SE. ***p < 0.001; **p < 0.01; p < 0.05. Data shown are mean ± S.E.M. Figure was modified from Niquet et al., 2017b, 2019, and Lumley et al., 2019a.
These findings extended to reduced seizure severity in soman-exposed animals treated with triple therapy. Rats exposed to soman and treated at 40 min after seizure onset with a combination of midazolam-ketamine-valproate spent an average (± SD) of 128.8 ± 172.2 min in seizure, which was significantly less than seizure duration in the midazolam (327.4 ± 179.2 min) and midazolam-ketamine (301.6 ± 248.6 min) groups. As increased seizure duration correlates with increased neuropathology (McDonough et al., 1995), this reduced time in initial seizure may contribute to reduced loss of neurons . Midazolam-ketamine-valproate triple therapy was also more effective than midazolam or midazolam-ketamine at reducing the EEG power integral when administered 40 min after soman-induced SE in rats (Figure 3E). At 1 h after treatment, the combination of midazolam-ketamine and the combination of midazolam-ketamine-valproate reduced the EEG power integral much better than midazolam monotherapy. The triple therapy significantly reduced the EEG power integral over the 6 h posttreatment when compared to midazolam monotherapy or midazolam-ketamine dual therapy.
3.1.4. Drug combinations based on the receptor trafficking hypothesis are synergistic and have a high efficacy/toxicity ratio
Isobolograms (Tallarida, 2000, 2006) suggest that the midazolam-ketamine-valproate combination potentiates the therapeutic response without potentiating drug toxicity (Figure 4) so that the therapeutic index (Muller and Milton, 2012) is improved by switching from mono- to polytherapy. The key to therapeutic success is the ratio of efficacy to toxicity. If a combination of drugs potentiates both efficacy and toxicity, the latter will reduce the amount of drug tolerated, and nothing is gained. Figure 4 shows that toxicity was simply additive between the three drugs of the combination, while the therapeutic response was synergistic and showed positive cooperativity. As a result of that synergy, this three-drug combination delivered a greater therapeutic response than monotherapy for the same level of drug-induced toxicity. The mechanism of that synergy is unknown, since the drugs involved have no known direct interactions at the molecular level, and the effect is too rapid to be due to pharmacokinetic interactions. This synergism was powerful enough to overcome pharmacoresistance at the times tested in this study: the diazepam/ketamine/valproate combination stopped seizures at a time when pharmacoresistance to high doses of diazepam (20 mg/kg) was clearly present.
Figure 4. Drug interactions increased the therapeutic effects, but not the toxic effects.

(A) Toxicity isobologram: the toxicity score for the combination of diazepam, ketamine and valproate (red dot) was precisely in the plane connecting monotherapy values, suggesting a simply additive effect. (B) Efficacy isobologram: the efficacy of diazepam-ketamine-valproate combination in reducing the time needed for EEG power to fall to two times the pre-seizure power, a good measure of seizure termination (red dot), was far below the plane connecting individual drug dosages producing a similar efficacy score (ED50s), suggesting a synergistic effect between drugs. Therefore, for the diazepam-ketamine-valproate combination, the therapeutic effects were increased by giving these drugs in combination, but the toxic effects were not. Figure is republished with permission from Niquet et al., 2017a.
3.2. Effect of GABAAR agonist-NMDAR antagonist combinations on long-term consequences of cholinergic SE
3.2.1. SRS after soman-induced SE in rats: Efficacy of combination therapy
We previously demonstrated that in rats the development of SRS was linked to more severe neuropathological damage following soman-induced SE (de Araujo Furtado et al., 2010). Rats that develop SE following acute exposure to soman and undergo epileptogenesis in the days following the toxic insult show a higher degree of neuronal degeneration in the CA1 region of the hippocampus than animals that developed SE without the appearance of SRS. It is hypothesized that the neuropathological damage caused by the initial excitatory drive resulting from soman-induced seizure results in plastic changes that may underlie the appearance of SRS (de Araujo Furtado et al., 2010). Rats were exposed SC to soman, and EEG recordings were analyzed for 36 days following exposure to identify and quantify SRS. Kaplan-Meier analysis revealed that the combination of midazolam with ketamine significantly increased the median latency to first SRS compared to the midazolam monotherapy group (Figure 5A). The number of SRS events that animals presented is also shown (Figure 5B).
Figure 5. Addition of ketamine to delayed midazolam treatment after soman (GD) exposure reduced incidence of epileptogenesis in rats following GD-induced seizure.

Rats were SC exposed to GD (132 µg/kg) and ketamine (KET; 30 mg/kg), midazolam (MDZ; 3 mg/kg) or KET/MDZ combination was administered IP at 40 min after seizure onset. (A) Treatment with the midazolam–ketamine combination (GD/MDZ/KET; 20%, n=2/10) reduced the incidence of SRS during the first 36 days after GD-induced SE, compared to midazolam monotherapy (GD/MDZ; 63.6%, n=7/11) and ketamine monotherapy (GD/KET; n=6/10). (B) Number of SRS is shown. *p < 0.05. Data modified from Lumley et al., 2019, and Niquet et al., 2019.
3.2.2. SRS after soman-induced SE in Es1−/− mice: Efficacy of combination therapy
The findings in rats that midazolam-ketamine combination reduced the development of SRS compared to midazolam monotherapy following soman exposure extended to the Es1−/−mouse model. In the days following soman exposure, the development of SRS was observed in a portion of Es1−/− mice that developed SE. A chi-square analysis with Fisher’s exact test detected a significant difference between treatment groups in the percentages of animals developing SRS, with midazolam monotherapy resulting in 11 out of 12 (91.7%) surviving animals which developed SRS, while 8 out of 17 (47.1%) animals in the midazolam-ketamine combination developed SRS (Figure 6A). Additionally, a significant effect of treatment on the median onset of SRS development was found by Cox regression analysis, without a main effect of sex (Figure 6A). A general linear model analysis did not detect main effects of treatment or sex in the average total number of SRS events, with the midazolam monotherapy group presenting an average (± SD) of 11.5 ± 9.6 SRS and the midazolam-ketamine group presenting an average (± SD) of 5.8 ± 9.0 SRS (Figure 6B).
Figure 6. Effect of delayed midazolam treatment combined with ketamine on epileptogenesis following soman (GD)-induced status epilepticus in Es1−/− mice.

Es1−/− mice were SC exposed to 80 μg/kg of GD and treated at 40 min after seizure onset with midazolam monotherapy (GD+MDZ) or midazolam-ketamine combination therapy (GD+MDZ/KET), and EEG activity was monitored for 14 days after exposure. (A) The onset of the first detected SRS for each surviving animal is graphed to indicate the percentage of mice in GD + MDZ (91.7%, n=11/12) and GD + MDZ/KET (47.1%, n=8/17) that developed SRS. (B) The average (± SD) number of SRS events is graphed for each group. One animal presented with SRS before dying at post-exposure day 10 and was not included in the total count of SRS during the 14 days after exposure. *P < 0.05, compared with the GD + MDZ group. Figure was previously published in Marrero-Rosado, et al., 2020.
3.2.3. The midazolam-ketamine-valproate combination is more effective than mono or dual therapy in reducing cholinergic-induced SRS.
We tested in the rat model of soman-induced SE the addition of valproate to the midazolam-ketamine combination, and compared the anti-seizure efficacy of the triple polytherapy to that of dual (midazolam-ketamine) and midazolam monotherapy. Rats in the midazolam-ketamine-valproate group showed a lower incidence of epileptogenesis (Figure 7A) and a smaller number of SRS (Figure 7B) throughout the 14 days following soman exposure. Although midazolam-ketamine combination reduces SRS in soman-exposed rodents, the addition of valproate significantly reduced the number of SRS compared to midazolam. Thus, this three-drug combination is synergistic at relatively low doses and delivers a greater therapeutic response than monotherapy.
