

Abstract
BACKGROUND:
Understanding biology, particularly at the level of actionable drug discovery, is often a matter of developing accurate stories about how proteins work. This requires understanding the physics of the system, and physics-based computer modeling is a prime tool for that. However, the computational molecular physics (CMP) of proteins has previously been much too expensive and slow. A large fraction of public supercomputing resources worldwide is currently running CMP simulations of biologically relevant systems. We review here the history and status of this large and diverse scientific enterprise. Among other things, protein modeling has driven major computer hardware advances, such as IBM’s Blue Gene and DE Shaw’s Anton computers. Further, protein modeling has advanced rapidly over 50 years, even slightly faster than Moore’s law. We also review an interesting scientific social construct that has arisen around protein modeling: community-wide blind competitions. They have transformed how we test, validate, and improve our computational models of proteins.
ADVANCES:
For 50 years, two approaches to computer modeling have been mainstays for developing stories about protein molecules and their biological actions. (i) Inferences from structure-property relations: Based on the principle that a protein’s action depends on its shape, it is possible to use databases of known proteins to learn about unknown proteins. (ii) Computational molecular physics uses force fields of atom-atom interactions, sampled by molecular dynamics (MD), to develop biological action stories that satisfy principles of chemistry and thermodynamics. CMP has traditionally been computationally costly, limited to studying only simple actions of small proteins. But CMP has recently advanced enormously. (i) Force fields and their corresponding solvent models are now sufficiently accurate at capturing the molecular interactions, and conformational searching and sampling methods are sufficiently fast, that CMP is able to model, fairly accurately, protein actions on time scales longer than microseconds, and sometimes milliseconds. So, we are now accessing important biological events, such as protein folding, unbinding, allosteric change, and assembly. (ii) Just as car races do for auto manufacturers, communal blind tests such as protein structure-prediction events are giving protein modelers a shared evaluation venue for improving our methods. CMP methods are now competing and often doing quite well. (iii) New methods are harnessing external information—like experimental structural data—to accelerate CMP, notably, while preserving proper physics.
What are we learning? For one thing, a long-standing hypothesis is that proteins fold by multiple different microscopic routes, a story that is too granular to learn from experiments alone. CMP recently affirmed this principle while giving accurate and testable microscopic details, protein by protein. In addition, CMP is now contributing to physico-chemical drug design. Structure-based methods of drug discovery have long been able to discern what small-molecule drug candidates might bind to a given target protein and where on the protein they might bind. However, such methods don’t reveal some all-important physical properties needed for drug discovery campaigns—the affinities and the on- and off-rates of the ligand binding to the protein. CMP is beginning to compute these properties accurately. A third example is shown in the figure. It shows the spike protein of severe acute respiratory syndrome coronavirus 2(SARS-CoV-2), the causative agent of today’s coronavirus disease 2019 (COVID-19) pandemic. A large, hinge-like movement of this sizable protein is the critical action needed for the virus to enter and infect the human cell. The only way to see the details of this motion—to attempt to block it with drugs—is by CMP. The figure shows CMP simulation results of three dynamical states of this motion.
OUTLOOK:
A cell’s behavior is due to the actions of its thousands of different proteins. Every protein has its own story to tell. CMP is a granular and principled tool that is able to discover those stories. CMP is now being tested and improved through blind communal validations. It is attacking ever larger proteins, exploring increasingly bigger and slower motions, and with ever more accurate physics. We are reaching a physical understanding of biology at the microscopic level as CMP reveals causations and forces, step-by-step actions in space and time, conformational distributions along the way, and important physical quantities such as free energies, rates, and equilibrium constants.■
Graphical Abstract
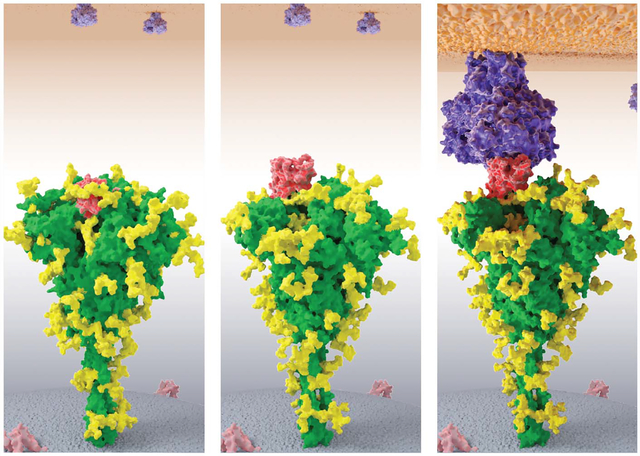
CMP modeling of COVID-19 infecting the human cell. SARS-CoV-2 spike glycoprotein (green, with its glycan shield in yellow) attaching to the human angiotensin-converting enzyme 2 (ACE2) receptor protein (purple) through its spike receptor-binding domain (red). (Left) The receptor binding domain (RBD) is hidden. (Middle) The RBD is open and accessible. (Right) The RBD binds human ACE2 receptor. This is followed by a cascade of larger conformational changes in the spike protein, leading to viral fusion to the human host cell.
Every protein has a story—how it folds, what it binds, its biological actions, and how it misbehaves in aging or disease. Stories are often inferred from a protein’s shape (i.e., its structure). But increasingly, stories are told using computational molecular physics (CMP). CMP is rooted in the principled physics of driving forces and reveals granular detail of conformational populations in space and time. Recent advances are accessing longer time scales, larger actions, and blind testing, enabling more of biology’s stories to be told in the language of atomistic physics.
Science…is the organized systematic enterprise that gathers knowledge about the world and condenses [it] into testable laws and principles.
—E. O. Wilson, Consilience
Scientists are storytellers. Biologists tell stories about biomolecules and their actions in the cell. Even the simplest cell has thousands of types of proteins. Based on the premise that structure determines function—the same principle that helps you discern how a knife and fork work—protein shapes are often the starting points for protein storytelling. Like a necklace of beads, a protein is a molecule of amino acids (the beads) chained together. The 20 different types of amino acids make up an alphabet for stringing the beads into different sequences that fold into different native structures (the compact shapes that proteins adopt in the cell). These shapes are known at atomic detail for more than 150,000 proteins and available in a public resource called the Protein DataBank (PDB) (1), thanks to x-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cryo–electron microscopy (cryo-EM) experiments from a large community of structural biologists over the past 60 years.
Computer modeling plays a big role in molecular storytelling. For one thing, two proteins having similar sequences often have similar shapes and perform similar actions. After a protein folds, it can go to work in the cell through binding to other molecules, or its motions, or working together in complex assemblies. Some computer algorithms are designed to get insights about poorly understood proteins by looking at proteins with known structures. For another thing, computing can leverage the laws of physics for understanding the motions and actions of biomolecules. The former approach is called structural bioinformatics (SBI) and the latter, computational molecular physics (CMP). Today’s computational modeling is often a combination of both. For protein storytelling, what’s usually asked of SBI is to reveal a native structure. But the native structure is a single static snapshot, often insufficient to tell the story of how the protein works. What’s missing in SBI is the physics—the driving forces, the causes and effects, the intermediate steps—the how’s, not just the what’s. What’s asked of CMP is to reveal the forces, motions, binding, and actions, in addition to structures. Atomistic physical modeling with force fields, often sampled by molecular dynamics (MD), draws upon our knowledge of covalent and noncovalent bonding in molecules, how those interactions are affected by the solvent water molecules, the considerable role of entropies, and how it all manifests in the complex environment inside the protein.
CMP helps to write the stories of biomolecules
Molecular physics aims to accomplish the following: (i) Reveal causalities, by allowing investigations of the driving forces, dynamics, and motions. (ii) Give the intermediate steps in time and space, angstrom by angstrom, nanosecond by nanosecond—narratives of molecular actions such as folding, binding, and rearrangements. (iii) Give conformational distributions, not just a single averaged structure. Proteins writhe and deform in ways that are needed for matching our stories to experiments. Drugs often don’t bind “key-in-lock” to preexisting protein cavities; they push the protein into a new shape that we seek to learn. (iv) Systematize, giving a common language for our storytelling. In principle, physical modeling is transferable: to different proteins, different ligands, different binding situations, or different experimental conditions, including those not yet measured. And physics-based models are less susceptible to errors of so-called hasty generalization (i.e., incorrect inferences from too little data), so their parameters can be systematically improved. (v) Go beyond limitations of databases. Physical modeling can be applied to classes of biomolecules that are sparsely represented in databases, such as membrane proteins, or molecules that are intrinsically disordered, large, or complexed. These are not well populated in databases because experiments are difficult. But they obey the same laws of physics.
CMP needs large computations. In practice, it means sacrificing some accuracy in the physical model, or limiting the time scale simulated to those shorter than the biological actions of interest. To reach for the deepest truths, we need to root our stories in our deepest understanding of nature. And though CMP modeling entails simplifications and approximations, it nevertheless provides today’s best achievable description of the underlying physics consistent with achievable computational costs.
CMP uses physical potentials and Boltzmann sampling
The properties of molecules are governed by quantum mechanics (QM). But QM computations are much too costly for large, flexible systems like proteins in water. So, in biomolecular modeling, true forces are approximated using force fields. Such models treat molecules as having preferred bond lengths and angles and describe other interatomic interactions as a combination of van der Waals and Coulomb forces, including the impact of solvation (2)—typically in water—and ultimately of proteins’ preferential conformations. Molecular force fields originated in the 1960s and ‘70s (3–13); they were pioneered by Allinger, Lifson, Kollman, and Scheraga, in addition to Levitt, Warshel, and Karplus, whose achievements were recognized by the 2013 Nobel Prize in Chemistry. They continue to undergo systematic improvement by a large community (14–20).
