

ABSTRACT
Fc galactosylation is a critical quality attribute for anti-tumor recombinant immunoglobulin G (IgG)-based monoclonal antibody (mAb) therapeutics with complement-dependent cytotoxicity (CDC) as the mechanism of action. Although the correlation between galactosylation and CDC has been known, the underlying structure–function relationship is unclear. Heterogeneity of the Fc N-glycosylation produced by Chinese hamster ovary (CHO) cell culture biomanufacturing process leads to variable CDC potency. Here, we derived a kinetic model of galactose transfer reaction in the Golgi apparatus and used this model to determine the correlation between differently galactosylated species from CHO cell culture process. The model was validated by a retrospective data analysis of more than 800 historical samples from small-scale and large-scale CHO cell cultures. Furthermore, using various analytical technologies, we discovered the molecular basis for Fc glycan terminal galactosylation changing the three-dimensional conformation of the Fc, which facilitates the IgG1 hexamerization, thus enhancing C1q avidity and subsequent complement activation. Our study offers insight into the formation of galactosylated species, as well as a novel three-dimensional understanding of the structure–function relationship of terminal galactose to complement activation in mAb therapeutics.
KEYWORDS: monoclona antibody; IgG1, galactosylation, CDC; CHO Cell Culture, CQA; mass Spectrometry, kinetics
Introduction
Recombinant monoclonal antibodies (mAbs) are the largest and fastest-growing segment of the biologic therapeutic market.1,2 Immunoglobulin G (IgG) is the dominant form of mAb drugs among all five classes of human antibodies. Chinese hamster ovary (CHO) cell fed-batch processes are the most commonly used expression system for full-length antibodies.3 The IgG mAbs manufactured by CHO cell culture undergo N-linked glycosylation in the CH2 domain, typically at N297. Fc glycosylation is a critical quality attribute because it influences the mechanism of action (MOA) involving effector functions, such as complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC),4–6 as well as immunogenicity and pharmacokinetics.7–9 During the CHO cell culture process, the Fc glycans of the IgG are pre-modified in the endoplasmic reticulum and finally formed in the Golgi apparatus through various pathways (Figure 1a). Despite a tremendous number of experimental and modeling efforts on understand N-glycan formation in CHO cell culture process,6,10–14 the heterogeneity and manufacturing inconsistency of N-glycans remain the fundamental challenges for CHO cell culture processes. Terminal galactose is one of the major contributors of IgG glycan variation, such as in the case of rituximab,15–17 but the kinetic and mechanistic understanding of terminal galactose formation during CHO cell culture process is still lacking.
Figure 1.
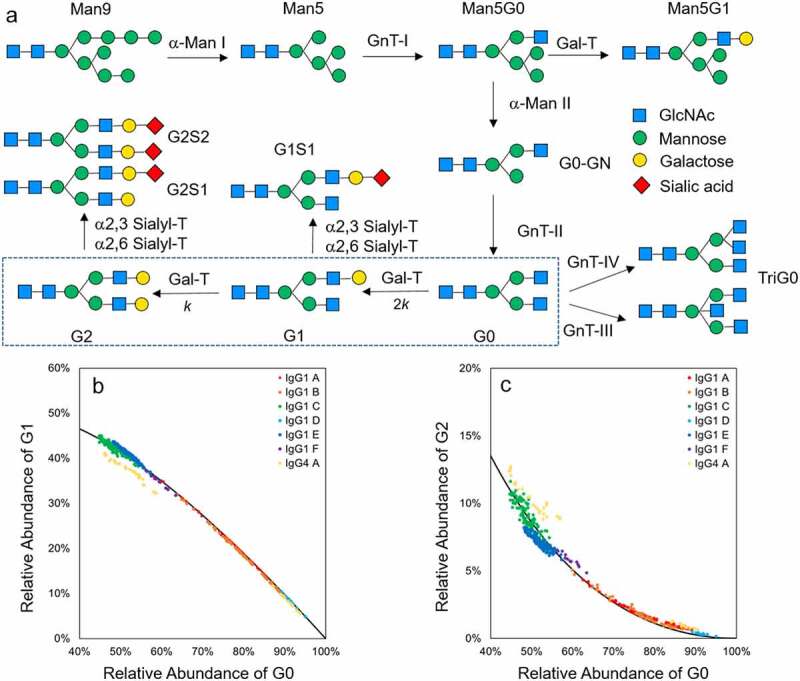
N-Glycan synthesis pathway and correlation between different galactosylation species. (a) IgG1 N-glycan synthesis pathway in Golgi apparatus. The IgG enters the Golgi apparatus from the endoplasmic reticulum (ER) with the Man9 glycan on Fc. The glycan is further modified with the pathway shown in the figure. α-Man I and II: α-mannosidase I and II; GnT I, II, III and IV: GlcNAc-transferase I, II, III and IV; Gal-T: β-1,4 galactosyltransferases; α2,3 Sialyl-T: α-2,3 sialyltransferase; α-2,6 Sialyl-T: α-2,6 sialyltransferase; (b) Correlation between G1 and G0(left), G2 and G0 (right) with 12,000 L drug substance batches data (N = 410). Each point represents one 12,000 L batch data. Each IgG molecule is represented by a different color. Solid lines represents the theoretical derived curve based on the consecutive kinetic model