Figure 7. Results of treating soman (GD)-induced SE with benzodiazepine monotherapy, with a GABAAR agonist-NMDAR antagonist combination, or with a GABAAR agonist-NMDAR antagonist-anti-seizure drug combination, 40 min after seizure onset.

A. Treatment with ketamine/midazolam (GD/MDZ/KET) or ketamine/midazolam/valproate acid (GD/MDZ/KET/VALP) 40 min following GD (118.1 µg/kg, SC) exposure significantly reduced the percentage of animals that developed SRS compared to midazolam monotherapy. B. Triple therapy (GD/MDZ/KET/VALP) significantly reduced the mean (± SD) number of SRS during the 2 weeks following GD-induced seizure compared to midazolam monotherapy (GD/MDZ). *p < 0.05; ** p < 0.01, compared to MDZ monotherapy. Data shown are mean ± S.E.M.
3.2.4. Soman-induced spatial memory deficits in rats: Efficacy of combination therapy
The Morris water maze was used to assess spatial learning one month after exposure. Soman-exposed rats treated only with midazolam or ketamine monotherapy displayed longer escape latencies over repeated training sessions, but soman-exposed rats treated with the combination of midazolam and ketamine did not differ from saline (No soman) controls (Figure 8A). The combination of midazolam and ketamine also protected against the soman-induced decrease in time searching the target quadrant (Figure 8B). Rats treated with only midazolam or ketamine spent less time in the target quadrant than controls. Soman-exposed rats treated with either ketamine or midazolam alone displayed increased thigmotaxis during training (Figure 8C), but rats treated with the combination of midazolam/ketamine did not differ from No soman controls. Soman-exposed rats treated with either midazolam or ketamine alone also traveled a longer distance (Figure 8D) to reach the platform over repeated sessions, but soman-exposed rats treated with a combination of midazolam/ketamine did not differ from unexposed controls. During the probe trials, rats that received midazolam or ketamine spent the trial further from the platform (higher Gallagher score) compared to control while those that received midazolam-ketamine combination spent the trial closer to the platform location (lower Gallagher score) (Figure 8E). These findings demonstrate long-term functional protection afforded by midazolam-ketamine combination treatment of soman-induced SE.
Figure 8. Delayed ketamine and midazolam combination therapy ameliorates spatial memory acquisition impairment following soman (GD)-induced seizure.

Ketamine (KET; 30 mg/kg) with or without midazolam (MDZ; 3 mg/kg) was administered at 40 min after GD-induced (132 µg/kg, SC) seizure onset and compared with no agent control rats (n = 10–11/group). Morris water maze testing was conducted one month after GD exposure. Soman-exposed rats treated with combination therapy (GD/MDZ/KET) performed similarly to No GD control rats. Rats treated with MDZ monotherapy (GD/MDZ) or KET monotherapy (GD/KET) had greater latency to locate the platform (A), spent less time in the target quad (B), spent more time in thigmotaxis (C), and traveled a greater distance (D). (E) The Gallagher score, which is a measurement of the cumulative distance between the rat and the platform for the entire trial, was used to assess memory retention. GD-exposed rats treated with KET or MDZ had significantly higher Gallagher scores than unexposed controls, whereas the scores of GD-exposed rats that had been treated with GD/KET/MDZ were comparable to those of unexposed control rats*p < 0.05. Figure was modified from Niquet et al., 2019, and Lumley et al., 2019. Data shown are mean ± SEM.
3.2.5. Lithium-pilocarpine-induced spatial memory in rats: Efficacy of combination therapy.
The midazolam-ketamine-valproate combination, which reduced the severity of SE, also reduced its long-term consequences in the lithium-pilocarpine model 6 weeks after SE. The performance was impaired in the midazolam group compared to sham controls. The triple therapy group performed better than the high-dose midazolam group (p < 0.05) and did not differ significantly from sham (no seizure) animals in both the acquisition (Figure 9A) and the retention tests (Figure 9B).
Figure 9. Delayed triple therapy reduces behavioral deficits in the Morris water maze after lithium-pilocarpine-induced seizures in rats.

In the lithium-pilocarpine model, midazolam-ketamine-valproate therapy (n=13) reduced behavioral deficits in the Morris water maze compared to midazolam-treated (n=14), which performed poorly compared to sham (no pilocarpine) control rats (n=16). (A) Graph A shows the latency to reach the hidden platform (y-axis) on each testing day (x-axis). Data are presented as mean ± SEM. * p < 0.05 vs Mz 9 mg/kg by 2-way-ANOVA. (B) Graph B shows the latency during the retention test. * p < 0.05 and ** p < 0.01 vs Mz 9 mg/kg by Kruskal-Wallis followed by Dunn’s test. Figure was modified from Niquet et al., 2017b.
3.3. Effect of GABAAR agonist-NMDAR antagonist combinations on neuronal injury and neuroinflammatory response
3.3.1. Soman-induced neurodegeneration and neuronal loss in rats: Efficacy of combination therapy
CWNA-induced SE has been associated with neuropathological damage in both human victims of sarin exposure (reviewed in Jett et al., 2020 and Figueiredo et al., 2018) and animal models of CWNA toxicity (Petras, 1981; McDonough et al., 1995; Schultz et al., 2012). This brain damage is often accompanied by long-term cognitive impairments (Grauer et al., 2008; Moffett et al., 2011; Langston et al., 2012; Schultz et al., 2014; reviewed in Aroniadou-Anderjaska et al., 2016; Lumley et al., 2019b). Thirty-six days following soman-induced SE, the density of NeuN-positive cells in the thalamus (mediodorsal, laterodorsal, and centromedial), reuniens, amygdala, and piriform cortex of soman-exposed rats treated with midazolam-ketamine combination was not significantly different from that seen in brain sections of unexposed control rats (Figure 10A). Rats treated with midazolam or ketamine monotherapy had significantly lower NeuN-positive profile densities than did unexposed controls in all of the aforementioned regions. Representative images of brain slices immunohistochemically processed for NeuN are shown in Figure 10B.
Figure 10. Delayed ketamine and midazolam combination therapy reduced neuronal cell death and neuronal fiber degeneration following soman (GD)-induced seizure.