But knowing accurate physical potentials is only part of the problem. To generate faithful protein stories, modeling must respect other physics: the nature of movement and change and the laws of thermodynamics. Dynamical processes must follow Newton’s laws of motion. Equilibrium modeling must give the Boltzmann distribution of conformational populations—and thus free energies, which are where models meet experiments. The latter can only be fulfilled by using special sampling approaches, such as Monte Carlo (MC) or MD, sometimes accelerated by enhanced sampling (21). MD computations are expensive because they require femtosecond timesteps, to avoid violating Newton’s laws (22). And even though such methods have now become relatively efficient, they still entail compromises, and the stories that are most faithful to nature are of the simplest proteins, the shortest time scales, or the smallest actions.
A key consideration is always speed versus accuracy. To gain speed, you can use coarse-grained models. Coarse-grained modeling lumps together atoms into larger rigid units (23–26). This approach is useful when you know in advance which degrees of freedom are relevant to the problem at hand. Conversely, some questions, involving mechanisms of enzyme reactions or spectroscopic observables, for example, require quantum-mechanical details, including the electrons on all or part of the protein. Including such details increases the cost of the calculation and is tractable only when protein movements are small. Although important advances have been made with both of these approaches, atomistic CMP modeling remains a popular compromise between speed and accuracy for much of biology and drug discovery and is the focus of this review.
CMP has been both a driver and beneficiary of many advances
As computer power has grown exponentially, so has CMP modeling power (Fig. 1). The first stories told were of how proteins fold and how they bind small molecules, both of which have relevance to drug discovery. The earliest computational physics of proteins, in the 1960s and ‘70s (27), was done largely on central mainframes. Then came labclusters in the ‘90s and supercomputing. Among the first protein-folding stories, in 1998, was one from van Gunsteren and co-workers on peptides (28) and one from Duan and Kollman and their colleagues, who applied supercomputing to attempt to fold the villin headpiece, one of the smallest foldable proteins (29).
Fig. 1. Molecular simulations have improved faster than Moore’s law.
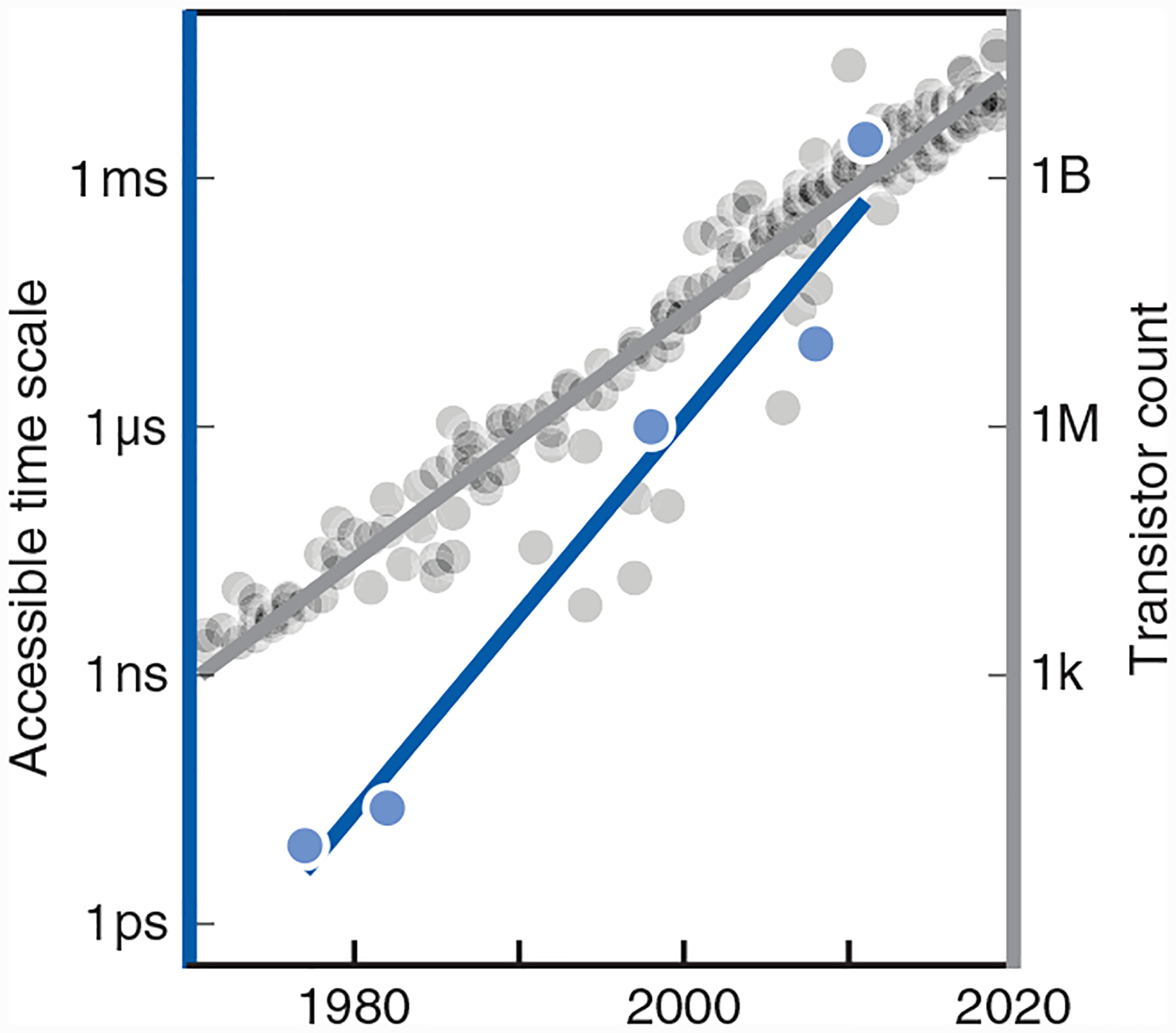
Blue: MD simulations have accessed exponentially longer time scales of molecular motions over the past 50 years. Gray: For reference, Moore’s law of increased densities of transistors on microchips (136).
Protein modeling is now a major activity of public supercomputers. The demand for better CMP modeling of biomolecules has driven advances in high-performance computing, including (i) the IBM Blue Gene computer (30); (ii) Folding@Home, a distributed grid network developed by Pande and Shirts at Stanford (31); and (iii) D. E. Shaw and colleagues’ special purpose Anton and Anton 2 supercomputer (32, 33).
CMP modeling has advanced at Moore’s law rates
Advances have come not only from computer hardware. CMP has also advanced from systematic improvements in atomistic force fields, solvation, sampling, and workflows. The first MD simulations, in 1977, sampled motions of a small protein representing a real time of only about 3 ps and necessarily left out an essential component, namely, the water solvent (27). Today’s simulations are run with quality force fields and models of surrounding solvent or membrane, and over much longer time scales, through developments described below. An important milestone, which was reached about a decade ago, was simulating time scales of milliseconds and longer. This has been transformative because these are the time scales on which large-scale, biologically interesting motions appear.
Enhanced sampling methods help to tell bigger stories
Today’s storytelling is often limited to small proteins, simple actions, or short time scales. To tell bigger stories, we need faster and more efficient conformational searching and sampling. Advances are coming from enhanced sampling methods, such as metadynamics (34), replica exchange molecular dynamics (REMD) (35), simulated annealing (36), adaptive force biasing (37), and umbrella sampling (38) [for reviews, see (39, 40), and for the basic concepts see (21, 41)]. Also important has been systematic improvement in measuring and controlling errors, such as through the multi-state Bennett acceptance ratio estimator (MBAR) (42) and the weighted histogram analysis method (WHAM) (43).
Protein structure modeling can benefit from harnessing external information, an approach called integrative modeling (44). Computer algorithms can be accelerated by leveraging additional SBI insights about protein structures. For example, Rosetta (45), Quark (46), Dock 1 (47), Cluspro (48), Haddock (49), MODELLER (50), and IMP (51) do this to outstanding effect for certain structure-based computations. However, methods for leveraging external information often require sacrificing the ability to give Boltzmann populations, free energies, or dynamics.
An alternative integrative approach is MELD (modeling employing limited data), an accelerator of MD that also retains the advantages of the physics (52, 53). MELD-accelerated MD (MELD × MD) melds external information or directives about the end state of interest with CMP, in a way that preserves Boltzmann populations. It uses Bayesian inference to leverage noisy, combinatoric, corrupted, sparse, or ambiguous information, to accelerate MD, often by orders of magnitude. It adds value in protein structure prediction from sequences (52, 54, 55), native structure determination in conjunction with problematic experimental data (53, 56), ligand binding to proteins (57–59), and finding dynamical routes of conformational change processes (60).
CMP can tell dynamical stories
Some protein stories are about dynamics and motions. In those cases, we seek the sequences of events (i.e., the pathways or routes) and their speeds (i.e., the transition rates). Pathways can be studied in CMP by using long simulations of individual trajectories (61). But long trajectories are expensive to compute and hard to converge, owing to many stochastic meanderings. Often, the story line is better obtained by dividing the possible routes into multiple short trajectories that can be computed in parallel, each of which traverses fewer states. Increasingly, studies of processes are done by stitching together many short simulations in parallel.
One strategy for computational parallelization is Markov state modeling (62–68). Markov state models (MSMs) use short trajectories in parallel to efficiently compute the rates and routes of protein motions and actions (69), protein-folding pathways (70), motions and dynamics (71), and ligand binding (72) and unbinding (73). MSMs are best when a pathway has a few dominant routes (“superhighways” in conformational space).
Another parallelization strategy is milestoning, which is best when a process has a dispersed diffusional route structure (74). In milestoning, a path along a predefined reaction coordinate is metaphorically “fenced off into different time zones,” called milestones. Each milestone boundary is a starting point for spawning independent parallel trajectories to the next milestone. First-passage times are computed, then combined, to give the time evolution of the whole process. Milestoning is effective in simulating binding (75), membrane dynamics (76), and transitions between protein conformations (77). Milestoning relies on the assumptions that the transition events between successive milestones and the time lags between these transitions are statistically independent.
