

Abstract
Background
To date, no genetic analysis of inherited retinal disease (IRD) using whole-exome sequencing (WES) has been conducted in a large-scale Korean cohort. The aim of this study was to characterise the genetic profile of IRD patients in Korea using WES.
Methods
We performed comprehensive molecular testing in 168 unrelated Korean IRD patients using WES. The potential pathogenicity of candidate variants was assessed using the American College of Medical Genetics and Genomics and the Association for Molecular Pathology variant interpretation guidelines, in silico prediction tools, published literature, and compatibility with known phenotypes or inheritance patterns.
Results
Causative variants were detected in 86/168 (51.2%) IRD patients, including 58/107 (54.2%) with retinitis pigmentosa, 7/15 (46.7%) with cone and cone-rod dystrophy, 2/3 (66.6%) with Usher syndrome, 1/2 (50.0%) with congenital stationary night blindness, 2/2 (100.0%) with Leber congenital amaurosis, 1/1 (100.0%) with Bietti crystalline dystrophy, 1/1 (100.0%) with Joubert syndrome, 9/10 (90.0%) with Stargardt macular dystrophy, 1/10 (10.0%) with vitelliform macular dystrophy, 1/11 (9.1%) with other forms of macular dystrophy, and 3/4 (75.0%) with choroideraemia. USH2A, ABCA4, and EYS were the most common causative genes associated with IRD. For retinitis pigmentosa, variants of USH2A and EYS were the most common causative gene mutations.
Conclusions
This study demonstrated the distribution of causative genetic mutations in Korean IRD patients. The data will serve as a reference for future genetic screening and development of treatment modalities for Korean IRD patients.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12920-021-00874-6.
Keywords: Whole-exome sequencing, Inherited retinal degeneration, Retinitis pigmentosa
Background
Inherited retinal degeneration (IRD) is a group of clinically and genetically heterogeneous diseases, including retinitis pigmentosa (RP) and allied diseases, characterised by the progressive loss of photoreceptors and/or retinal pigment epithelial cells due to genetic anomalies related to the phototransduction cascade, retinal transcription factor-related pathway, RNA splicing machinery, retinal metabolism, retinal cell structure, and ciliary structure and function [1, 2]. To date, more than 270 genes have been identified for IRD, as listed in the Retinal Information Network [3].
Presently, a diagnosis of IRD is based mainly on clinical findings, including the primarily involved anatomical location or cell types, disease progression, and involvement of additional organs [1, 4]. However, the variable age of onset, genotypic heterogeneity (one phenotype can be caused by multiple genes), phenotypic heterogeneity (various mutations in a single gene result in numerous phenotypes), incomplete penetrance, unclear inheritance, and progressive nature of IRD, impede a definitive diagnosis [1]. Thus, molecular genetic testing is imperative for a definitive IRD diagnosis.
We previously reported that the hereditary features and mutation profile of Korean patients with IRD differed from those of Chinese and Japanese patients, as well as patients of other ethnicities [5–7]. However, no genetic analysis of IRD using whole-exome sequencing (WES) has been conducted in a large-scale Korean cohort. The aim of this study was to investigate the mutation spectrum and frequency in a large-scale Korean IRD cohort using WES.
Methods
Patients
A cohort of 168 unrelated Korean patients with IRD was recruited from the Department of Ophthalmology, Seoul National University Hospital (SNUH), from October 2008 to January 2019. A detailed family history was obtained from each patient to construct a pedigree. All patients underwent a detailed ophthalmic history and ophthalmic examinations, including best-corrected visual acuity, slit-lamp biomicroscopy, fundus photography, optical coherence tomography, fluorescein angiography, visual field tests, and full-field electroretinograms. Clinical diagnosis was determined by at least two retinal specialists (DJM and UCP).
Comparison of exome capture products
In preparation for higher-throughput exome sequencing using the NextSeq500 (Illumina, San Diego, CA, USA), we evaluated the evenness, depth, duplication rate, and fraction sequenced to ≥ 20 × depth of three exome capture products: SureSelect Human All Exon v6 (Agilent Technologies, Santa Clara, CA, USA) with 150 bp paired-end reads or with 350 bp paired-end reads, and xGen Lockdown panel (Integrated DNA Technologies, Coralville, IA, USA) with 150 bp paired-end reads. The evenness score describes the uniformity of the base coverage over the target regions, and was calculated according to the method described by Mokry et al. [8]. In addition, sensitivity and positive predictive value of variant calling in comparison to a reference genome were calculated. To reduce experimental variables and minimise bias between libraries, all libraries were prepared using the Topomize DNA LT Library Prep Kit (MCLAB, South San Francisco, CA, USA). Agilent captures were hybridised as single sample reactions using 500 ng of library as input. Integrated DNA Technologies captures were hybridised as pools of three samples using 500 ng of library input. All hybridisation and post-hybridisation captures, and washes were performed according to each respective manufacturer’s protocol. The same samples were used for all exon capture methods (three sets of three identical samples).
WES
WES was performed at the Genomics core facility of the Center for BioMarkers (SNUH, South Korea) using an Illumina NextSeq500. For sequencing on the NextSeq500 platform, libraries were generated using the Topomize DNA LT Library Prep Kit and the hybridisation capture of DNA libraries was performed with xGen Lockdown panels to generate 150 bp paired-end reads. Sequenced reads were aligned to UCSC hg19 human reference genome downloaded from the GATK website (https://gatk.broadinstitute.org). Alignment of the sequence reads, indexing of the reference genome, variant calling, and annotation were performed with a pipeline based on Burrows-Wheeler Alignment [9] using BaseSpace Onsite (Illumina). Variants were annotated using Alamut-HT and visualised on Alamut Viewer 2.2 (Interactive Biosoftware, Rouen, France).
Bioinformatic analysis
After read mapping, the output alignment file was sorted using the Genome Analysis Tool Kit (GATK) AddOrReplaceReadGroups [10]. Potential polymerase chain reaction duplicates were removed using GATK MarkDuplicates. GATK BaseRecalibrator and ApplyBQSR were further used to recalibrate the base quality scores. After these pre-processing steps, germline variants were called using GATK HaplotypeCaller, while single nucleotide polymorphisms and insertion-deletion polymorphisms were annotated using Annotate Variation software [11]. After variant annotation, low-quality variants that had a read depth < 10, variant allele fraction < 10%, or variant read count < 2 were removed.