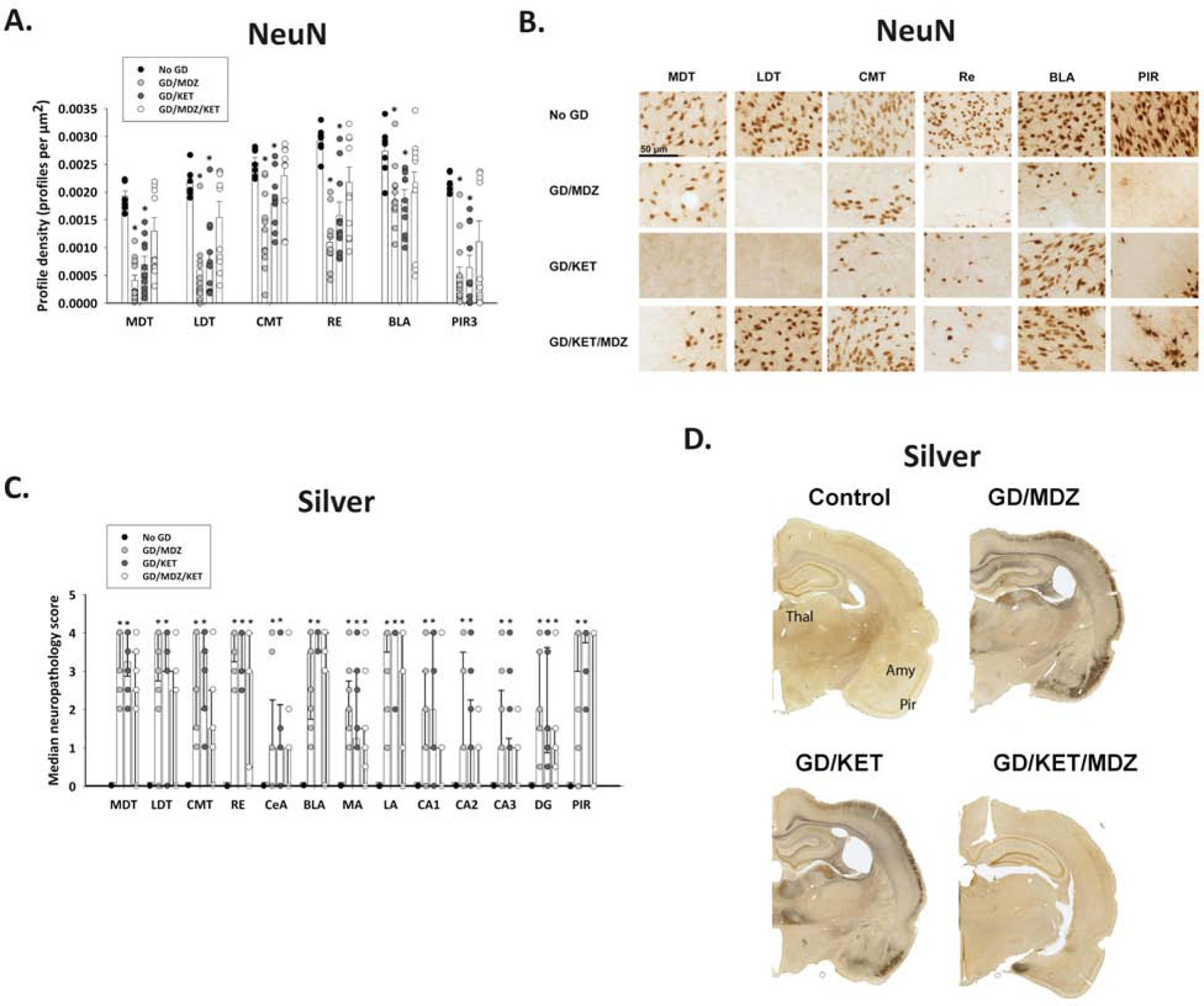
Ketamine (KET; 30 mg/kg) with or without midazolam (MDZ; 3 mg/kg) was administered at 40 min after GD-induced (132 µg/kg, SC) seizure onset and compared with no agent control rats (n = 10–11/group). Brain samples were processed for immunohistochemistry with NeuN antibody to visualize mature neurons (A, B) and a silver stain to visualize neuronal fiber degeneration (C, D). (A) Rats treated with MDZ monotherapy (GD/MDZ) or KET monotherapy (GD/KET) had significantly reduced NeuN-positive (NeuN+) cells compared to No GD control in the dorsomedial thalamus (MDT), dorsolateral thalamus (LDT), central medial thalamus (CMT), reuniens (RE) nucleus of the thalamus, basolateral amygdala (BLA), and layer 3 of the piriform cortex (PIR3); Data shown are mean ± SEM. (B) Representative images are shown for brain tissue stained with NeuN. (C) Rats in GD/MDZ and GD/KET groups had significantly higher neuropathology scores shown via silver stain compared to No GD control in the MDT, LDT, CMT, RE, central amygdala (CeA), BLA, medial amygdala (MA), lateral amygdala (LA), CA1, CA2, CA3, and dentate gyrus (DG) regions of the hippocampus, and the piriform cortex (PIR). (D) Representative images of silver-stained brain tissue are shown. Data shown are median ± SD. *p < 0.05, compared to No GD group.
Intense silver staining, an indicator of neuronal degeneration, was also present in the thalamus, amygdala, hippocampus and the piriform cortex of soman-exposed rats treated with midazolam or ketamine monotherapies (Figure 10C). Soman-exposed rats treated only with midazolam or ketamine had higher neuropathology scores than unexposed controls in all regions. Soman-exposed rats treated with the combination of midazolam and ketamine had significantly lower neuropathology scores in all regions except reuniens, medial and lateral amygdala, and the dentate gyrus, suggesting that partial protection against neurodegeneration was afforded by the combination treatment. Representative images of brain slices stained with silver are shown in Figure 10D.
3.3.2. Soman-induced neuronal loss and neuroinflammation in Es1−/− mice: Efficacy of combination therapy
Similar to findings in rats exposed to soman, male and female Es1−/− mice exposed SC to a seizure-inducing dose of soman had widespread severe neuropathology at two weeks following exposure. Irrespective of sex, a significant effect of treatment on neuronal cell density was detected. In soman-exposed Es1−/− mice treated with midazolam monotherapy a significant decrease in neuronal (NeuN-positive) cell density was detected in the dorsomedial thalamus, dorsolateral thalamus, basolateral amygdala, layer 3 of the piriform cortex, and the hilus and CA1 regions of the hippocampus compared with the control (No soman) group (Figure 11A). In contrast, neuronal cell density was significantly higher in the thalamic nuclei, basolateral amygdala, and CA1 of the hippocampus in the midazolam and ketamine combination treatment group compared with the midazolam group, suggesting that the combination therapy conferred better neuroprotection than the standard monotherapy. In the piriform cortex and the CA1 region of the hippocampus of animals treated with midazolam and ketamine neuronal cell density was not significantly different from the control group, demonstrating that the combination therapy fully protected in these regions. Representative images of brain slices immunohistochemically processed for NeuN are shown in Figure 11D.
Figure 11. Effect of soman (GD)-induced SE and delayed midazolam monotherapy or midazolamketamine combination treatment on mature neuronal cell populations and neuroinflammation of Es1−/− mice.

At 2 weeks following SC exposure to 80 µg/kg of GD and delayed midazolam (GD+MDZ) or midazolam-ketamine (GD+MDZ/KET) treatment, Es1−/− mice were perfused and brains collected and processed for immunohistochemistry NeuN and Iba1 to visualize mature neurons and microglia, respectively. (A) NeuN-positive (NeuN+) cells and (B) Iba1-positive (Iba1+) cells were counted in the bregma range of −1.28 to −1.64 mm, and cell densities estimated in the dorsomedial thalamus, dorsolateral thalamus, basolateral amygdala, hilus (NeuN only), and layer 3 of the piriform cortex. NeuN+ cell density in the CA1 region of the hippocampus was quantified following stereology methods in the region of −1.22 to −3.88 mm from bregma. (C) The cell-body-to-cell-size ratio was also estimated for Iba1+ cells as a measure of activated microglia. Representative images are shown for brain tissue stained with (D) NeuN and (E) Iba1. *p < 0.05, **p < 0.01, ***p < 0.001, compared with the control (No GD) group. ^p < 0.05, ^^p < 0.01, ^^^p < 0.001, GD + MDZ compared with the GD + MDZ/KET group. Modified from Marrero-Rosado et al., 2020.
Our results of neuroprotective effects in brain regions associated with seizure initiation and propagation in rats and Es1−/− mice that received midazolam and ketamine combination following soman exposure are consistent with findings in rats that ketamine and diazepam treatment given at 40 min after soman-induced seizure is neuroprotective in the piriform cortex and thalamic nuclei (reviewed in Ballough et al., 2008). Similar neuroprotective effects occur in the hippocampus following sarin-induced SE in rats treated with midazolam and ketamine combination therapy at 50 min after exposure (Lewine et al., 2018). The neuroprotection of the midazolam and ketamine combination therapy in soman-exposed rats and Es1−/− mice could be the result of a lower incidence of SRS development, previously shown to exacerbate neuropathology (de Araujo Furtado et al., 2010), but also may relate to decreased seizure severity. However, the neuroprotective effect of midazolam and ketamine combination therapy against soman exposure was incomplete, suggesting that additional anti-seizure drugs may be needed.