The weighted ensemble method (78) also fences off conformational space into zones but then spawns new daughter trajectories whenever a trajectory hits a fence that leads to an “interesting” area of the phase space. This enhances the sampling of interesting events while providing a rigorous nonequilibrium reweighting scheme to learn the unbiased time evolution of the trajectory (79).
Blind competitions are validating our storytelling tools
How do we know if our stories are true? This question applies to both types of stories—those from structural biology and those from CMP. It has long been difficult to model the native structures—and tell the stories—of most proteins. It still is. But the field was advanced enormously in 1994 when John Moult introduced a new type of community-wide blind-test event (80). In CASP (critical assessment of protein structure prediction), a core team of assessors releases various amino acid sequences onto a website. Modelers then have a short, fixed time to submit their predictions of that sequence’s three-dimensional (3D) structure. Subsequently, the true experimentally determined 3D structures are released. The assessors then compare and evaluate all the predictions. This event has brought considerable value. Blind testing helps eliminate our biases, accelerate our learning, and enhance our communal interests.
Until recently, CMP modeling has been too slow to compete, so CASP has given insights mostly about SBI modeling. Because SBI methods draw inferences from other known protein structures, SBI predictors are most successful when a closely related PDB template protein can be found. And SBI successes at CASP have benefited from (i) rapid growth of the PDB database; (ii) algorithms for improved alignments of the target with the PDB template protein (such as PSIBLAST (81)); (iii) using protein fragments rather than individual amino acids as units of structure; (iv) using information fromsequence coevolution (82) to identify native contacts; and recently, (v) deep learning (DL) modeling; see Fig. 2.
Fig. 2. CASP success rates versus difficulty of the target protein, over the years.
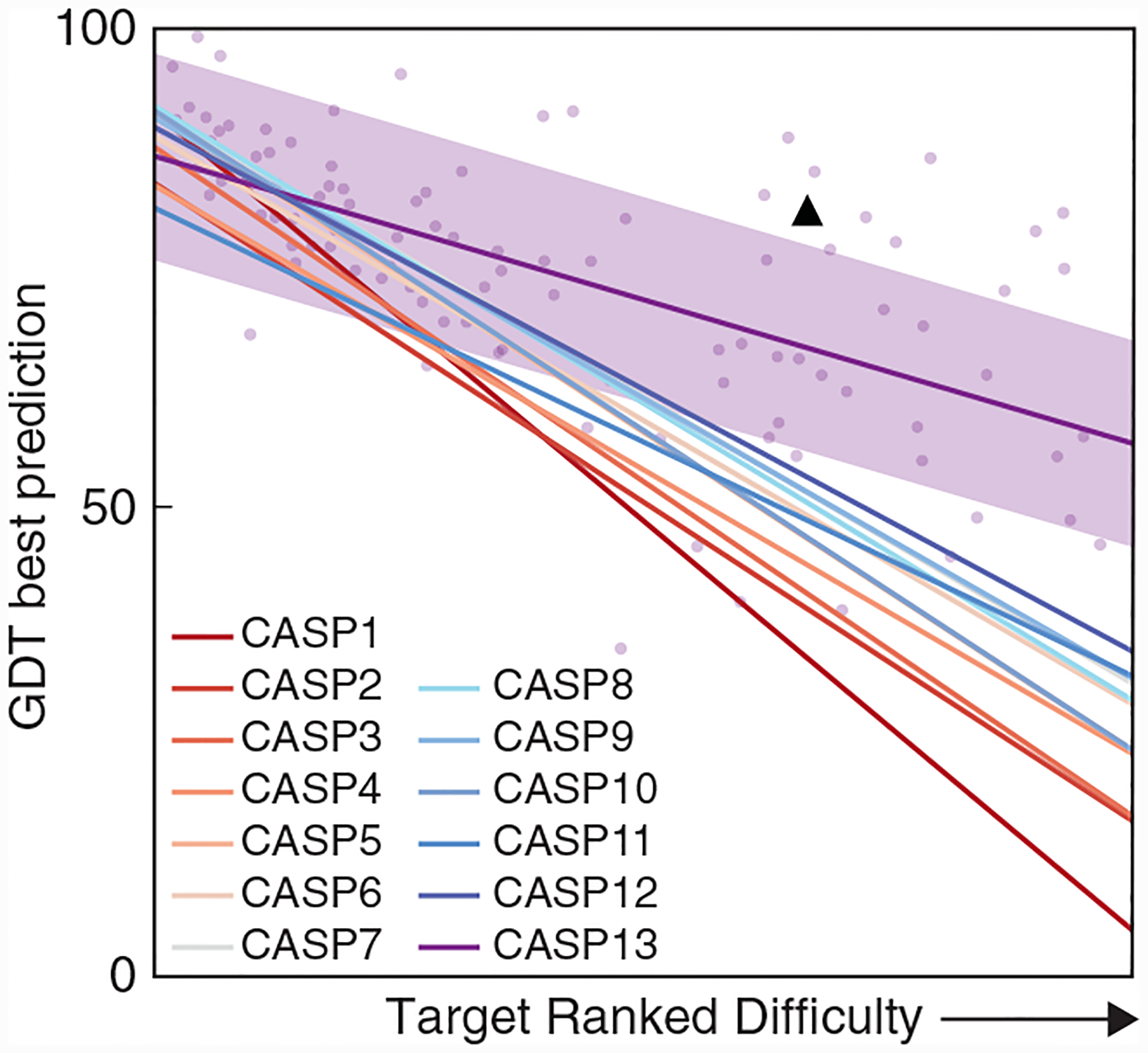
“Difficulty” is defined by how similar a template sequence that can be found in the PDB is to the target protein being predicted. Data points show the best predictions for each target protein in CASP 13; the purple shading shows the variance. Lines show the mean for each CASP. Main conclusions: (i) SBI requires good templates (all lines slope down). (ii) Predictions are improving over time (lines are higher in later events). (iii) Coevolutionary data and deep learning are adding value (step from CASP 12 to 13). (iv) CMP is now competitive in CASP (black triangle).
CASP has spawned other protein computational competitions. Among the newest is the EM validation challenge, started in 2019, which tests protein structures computed from limited EM data (83). Other events concern binding actions [CAPRI (Critical Assessment of Prediction of Interactions], which began in 2001, addresses multiple proteins binding to each other. SAMPL (Statistical Assessment of the Modeling of Proteins and Ligands), initiated in 2008 (84), is about predicting solvation free energies, binding affinities, host-guest conformational sampling, and pKa’s (acid dissociation constants) of small molecules. D3R (Drug Design Data Resource), first run in 2015, evaluates small-molecule and drug-like ligands bound to proteins (85).] And still other events focus on the functions of proteins [CAFA (Critical Assessment of protein Function Annotation), initiated in 2013, assesses protein function predicted from amino acid sequences (86) and CAGI (Critical Assessment of Genome Interpretation), now in its fifth iteration, assesses effects of mutations on the stabilities and functions of proteins (87).] DREAM (Dialogue on Reverse Engineering Assessment and Methods), established in 2006, is crowdsourcing competitions in computational biomedicine for disease and drug discovery (88). These communal blind events have advanced our understanding of methodology through systematic comparative evaluations. They provide benchmarks of the year-to-year progress in computational protein modeling, illuminating the best methods and indicating where improvements are needed.
CMP is now becoming competitive in blind communal tests
Over the past half-decade, physics-based methods have become fast enough to enter the protein modeling competitions (89, 90). Physical methods predict more than is tested by these competitions, but these events provide quantitative touchstones that any storytelling tool must get right. If our stories don’t have the correct ending (native structure), how can we trust the rest of the story? And, whereas blind competitions help advance CMP, conversely, CMP adds value to protein structure prediction. CMP can circumvent an Achilles heel of SBI, namely, the need for structure databases, homology modeling, and sequence alignments. CMP can tackle proteins that don’t resemble known proteins. Some successes from the 2018 events are reported below.
Predicting native structures, assisted by data
Experimental structural biology provides data, but computations are needed to leverage that data to give an atomically detailed structure. CASP’s structure refinement category asks: Are approximately correct structures improvable? Early CASP results showed that trying to improve one SBI algorithm by another SBI method was mostly unsuccessful (91). However, recently, Feig and others have shown good success in improving SBI predictions by CMP approaches; see Fig. 3A (i) (92). In one CASP event category, predictors are given sparse NMR data from nuclear Overhauser effect spectroscopy and dipolar couplings. On its own, these data are not sufficient to determine a native structure. CASP asks whether computations can augment this data to give correct structures. The best predictor in this CASP category in 2018 was a CMP method, MELD × MD; see Fig. 3A (ii) (56, 93). The 2019 first EM validation challenge provided data from cryo-EM for predicting the structure of a protein at different EM map resolutions, and the structure of a protein-ligand complex at a single resolution (94). By several metrics, including root mean square deviation (RMSD), all predictors gave fairly accurate structures. But a CMP method (MELD × MDFF) also gave the best-fitting map, at a resolution of 2.3 Å; see Fig. 3A (iii). Structure 3AJO in Fig. 3B shows good agreement between the predicted (blue) and true (orange) native structure.
Fig. 3. Successes of CMP in blind communal events.