CDC, an effector function of IgG from the classical complement pathway, plays a critical role in the MOA of several US Food and Drug Administration-approved mAb therapeutics, including rituximab, ofatumumab, and I-tositumomAb.18,19 When IgGs bind to the antigen on the surface of the target cell, the classical complement pathway is triggered by binding protein Complement component 1q (C1q) to the antibody, forming a membrane attack complex and leading to tumor cell lysis.18–20 Diebolder et al. discovered IgG1s hexamerize orderly through Fc interaction after antigen binding at the cell surface, and then bind to C1q, which leads to effective complement activation.21–23 The binding sites between IgG1 hexamer and C1q in the structure complex were identified recently.24 Van den Bremer et al. demonstrated charge variants at the C terminus, such as C-terminal lysine, may hinder hexamer formation.25 Wang et al. found the presence of asparagine (N)-linked glycans affect hexamerization, which implies the conformation of the Fc plays a key role in hexamerization.26 The post-translational modifications of IgG1s, especially the modifications on Fc, could affect the formation of IgG1 hexamerization, and hence affect C1q binding and CDC potency. As an important part of the Fc glycan, terminal galactose was found to affect protein conformation27 and promote the activation of the complement system by increasing C1q binding affinities.17,28,29 However, the structural and functional correlations between Fc galactosylation and CDC potency remain unclear.
In this study, we investigated the kinetics of terminal galactose formation during CHO cell culture process, galactosylation-mediated protein conformation, and CDC potency variation to understand the structural, functional and mechanistic aspect of Fc galactosylation and CDC potency. We derived a novel kinetic model for galactosylation in CHO cell culture process with the assumption of the galactose transfer reactions following a two-step consecutive reaction hypothesis and validated the model with small-scale and large-scale cell culture batches across five years manufacturing experiences and 11 different molecules. Various analytical and biological tools were used to characterize the glycoengineered IgGs to unveil the structural and functional impact of Fc galactosylation on CDC potency.
Results
The Fc glycan distribution of the IgG was measured by applying hydrophilic interaction chromatography (HILIC) to enzymatically released glycans. In a typical HILIC chromatogram of CHO-derived mAbs, high mannose (Man5 – Man9), sialylated (G1S1, G2S2 and G2S1) and bisecting N-acetylglucosamine (TriG0) species are present at relatively low levels (<2%). The majority of the Fc glycans belong to three categories: zero (G0), one (G1) and two (G2) terminal galactoses (Figure S1). The galactose transfer reaction (Box in Figure 1a) is the major rate-limiting step during the Fc glycosylation.6 The extent of mAb A galactosylation were measured with both HILIC released glycan method at the released glycan level and time-of-flight mass spectrometry (TOF-MS) at the intact protein level. As shown in Figure S2, the intact protein level Fc galactosylation distribution reconstructed by the released glycan data following binominal distribution agrees well with the experimental results of TOF-MS. The results demonstrate the galactose transfer reaction is independent on the glycan structure on the other arm. Based on these observations, we hypothesize that: 1) the galactose transfer reaction in the Golgi apparatus follows consecutive reaction kinetics, and 2) the reaction rate constant of the galactose transfer reaction for G0 is twice that of G1 based on number of reactions sites (Box in Figure 1a). With the hypothesis, the equation of the time dependence of the concentration of each galactosylation species is derived as follows:
In these equations, the k is the rate constant of the galactosylation reaction, t is the reaction time and [G0]0 is the starting concentration of G0. Using these formulas, we can derive the correlations between the relative abundance of different galactosylated species (see supplemental information for the detailed derivation process).