Persistent neuroinflammation may play an important role in epileptogenesis and secondary neuronal degeneration (reviewed in Clossen and Reddy, 2017). We therefore assessed the degree of microgliosis and microglial activation in soman-exposed Es1−/− mice. A robust neuroinflammatory response, indicated by the presence of Iba1-positive cells (microglia), was observed at two weeks following soman-induced seizures and delayed anticonvulsant therapy. A significant effect of treatment on Iba1-positive cell density was detected, without an effect of sex. Exposure to soman and treatment with midazolam monotherapy resulted in an increase in Iba1-positive cell density in the dorsomedial thalamus, dorsolateral thalamus, basolateral amygdala, layer 3 of the piriform cortex, and the CA1 region of the hippocampus compared with the control group (Figure 11B). Treatment with midazolam and ketamine combination therapy resulted in significantly reduced Iba1-positive cell density in the piriform cortex and the CA1 region of the hippocampus compared with midazolam monotherapy; these densities were not significantly different from those in the control group, demonstrating the effective anti-neuroinflammatory properties of adding ketamine to a benzodiazepine treatment. Furthermore, Iba1-positive cell densities in the basolateral amygdala and dorsomedial thalamus of soman-exposed mice treated with midazolam-ketamine combination were not significantly different from those of the control group. We also performed a quantitative analysis of the average cell-body-to-cell-size ratio to assess the state of microglia present in the brain regions of interest. A significant increase in the average cell-body-to-cell-size ratio was detected in the dorsomedial thalamus, dorsolateral thalamus, basolateral amygdala, layer 3 of the piriform cortex, and the CA1 region of the hippocampus of soman-exposed mice treated with midazolam only (Figure 11C). The midazolam and ketamine combination therapy significantly reduced the average cell-body-to-cell-size ratio in all brain regions of interest when compared with the midazolam monotherapy group. The average cell-body-to-cell-size ratio in Iba1-positive cells in the piriform cortex and CA1 was not significantly different from that in the control group. Representative images of brain slices immunohistochemically processed for Iba1 are shown in Figure 11E.
Microglia are partly responsible for the release of some cytokines associated with the epileptic brain (reviewed in Hiragi et al. 2018, and Devinsky et al., 2013). We and others observe that these cytokines are also present in animal models following CWNA exposure (Dhote et al., 2007; Spradling et al., 2011). We have previously reported on the robust neuroinflammatory response resulting from soman-induced seizures and delayed midazolam treatment in the Es1−/− mouse (Kundrick et al., 2020; Marrero-Rosado et al., 2018). The reduction of microgliosis was only observed in animals that received 9 mg/kg of midazolam monotherapy (Kundrick et al., 2020). Consistent with a synergistic effect, the combination of ketamine with a lower dose of midazolam (3 mg/kg) was sufficient to reduce microgliosis and microglia activation. Interestingly, the CA1 and piriform cortex, where the least neuroinflammation was observed, are also the locations of the highest level of neuroprotection in the group that received the midazolam and ketamine combination therapy, indicated by neuronal cell densities that were not significantly different from those in the control group. Similar anti-inflammatory properties have also been observed in a mouse model of acute soman poisoning where ketamine in combination with atropine administered at a delayed time point was shown to reduce glial activation (Dhote et al., 2012).
3.3.3. Triple anti-epileptic drug efficacy against soman-induced neuronal loss
Given the successful amelioration of acute seizure severity and the reduction in the incidence and frequency of epileptogenesis, we examined the effect of treatment with midazolam/ketamine/valproate combination on neuronal cell density after soman-induced SE. Rats were exposed to a seizure-inducing dose of soman and given midazolam monotherapy, valproate monotherapy, midazolam/valproate combination, or midazolam/ketamine/valproate combination therapy at 40 min after SE onset. At 2 weeks following soman exposure, animals were euthanized and brains collected and sectioned for immunohistochemical processing with NeuN to visualize mature neurons. The therapeutic combination of midazolam/ketamine/valproate was able to protect from neuronal cell degeneration in the medial thalamus and piriform cortex as demonstrated by NeuN-positive cell densities that were not significantly different from the control (no agent) group. In contrast, neither midazolam nor valproate monotherapy, nor midazolam/valproate combination, was able to prevent the severe neuronal loss in the medial thalamus and piriform cortex that results from prolonged seizure activity after acute exposure to soman. Ketamine monotherapy is not included since we previously reported poor survival to soman exposure at doses 10, 30, and 90 mg/kg (Lumley et al., 2019a) and the goal was to identify adjunct therapy to midazolam. These results are consistent with observations that a combination of diazepam (10 mg/kg) and valproate (25 mg/kg, 50 mg/kg) were ineffective at reducing time in SE, SRS or brain pathology compared to diazepam monotherapy in soman exposed rats when administered 30 min after GD exposure (Rossetti et al., 2012). However, a higher dose of valproate (100 mg/kg) in combination with diazepam reduced time in SE and cell death in the piriform cortex, thalamus and ventral hippocampus compared to diazepam alone. The ventral hippocampus is typically more damaged following soman exposure compared to the dorsal hippocampus (Rossetti et al., 2012; Apland et al., 2010). The additional neuroprotection afforded by the triple therapy over monotherapy or dual therapy may be related to the reduced time in initial seizure that occurs with the triple therapy since reduced seizure duration is associated with reduced neuronal damage (McDonough and Shih, 1995). However, both ketamine and valproate have neuroprotective effects in animal models of seizure as well as traumatic brain injury, so there may be neuroprotectant effects above that provided by reduction of initial seizure. In addition to its anticonvulsive effects, valproate is a histone deacetylase inhibitor (HDAC) and, thus, through modification of gene expression has long-term effects (e.g., modulation of neurogenesis, neuroprotection, etc.) that may translate to reduced epileptogenesis and neuroprotection (reviewed in Romoli et al., 2019).
3.4. Timing of drug delivery is key
Standard practice in treating SE is sequential polytherapy, in which the second drug is only administered when the first drug fails, and wait for the second drug to fail before giving the third drug (Glauser et al., 2016). This has the advantage of minimizing the number of drugs delivered to responders, but the disadvantage of delaying delivery of the second drug by at least 30 min, and delivery of the third drug by at least 1 h. To mimic clinical situations where drugs are only injected after the previous treatment fails, SE was treated with the same drugs at the same dose in two groups of rats. In one group the three drugs were injected simultaneously. In the second group the second drug was injected 30 min after the first, and the third drug was delivered 30 min after the second drug (Figure 13A). Simultaneous polytherapy was far more effective than sequential monotherapies in reducing the post-treatment EEG power integral during the first hour (Figure 13B), or the first 6 h after treatment; in reducing the time needed for EEG amplitude to decline to twice the pre-seizure baseline (Figure 13C); and in reducing the number of post-treatment seizures (not shown). By those measures, sequential monotherapies were not significantly different from high-dose benzodiazepine monotherapy, matching the clinical experience where the second and third drug used in sequential polytherapy have a low success rate (Treiman et al., 1998). The far greater efficacy of simultaneous polytherapy over sequential monotherapies at the same dose is compatible with increasing pharmacoresistance, associated with seizure-induced increases in receptor trafficking, during the delay between sequential drug injections. The RAMPART study (Silbergleit et al., 2012, 2013) showed that earlier treatment leads to better results, and that a brief gain of time (4 min) can make a significant difference in outcome. Figure 13 confirms that principle in the treatment of cholinergic RSE.