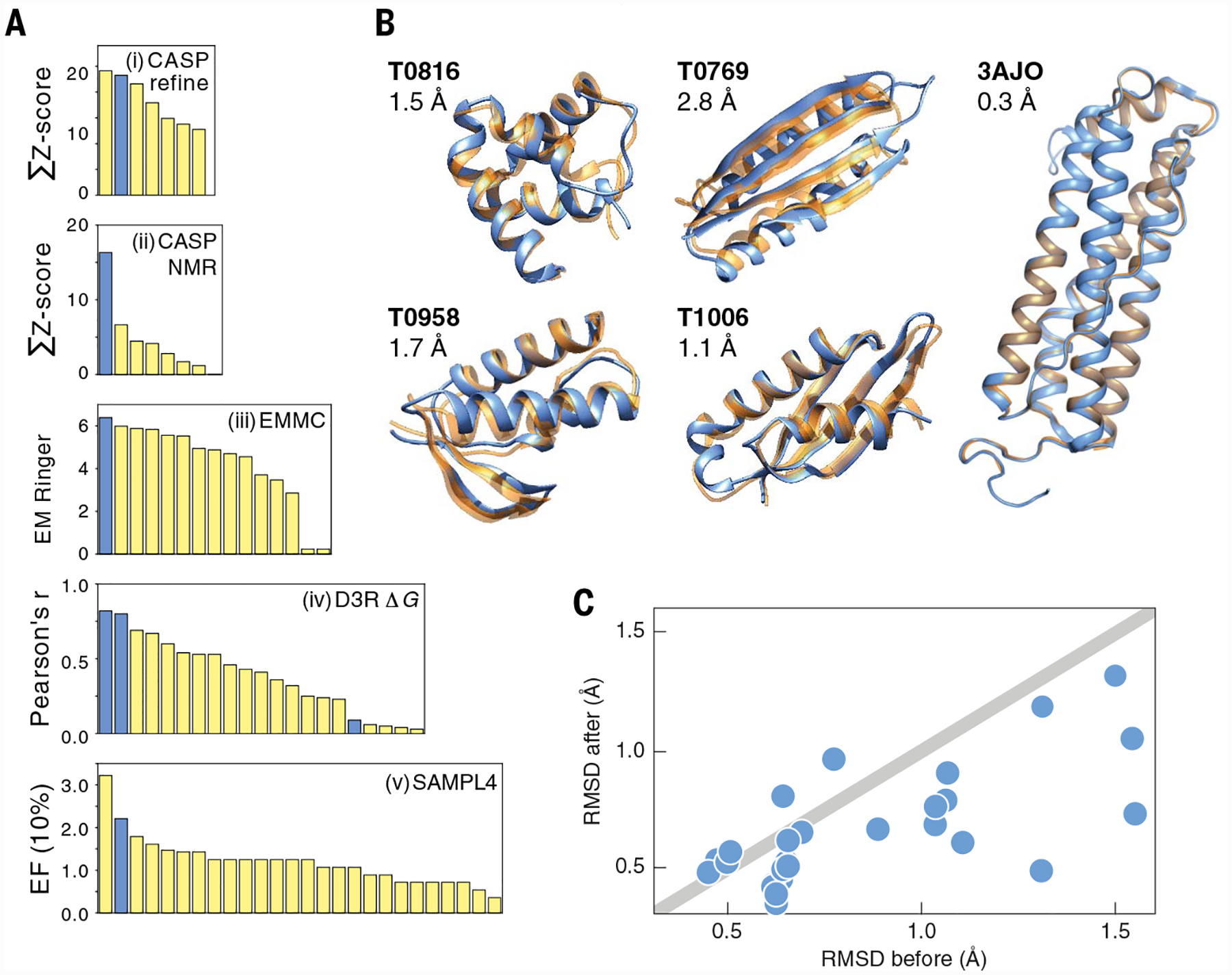
(A) Comparisons with other predictions, in five events. Blue bars, CMP predictions; yellow bars, other. Left to right: Most- to least-successful predictions, based on each y axis metric. From top to bottom: CASP 13 structure refinement, CASP 13 leveraging NMR data to determine protein structures, Cryo-Electron Microscopy Model Challenge (2019 EMMC), D3R binding affinity of 300 ligands to cathepsin S, SAMPL 4 virtual screening of ligands to HIV integrase. (B) Predicted versus true structures from CMP in CASP 13 ab initio folding and 2019 EMMC cryo-EM refinement. (C) CMP-based protein-protein docking structures, in the D3R Grand Challenge 4 stage 2b event (59), after a rigid-body ClusPro first step. y axis, RMSD error of MELD + ClusPro refined protein docked structures; x axis, RMSD error of ClusPro prediction alone. Points below the line indicate successful refinements.
Predicting a native structure from sequence alone
More challenging than predicting a structure by using ancillary data is CASP’s T0 category, for predicting structure from the amino acid sequence alone. For a few small, simple proteins, CMP methods succeed at these ab initio folding tests, from amino acid sequence alone (Fig. 3B). Each image in that figure gives its CASP target ID number (Txxx) and the RMSD error between the predicted (blue) and true (orange) native structure. For protein target T0958, a CMP model was the best prediction in CASP among all submitted (black triangle, Fig. 2). Although CMP modeling in CASP does not require externally supplied structural knowledge, the result in Fig. 3B was accelerated by seeding SBI server–predicted structures as initial structures for the simulation.
Predicting the tightness of binding
In the 2018 D3R grand challenge 4, predictors were given a target protein, cathepsin S, and 39 ligands that bind to it, spanning three orders of magnitude in binding affinity. The challenge was to compute the binding affinities (95). In this test, CMP free-energy calculations outperformed all other methods (including DL) to achieve an RMSE (root mean square energy error) of 0.49 kcal/mol over all ligands; see Fig. 3A (iv) (96). A second test, of protein BACE1 bound to a challenging set of ligands, which have multiple scaffolds and high chemical and structural diversity, was not successful (95). However, CMP was partially successful in finding correct docking orientations; see Fig. 3C (59). A 2013 SAMPL 4 challenge sought small-molecule inhibitors of the HIV integrase catalytic core domain (97). CMP performed significantly better than control and null models (98); see Fig. 3A (v).
Predicting the shape fitting of two proteins binding together
It is currently challenging to predict the structure and relative orientations of two proteins that are bound to each other, starting from knowledge of only their amino acid sequences. If successful, it would pave the way to atomistic modeling of whole biochemical pathways, the elementary units of which are pairs of interacting proteins. Today’s successes require much more input knowledge. If the conformation of the bound form of both proteins are already known, then existing algorithms can often find the right docked structure by rigid body rotations of one relative to the other (99–101). A step in the direction of the grander challenge was protein T121 in CAPRI round 38: seeking the binding conformation of a protein of known shape to a peptide that is unstructured prior to binding. The challenge is to find the induced structure of the peptide in its bound state. In this case, combined with a rigid-body front-end step, MELD × MD gives a medium-quality model (58, 101).
Predicting conformational populations, foretelling when structures are right
Here is a common problem in predictions: An algorithm outputs several plausible native structures, but we don’t know which one is right. SBI methods don’t provide a principle for choosing and have traditionally not been able to tell (89). The physically principled way to choose is to know the free energies (i.e., the relative populations). CMP methods can compute these. Indeed, as MD modeling has recently entered CASP, its predictions of relative populations have been shown to correctly foretell when its methods have found the true native structure (52–54).
MD can predict structures without assistance from homology models
For some proteins, there are no templates in the PDB that are good starting points for bioinformatics predictions of native structures. Because CMP methods make predictions from physical principles and don’t require starting structural knowledge, they can often predict protein structures that database methods cannot (55). At the moment, however, the greatest power is still achieved by combining CMP with external structural information (52–54, 56). CMP plus information is still CMP, provided it satisfies the physics of Newton and Boltzmann.
In short, CMP methods have now reached two key mileposts. (i) They are computationally fast enough to enter communal blind competitions, and (ii) they add value in some cases. However, CMP is still computationally expensive; does not yet have turn-key reliability across challenges; is best when some constraints are known; and is limited to small, simple proteins and ligands. But these are early days. Below are some stories being told; see also (21, 102).
A few of the stories being told by CMP
CMP modeling has several roles. It helps to turn NMR, x-ray, and cryo-EM data into protein structures; to compute binding affinities that can be actionable in discovering new drugs; and to establish detailed principled narratives about biomolecule behaviors and their actions in the cell. Below are a few examples.
Resolving the paradox of protein-folding route heterogeneity
A long-standing puzzle entails the routes of protein folding. Is there a general kinetic principle by which all proteins fold from their unfolded state to each one’s unique native state? The issue has hinged on the meaning of the word “pathway.” For experimentalists, a pathway is often defined on the experimental macroscale as a single predominant sequence of events. For theorists, a pathway is often defined by the simulational microscale watching the individual wiggling of each trajectory. State-of-the-art MD simulations over the past decade have shown that individual molecules reflect the heterogeneity expected from statistical mechanical theories, yet with average behaviors that reflect the routes that experimentalists see (103–105), resolving such paradoxes. An important side consequence of modeling pathways is the demonstration that the molecular physics alone can accurately predict the pathway end states, namely, the native structures of (small, simple) proteins, without the need for homology modeling of other reference proteins (55, 104, 105).
Computing drug–protein affinities
A major objective has been to compute how tightly a chemical (natural ligand or drug, for example) can bind to a protein. So-called docking methods can often estimate a pose, what protein crevice the molecule might fit, and how it orients inside it. But more crucial for understanding biological mechanisms and protein stories is how tightly it binds. This is becoming increasingly possible through CMP. Here are some of the advances (106–110): (i) A test of small ligands in different proteins. In 2015, the Schrodinger group modeled 200 ligands in eight proteins (108), giving the results in Fig. 4A. (ii) Computing selectivities. Aldeghi et al. computed the selectivities of ligand binding across a family of related proteins (111); see Fig 4B. (iii) Complex flexible ligands. Morrone et al. (57) computed the relative binding affinities of highly flexible peptide ligands (P53 mutants) to MDM2 and MDMx proteins; see Fig. 4C. (iv) Mutations in protein-protein binding. Free energies of binding one protein to another can change upon mutation. Some are now accurately captured by CMP (112); see Fig. 4E. The average errors are very small, 0.27 kcal/mol. Similarly small average errors are found for CMP predictions of mutational effects on protein stabilities (113). The high accuracy likely reflects the relative maturity of today’s force fields designed to handle proteins. Force fields for more diverse molecules, such as small-molecule drugs and ligands, are not as accurate, but communal efforts (114) are improving them. (v) Thermodynamic components. MD simulations are now also giving the thermodynamic components, enthalpy and entropy, of binding, which give additional insights for storytelling about molecular recognition (115, 116).