Variants were prioritised based on their presence amongst the 271 genes associated with IRD in RetNet [3]. Variants were classified according to the recent recommendations of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) using InterVar, as follows: pathogenic, likely pathogenic, uncertain significance, likely benign, or benign variant [12–14].
Variants were considered pathogenic when one of the following criteria was met: (1) mutation was previously described as disease-causing in the Human Gene Mutation Database or published literature [15]; (2) mutation was classified as pathogenic or likely pathogenic according to the ACMG/AMP guidelines; (3) mutation was predicted as likely damaging, deleterious, disease-causing, and medium or high impact by more than half of the six pathogenicity prediction tools, including SIFT [16], PolyPhen‐2 HDIV and PolyPhen‐2 Hvar [17], MutationTaster [18], MutationAssesor [19], and FATHMM [20]; or (4) a resultant protein truncation mutation, such as a nonsense or frameshift (insertion or deletion) mutation, was identified. All variants not compatible with known phenotypes or inheritance patterns were excluded.
Patients were determined to have causative variant(s) when (1) one pathogenic variant was present in a gene with autosomal dominant (AD) or X-linked (XL) inheritance; (2) two heterozygous variants or one homozygous pathogenic variant was present in a gene with autosomal recessive (AR) inheritance. Patients were determined to have possible causative variant(s) when one heterozygous pathogenic variant was present in a gene with AR inheritance. Patients were determined to have no causative variants when no (possible) pathogenic variant was detected.
Variant validation
Additional Sanger sequencing was performed for all pathogenic variants with a coverage < 20 reads. Only 5/177 variants (2.8%) have coverage < 20 reads, all of which were successfully sequenced. The concordance rate was 100.0% (5/5).
Statistical analysis
To compare the performance of the exon capture products, we applied the Mann–Whitney U-test. All statistical analyses were performed using SPSS software for Windows, version 22.0 (IBM, Armonk, NY, USA). A value of P < 0.05 was regarded as statistically significant.
Results
Patients
We analysed samples from 168 unrelated Korean patients with IRD (77 females, 91 males). The mean patient age was 42.8 ± 15.9 years (range: 10.4–85.4; median: 37.1). Among these patients, 133 (79.2%) had photoreceptor disease, 33 (19.6%) had macular disease, and 2 (1.2%) had choroideraemia (Table 1). RP was the most common form of IRD (60.7%).
Table 1.
Clinical details of 168 Korean patients with inherited retinal degeneration
| Clinical details | n (%) | ||
|---|---|---|---|
| Clinical diagnosis | Photoreceptor disease | Retinitis pigmentosa | 102 (60.4%) |
| Cone and cone-rod dystrophy | 22 (13.1%) | ||
| Usher syndrome | 3 (1.8%) | ||
| Congenital stationary night blindness | 2 (1.2%) | ||
| Leber congenital amaurosis | 2 (1.2%) | ||
| Bardet–Biedl syndrome | 1 (0.6%) | ||
| Joubert syndrome | 1 (0.6%) | ||
| Macular disease | Stargardt macular dystrophy | 13 (7.7%) | |
| Vitelliform macular dystrophy | 11 (6.5%) | ||
| North Carolina macular dystrophy | 1 (0.6%) | ||
| Other macular dystrophy | 8 (4.8%) | ||
| Choroideraemia | 2 (1.2%) | ||
| Inheritance pattern | Autosomal recessive | 67 (39.9%) | |
| Autosomal dominant | 35 (20.8%) | ||
| X-linked | 6 (3.6%) | ||
| Simplex | 60 (35.7%) |
Based on their family pedigrees, 67 (39.9%) of the patients were presumed to have IRD with AR inheritance, 35 (20.8%) with AD inheritance, and 6 (3.6%) with XL inheritance. Sixty (35.7%) patients were the only affected individuals in their family (simplex cases).
Comparison of exome capture products
The evenness score and sensitivity were significantly higher for the SureSelect Human All Exon v6 with 350 bp paired-end reads than for the other capture products (Table 2). The SureSelect Human All Exon v6 with 150 bp paired-end reads and xGen Lockdown panel produced statistically equivalent results. The depth and duplication rate were highest for the SureSelect Human All Exon v6 with 150 bp paired-end reads, then for the xGen Lockdown panel, and lowest for the SureSelect Human All Exon v6 with 350 bp paired-end reads. The positive predictive value was highest for the xGen Lockdown panel, then for the SureSelect Human All Exon v6 with 150 bp paired-end reads, and lowest for the SureSelect Human All Exon v6 with 350 bp paired-end reads.
Table 2.
Comparison of exome capture products
| SureSelect Human All Exon v6 | xGen Lockdown panel (n = 9a) | P valueb | P valuec | P valued | ||
|---|---|---|---|---|---|---|
| 150-bp paired-end reads (n = 9a) | 350-bp paired-end reads (n = 9a) | |||||
| Mean ± SD | Mean ± SD | Mean ± SD | ||||
| Evenness (%) | 75.3 ± 5.7 | 79.9 ± 0.4 | 76.6 ± 0.1 | < 0.001 | 0.258 | < 0.001 |
| Depth (x) | 61.3 ± 3.8 | 47.6 ± 0.5 | 56.3 ± 1.1 | < 0.001 | < 0.001 | < 0.001 |
| Duplication rate (%) | 33.4 ± 2.4 | 17.5 ± 0.2 | 22.9 ± 0.4 | < 0.001 | < 0.001 | < 0.001 |
| ≥ 20 × depth fraction (%) | 87.9 ± 7.4 | 90.8 ± 0.6 | 91.3 ± 0.2 | 0.258 | 0.258 | 0.258 |
| Sensitivity (%) | 94.9 ± 4.0 | 97.8 ± 0.1 | 96.5 ± 0.2 | < 0.001 | 0.258 | < 0.001 |
| Positive predictive value (%) | 74.9 ± 0.7 | 73.8 ± 0.2 | 76.1 ± 0.2 | < 0.001 | < 0.001 | < 0.001 |
SD standard deviation
aThree sets of three identical samples
bP value comparing SureSelect Human All Exon v6 150-bp paired-end reads with 350-bp paired-end reads
cP value comparing SureSelect Human All Exon v6 150-bp paired-end reads with xGen Lockdown panel
dP value comparing SureSelect Human All Exon v6 350-bp paired-end reads with xGen Lockdown panel
The fraction of nucleotides covered to a depth ≥ 20 × did not differ among the exome capture products. In summary, the xGen Lockdown panel demonstrated noninferiority to the SureSelect Human All Exon v6, and further exome capture for WES was performed using the xGen Lockdown panel.