Figure 13. In the lithium-pilocarpine model, simultaneous polytherapy was far more effective in reducing EEG power and stopping SE than sequential monotherapies.

A) Experimental flow: In the simultaneous group, the combination of midazolam 3 mg/kg, ketamine 30 mg/kg and valproate 90 mg/kg was administered simultaneously 40 min after SE onset. In the sequential group, the same drugs at the same dose were injected 30 min apart. B-E) The graphs show the ratio of EEG power integral to initial EEG power at baseline over the first hour (B) or the first six hours (C), the time needed for EEG amplitude to decline to twice the pre-seizure baseline (D) and the number of computer-detected seizures per 24 hours (E). Simultaneous polytherapy (n=10) was far more effective than sequential monotherapies (n=8–9) or higher-dose midazolam (n=10) in reducing EEG power, stopping SE (as indicated by EEG amplitude declining to twice pre-seizure baseline) and reducing the number of seizures. In graphs B-C, * p <0.05 or **** p <0.0001 by ANOVA followed by Tukey’s multiple comparison. In graphs D-E, * p <0.05, ** p <0.01, *** p <0.001 by Kruskal-Wallis analysis followed by Dunn’s test. Figure was modified from Niquet et al., 2017.
4. Conclusions
When treatment is delayed to cholinergic-induced SE, pharmacoresistance develops. Our improving knowledge of the mechanisms of seizure-induced pharmacoresistance should guide treatment. Cholinergic-induced SE involves key changes in GABA and glutamate receptors, yet treatment continues with a single drug that targets the GABAergic system, leaving changes in excitatory networks untreated. Current treatment guidance is to delay administration of a second-line anti-convulsant drug until after the first-line anticonvulsant has failed, and administration of a third drug until after the second drug has failed, ignoring evidence that pharmacoresistance increases with time and with seizure burden. Although benzodiazepines are effective when administered early after seizure onset, their efficacy is drastically reduced with time and inversely related to seizure duration and the main mechanism for benzodiazepine pharmacoresistance, the loss of synaptic GABAAR, remains unaddressed. Clinical trials comparing monotherapies are bound to show only minor differences between drugs, since each one treats only part of the problem, unless one practices “polytherapy” with a single drug that has multiple mechanisms of action. Our results support our hypothesis that a decrease in synaptic GABAAR and an increase in synaptic glutamate receptors are key elements of the initiation and maintenance of cholinergic-induced SE, and thus, both systems need to be treated to overcome seizure-induced pharmacoresistance when treatment is delayed. In the treatment of severe benzodiazepine-refractory cholinergic-induced SE, drug combinations that include a GABAAR agonist and a NMDAR antagonist are far more effective at reducing seizure severity than higher-dose monotherapies or than other drug combinations, supporting a key role of GABAA and glutamate receptor trafficking in the pathophysiology of cholinergic-induced SE. The observed polytherapeutic synergism occurs in rat models of soman-induced SE and lithium/pilocarpine-induced SE. The improved efficacy when anti-seizure drugs are administered in combination was also validated following soman exposure in Es1−/− mice, which similar to humans lack plasma CaE and may better predict response compared to wild-type mice. Not only do these drugs terminate cholinergic-induced RSE, but they also reduce or abolish its long-term consequences: neuronal loss, neuroinflammatory responses, spatial memory deficits, and epileptogenesis. Adding in a third anti-epileptic drug (valproate) had even greater efficacy than dual therapy in reducing seizure severity, epileptogenesis and brain pathology. Unlike benzodiazepines, the drug combinations are effective when treatment is delayed by 40 min after seizure onset. Furthermore, the simultaneous administration of the three anti-epileptic drugs (midazolam-ketamine-valproate) was far more efficient in stopping seizures than the standard practice of injecting the drugs sequentially, suggesting that the timing of treatment is essential and that an early polytherapy arm should be included in future clinical trials. Their anticonvulsive effects showed positive cooperativity between drugs so that their therapeutic index was improved by their synergistic interaction.
Figure 12. Triple anti-epileptic drug combination therapy (midazolam-ketamine-valproate) reduced neuronal cell death following soman (GD)-induced seizure.

Valproate (VALP; 90 mg/kg; IP) was administered alone or combination with midazolam (MDZ; 3 mg/kg), ketamine (KET; 30 mg/kg), or KET+MDZ 40 min after GD-induced (118.1 µg/kg, SC) seizure onset and compared with no agent (No GD) control rats. Brain samples were processed for immunohistochemistry with NeuN antibody to visualize mature neurons. (A) Rats treated with triple therapy (GD+MDZ/KET/VALP) had significantly greater NeuN-positive (NeuN+) cells compared to rats treated with MDZ, VALP, MDZ/VALP, or KET/VALP in both the medial thalamus and the piriform cortex. Data shown are mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, compared to No GD (B) Representative images in the thalamus (left) and piriform cortex (right) are shown for brain tissue stained with NeuN.
Highlights.
GABAA and NMDA receptor trafficking and pharmacoresistance to cholinergic-induced SE
Drug combinations to counter SE-induced receptor changes in experimental models of SE
Midazolam-ketamine-valproate therapy synergistically stopped cholinergic-induced SE
Triple therapy reduced epileptogenesis, spatial memory deficits and neuronal loss
Simultaneous triple therapy is more effective than sequential triple therapy against SE
Acknowledgements
This research was supported in part by grants from the National Institute of Neurological Disorders and Stroke (R21 NS103820 and U01 NS074926), by Merit Review Award#101 BX000273–07 from the United States Department of Veterans Affairs (Biomedical Laboratory Research and Development Service) and by the Debbie and James Cho Foundation. We confirm that we have read the journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The views expressed in this manuscript are those of the authors and do not reflect the official policy of the Department of Army, Department of Defense, or the U.S. Government.
Declaration of Competing Interest
Jerome Niquet and Claude Wasterlain have a patent on polytherapy of cholinergic seizures (UC Case No. 2012–172-2). Other authors have no conflict of interest to disclose.