Fig. 4. CMP can predict relatively accurate experimental binding affinities, across multiple ligands and proteins.
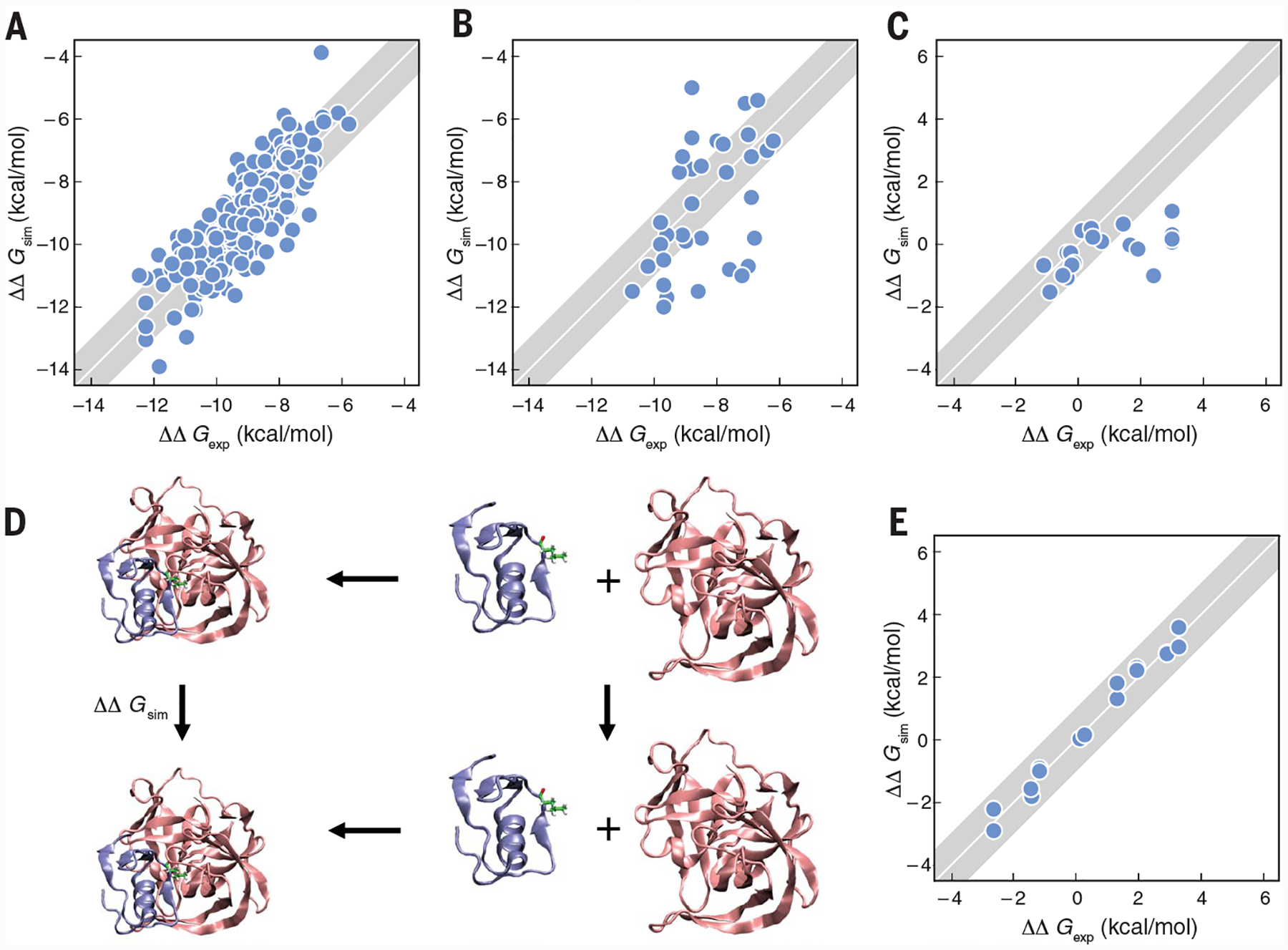
The diagonal line represents perfect agreement with experiments. The shaded area indicate 1 kcal-mol−1 error bars. (A) Schrodinger free-energy perturbation calculations of 200 ligands in eight proteins (108). (B) Algedhi et al. prediction of binding affinities of ligands across protein families (111). (C) MELD × MD relative binding affinities of various P53 mutant peptides, which are highly flexible, to the MDM2/x protein (57). (D) The thermodynamic cycle of Zou et al. for computing mutational effects on protein-protein binding (112). (E) Predicted affinities versus experiments for (D) (112).
Mechanisms of ligand dissociation and off-rate estimation
There is interest in knowing not just the affinity of a ligand molecule for a protein, but also its off-rate. The off-rate (koff) is a measure of the residence time that the ligand spends in the site and is sometimes a better predictor of biological efficacy than is the equilibrium binding affinity (K = kon/koff) (117). Even though some off-rates are known to be as slow as minutes, advanced-sampling MD simulations give good agreement with experiments (118, 119), including over these slow time scales (120). MD simulations identified the mechanisms underlying slow off-rates in a tuberculosis drug target, guiding the rational design of new ligands with even longer residence times. Subsequent synthesis, crystallography, and kinetic assays have confirmed the predictions (121, 122).
Mechanisms of amyloid aggregation
No experiment is yet granular enough to show the molecular events as protein chains come together to form the amyloid fibers that are relevant to diseases of neurodegeneration. MD simulations are elucidating the kinetic steps (123–126) and giving insights into new drug discovery approaches (127).
Space here does not allow us to review the many other important emerging stories, about how transducing proteins convert the chemical energy of adenosine 5′-triphosphate hydrolysis into mechanical work and forces, how allosteric “action-at-a-distance” conformational changes transmit chemical signals across large molecules, how kinase proteins send signals in cancer cells, and how molecules cause biological motions by “walking” along tracks made of other proteins.
What lies ahead?
How can we model bigger proteins, bigger motions, and longer time scales?
We need continued improvements in force fields and solvent models. We need faster and more targeted searching and sampling, to reduce the combinatorial nightmare of conformational space. One innovative idea is to estimate entropies by using ideas based on computer compression algorithms (128). One new accelerator of molecular dynamics is Boltzmann generators, which use deep learning methods to learn where to find the deepest free-energy wells on landscapes (129) and improve sampling (130). And quantum computing might ultimately help us tackle these big stochastic optimization challenges. But we also need more than better hardware alone, because the scaling to larger proteins is a bigger problem than even continued advances based on Moore’s law can solve.
The role of deep learning
DL can bring insights into protein structures that can complement the CMP’s insights into the physics. In December 2018, the CASP evaluators reported notable success from AlphaFold, a new DL method from Google’s London-based DeepMind group and other groups (131). The superior ability of DL methods (131, 132) in harnessing data relative to earlier statistical methods led to the best results in CASP’s free modeling category (90). The advance from DL was about twofold that of the average natural biennial advance in CASP. DL is powerful when given either large databases to learn from, or when some rules—such as in games—can be used to generate input. Alpha-Fold learned from the protein structures in the PDB. But DL does not know what it cannot see. In drug discovery, what’s most needed is to generate diversity, i.e., new classes of molecules that are not already known drugs (133, 134). Protein storytelling can benefit from both DL for insights from knowledge bases and from CMP for causation and driving forces, motions, and binding, where structures are heterogeneous, and where we need to understand the role of the protein’s external environment.
Summary
CMP is an increasingly powerful tool for telling the stories of protein molecule actions. Systematic improvements in force fields, enhanced sampling methods, and accelerators have enabled CMP to reach time scales of important biological actions and opened the door to evaluation in communal blind events, like CASP. These time-scale successes were forecast a quarter-century ago (135). At this rate, in the next quarter-century, we’ll be telling stories of protein molecules over the whole life span—tens of minutes—of a bacterial cell; perhaps the updated version of this article will be called “Cellular storytelling through physics.” CMP is increasingly grounding our narrative stories of the biological actions of proteins in principled physics.
ACKNOWLEDGMENTS
We thank D. Case, M. Feig, M. Gilson, D. Kozakov, A. Kryshtafovych,A. MacKerrell, D. Mobley, J. Moult, R. Nassar, D. Padhorny, R. Read, and B. Sharma for valuable comments and insights. We are grateful to S. Bromberg for help with figures and her deeply insightful comments and editing. Thanks also to the anonymous referees, who provided excellent feedback and constructive criticism.
Funding: We are grateful for the support from NIH grants 5R01GM107104, 5R01GM125813, and RM1GM135136 and the Stony Brook Laufer Center for Physical and Quantitative Biology.
Footnotes
Competing interests: The authors have no competing interests to declare.