Identification of pathogenic variants
We identified disease-causing variants in 86 (51.2%) patients (Additional file 1: Table S1). In all, 147 mutant alleles were identified in 35 known IRD-related genes. Among these mutant alleles, 96 (65.3%) had a previously reported association with IRD, and 51 (34.7%) were novel variants in defined IRD genes. Amongst the identified disease-associated variants, 89 (60.5%) were missense variants, 26 (17.7%) were nonsense variants, 25 (17.0%) were frameshift deletions or insertions, and 7 (4.8%) were predicted to affect splicing. USH2A was the most common causative gene amongst the known disease-associated genes (15.1% of cases), followed by ABCA4 (14.0%) (Fig. 1).
Fig. 1.

USH2A and ABCA4 mutations were the most common causative variants among inherited retinal degeneration patients. All data are from the present study
Molecular diagnosis redefined inheritance pattern and clinical diagnosis
All patients who received a definite molecular diagnosis had their clinical diagnosis and inheritance pattern redefined based on the genetic findings (Additional file 1: Table S1). Figure 2 shows a comparison of the mutation detection rates for various IRDs after reclassification. The following are two representative cases.
Fig. 2.
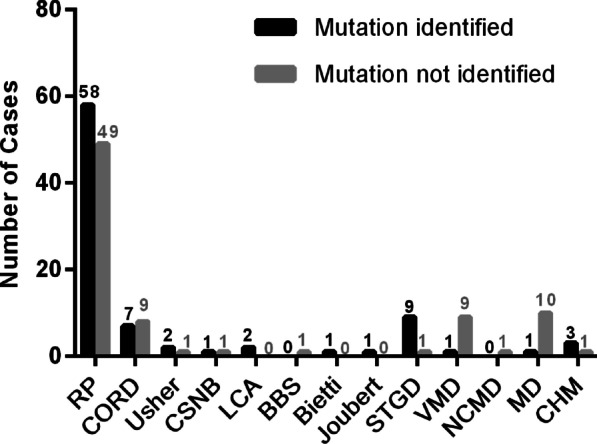
The number of detected pathogenic mutations for each disease phenotype. BBS Bardet–Biedl syndrome, Bietti Bietti crystalline dystrophy, CHM choroideraemia, CSNB congenital stationary night blindness, CORD cone and cone-rod dystrophy, Joubert Joubert syndrome, LCA Leber congenital amaurosis, MD other macular dystrophy, NCMD North Carolina macular dystrophy, RP retinitis pigmentosa, STGD Stargardt macular dystrophy, Usher Usher syndrome, VMD vitelliform macular dystrophy
Case 55, who had been clinically diagnosed with RP, had macular-sparing photoreceptor degeneration with diffuse bone spicule pigmentation and degeneration of the retinal pigment epithelium and choriocapillaris (Fig. 3). WES identified causative variants in the CHM gene, and the clinical diagnosis for case 55 was redefined as choroideraemia. Case 147 was clinically diagnosed with RP with generalised photoreceptor, retinal pigment epithelium, and choriocapillaris degeneration with diffuse bone spicule pigmentation (Fig. 4). WES identified causative variants in the CYP4V2 gene, and the clinical diagnosis for case 147 was redefined as Bietti crystalline dystrophy. Fundus photographs from 9 years prior to sequencing showed many shiny yellow deposits in the posterior pole, which are consistent with Bietti crystalline dystrophy.
Fig. 3.

Clinical phenotypes of case 55, who carries a hemizygous mutation in the CHM gene. a, b Colour fundus photograph. c, d Optical coherence tomography images. e, f Vision field diagram. g Electroretinography recording
Fig. 4.

Clinical phenotypes of case 147, who carries compound heterozygous mutations in the CYP4V2 gene. a, b Colour fundus photographs taken at the time of genetic analysis. c, d Colour fundus photographs from 9 years prior to the study
Fifty-five patients (32.7%) had their inheritance patterns redefined based on the genetic findings. Of the 37 simplex cases, 35 patients (94.6%) were reclassified as having AR inheritance, while AD inheritance and XL inheritance were diagnosed in one patient each (2.7%).
Non-syndromic RP
After refinement of clinical diagnosis, 107 patients received molecular diagnosis of RP, comprising 63.7% of the total cohort. We identified causative variants for 58 of the 107 (54.2%) patients, including 96 variants in 26 genes (Additional file 1: Table S1). Variants in USH2A were the most common, which were carried by 13 patients (22.4%), followed by EYS variants in six patients (10.3%; Fig. 5).
Fig. 5.

Percentages of causative genes identified in 58 retinitis pigmentosa patients. Mutations in EYS and USH2A were the most common in the population (a). The proportion of causative genes based on inheritance pattern is shown in b. All data are from the present study
RHO (3, 25.0%) was the most commonly identified mutated gene in 12 RP patients with AD inheritance (adRP; 20.7%), USHA2A (13, 31.0%) and EYS (6, 14.3%) were the most commonly identified mutated genes in 42 RP patients with AR inheritance (arRP; 72.4%), and RPGR (2, 50%) was the most commonly identified mutated gene in four RP patients with XL inheritance (xlRP, 6.9%).
Cone and cone-rod dystrophy
Amongst the 15 cone and cone-rod dystrophy (CORD) patient cases, we identified causative variants in seven patients (46.7%) (Additional file 1: Table S1). CRX (2, 28.6%) was the most commonly identified mutated gene.
Stargardt macular dystrophy
Of the 13 patients with an initial clinical diagnosis of Stargardt macular dystrophy (STGD), three patients were reclassified as having CORD or other macular dystrophy based on the genetic findings. WES revealed causative variants for 9 out of the 10 remaining STGD patients and a possible causative variant for the tenth patient. All identified causative variants were of the ABCA4 gene (Additional file 1: Table S1).
Possible pathogenic variants were identified
A further 17.9% (30/168) of the patients received a possible molecular diagnosis based on identification of a single pathogenic variant in a gene known to be associated with AR IRD that was compatible with known phenotypes or inheritance patterns (Additional file 2: Table S2). Overall, 69.0% (116/168) of study participants received a definite or possible molecular diagnosis after WES testing.
Discussion
In this study, we employed a single genetic test, WES, for the genetic diagnosis of 168 unrelated Korean patients with different types of IRD. Using this single test, we molecularly diagnosed 86 (51.2%) patients. To the best of our knowledge, this is the first study to evaluate clinical diagnostic accuracy and causative genes in Korean patients with various IRD using a WES approach.