References
- Acon-Chen C, Koenig JA, Smith GR, Truitt AR, Thomas TP, & Shih TM, 2016. Evaluation of acetylcholine, seizure activity and neuropathology following high-dose nerve agent exposure and delayed neuroprotective treatment drugs in freely moving rats. Toxicol Mech Methods, 26(5), 378–388. [DOI] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Figueiredo TH, Apland JP, Prager EM, Pidoplichko VI, Miller SL, & Braga MF, 2016. Long-term neuropathological and behavioral impairments after exposure to nerve agents. Ann N Y Acad Sci, 1374(1), 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballough GP, Newmark J, Levine ES, & Filbert MG, 2008. Neuroprotection as a Treatment. In Medical aspects of chemical warfare, edited by Tuorinsky Shirley D.. In Textbook of Military Medicine Washington, D.C.: Department of the Army, Office of the Surgeon General and Borden Institute: 221–242. [Google Scholar]
- Barbier L, Canini F, Giroud C, Beaup C, Foquin A, Maury R, Denis J, Peinnequin A, & Dorandeu F, 2015. Beneficial effects of a ketamine/atropine combination in soman-poisoned rats under a neutral thermal environment. Neurotoxicology, 50, 10–19. [DOI] [PubMed] [Google Scholar]
- Chamberlain JM, Kapur J, Shinnar S, Elm J, Holsti M, Babcock L, Rogers A, Barsan W, Cloyd J, Lowenstein D, Bleck TP, Conwit R, Meinzer C, Cock H, Fountain NB, Underwood E, Connor JT, Silbergleit R, Neurological Emergencies Treatment T, & Pediatric Emergency Care Applied Research Network, i., 2020. Efficacy of levetiracetam, fosphenytoin, and valproate for established status epilepticus by age group (ESETT): a double-blind, responsive-adaptive, randomised controlled trial. Lancet, 395(10231), 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JW, & Wasterlain CG, 2006. Status epilepticus: pathophysiology and management in adults. Lancet Neurol, 5(3), 246–256. [DOI] [PubMed] [Google Scholar]
- Chou TC, 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev, 58(3), 621–681. [DOI] [PubMed] [Google Scholar]
- Clement JG, Hand BT, & Shiloff JD, 1981. Differences in the toxicity of soman in various strains of mice. Fundam Appl Toxicol, 1(6), 419–420. [DOI] [PubMed] [Google Scholar]
- Clossen BL, & Reddy DS, 2017. Novel therapeutic approaches for disease-modification of epileptogenesis for curing epilepsy. Biochim Biophys Acta Mol Basis Dis, 1863(6), 1519–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo Furtado M, Lumley LA, Robison C, Tong LC, Lichtenstein S, & Yourick DL, 2010. Spontaneous recurrent seizures after status epilepticus induced by soman in Sprague-Dawley rats. Epilepsia, 51(8), 1503–1510. [DOI] [PubMed] [Google Scholar]
- de Araujo Furtado M, Zheng A, Sedigh-Sarvestani M, Lumley L, Lichtenstein S, & Yourick D, 2009. Analyzing large data sets acquired through telemetry from rats exposed to organophosphorous compounds: an EEG study. J Neurosci Methods, 184(1), 176–183. [DOI] [PubMed] [Google Scholar]
- DeLorenzo RJ, Hauser WA, Towne AR, Boggs JG, Pellock JM, Penberthy L, Garnett L, Fortner CA, & Ko D, 1996. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology, 46(4), 1029–1035. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, & Rogawski MA, 2013. Glia and epilepsy: excitability and inflammation. Trends Neurosci, 36(3), 174–184. [DOI] [PubMed] [Google Scholar]
- Dhote F, Carpentier P, Barbier L, Peinnequin A, Baille V, Pernot F, Testylier G, Beaup C, Foquin A, & Dorandeu F, 2012. Combinations of ketamine and atropine are neuroprotective and reduce neuroinflammation after a toxic status epilepticus in mice. Toxicol Appl Pharmacol, 259(2), 195–209. [DOI] [PubMed] [Google Scholar]
- Dhote F, Peinnequin A, Carpentier P, Baille V, Delacour C, Foquin A, Lallement G, & Dorandeu F, 2007. Prolonged inflammatory gene response following soman-induced seizures in mice. Toxicology, 238(2–3), 166–176. [DOI] [PubMed] [Google Scholar]
- Dorandeu F, Baille V, Mikler J, Testylier G, Lallement G, Sawyer T, & Carpentier P, 2007. Protective effects of S+ ketamine and atropine against lethality and brain damage during soman-induced status epilepticus in guinea-pigs. Toxicology, 234(3), 185–193. [DOI] [PubMed] [Google Scholar]
- Dorandeu F, Carpentier P, Baubichon D, Four E, Bernabe D, Burckhart MF, & Lallement G, 2005. Efficacy of the ketamine-atropine combination in the delayed treatment of soman-induced status epilepticus. Brain Res, 1051(1–2), 164–175. [DOI] [PubMed] [Google Scholar]
- Dorandeu F, Dhote F, Barbier L, Baccus B, & Testylier G, 2013. Treatment of status epilepticus with ketamine, are we there yet? CNS Neurosci Ther, 19(6), 411–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux ME, 1995. Inhibition by ketamine of muscarinic acetylcholine receptor function. Anesth Analg, 81(1), 57–62. [DOI] [PubMed] [Google Scholar]
- Duysen EG, Cashman JR, Schopfer LM, Nachon F, Masson P, & Lockridge O, 2012. Differential sensitivity of plasma carboxylesterase-null mice to parathion, chlorpyrifos and chlorpyrifos oxon, but not to diazinon, dichlorvos, diisopropylfluorophosphate, cresyl saligenin phosphate, cyclosarin thiocholine, tabun thiocholine, and carbofuran. Chem Biol Interact, 195(3), 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddleston M, 2000. Patterns and problems of deliberate self-poisoning in the developing world. QJM, 93(11), 715–731. [DOI] [PubMed] [Google Scholar]
- Feder PI, Hobson DW, Olson CT, Joiner RL, & Matthews MC, 1991. Stagewise, adaptive dose allocation for quantal response dose-response studies. Neurosci Biobehav Rev, 15(1), 109–114. [DOI] [PubMed] [Google Scholar]
- Figueiredo TH, Apland JP, Braga MFM, & Marini AM, 2018. Acute and long-term consequences of exposure to organophosphate nerve agents in humans. Epilepsia, 59 Suppl 2, 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friederich P, Dybek A, & Urban BW, 2000. Stereospecific interaction of ketamine with nicotinic acetylcholine receptors in human sympathetic ganglion-like SH-SY5Y cells. Anesthesiology, 93(3), 818–824. [DOI] [PubMed] [Google Scholar]
- Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J, Bare M, Bleck T, Dodson WE, Garrity L, Jagoda A, Lowenstein D, Pellock J, Riviello J, Sloan E, & Treiman DM, 2016. Evidence-Based Guideline: Treatment of Convulsive Status Epilepticus in Children and Adults: Report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr, 16(1), 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, & Kapur J, 2008. Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J Neurosci, 28(10), 2527–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Liu X, & Holmes GL, 2003. Diazepam terminates brief but not prolonged seizures in young, naive rats. Epilepsia, 44(8), 1109–1112. [DOI] [PubMed] [Google Scholar]
- Grauer E, Chapman S, Rabinovitz I, Raveh L, Weissman BA, Kadar T, & Allon N, 2008. Single whole-body exposure to sarin vapor in rats: long-term neuronal and behavioral deficits. Toxicology and applied pharmacology, 227(2), 265–274. [DOI] [PubMed] [Google Scholar]
- Gunnell D, Eddleston M, Phillips MR, & Konradsen F, 2007. The global distribution of fatal pesticide self-poisoning: systematic review. BMC Public Health, 7, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevers W, Hadley SH, Luddens H, & Amin J, 2008. Ketamine, but not phencyclidine, selectively modulates cerebellar GABA(A) receptors containing alpha6 and delta subunits. J Neurosci, 28(20), 5383–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CE, Parikh AO, Ellis C, Myers JS, & Litt B, 2017. Timing is everything: Where status epilepticus treatment fails. Ann Neurol, 82(2), 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiragi T, Ikegaya Y, & Koyama R, 2018. Microglia after Seizures and in Epilepsy. Cells, 7(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jett DA, Sibrizzi CA, Blain RB, Hartman PA, Lein PJ, Taylor KW, & Rooney AA, 2020. A national toxicology program systematic review of the evidence for long-term effects after acute exposure to sarin nerve agent. Critical reviews in toxicology, 50(6), 474–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur J, & Lothman EW, 1990. NMDA receptor activation mediates the loss of GABAergic inhibition induced by recurrent seizures. Epilepsy Res, 5(2), 103–111. [DOI] [PubMed] [Google Scholar]