REFERENCES AND NOTES
- 1.Berman HM et al. , The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr 58, 899–907 (2002). doi: 10.1107/S0907444902003451; [DOI] [PubMed] [Google Scholar]
- 2.Brini E et al. , How Water’s Properties Are Encoded in Its Molecular Structure and Energies. Chem. Rev 117, 12385–12414 (2017). doi: 10.1021/acs.chemrev.7b00259; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scott RA, Scheraga HA, Conformational Analysis of Macromolecules. III. Helical Structures of Polyglycine and Poly-L-Alanine. J. Chem. Phys 45, 2091–2101 (1966). doi: 10.1063/1.1727894 [DOI] [Google Scholar]
- 4.Brant DA, Miller WG, Flory PJ, Conformational energy estimates for statistically coiling polypeptide chains. J. Mol. Biol 23, 47–65 (1967). doi: 10.1016/S0022-2836(67)80066-4 [DOI] [Google Scholar]
- 5.Levitt M, Lifson S, Refinement of protein conformations using a macromolecular energy minimization procedure. J. Mol. Biol 46, 269–279 (1969). doi: 10.1016/0022-2836(69)90421-5; [DOI] [PubMed] [Google Scholar]
- 6.Hagler AT, Huler E, Lifson S, Energy functions for peptides and proteins. I. Derivation of a consistent force field including the hydrogen bond from amide crystals. J. Am. Chem. Soc 96, 5319–5327 (1974). doi: 10.1021/ja00824a004; [DOI] [PubMed] [Google Scholar]
- 7.Momany F, McGuire RF, Burgess A, Scheraga HA, Energy parameters in polypeptides. VII. Geometric parameters, partial atomic charges, nonbonded interactions, hydrogen bond interactions, and intrinsic torsional potentials for the naturally occurring amino acids. J. Phys. Chem 79, 2361–2381 (1975). doi: 10.1021/j100589a006 [DOI] [Google Scholar]
- 8.Brooks BR et al. , CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem 4, 187–217 (1983). doi: 10.1002/jcc.540040211 [DOI] [Google Scholar]
- 9.Weiner SJ et al. , A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc 106, 765–784 (1984). doi: 10.1021/ja00315a051 [DOI] [Google Scholar]
- 10.van Gunsteren WF, Berendsen HJ, Groningen Molecular Simulation (GROMOS) Library Manual (Biomos, Groningen, Netherlands, 1987), pp. 1–221. [Google Scholar]
- 11.Jorgensen WL, Tirado-Rives J, The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc 110, 1657–1666 (1988). doi: 10.1021/ja00214a001; [DOI] [PubMed] [Google Scholar]
- 12.Cornell WD et al. , A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc 117, 5179–5197 (1995). doi: 10.1021/ja00124a002 [DOI] [Google Scholar]
- 13.Case DA et al. , The Amber biomolecular simulation programs. J. Comput. Chem 26, 1668–1688 (2005). doi: 10.1002/jcc.20290; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tian C et al. , ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput 16, 528–552 (2020). doi: 10.1021/acs.jctc.9b00591; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Debiec KT et al. , Further along the Road Less Traveled: AMBER ff15ipq, an Original Protein Force Field Built on a Self-Consistent Physical Model. J. Chem. Theory Comput 12, 3926–3947 (2016). doi: 10.1021/acs.jctc.6b00567; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L-P et al. , Building a More Predictive Protein Force Field: A Systematic and Reproducible Route to AMBER-FB15. J. Phys. Chem. B 121, 4023–4039 (2017). doi: 10.1021/acs.jpcb.7b02320; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang J et al. , CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). doi: 10.1038/nmeth.4067; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harder E et al. , OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput 12, 281–296 (2016). doi: 10.1021/acs.jctc.5b00864; [DOI] [PubMed] [Google Scholar]
- 19.Robustelli P, Piana S, Shaw DE, Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U.S.A 115, E4758–E4766 (2018). doi: 10.1073/pnas.1800690115; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopes PEM et al. , Polarizable Force Field for Peptides and Proteins based on the Classical Drude Oscillator. J. Chem. Theory Comput 9, 5430–5449 (2013). doi: 10.1021/ct400781b; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bahar I, Jernigan RL, Dill K, Protein actions: Principles and modeling (Garland Science, 2017). [Google Scholar]
- 22. By timestep, we mean that inside the computer, over a discrete unit of time, the algorithm is representing a physical process happening over that unit of time. The computer is approximating Newton’s laws and the timesteps must be extremely short to keep the errors acceptably small.
- 23.Izvekov S, Voth GA, A multiscale coarse-graining method for biomolecular systems. J. Phys. Chem. B 109, 2469–2473 (2005). doi: 10.1021/jp044629q; [DOI] [PubMed] [Google Scholar]
- 24.Carmichael SP, Shell MS, A new multiscale algorithm and its application to coarse-grained peptide models for self-assembly. J. Phys. Chem. B 116, 8383–8393 (2012). doi: 10.1021/jp2114994; [DOI] [PubMed] [Google Scholar]
- 25.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP,de Vries AH, The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 (2007). doi: 10.1021/jp071097f; [DOI] [PubMed] [Google Scholar]
- 26.Kmiecik S et al. , Coarse-Grained Protein Models and Their Applications. Chem. Rev 116, 7898–7936 (2016).doi: 10.1021/acs.chemrev.6b00163; [DOI] [PubMed] [Google Scholar]
- 27.McCammon JA, Gelin BR, Karplus M, Dynamics of folded proteins. Nature 267, 585–590 (1977). doi: 10.1038/267585a0; [DOI] [PubMed] [Google Scholar]
- 28.Daura X et al. , Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed 38, 236–240 (1999). doi: 10.1002/(SICI)1521-3773(19990115)38:1/2<236::AIDANIE236>3.0.CO;2-M [DOI] [Google Scholar]
- 29.Duan Y, Wang L, Kollman PA, The early stage of folding of villin headpiece subdomain observed in a 200-nanosecond fully solvated molecular dynamics simulation. Proc. Natl. Acad. Sci. U.S.A 95, 9897–9902 (1998). doi: 10.1073/pnas.95.17.9897; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allen F et al. , Blue Gene: A vision for protein science using a petaflop supercomputer. IBM Syst. J 40, 310–327 (2001). doi: 10.1147/sj.402.0310 [DOI] [Google Scholar]
- 31.Shirts M, Pande VS, COMPUTING: Screen Savers of the World Unite! Science 290, 1903–1904 (2000). doi: 10.1126/science.290.5498.1903; [DOI] [PubMed] [Google Scholar]
- 32.Shaw DE et al. , Anton, a special-purpose machine for molecular dynamics simulation. Commun. ACM 51, 91–97 (2008). doi: 10.1145/1364782.1364802 [DOI] [Google Scholar]
- 33.Shaw DE et al. , Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis (IEEE, 2014), pp. 41–53. [Google Scholar]
- 34.Laio A, Parrinello M, Escaping free-energy minima. Proc. Natl. Acad. Sci. U.S.A 99, 12562–12566 (2002). doi: 10.1073/pnas.202427399; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sugita Y, Okamoto Y, Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett 314, 141–151 (1999). doi: 10.1016/S0009-2614(99)01123-9 [DOI] [Google Scholar]
- 36.Tsallis C, Stariolo DA, Generalized simulated annealing. Physica A 233, 395–406 (1996). doi: 10.1016/S0378-4371(96)00271-3 [DOI] [Google Scholar]
- 37.Darve E, Pohorille A, Calculating free energies using average force. J. Chem. Phys 115, 9169–9183 (2001). doi: 10.1063/1.1410978 [DOI] [Google Scholar]
- 38.Torrie GM, Valleau JP, Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys 23, 187–199 (1977). doi: 10.1016/0021-9991(77)90121-8 [DOI] [Google Scholar]
- 39.Bernardi RC, Melo MC, Schulten K, Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta Gen. Subj 1850, 872–877 (2015). doi: 10.1016/j.bbagen.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang A, Zhang Z, Li G, Advances in enhanced sampling molecular dynamics simulations for biomolecules. Chin. J. Chem. Phys 32, 277–286 (2019). doi: 10.1063/1674-0068/cjcp1905091 [DOI] [Google Scholar]
- 41.Chipot C, Pohorille A, Free Energy Calculations (Springer, 2007). [Google Scholar]
- 42.Shirts MR, Chodera JD, Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys 129, 124105 (2008). doi: 10.1063/1.2978177; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH,Kollman PA, THE weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem 13, 1011–1021 (1992). doi: 10.1002/jcc.540130812 [DOI] [Google Scholar]
- 44.Rout MP, Sali A, Principles for Integrative Structural Biology Studies. Cell 177, 1384–1403 (2019). doi: 10.1016/j.cell.2019.05.016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leaver-Fay A et al. , ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 (2011). doi: 10.1016/B978-0-12-381270-4.00019-6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu D, Zhang Y, Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80, 1715–1735 (2012).doi: 10.1002/prot.24065; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuntz ID, Blaney JM, Oatley SJ, Langridge R,Ferrin TE, A geometric approach to macromolecule-ligand interactions. J. Mol. Biol 161, 269–288 (1982). doi: 10.1016/0022-2836(82)90153-X; [DOI] [PubMed] [Google Scholar]
- 48.Kozakov D et al. , The ClusPro web server for protein-protein docking. Nat. Protoc 12, 255–278 (2017). doi: 10.1038/nprot.2016.169; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Zundert GCP et al. , The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol 428, 720–725 (2016). doi: 10.1016/j.jmb.2015.09.014; [DOI] [PubMed] [Google Scholar]