Targeted sequence capture is used to isolate and enrich specific genomic regions prior to massively parallel sequencing. Although the cost of WES is gradually decreasing, it remains expensive for clinical use. Herein, we used pre-capture pooling with targeted sequence capture to reduce the reagent cost and hands-on time. As pre-capture pooling using xGen Lockdown panel was not inferior to routine post-capture pooling using SureSelect Human All Exon v6, we successfully utilised the xGen panel for WES in Korean IRD patients. We identified 147 causative variants in 35 known IRD-related genes, including 51 (34.7%) novel variants. However, most of these novel variants were potentially causative which were annotated by computational tools. Functional validation would be required to derive a definite causality of these potentially causative variant.
Several studies have reported that the hereditary features and causative genes of IRD vary among ethnicities, even in geopolitically close Asian countries [5–7, 29, 71–73]. However, only a few reports have been published on the mutation spectrum of a large-scale Korean cohort with IRD. Our previous study using targeted exome sequencing (TES) of 53 RP-related genes in 62 Korean patients with non-syndromic RP revealed causal variants in 50.0% of the patients [7]. PRPF31 mutations (17.6%) were the most frequently found causative variants, followed by mutations in EYS, PDE6B, RHO, RP1, and RP2 (11.8% each). A recent study reported the TES results for 204 IRD-related genes in 86 Korean patients with IRD [74], as well as the molecular diagnoses rate for 44.2% of the patients. In RP, EYS mutations (22.2%) were the most frequent causative variants, followed by mutations in PED6B (16.7%), PED6A (11.1%), and USH2A (11.1%). In the present study, USH2A mutations (22.4%) were the most common causative variants in RP, followed by variants in EYS (10.3%), RP1 (6.9%), and ABCA4, PED6B, and RHO (5.2% each) mutations. The differences in the mutation spectra amongst the Korean studies appears to largely originate from selection of the sample population.
Moreover, the sample sizes of the two previous studies were much smaller than that of the present study making them more vulnerable to sampling error. In addition, the inclusion of many subjects from a single family can result in increased representation of a specific genetic mutation, thereby skewing the mutational spectrum. In the present study, we only included one proband from each family to maximise the representativeness of the cohort for the total population and to reduce sampling error. In addition, differing inheritance pattern distributions in the sampled populations may result in different distributions of causative mutations. Our previous study included a large proportion of subjects with AD inheritance, which resulted in a higher proportion of mutations in PRPF31, an AD inheritance gene [7]. In the present study, the distribution of the inheritance pattern for RP resembles that in a previous report on a large cohort of Korean RP patients [6].
The mutation spectrum of the patients in the present study resembles those established in recent large-scale studies in other Asian countries [21, 75]. For instance, the USH2A mutation was the most common in the present study, as in the Chinese study, and it was the second most common in the Japanese study. The second most common mutation was in the EYS gene, which was the most common in the Japanese study and the third most common in the Chinese study. These findings were similar to those of previous studies demonstrating ethnic differences between Asian and Caucasian populations, with a higher incidence of EYS mutations in arRP patients of an Asian background than Caucasian background [76]. The most common EYS mutation in our cohort was c.4957dupA; it was also common in Japanese RP patients, however, was rare, or not detected in European RP patients and Chinese RP patients [66, 77]. Meanwhile, the c.C8805A and c.C7394G EYS mutations, which were frequently observed in the Japanese RP cohort, were not detected in Korean studies [7, 74, 78]. This suggests that differences in the mutational spectra of IRD patients exist among East Asian countries, despite their geographical proximity, and emphasises the importance of obtaining reference data for individual nations or regions to determine the local genomic IRD landscape.
The prevalence of the RP1 mutation was 6.9% in arRP cases in this study, which was higher than those among Japanese and Chinese arRP patients (1.7–2%) [29, 73]. Mutations in the RP1 gene cause both AR and AD forms of RP, accounting for 5.5% of adRP and less than 1% of arRP [76]. Different explanations for the dominant/recessive mutation effect of the RP1 gene have been proposed, however, the precise mechanism remains unclear. In addition, a recent study suggested that the phenotypic spectrum associated with RP1 mutations should be expanded to CORD and macular dystrophy [79, 80]. In our cohort, Case 103, who showed well-demarcated macular atrophy with normal electroretinography findings (see Additional file 3) carried compound heterozygous nonsense (c.C5797T) and frameshift (c.649delG) mutations, and received a molecular diagnosis of RP1-associated AR macular dystrophy.
WES has clear advantages over TES for the molecular diagnosis of IRD. The heterogeneity of the genotype and phenotype, as well as the unclear inheritance patterns of IRD, make it difficult to select target genes for TES. In addition, more than 270 causative genes have been identified for IRD to date, and new causative genes continue to be discovered. To keep pace with the literature, researchers must redesign panels to incorporate new genomic regions at additional expense. In contrast, WES provides the advantage of re-evaluating previously analysed datasets when a novel gene associated with IRD is reported. In a study comparing WES and three commercial gene panels, WES discovered causative gene mutations in 42% of cases, which were not included in at least one commercial panel [81]. In the present study, six genes (CYP4V2, NMNAT1, RP1L1, CACNA1F, BBS2, and REEP6) out of 35 causative genes detected in our cohort were not included in at least one of the TES studies in Korean IRD patients [7, 74].
Among patients with macular disease, patients with STGD had the highest detection rate for causative variants (90.0%), however, patients with other forms had a poorer detection rate (9.5%). This is likely due to the presence of pathognomonic findings for the accurate clinical diagnosis of STGD. Initially, 13 patients with clinical diagnosis of STGD were included in this study. Three patients who were redefined as having another disease based on their molecular diagnosis did not have dark choroid rings on fluorescein angiography. In contrast, the remaining ten patients had dark choroid signs and a (probable) molecular diagnosis of STGD. This pathognomonic finding, the dark choroid sign, can differentiate STGD from other similar conditions including non-hereditary diseases [82]. Meanwhile, all vitelliform macular dystrophy cases in our cohort were clinically diagnosed as adult-onset vitelliform macular dystrophy with clinical findings of submacular vitelliform material and normal electrooculogram. The mean age of the vitelliform macular dystrophy patients was 64.5 years. These clinical features are similar to those of exudative age-related macular degeneration, choroidal neovascularisation, or central serous chorioretinopathy, which do not belong to the category of IRD. There is a possibility that some of our vitelliform macular dystrophy cases may arise from non-genetic conditions. This finding suggests that accurate clinical diagnosis can increase the efficiency of molecular diagnosis.