- Kapur J, Stringer JL, & Lothman EW, 1989. Evidence that repetitive seizures in the hippocampus cause a lasting reduction of GABAergic inhibition. J Neurophysiol, 61(2), 417–426. [DOI] [PubMed] [Google Scholar]
- Kapur J, Elm J, Chamberlain JM, Barsan W, Cloyd J, Lowenstein D, Shinnar S, Conwit R, Meinzer C, Cock H, Fountain N, Connor JT, Silbergleit R, Nett, & Investigators, P., 2019. Randomized Trial of Three Anticonvulsant Medications for Status Epilepticus. N Engl J Med, 381(22), 2103–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm WR, 1985. Comparison of known sodium-channel blockers in DFP toxicity. Toxicol Lett, 25(3), 307–312. [DOI] [PubMed] [Google Scholar]
- Kundrick E, Marrero-Rosado B, Stone M, Schultz C, Walker K, Lee-Stubbs RB, de Araujo Furtado M, & Lumley LA, 2020. Delayed midazolam dose effects against soman in male and female plasma carboxylesterase knockout mice. Ann N Y Acad Sci [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston JL, Wright LK, Connis N, & Lumley LA, 2012. Characterizing the behavioral effects of nerve agent-induced seizure activity in rats: increased startle reactivity and perseverative behavior. Pharmacol Biochem Behav, 100(3), 382–391. [DOI] [PubMed] [Google Scholar]
- Lewine JD, Weber W, Gigliotti A, McDonald JD, Doyle-Eisele M, Bangera N, Paulson K, Magcalas C, Hamilton DA, Garcia E, Raulli R, & Laney J, 2018. Addition of ketamine to standard-of-care countermeasures for acute organophosphate poisoning improves neurobiological outcomes. Neurotoxicology, 69, 37–46. [DOI] [PubMed] [Google Scholar]
- Lipp JA, 1972. Effect of diazepam upon soman-induced seizure activity and convulsions. Electroencephalogr Clin Neurophysiol, 32(5), 557–560. [DOI] [PubMed] [Google Scholar]
- Loss CM, Cordova SD, & de Oliveira DL, 2012. Ketamine reduces neuronal degeneration and anxiety levels when administered during early life-induced status epilepticus in rats. Brain Res, 1474, 110–117. [DOI] [PubMed] [Google Scholar]
- Lowenstein DH, 2015. Decade in review-epilepsy: edging toward breakthroughs in epilepsy diagnostics and care. Nat Rev Neurol, 11(11), 616–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumley LA, Rossetti F, de Araujo Furtado M, Marrero-Rosado B, Schultz CR, Schultz MK, Niquet J, & Wasterlain CG, 2019a. Dataset of EEG power integral, spontaneous recurrent seizure and behavioral responses following combination drug therapy in soman-exposed rats. Data Brief, 27, 104629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumley L, Miller D, Muse WT, Marrero‐Rosado B, de Araujo Furtado M, Stone M, McGuire J, & Whalley C, 2019b. Neurosteroid and benzodiazepine combination therapy reduces status epilepticus and long‐term effects of whole‐body sarin exposure in rats. Epilepsia open, 4(3), 382–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madzar D, Reindl C, Giede-Jeppe A, Bobinger T, Sprugel MI, Knappe RU, Hamer HM, & Huttner HB, 2018. Impact of timing of continuous intravenous anesthetic drug treatment on outcome in refractory status epilepticus. Crit Care, 22(1), 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrero-Rosado B, de Araujo Furtado M, Schultz CR, Stone M, Kundrick E, Walker K, O’Brien S, Du F, & Lumley LA, 2018. Soman-induced status epilepticus, epileptogenesis, and neuropathology in carboxylesterase knockout mice treated with midazolam. Epilepsia, 59(12), 2206–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrero-Rosado BM, de Araujo Furtado M, Kundrick ER, Walker KA, Stone MF, Schultz CR, Nguyen DA, & Lumley LA, 2020. Ketamine as adjunct to midazolam treatment following soman-induced status epilepticus reduces seizure severity, epileptogenesis, and brain pathology in plasma carboxylesterase knockout mice. Epilepsy Behav, 111, 107229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BS, & Kapur J, 2008. A combination of ketamine and diazepam synergistically controls refractory status epilepticus induced by cholinergic stimulation. Epilepsia, 49(2), 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell DM, Brecht KM, & O’Neill BL, 1987. The effect of carboxylesterase inhibition on interspecies differences in soman toxicity. Toxicol Lett, 39(1), 35–42. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Baldwin RA, Sankar R, & Wasterlain CG, 1998. Time-dependent decrease in the effectiveness of antiepileptic drugs during the course of self-sustaining status epilepticus. Brain Res, 814(1–2), 179–185. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, & Wasterlain CG, 1999. N-methyl-D-asparate receptor antagonists abolish the maintenance phase of self-sustaining status epilepticus in rat. Neurosci Lett, 265(3), 187–190. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Wasterlain CG, Sankar R, & Shin D, 1998. Self-sustaining status epilepticus after brief electrical stimulation of the perforant path. Brain Res, 801(1–2), 251–253. [DOI] [PubMed] [Google Scholar]
- McDonough JH Jr., & Shih TM, 1993. Pharmacological modulation of soman-induced seizures. Neurosci Biobehav Rev, 17(2), 203–215. [DOI] [PubMed] [Google Scholar]
- McDonough JH Jr., Dochterman LW, Smith CD, & Shih TM, 1995. Protection against nerve agent-induced neuropathology, but not cardiac pathology, is associated with the anticonvulsant action of drug treatment. Neurotoxicology, 16(1), 123–132. [PubMed] [Google Scholar]
- McDonough JH Jr., Clark TR, Slone TW Jr., Zoeffel D, Brown K, Kim S and Smith CD, 1998. Neural lesions in the rat and their relationship to EEG delta activity following seizures induced by the nerve agent soman. Neurotoxicology, 19(3): 381–391 [PubMed] [Google Scholar]
- McDonough JH Jr., Zoeffel LD, McMonagle J, Copeland TL, Smith CD and Shih TM, 2000. Anticonvulsant treatment of nerve agent seizures: anticholinergics versus diazepam in soman-intoxicated guinea pigs. Epilepsy Res 38(1): 1–14. [DOI] [PubMed] [Google Scholar]
- Moffett MC, Schultz MK, Schwartz JE, Stone MF, & Lumley LA, 2011. Impaired auditory and contextual fear conditioning in soman-exposed rats. Pharmacol Biochem Behav, 98(1), 120–129. [DOI] [PubMed] [Google Scholar]
- Morrisett RA, Jope RS, & Snead OC 3rd, 1987. Effects of drugs on the initiation and maintenance of status epilepticus induced by administration of pilocarpine to lithium-pretreated rats. Exp Neurol, 97(1), 193–200. [DOI] [PubMed] [Google Scholar]
- Muller PY, & Milton MN, 2012. The determination and interpretation of the therapeutic index in drug development. Nat Rev Drug Discov, 11(10), 751–761. [DOI] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Niquet J, & Wasterlain CG, 2013. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol Dis, 54, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor DE, Liu H and Wasterlain CG, 2005a. Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci, 25(34): 7724–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor DE, Wasterlain CG, 2005b. GABA synapses and the rapid loss of inhibition to dentate gyrus granule cells after brief perforant-path stimulation. Epilepsia, 46 (Suppl. 5), 142–147. [DOI] [PubMed] [Google Scholar]