- 50.Eswar N et al. , Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics 15, 5.6.1–5.6.30 (2006). doi: 10.1002/0471250953.bi0506s15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Russel D et al. , Putting the pieces together: Integrative modeling platform software for structure determination of macromolecular assemblies. PLOS Biol. 10, e1001244 (2012). doi: 10.1371/journal.pbio.1001244; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perez A, MacCallum JL, Dill KA, Accelerating molecular simulations of proteins using Bayesian inference on weak information. Proc. Natl. Acad. Sci. U.S.A 112, 11846–11851 (2015). doi: 10.1073/pnas.1515561112; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.MacCallum JL, Perez A, Dill KA, Determining protein structures by combining semireliable data with atomistic physical models by Bayesian inference. Proc. Natl. Acad. Sci. U.S.A 112, 6985–6990 (2015). doi: 10.1073/pnas.1506788112; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perez A, Morrone JA, Brini E, MacCallum JL, Dill KA, Blind protein structure prediction using accelerated free-energy simulations. Sci. Adv 2, e1601274 (2016).doi: 10.1126/sciadv.1601274; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robertson JC, Perez A, Dill KA, MELD × MD Folds Nonthreadables, Giving Native Structures and Populations. J. Chem. Theory Comput 14, 6734–6740 (2018). doi: 10.1021/acs.jctc.8b00886; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robertson JC et al. , NMR-assisted protein structure prediction with MELDxMD. Proteins 87, 1333–1340 (2019). doi: 10.1002/prot.25788; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morrone JA et al. , Molecular Simulations Identify Binding Poses and Approximate Affinities of Stapled α-Helical Peptides to MDM2 and MDMX. J. Chem. Theory Comput 13, 863–869 (2017). doi: 10.1021/acs.jctc.6b00978; [DOI] [PubMed] [Google Scholar]
- 58.Khramushin A et al. , Modeling beta-sheet peptide-protein interactions: Rosetta FlexPepDock in CAPRI rounds 38–45. Proteins 88, 1037–1049 (2020). doi: 10.1002/prot.25871; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kotelnikov S et al. , Sampling and refinement protocols for template-based macrocycle docking: 2018 D3R Grand Challenge 4. J. Comput. Aided Mol. Des 34, 179–189 (2020). doi: 10.1007/s10822-019-00257-1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perez A, Sittel F, Stock G, Dill K, MELD-Path Efficiently Computes Conformational Transitions, Including Multiple and Diverse Paths. J. Chem. Theory Comput 14, 2109–2116 (2018). doi: 10.1021/acs.jctc.7b01294; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shan Y et al. , How does a drug molecule find its target binding site? J. Am. Chem. Soc 133, 9181–9183 (2011). doi: 10.1021/ja202726y; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schütte C, Fischer A, Huisinga W, Deuflhard P, A Direct Approach to Conformational Dynamics Based on Hybrid Monte Carlo. J. Comput. Phys 151, 146–168 (1999).doi: 10.1006/jcph.1999.6231 [DOI] [Google Scholar]
- 63.Swope WC, Pitera JW, Suits F, Describing Protein Folding Kinetics by Molecular Dynamics Simulations. 1. Theory †. J. Phys. Chem. B 108, 6571–6581 (2004). doi: 10.1021/jp037421y [DOI] [Google Scholar]
- 64.Noé F, Horenko I, Schütte C, Smith JC, Hierarchical analysis of conformational dynamics in biomolecules: Transition networks of metastable states. J. Chem. Phys 126, 155102 (2007). doi: 10.1063/1.2714539; [DOI] [PubMed] [Google Scholar]
- 65.Chodera JD, Singhal N, Pande VS, Dill KA, Swope WC, Automatic discovery of metastable states for the construction of Markov models of macromolecular conformational dynamics. J. Chem. Phys 126, 155101 (2007). doi: 10.1063/1.2714538; [DOI] [PubMed] [Google Scholar]
- 66.Buchete N-V, Hummer G, Coarse master equations for peptide folding dynamics. J. Phys. Chem. B 112, 6057–6069 (2008). doi: 10.1021/jp0761665; [DOI] [PubMed] [Google Scholar]
- 67.Bowman GR, Huang X, Pande VS, Using generalized ensemble simulations and Markov state models to identify conformational states. Methods 49, 197–201 (2009).doi: 10.1016/j.ymeth.2009.04.013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prinz J-H et al. , Markov models of molecular kinetics: Generation and validation. J. Chem. Phys 134, 174105 (2011). doi: 10.1063/1.3565032; [DOI] [PubMed] [Google Scholar]
- 69.Noé F, Rosta E, Markov Models of Molecular Kinetics. J. Chem. Phys 151, 190401 (2019). doi: 10.1063/1.5134029 [DOI] [PubMed] [Google Scholar]
- 70.Voelz VA et al. , Slow unfolded-state structuring in Acyl-CoA binding protein folding revealed by simulation and experiment. J. Am. Chem. Soc 134, 12565–12577 (2012). doi: 10.1021/ja302528z; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sadiq SK, Noé F, De Fabritiis G, Kinetic characterization of the critical step in HIV-1 protease maturation. Proc. Natl. Acad. Sci. U.S.A 109, 20449–20454 (2012). doi: 10.1073/pnas.1210983109; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Buch I, Giorgino T, De Fabritiis G, Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. U.S.A 108, 10184–10189 (2011). doi: 10.1073/pnas.1103547108; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang D, Caflisch A, The free energy landscape of small molecule unbinding. PLOS Comput. Biol 7, e1002002 (2011). doi: 10.1371/journal.pcbi.1002002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Faradjian AK, Elber R, Computing time scales from reaction coordinates by milestoning. J. Chem. Phys 120, 10880–10889 (2004). doi: 10.1063/1.1738640; [DOI] [PubMed] [Google Scholar]
- 75.Votapka LW, Amaro RE, Multiscale Estimation of Binding Kinetics Using Brownian Dynamics, Molecular Dynamicsand Milestoning. PLOS Comput. Biol 11, e1004381 (2015). doi: 10.1371/journal.pcbi.1004381; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cardenas AE, Elber R, Markovian and Non-Markovian Modeling of Membrane Dynamics with Milestoning. J. Phys. Chem. B 120, 8208–8216 (2016). doi: 10.1021/acs.jpcb.6b01890; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Narayan B et al. , The transition between active and inactive conformations of Abl kinase studied by rock climbing and Milestoning. Biochim. Biophys. Acta, Gen. Subj 1864, 129508 (2020). doi: 10.1016/j.bbagen.2019.129508; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huber GA, Kim S, Weighted-ensemble Brownian dynamics simulations for protein association reactions. Biophys. J 70, 97–110 (1996). doi: 10.1016/S0006-3495(96)79552-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zuckerman DM, Chong LT, Weighted Ensemble Simulation: Review of Methodology, Applications, and Software. Annu. Rev. Biophys 46, 43–57 (2017). doi: 10.1146/annurev-biophys-070816-033834; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moult J, Pedersen JT, Judson R, Fidelis K, A large-scale experiment to assess protein structure prediction methods. Proteins 23, ii–v (1995). doi: 10.1002/prot.340230303; [DOI] [PubMed] [Google Scholar]
- 81.Altschul SF et al. , Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). doi: 10.1093/nar/25.17.3389; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. If a given type of protein has two amino-acid positions that co-vary together across many different organisms, it can indicate they are in contact in their similar native structures.
- 83.Lawson CL, Chiu W, Comparing cryo-EM structures. J. Struct. Biol 204, 523–526 (2018). doi: 10.1016/j.jsb.2018.10.004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nicholls A et al. , Predicting small-molecule solvation free energies: An informal blind test for computational chemistry. J. Med. Chem 51, 769–779 (2008). doi: 10.1021/jm070549+; [DOI] [PubMed] [Google Scholar]
- 85.Gathiaka S et al. , D3R grand challenge 2015: Evaluation of protein-ligand pose and affinity predictions. J. Comput. Aided Mol. Des 30, 651–668 (2016). doi: 10.1007/s10822-016-9946-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Radivojac P et al. , A large-scale evaluation of computational protein function prediction. Nat. Methods 10, 221–227 (2013). doi: 10.1038/nmeth.2340; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Andreoletti G, Pal LR, Moult J, Brenner SE, Reports from the fifth edition of CAGI: The Critical Assessment of Genome Interpretation. Hum. Mutat 40, 1197–1201 (2019).doi: 10.1002/humu.23876; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stolovitzky G, Monroe D, Califano A, Dialogue on reverse-engineering assessment and methods: The DREAM of high-throughput pathway inference. Ann. N. Y. Acad. Sci 1115, 1–22 (2007). doi: 10.1196/annals.1407.021; [DOI] [PubMed] [Google Scholar]
- 89.Abriata LA, Tamò GE, Monastyrskyy B, Kryshtafovych A,Dal Peraro M, Assessment of hard target modeling in CASP12 reveals an emerging role of alignment-based contact prediction methods. Proteins 86 (Suppl 1), 97–112 (2018). doi: 10.1002/prot.25423; [DOI] [PubMed] [Google Scholar]
- 90.Croll TI, Sammito MD, Kryshtafovych A, Read RJ, Evaluation of template-based modeling in CASP13. Proteins 87, 1113–1127 (2019). doi: 10.1002/prot.25800; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.MacCallum JL et al. , Assessment of protein structure refinement in CASP9. Proteins 79 (suppl. 10), 74–90 (2011). doi: 10.1002/prot.23131; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Read RJ, Sammito MD, Kryshtafovych A, Croll TI, Evaluation of model refinement in CASP13. Proteins 87, 1249–1262 (2019). doi: 10.1002/prot.25794; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sala D et al. , Protein structure prediction assisted with sparse NMR data in CASP13. Proteins 87, 1315–1332 (2019). doi: 10.1002/prot.25837; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.EMDataResource Validation Challenges, Em validation challenge, https://challenges.emdataresource.org/ (2019); accessed 22 February 2020.