The 17.9% (30/168) of patients who received a possible molecular diagnosis may carry a second pathogenic variant in the same gene, which occurs in no- or low-coverage genetic regions [73]. In the exome sequencing approach, some genetic regions have low or no coverage. For example, repetitive regions, GC-rich regions, and regions with high homology are difficult to enrich. In addition, small copy number variations and deep-intronic regions cannot be detected via exome sequencing [83, 84]. In these patients, further analysis, such as direct sequence analysis or whole genome sequencing, is required.
In this study, we were unable to identify any causative variants in 31.0% (52/168) of the cases. Several explanations may account for these. The first explanation is the limitation of WES. Approximately 85% of known causative mutations occur in exonic regions that encode proteins, indicating that WES is unable to discover the cause of the remaining ~ 15% of causative mutations. In addition, some genetic regions have low or no coverage in the exome sequencing approach, as described above. The second explanation is potentially inaccurate clinical diagnosis. It is possible that some of our cohort have non-hereditary retinal conditions that phenotypically resemble IRD. Therefore, further study via direct sequence analysis or whole-genome sequencing with detailed clinical diagnosis is required to achieve an optimal detection rate.
Conclusion
The present study screened the largest sample of Korean IRD patients to date and described the genetic characteristics of the cohort. Our data will serve as a basis for genetic counselling of Korean IRD patients and lay the groundwork for the upcoming era of gene therapy.
Supplementary Information
Additional file 1. Table S1: Causative variants identified in 86 of 168 Korean inherited retinal degeneration probands.
Additional file 2. Table S2: Possible causative variants in 30 of 168 Korean inherited retinal degeneration probands.
Additional file 3. Figure S1: Phenotypes of case 103 who carries compound heterozygous mutations in the RP1 gene.
Acknowledgements
The biospecimens and data used in this study were provided by the Biobank of Seoul National University Hospital, a member of Korea Biobank Network, which is supported by the Ministry of Health and Welfare.
Abbreviations
- IRD
Inherited retinal disease
- WES
Whole-exome sequencing
- RP
Retinitis pigmentosa
- SNUH
Seoul National University Hospital
- GATK
Genome Analysis Tool Kit
- ACMG/AMP
American College of Medical Genetics and Genomics and the Association for Molecular Pathology
- AR
Autosomal recessive
- AD
Autosomal dominant
- XL
X-linked
- CORD
Cone and cone-rod dystrophy
- Usher
Usher syndrome
- LCA
Leber congenital amaurosis
- BBS
Bardet–Biedl syndrome
- Bietti
Bietti crystalline dystrophy
- CSNB
Congenital stationary night blindness
- Joubert
Joubert syndrome
- VMD
Vitelliform macular dystrophy
- STGD
Stargardt macular dystrophy
- NCMD
North Carolina macular dystrophy
- MD
Other macular dystrophy
- CHM
Choroideraemia
- adRP
RP with AD inheritance
- arRP
RP with AR inheritance
- xlRP
RP with XL inheritance
- TES
Targeted exome sequencing
Authors’ contributions
DJM participated in study coordination, bioinformatics analysis and clinical assessment and drafted the manuscript. HSL participated in WES and Sanger sequencing. KK, SC, and IJ participated in bioinformatics analysis of WES data and wrote the manuscript. SHC, CKY, and EKL participated in clinical analysis and helped to draft the manuscript. HGY conceived the study and participated in its design and coordination, as well as helping to draft the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the Korean Association of Retinal Degeneration, by a Grant Number 2620170060 from the SNUH Research Fund, and by a grant of the Korea Research-Driven Hospital (Grant Number: HI14C1277) through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare (MHW), Republic of Korea. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the NCBI Sequence Read Archive (SRA) repository, Accession Number PRJNA690657.
Declarations
Ethics approval and consent to participate
This study adhered to the tenets of the Declaration of Helsinki. All experiments and study protocols were approved by the Institutional Review Board (IRB) of Seoul National University Hospital (SNUH). Written informed consent was obtained from all participants, or from their guardians on behalf of minors or child participants. The biobanking protocols, including the deposition and the collection procedures, for the samples were approved by the IRB of SNUH. All participants, parents or legal guardians provided a written informed biorepository consent, including the consent for future genomics research.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Dias MF, Joo K, Kemp JA, Fialho SL, da Silva Cunha A, Jr, Woo SJ, et al. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog Retin Eye Res. 2018;63:107–131. doi: 10.1016/j.preteyeres.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 2.Hafler BP. Clinical progress in inherited retinal degenerations: gene therapy clinical trials and advances in genetic sequencing. Retina. 2017;37:417–423. doi: 10.1097/IAE.0000000000001341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Retinal Information Network. http://www.sph.uth.tmc.edu/retnet/. Accessed 14 Feb 2020.