- Newmark J, 2019. Therapy for acute nerve agent poisoning: An update. Neurol Clin Pract, 9(4), 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niquet J, Baldwin R, Norman K, Suchomelova L, Lumley L, & Wasterlain CG, 2016. Midazolam-ketamine dual therapy stops cholinergic status epilepticus and reduces Morris water maze deficits. Epilepsia, 57(9), 1406–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niquet J, Baldwin R, Suchomelova L, Lumley L, Eavey R and Wasterlain CG, 2017a. Treatment of experimental status epilepticus with synergistic drug combinations. Epilepsia, 58(4): e49–e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niquet J, Baldwin R, Norman K, Suchomelova L, Lumley L, Wasterlain CG, 2017b. Simultaneous triple therapy for the treatment of status epilepticus. Neurobiol Dis, 104:41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niquet J, Lumley L, Baldwin R, Rossetti F, Schultz M, de Araujo Furtado M, Suchomelova L, Naylor D, Franco-Estrada I, Wasterlain CG, 2019. Early polytherapy for benzodiazepine-refractory status epilepticus. Epilepsy Behav, 101(Pt B):106367. [DOI] [PubMed] [Google Scholar]
- Nissinen J, Halonen T, Koivisto E and Pitkanen A, 2000. A new model of chronic temporal lobe epilepsy induced by electrical stimulation of the amygdala in rat. Epilepsy Res, 38(2–3): 177–205. [DOI] [PubMed] [Google Scholar]
- Nissinen J, Lukasiuk K, & Pitkanen A, 2001. Is mossy fiber sprouting present at the time of the first spontaneous seizures in rat experimental temporal lobe epilepsy? Hippocampus, 11(3), 299–310. [DOI] [PubMed] [Google Scholar]
- Petras JM, 1981. Soman neurotoxicity. Fundam Appl Toxicol, 1(2), 242. [DOI] [PubMed] [Google Scholar]
- Philippens IH, Melchers BP, de Groot DM and Wolthuis OL, 1992. Behavioral performance, brain histology, and EEG sequela after immediate combined atropine/diazepam treatment of soman-intoxicated rats. Pharmacol Biochem Behav 42(4): 711–719. [DOI] [PubMed] [Google Scholar]
- Prasad M, Krishnan PR, Sequeira R, & Al‐Roomi K, 2014. Anticonvulsant therapy for status epilepticus. Cochrane database of systematic reviews(9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran K, Todorovic M, & Kapur J, 2012. Calcium-permeable AMPA receptors are expressed in a rodent model of status epilepticus. Ann Neurol, 72(1), 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy SD, & Reddy DS, 2015. Midazolam as an anticonvulsant antidote for organophosphate intoxication--A pharmacotherapeutic appraisal. Epilepsia, 56(6), 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AC, & DeLorenzo RJ, 1999. N-methyl-D-aspartate receptor activation regulates refractoriness of status epilepticus to diazepam. Neuroscience, 93(1), 117–123. [DOI] [PubMed] [Google Scholar]
- Romoli M, Mazzocchetti P, D’Alonzo R, Siliquini S, Rinaldi VE, Verrotti A, Calabresi P and Costa C, 2019. Valproic Acid and Epilepsy: From Molecular Mechanisms to Clinical Evidences. Curr Neuropharmacol, 17(10): 926–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti F, de Araujo Furtado M, Pak T, Bailey K, Shields M, Chanda S, Addis M, Robertson BD, Moffett M, Lumley LA, Yourick DL, 2012. Combined diazepam and HDAC inhibitor treatment protects against seizures and neuronal damage caused by soman exposure. Neurotoxilcology, 33(3) 500–511. [DOI] [PubMed] [Google Scholar]
- Sanchez Fernandez I, Goodkin HP, & Scott RC, 2019. Pathophysiology of convulsive status epilepticus. Seizure, 68, 16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz MK, 2015. Evaluation of multifunctional compounds possessing anticholinergic and antiglutamatergic properties for efficacy against soman toxicity in rats (Ph.D. Doctoral Dissertation), University of Maryland, Baltimore, MD. [Google Scholar]
- Schultz MK, Wright LK, de Araujo Furtado M, Stone MF, Moffett MC, Kelley NR, Bourne AR, Lumeh WZ, Schultz CR, Schwartz JE, & Lumley LA, 2014. Caramiphen edisylate as adjunct to standard therapy attenuates soman-induced seizures and cognitive deficits in rats. Neurotoxicol Teratol, 44, 89–104. [DOI] [PubMed] [Google Scholar]
- Schultz MK, Wright LK, Stone MF, Schwartz JE, Kelley NR, Moffett MC, Lee RB, & Lumley LA, 2012. The anticholinergic and antiglutamatergic drug caramiphen reduces seizure duration in soman-exposed rats: synergism with the benzodiazepine diazepam. Toxicol Appl Pharmacol, 259(3), 376–386. [DOI] [PubMed] [Google Scholar]
- Shih T, McDonough JH Jr., & Koplovitz I, 1999. Anticonvulsants for soman-induced seizure activity. J Biomed Sci, 6(2), 86–96. [DOI] [PubMed] [Google Scholar]
- Shih T, Whalley CE, & Valdes JJ, 1991. A comparison of cholinergic effects of HI-6 and pralidoxime-2-chloride (2-PAM) in soman poisoning. Toxicol Lett, 55(2), 131–147 [DOI] [PubMed] [Google Scholar]
- Shih TM, Duniho SM, & McDonough JH, 2003. Control of nerve agent-induced seizures is critical for neuroprotection and survival. Toxicol Appl Pharmacol, 188(2), 69–80. [DOI] [PubMed] [Google Scholar]
- Silbergleit R, Durkalski V, Lowenstein D, Conwit R, Pancioli A, Palesch Y, Barsan W, & Investigators N, 2012. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med, 366(7), 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbergleit R, Lowenstein D, Durkalski V, Conwit R, & Investigators N, 2013. Lessons from the RAMPART study – and which is the best route of administration of benzodiazepines in status epilepticus. Epilepsia, 54 Suppl 6, 74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sills GJ, & Rogawski MA, 2020. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology, 168, 107966. [DOI] [PubMed] [Google Scholar]
- Sivilotti ML, Bird SB, Lo JC, & Dickson EW, 2006. Multiple centrally acting antidotes protect against severe organophosphate toxicity. Acad Emerg Med, 13(4), 359–364. [DOI] [PubMed] [Google Scholar]
- Spradling KD, Lumley LA, Robison CL, Meyerhoff JL, & Dillman JF 3rd, 2011. Transcriptional responses of the nerve agent-sensitive brain regions amygdala, hippocampus, piriform cortex, septum, and thalamus following exposure to the organophosphonate anticholinesterase sarin. J Neuroinflammation, 8, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchomelova L, Baldwin RA, Kubova H, Thompson KW, Sankar R, & Wasterlain CG, 2006. Treatment of experimental status epilepticus in immature rats: dissociation between anticonvulsant and antiepileptogenic effects. Pediatr Res, 59(2), 237–243. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ, 2000. Drug synergism and dose-effect data analysis: CRC Press. [Google Scholar]
- Towne AR, & DeLorenzo RJ, 1999. Use of intramuscular midazolam for status epilepticus. J Emerg Med, 17(2), 323–328. [DOI] [PubMed] [Google Scholar]
- Treiman DM, Meyers PD, Walton NY, Collins JF, Colling C, Rowan AJ, Handforth A, Faught E, Calabrese VP, Uthman BM, Ramsay RE, & Mamdani MB, 1998. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med, 339(12), 792–798. [DOI] [PubMed] [Google Scholar]
- Wang S, Levesque M, & Avoli M, 2019. Effects of Diazepam and Ketamine on Pilocarpine-Induced Status Epilepticus in Mice. Neuroscience, 421, 112–122. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Liu H, Mazarati AM, Baldwin RA, Shirasaka Y, Katsumori H, Thompson KW, Sankar R, Pereira de Vasconselos A, & Nehlig A, 2000. Self-sustaining status epilepticus: a condition maintained by potentiation of glutamate receptors and by plastic changes in substance P and other peptide neuromodulators. Epilepsia, 41 Suppl 6, S134–143. [DOI] [PubMed] [Google Scholar]
- Wright LK, Lee RB, Vincelli NM, Whalley CE, & Lumley LA, 2016. Comparison of the lethal effects of chemical warfare nerve agents across multiple ages. Toxicol Lett, 241, 167–174. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU, Swandulla D, Geisslinger G, & Brune K, 1992. Differential effects of ketamine enantiomers on NMDA receptor currents in cultured neurons. Eur J Pharmacol, 213(1), 155–158 [DOI] [PubMed] [Google Scholar]