- 95.Parks CD et al. , D3R grand challenge 4: Blind prediction of protein-ligand poses, affinity rankings, and relative binding free energies. J. Comput. Aided Mol. Des 34, 99–119 (2020). doi: 10.1007/s10822-020-00289-y; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zou J, Tian C, Simmerling C, Blinded prediction of protein-ligand binding affinity using Amber thermodynamic integration for the 2018 D3R grand challenge 4. J. Comput. Aided Mol. Des 33, 1021–1029 (2019). doi: 10.1007/s10822-019-00223-x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mobley DL et al. , Blind prediction of HIV integrase binding from the SAMPL4 challenge. J. Comput. Aided Mol. Des 28, 327–345 (2014). doi: 10.1007/s10822-014-9723-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gallicchio E et al. , Virtual screening of integrase inhibitors by large scale binding free energy calculations: The SAMPL4 challenge. J. Comput. Aided Mol. Des 28, 475–490 (2014). doi: 10.1007/s10822-014-9711-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dapkūnas J, Olechnovič K, Venclovas Č, Structural modeling of protein complexes: Current capabilities and challenges. Proteins 87, 1222–1232 (2019). doi: 10.1002/prot.25774; [DOI] [PubMed] [Google Scholar]
- 100.Lensink MF et al. , Blind prediction of homo- and hetero-protein complexes: The CASP13-CAPRI experiment. Proteins 87, 1200–1221 (2019). doi: 10.1002/prot.25838; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lensink MF, Nadzirin N, Velankar S, Wodak SJ, Modeling protein-protein, protein-peptide, and protein-oligosaccharide complexes: CAPRI 7th edition. Proteins 88, 916–938 (2020). doi: 10.1002/prot.25870; [DOI] [PubMed] [Google Scholar]
- 102.Huggins DJ et al. , Biomolecular simulations: From dynamics and mechanisms to computational assays of biological activity. Wiley Interdiscip. Rev. Comput. Mol. Sci 9, e1393 (2019). doi: 10.1002/wcms.1393 [DOI] [Google Scholar]
- 103.Shaw DE et al. , Atomic-level characterization of the structural dynamics of proteins. Science 330, 341–346 (2010). doi: 10.1126/science.1187409; [DOI] [PubMed] [Google Scholar]
- 104.Lindorff-Larsen K, Piana S, Dror RO, Shaw DE, How fast-folding proteins fold. Science 334, 517–520 (2011). doi: 10.1126/science.1208351; [DOI] [PubMed] [Google Scholar]
- 105.Nguyen H, Maier J, Huang H, Perrone V, Simmerling C, Folding simulations for proteins with diverse topologies are accessible in days with a physics-based force field and implicit solvent. J. Am. Chem. Soc 136, 13959–13962 (2014). doi: 10.1021/ja5032776; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mobley DL et al. , Predicting absolute ligand binding free energies to a simple model site. J. Mol. Biol 371, 1118–1134 (2007). doi: 10.1016/j.jmb.2007.06.002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Boyce SE et al. , Predicting ligand binding affinity with alchemical free energy methods in a polar model binding site. J. Mol. Biol 394, 747–763 (2009). doi: 10.1016/j.jmb.2009.09.049; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang L et al. , Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J. Am. Chem. Soc 137, 2695–2703 (2015). doi: 10.1021/ja512751q; [DOI] [PubMed] [Google Scholar]
- 109.Matos GDR et al. , Approaches for calculating solvation free energies and enthalpies demonstrated with an update of the FreeSolv database. J. Chem. Eng. Data 62, 1559–1569 (2017). doi: 10.1021/acs.jced.7b00104; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Barros EP et al. , Improving the Efficiency of Ligand-Binding Protein Design with Molecular Dynamics Simulations. J. Chem. Theory Comput 15, 5703–5715 (2019).doi: 10.1021/acs.jctc.9b00483; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aldeghi M, Heifetz A, Bodkin MJ, Knapp S, Biggin PC, Predictions of Ligand Selectivity from Absolute Binding Free Energy Calculations. J. Am. Chem. Soc 139, 946–957 (2017). doi: 10.1021/jacs.6b11467; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zou J, Simmerling C, Raleigh DP, Dissecting the Energetics of Intrinsically Disordered Proteins via a Hybrid Experimental and Computational Approach. J. Phys. Chem. B 123, 10394–10402 (2019). doi: 10.1021/acs.jpcb.9b08323; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zou J, Song B, Simmerling C, Raleigh D, Experimental and Computational Analysis of Protein Stabilization by Gly-to-d Ala Substitution: A Convolution of Native State and Unfolded State Effects. J. Am. Chem. Soc 138, 15682–15689 (2016). doi: 10.1021/jacs.6b09511; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mobley DL et al. , Escaping Atom Types in Force Fields Using Direct Chemical Perception. J. Chem. Theory Comput 14, 6076–6092 (2018). doi: 10.1021/acs.jctc.8b00640; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fenley AT, Muddana HS, Gilson MK, Entropy-enthalpy transduction caused by conformational shifts can obscure the forces driving protein-ligand binding. Proc. Natl. Acad. Sci. U.S.A 109, 20006–20011 (2012). doi: 10.1073/pnas.1213180109; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Li A, Gilson MK, Protein-ligand binding enthalpies from near-millisecond simulations: Analysis of a preorganization paradox. J. Chem. Phys 149, 072311 (2018). doi: 10.1063/1.5027439; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Copeland RA, The drug-target residence time model: A 10-year retrospective. Nat. Rev. Drug Discov 15, 87–95 (2016). doi: 10.1038/nrd.2015.18; [DOI] [PubMed] [Google Scholar]
- 118.Re S, Oshima H, Kasahara K, Kamiya M, Sugita Y, Encounter complexes and hidden poses of kinase-inhibitor binding on the free-energy landscape. Proc. Natl. Acad. Sci. U.S.A 116, 18404–18409 (2019). doi: 10.1073/pnas.1904707116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tiwary P, Mondal J, Berne BJ, How and when does an anticancer drug leave its binding site? Sci. Adv 3, e1700014 (2017). doi: 10.1126/sciadv.1700014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lotz SD, Dickson A, Unbiased Molecular Dynamics of 11 min Timescale Drug Unbinding Reveals Transition State Stabilizing Interactions. J. Am. Chem. Soc 140, 618–628 (2018). doi: 10.1021/jacs.7b08572; [DOI] [PubMed] [Google Scholar]
- 121.Lai C-T et al. , Rational Modulation of the Induced-Fit Conformational Change for Slow-Onset Inhibition in Mycobacterium tuberculosis InhA. Biochemistry 54, 4683–4691 (2015). doi: 10.1021/acs.biochem.5b00284; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li H-J et al. , A structural and energetic model for the slow-onset inhibition of the Mycobacterium tuberculosis enoyl-ACP reductase InhA. ACS Chem. Biol 9, 986–993 (2014). doi: 10.1021/cb400896g; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Klimov DK, Thirumalai D, Dissecting the assembly of Abeta16–22 amyloid peptides into antiparallel β sheets. Structure 11, 295–307 (2003). doi: 10.1016/S0969-2126(03)00031-5; [DOI] [PubMed] [Google Scholar]
- 124.Thirumalai D, Reddy G, Straub JE, Role of water in protein aggregation and amyloid polymorphism. Acc. Chem. Res 45, 83–92 (2012). doi: 10.1021/ar2000869; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Buchanan LE et al. , Mechanism of IAPP amyloid fibril formation involves an intermediate with a transient β-sheet. Proc. Natl. Acad. Sci. U.S.A 110, 19285–19290 (2013).doi: 10.1073/pnas.1314481110; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nasica-Labouze J et al. , Amyloid β Protein and Alzheimer’s Disease: When Computer Simulations Complement Experimental Studies. Chem. Rev 115, 3518–3563 (2015). doi: 10.1021/cr500638n; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang T, Xu W, Mu Y, Derreumaux P, Atomic and dynamic insights into the beneficial effect of the 1,4-naphthoquinon-2-yl-L-tryptophan inhibitor on Alzheimer’s Aβ1–42 dimer in terms of aggregation and toxicity. ACS Chem. Neurosci 5, 148–159 (2014). doi: 10.1021/cn400197x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Avinery R, Kornreich M, Beck R, Universal and Accessible Entropy Estimation Using a Compression Algorithm. Phys. Rev. Lett 123, 178102 (2019). doi: 10.1103/PhysRevLett.123.178102; [DOI] [PubMed] [Google Scholar]
- 129.Noé F, Olsson S, Köhler J, Wu H, Boltzmann generators: Sampling equilibrium states of many-body systems with deep learning. Science 365, eaaw1147 (2019). doi: 10.1126/science.aaw1147; [DOI] [PubMed] [Google Scholar]
- 130.Tuckerman ME, Machine learning transforms how microstates are sampled. Science 365, 982–983 (2019). doi: 10.1126/science.aay2568; [DOI] [PubMed] [Google Scholar]
- 131.Senior AW et al. , Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710 (2020). doi: 10.1038/s41586-019-1923-7; [DOI] [PubMed] [Google Scholar]
- 132.Xu J, Distance-based protein folding powered by deep learning. Proc. Natl. Acad. Sci. U.S.A 116, 16856–16865 (2019). doi: 10.1073/pnas.1821309116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jia X et al. , Anthropogenic biases in chemical reaction data hinder exploratory inorganic synthesis. Nature 573, 251–255 (2019). doi: 10.1038/s41586-019-1540-5; [DOI] [PubMed] [Google Scholar]
- 134.Jarvis LM, Genentech’s R&D chief Michael Varney on the future of drug discovery. C&EN 97 (2019); https://cen.acs.org/biological-chemistry/biotechnology/Genentechs-RD-chief-Michael-Varney/97/i31. [Google Scholar]
- 135.Chan HS, Dill KA, The Protein Folding Problem. Phys. Today 46, 24–32 (1993). doi: 10.1063/1.881371 [DOI] [Google Scholar]
- 136.Perez A, Morrone JA, Simmerling C, Dill KA, Advances in free-energy-based simulations of protein folding and ligand binding. Curr. Opin. Struct. Biol 36, 25–31 (2016).doi: 10.1016/j.sbi.2015.12.002; [DOI] [PMC free article] [PubMed] [Google Scholar]