- 4.Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29:335–375. doi: 10.1016/j.preteyeres.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Kim KJ, Kim C, Bok J, Kim KS, Lee EJ, Park SP, et al. Spectrum of rhodopsin mutations in Korean patients with retinitis pigmentosa. Mol Vis. 2011;17:844–853. [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SH, Yu HG, Seo JM, Moon SW, Moon JW, Kim SJ, et al. Hereditary and clinical features of retinitis pigmentosa in Koreans. J Korean Med Sci. 2010;25:918–923. doi: 10.3346/jkms.2010.25.6.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoon CK, Kim NK, Joung JG, Shin JY, Park JH, Eum HH, et al. The diagnostic application of targeted re-sequencing in Korean patients with retinitis pigmentosa. BMC Genom. 2015;16:515. doi: 10.1186/s12864-015-1723-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mokry M, Feitsma H, Nijman IJ, de Bruijn E, van der Zaag PJ, Guryev V, et al. Accurate SNP and mutation detection by targeted custom microarray-based genomic enrichment of short-fragment sequencing libraries. Nucleic Acids Res. 2010;38:e116. doi: 10.1093/nar/gkq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. Am J Hum Genet. 2016;98:1067–1076. doi: 10.1016/j.ajhg.2016.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017;100:267–280. doi: 10.1016/j.ajhg.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stenson PD, Ball EV, Mort M, Phillips AD, Shaw K, Cooper DN. The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr Protoc Bioinform. 2012;Chapter 1:Unit1.13. doi: 10.1002/0471250953.bi0113s39. [DOI] [PubMed] [Google Scholar]
- 16.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 17.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 19.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koyanagi Y, Akiyama M, Nishiguchi KM, Momozawa Y, Kamatani Y, Takata S, et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. J Med Genet. 2019;56:662–670. doi: 10.1136/jmedgenet-2018-105691. [DOI] [PubMed] [Google Scholar]
- 22.Payne AM, Morris AG, Downes SM, Johnson S, Bird AC, Moore AT, et al. Clustering and frequency of mutations in the retinal guanylate cyclase (GUCY2D) gene in patients with dominant cone-rod dystrophies. J Med Genet. 2001;38:611–614. doi: 10.1136/jmg.38.9.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sung Y, Choi SW, Shim SH, Song WK. Clinical and genetic characteristics analysis of Korean patients with Stargardt disease using targeted exome sequencing. Ophthalmologica. 2019;241:38–48. doi: 10.1159/000490073. [DOI] [PubMed] [Google Scholar]
- 24.Jiang F, Pan Z, Xu K, Tian L, Xie Y, Zhang X, et al. Screening of ABCA4 gene in a Chinese cohort with Stargardt disease or cone-rod dystrophy with a report on 85 novel mutations. Investig Ophthalmol Vis Sci. 2016;57:145–152. doi: 10.1167/iovs.15-18190. [DOI] [PubMed] [Google Scholar]
- 25.Fujinami K, Zernant J, Chana RK, Wright GA, Tsunoda K, Ozawa Y, et al. ABCA4 gene screening by next-generation sequencing in a British cohort. Investig Ophthalmol Vis Sci. 2013;54:6662–6674. doi: 10.1167/iovs.13-12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zernant J, Collison FT, Lee W, Fishman GA, Noupuu K, Yuan B, et al. Genetic and clinical analysis of ABCA4-associated disease in African American patients. Hum Mutat. 2014;35:1187–1194. doi: 10.1002/humu.22626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joo K, Seong MW, Park KH, Park SS, Woo SJ. Genotypic profile and phenotype correlations of ABCA4-associated retinopathy in Koreans. Mol Vis. 2019;25:679–690. [PMC free article] [PubMed] [Google Scholar]
- 28.Gao FJ, Qi YH, Hu FY, Wang DD, Xu P, Guo JL, et al. Mutation spectrum of the bestrophin-1 gene in a large Chinese cohort with bestrophinopathy. Br J Ophthalmol. 2020;104:846–851. doi: 10.1136/bjophthalmol-2019-314679. [DOI] [PubMed] [Google Scholar]
- 29.Oishi M, Oishi A, Gotoh N, Ogino K, Higasa K, Iida K, et al. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Investig Ophthalmol Vis Sci. 2014;55:7369–7375. doi: 10.1167/iovs.14-15458. [DOI] [PubMed] [Google Scholar]
- 30.Katagiri S, Akahori M, Sergeev Y, Yoshitake K, Ikeo K, Furuno M, et al. Whole exome analysis identifies frequent CNGA1 mutations in Japanese population with autosomal recessive retinitis pigmentosa. PLoS ONE. 2014;9:e108721. doi: 10.1371/journal.pone.0108721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li S, Yang M, Liu W, Liu Y, Zhang L, Yang Y, et al. Targeted next-generation sequencing reveals novel RP1 mutations in autosomal recessive retinitis pigmentosa. Genet Test Mol Biomark. 2018;22:109–114. doi: 10.1089/gtmb.2017.0223. [DOI] [PubMed] [Google Scholar]
- 32.Liu T, Jin X, Zhang X, Yuan H, Cheng J, Lee J, et al. A novel missense SNRNP200 mutation associated with autosomal dominant retinitis pigmentosa in a Chinese family. PLoS ONE. 2012;7:e45464. doi: 10.1371/journal.pone.0045464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshimura H, Miyagawa M, Kumakawa K, Nishio SY, Usami S. Frequency of Usher syndrome type 1 in deaf children by massively parallel DNA sequencing. J Hum Genet. 2016;61:419–422. doi: 10.1038/jhg.2015.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonnet C, Riahi Z, Chantot-Bastaraud S, Smagghe L, Letexier M, Marcaillou C, et al. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur J Hum Genet EJHG. 2016;24:1730–1738. doi: 10.1038/ejhg.2016.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Esposito G, De Falco F, Tinto N, Testa F, Vitagliano L, Tandurella IC, et al. Comprehensive mutation analysis (20 families) of the choroideremia gene reveals a missense variant that prevents the binding of REP1 with Rab geranylgeranyl transferase. Hum Mutat. 2011;32:1460–1469. doi: 10.1002/humu.21591. [DOI] [PubMed] [Google Scholar]
- 36.Dryja TP, Rucinski DE, Chen SH, Berson EL. Frequency of mutations in the gene encoding the α subunit of rod cGMP-phosphodiesterase in autosomal recessive retinitis pigmentosa. Investig Ophthalmol Vis Sci. 1999;40:1859–1865. [PubMed] [Google Scholar]
- 37.Dockery A, Stephenson K, Keegan D, Wynne N, Silvestri G, Humphries P, et al. Target 5000: target capture sequencing for inherited retinal degenerations. Genes (Basel) 2017;8:E304. doi: 10.3390/genes8110304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis RA, Shroyer NF, Singh N, Allikmets R, Hutchinson A, Li Y, et al. Genotype/phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am J Hum Genet. 1999;64:422–434. doi: 10.1086/302251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poloschek CM, Bach M, Lagrèze WA, Glaus E, Lemke JR, Berger W, et al. ABCA4 and ROM1: implications for modification of the PRPH2-associated macular dystrophy phenotype. Investig Ophthalmol Vis Sci. 2010;51:4253–4265. doi: 10.1167/iovs.09-4655. [DOI] [PubMed] [Google Scholar]
- 40.Jespersgaard C, Fang M, Bertelsen M, Dang X, Jensen H, Chen Y, et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci Rep. 2019;9:1219. doi: 10.1038/s41598-018-38007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foote KG, De la Huerta I, Gustafson K, Baldwin A, Zayit-Soudry S, Rinella N, et al. Cone spacing correlates with retinal thickness and microperimetry in patients with inherited retinal degenerations. Investig Ophthalmol Vis Sci. 2019;60:1234–1243. doi: 10.1167/iovs.18-25688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hosono K, Nishina S, Yokoi T, Katagiri S, Saitsu H, Kurata K, et al. Molecular diagnosis of 34 Japanese families with leber congenital amaurosis using targeted next generation sequencing. Sci Rep. 2018;8:8279. doi: 10.1038/s41598-018-26524-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thompson DA, Janecke AR, Lange J, Feathers KL, Hübner CA, McHenry CL, et al. Retinal degeneration associated with RDH12 mutations results from decreased 11-cis retinal synthesis due to disruption of the visual cycle. Hum Mol Genet. 2005;14:3865–3875. doi: 10.1093/hmg/ddi411. [DOI] [PubMed] [Google Scholar]
- 44.Maeda A, Yoshida A, Kawai K, Arai Y, Akiba R, Inaba A, et al. Development of a molecular diagnostic test for retinitis pigmentosa in the Japanese population. Jpn J Ophthalmol. 2018;62:451–457. doi: 10.1007/s10384-018-0601-x. [DOI] [PubMed] [Google Scholar]
- 45.Chafai-Elalaoui S, Chalon M, Elkhartoufi N, Kriouele Y, Mansouri M, Attie-Bitach T, et al. A homozygous AHI1 gene mutation (p.Thr304AsnfsX6) in a consanguineous Moroccan family with Joubert syndrome: a case report. J Med Case Rep. 2015;9:254. doi: 10.1186/s13256-015-0732-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen TT, Hull S, Roepman R, van den Born LI, Oud MM, de Vrieze E, et al. Missense mutations in the WD40 domain of AHI1 cause non-syndromic retinitis pigmentosa. J Med Genet. 2017;54:624–632. doi: 10.1136/jmedgenet-2016-104200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dreyer B, Brox V, Tranebjaerg L, Rosenberg T, Sadeghi AM, Möller C, et al. Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum Mutat. 2008;29:451. doi: 10.1002/humu.9524. [DOI] [PubMed] [Google Scholar]
- 48.Kanda A, Friedman JS, Nishiguchi KM, Swaroop A. Retinopathy mutations in the bZIP protein NRL alter phosphorylation and transcriptional activity. Hum Mutat. 2007;28:589–598. doi: 10.1002/humu.20488. [DOI] [PubMed] [Google Scholar]
- 49.Huang L, Zhang Q, Huang X, Qu C, Ma S, Mao Y, et al. Mutation screening in genes known to be responsible for retinitis pigmentosa in 98 Small Han Chinese families. Sci Rep. 2017;7:1948. doi: 10.1038/s41598-017-00963-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sankila EM, Joensuu TH, Hämäläinen RH, Raitanen N, Valle O, Ignatius J, et al. A CRX mutation in a Finnish family with dominant cone-rod retinal dystrophy. Hum Mutat. 2000;16:94. doi: 10.1002/1098-1004(200007)16:1<94::AID-HUMU25>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 51.Oishi M, Oishi A, Gotoh N, Ogino K, Higasa K, Iida K, et al. Next-generation sequencing-based comprehensive molecular analysis of 43 Japanese patients with cone and cone-rod dystrophies. Mol Vis. 2016;22:150–160. [PMC free article] [PubMed] [Google Scholar]
- 52.Alapati A, Goetz K, Suk J, Navani M, Al-Tarouti A, Jayasundera T, et al. Molecular diagnostic testing by eyeGENE: analysis of patients with hereditary retinal dystrophy phenotypes involving central vision loss. Investig Ophthalmol Vis Sci. 2014;55:5510–5521. doi: 10.1167/iovs.14-14359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao FJ, Wang DD, Chen F, Sun HX, Hu FY, Xu P, et al. Prevalence and genetic-phenotypic characteristics of patients with USH2A mutations in a large cohort of Chinese patients with inherited retinal disease. Br J Ophthalmol. 2021;105:87–92. doi: 10.1136/bjophthalmol-2020-315878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bareil C, Delague V, Arnaud B, Demaille J, Hamel C, Claustres M. W179R: a novel missense mutation in the peripherin/RDS gene in a family with autosomal dominant retinitis pigmentosa. Hum Mutat. 2000;15:583–584. doi: 10.1002/1098-1004(200006)15:6<583::AID-HUMU24>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 55.Xu W, Dai H, Lu T, Zhang X, Dong B, Li Y. Seven novel mutations in the long isoform of the USH2A gene in Chinese families with nonsyndromic retinitis pigmentosa and Usher syndrome Type II. Mol Vis. 2011;17:1537–1552. [PMC free article] [PubMed] [Google Scholar]
- 56.Nakanishi H, Ohtsubo M, Iwasaki S, Hotta Y, Mizuta K, Mineta H, et al. Identification of 11 novel mutations in USH2A among Japanese patients with Usher syndrome type 2. Clin Genet. 2009;76:383–391. doi: 10.1111/j.1399-0004.2009.01257.x. [DOI] [PubMed] [Google Scholar]
- 57.Nakano M, Kelly EJ, Wiek C, Hanenberg H, Rettie AE. CYP4V2 in Bietti's crystalline dystrophy: ocular localization, metabolism of ω-3-polyunsaturated fatty acids, and functional deficit of the p.H331P variant. Mol Pharmacol. 2012;82:679–686. doi: 10.1124/mol.112.080085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jin X, Chen L, Wang D, Zhang Y, Chen Z, Huang H. Novel compound heterozygous mutation in the POC1B gene underlie peripheral cone dystrophy in a Chinese family. Ophthalmic Genet. 2018;39:300–306. doi: 10.1080/13816810.2018.1430239. [DOI] [PubMed] [Google Scholar]
- 59.Hosono K, Ishigami C, Takahashi M, Park DH, Hirami Y, Nakanishi H, et al. Two novel mutations in the EYS gene are possible major causes of autosomal recessive retinitis pigmentosa in the Japanese population. PLoS ONE. 2012;7:e31036. doi: 10.1371/journal.pone.0031036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu Y, Guan L, Xiao X, Zhang J, Li S, Jiang H, et al. Mutation analysis in 129 genes associated with other forms of retinal dystrophy in 157 families with retinitis pigmentosa based on exome sequencing. Mol Vis. 2015;21:477–486. [PMC free article] [PubMed] [Google Scholar]
- 61.Wang F, Wang H, Tuan HF, Nguyen DH, Sun V, Keser V, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. 2014;133:331–345. doi: 10.1007/s00439-013-1381-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin Y, Xu CL, Velez G, Yang J, Tanaka AJ, Breazzano MP, et al. Novel REEP6 gene mutation associated with autosomal recessive retinitis pigmentosa. Doc Ophthalmol. 2020;140:67–75. doi: 10.1007/s10633-019-09719-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fujinami K, Kameya S, Kikuchi S, Ueno S, Kondo M, Hayashi T, et al. Novel RP1L1 variants and genotype-photoreceptor microstructural phenotype associations in cohort of Japanese patients with occult macular dystrophy. Investig Ophthalmol Vis Sci. 2016;57:4837–4846. doi: 10.1167/iovs.16-19670. [DOI] [PubMed] [Google Scholar]
- 64.Jin ZB, Mandai M, Yokota T, Higuchi K, Ohmori K, Ohtsuki F, et al. Identifying pathogenic genetic background of simplex or multiplex retinitis pigmentosa patients: a large scale mutation screening study. J Med Genet. 2008;45:465–472. doi: 10.1136/jmg.2007.056416. [DOI] [PubMed] [Google Scholar]
- 65.Jin ZB, Huang XF, Lv JN, Xiang L, Li DQ, Chen J, et al. SLC7A14 linked to autosomal recessive retinitis pigmentosa. Nat Commun. 2014;5:3517. doi: 10.1038/ncomms4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gu S, Tian Y, Chen X, Zhao C. Targeted next-generation sequencing extends the phenotypic and mutational spectrums for EYS mutations. Mol Vis. 2016;22:646–657. [PMC free article] [PubMed] [Google Scholar]
- 67.Tajiguli A, Xu M, Fu Q, Yiming R, Wang K, Li Y, et al. Next-generation sequencing-based molecular diagnosis of 12 inherited retinal disease probands of Uyghur ethnicity. Sci Rep. 2016;6:21384. doi: 10.1038/srep21384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ge Z, Bowles K, Goetz K, Scholl HP, Wang F, Wang X, et al. NGS-based molecular diagnosis of 105 eyeGENE(®) probands with retinitis pigmentosa. Sci Rep. 2015;5:18287. doi: 10.1038/srep18287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang L, Zhang J, Chen N, Wang L, Zhang F, Ma Z, et al. Application of whole exome and targeted panel sequencing in the clinical molecular diagnosis of 319 Chinese families with inherited retinal dystrophy and comparison study. Genes (Basel) 2018;9:360. doi: 10.3390/genes9070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoshimura H, Iwasaki S, Nishio SY, Kumakawa K, Tono T, Kobayashi Y, et al. Massively parallel DNA sequencing facilitates diagnosis of patients with Usher syndrome type 1. PLoS ONE. 2014;9:e90688. doi: 10.1371/journal.pone.0090688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu Y, Guan L, Shen T, Zhang J, Xiao X, Jiang H, et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum Genet. 2014;133:1255–1271. doi: 10.1007/s00439-014-1460-2. [DOI] [PubMed] [Google Scholar]
- 72.Tiwari A, Bahr A, Bahr L, Fleischhauer J, Zinkernagel MS, Winkler N, et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci Rep. 2016;6:28755. doi: 10.1038/srep28755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haer-Wigman L, van Zelst-Stams WA, Pfundt R, van den Born LI, Klaver CC, Verheij JB, et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur J Hum Genet. 2017;25:591–599. doi: 10.1038/ejhg.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim MS, Joo K, Seong MW, Kim MJ, Park KH, Park SS, et al. Genetic mutation profiles in Korean patients with inherited retinal diseases. J Korean Med Sci. 2019;34:e161. doi: 10.3346/jkms.2019.34.e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao FJ, Li JK, Chen H, Hu FY, Zhang SH, Qi YH, et al. Genetic and clinical findings in a large cohort of Chinese patients with suspected retinitis pigmentosa. Ophthalmology. 2019;126:1549–1556. doi: 10.1016/j.ophtha.2019.04.038. [DOI] [PubMed] [Google Scholar]
- 76.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 77.Oishi M, Oishi A, Yoshimura N. Targeted exome sequencing in Japanese patients with retinitis pigmentosa. In: Prakash G, Iwata T, editors. Advances in vision research. Tokyo: Springer; 2017. pp. 31–39. [Google Scholar]
- 78.Yang L, Fujinami K, Ueno S, Kuniyoshi K, Hayashi T, Kondo M, et al. Genetic spectrum of EYS-associated retinal disease in a large Japanese cohort: identification of disease-associated variants with relatively high allele frequency. Sci Rep. 2020;10:5497. doi: 10.1038/s41598-020-62119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Riera M, Abad-Morales V, Navarro R, Ruiz-Nogales S, Méndez-Vendrell P, Corcostegui B, et al. Expanding the retinal phenotype of RP1: from retinitis pigmentosa to a novel and singular macular dystrophy. Br J Ophthalmol. 2020;104:173–181. doi: 10.1136/bjophthalmol-2018-313672. [DOI] [PubMed] [Google Scholar]
- 80.Verbakel SK, van Huet RAC, den Hollander AI, Geerlings MJ, Kersten E, Klevering BJ, et al. Macular dystrophy and cone-rod dystrophy caused by mutations in the RP1 gene: extending the RP1 disease spectrum. Investig Ophthalmol Vis Sci. 2019;60:1192–1203. doi: 10.1167/iovs.18-26084. [DOI] [PubMed] [Google Scholar]
- 81.Dillon OJ, Lunke S, Stark Z, Yeung A, Thorne N, Melbourne Genomics Health A et al. Exome sequencing has higher diagnostic yield compared to simulated disease-specific panels in children with suspected monogenic disorders. Eur J Hum Genet. 2018;26:644–651. doi: 10.1038/s41431-018-0099-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Uliss AE, Moore AT, Bird AC. The dark choroid in posterior retinal dystrophies. Ophthalmology. 1987;94:1423–1427. doi: 10.1016/S0161-6420(87)33270-1. [DOI] [PubMed] [Google Scholar]
- 83.Jin Z-B. Whole genome sequencing in genetic eye diseases. In: Prakash G, Iwata T, editors. Advances in vision research. Tokyo: Springer; 2017. pp. 21–29. [Google Scholar]
- 84.Sikkema-Raddatz B, Johansson LF, de Boer EN, Almomani R, Boven LG, van den Berg MP, et al. Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum Mutat. 2013;34:1035–1042. doi: 10.1002/humu.22332. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Table S1: Causative variants identified in 86 of 168 Korean inherited retinal degeneration probands.
Additional file 2. Table S2: Possible causative variants in 30 of 168 Korean inherited retinal degeneration probands.
Additional file 3. Figure S1: Phenotypes of case 103 who carries compound heterozygous mutations in the RP1 gene.
Data Availability Statement
The datasets generated and/or analysed during the current study are available in the NCBI Sequence Read Archive (SRA) repository, Accession Number PRJNA690657.