In this equation, %G0, %G1 and %G2 are the relative abundance of the particular galactosylation species relative to the total number of mAb measured by HILIC. These correlations between various galactosylation species are independent from the rate constant and cultural time, and should apply to any type of CHO cell culture process based on the derivation process. To test this hypothesized correlation, a retrospective analysis on the batch release data of 410 historical drug substance batches manufactured at 12,000 L scale under the Good Manufacturing Practice (GMP) environment, including six different IgG1s, one IgG4 (Figure 1b and 1c) and the process validation data of 426 historical small-scale cell culture research and development (R&D) samples at 2 L scale, including four different IgG1s and three different IgG4s (Figure S3) were performed. All the IgG1s with various levels of galactosylation under both GMP and R&D environments show good fit with the hypothesized correlations, but the IgG4s show some deviations from the model.
To establish the correlation between the CDC potency and Fc galactosylation, a glycoengineering method was developed to generate IgG galactosylation variants predictively. Compared with the previous method,30 the new method controls the rate of the reaction with a lower enzyme concentration, manganese concentration, and temperature, and includes a quench step using ethylenediaminetetraacetic acid (EDTA) to chelate the manganese cofactor in the solution. The level of Fc galactosylation is well controlled by the reaction time (Figure 2a). A comprehensive library of mAb A galactosylation variants were produced using this glycoengineering method. In addition, a linear correlation was found between the CDC potency and total galactose per heavy chain using this set of galactosylation variants, which aligns well with the previous studies.17 (Figure 2b)
Figure 2.
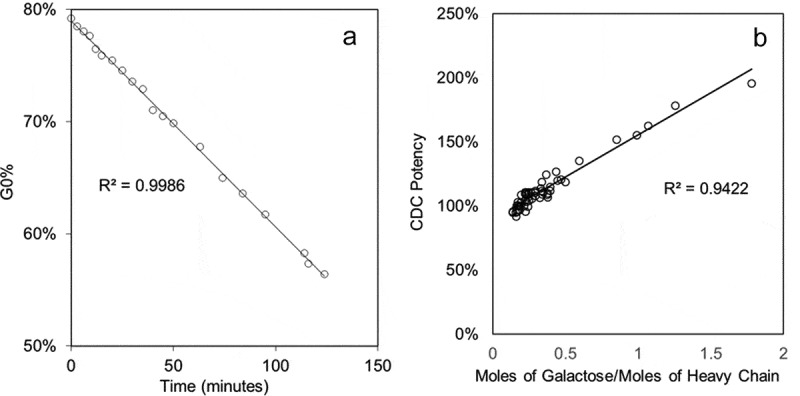
(a) The linear regression analysis of total %G0 correlation and reaction time with rate-controlled galactose transfer reaction of mAb A; (b) The linear regression analysis of the CDC potency of mAb A and moles of galactose per heavy chain
We further investigated the mechanism by which terminal galactose affects CDC potency. The hypogalactosylated mAb A (mAb A with a low level of galactosylation (>85% G0)) and hypergalactosylated mAb A (mAb A with a high level of galactosylation (>85% G2)) were generated through enzymatic reactions. The CDC potency of these two samples was tested with a cell-based potency assay and the relative binding activity of C1q was evaluated through surface plasma resonance (SPR). The C1q binding activity of hypergalactosylated mAb A was 20% higher than that of the hypogalactosylated mAb A. The relative CDC potency of hypergalactosylated mAb A was approximately double that of the hypogalactosylated mAb A (Figure 3a).
Figure 3.
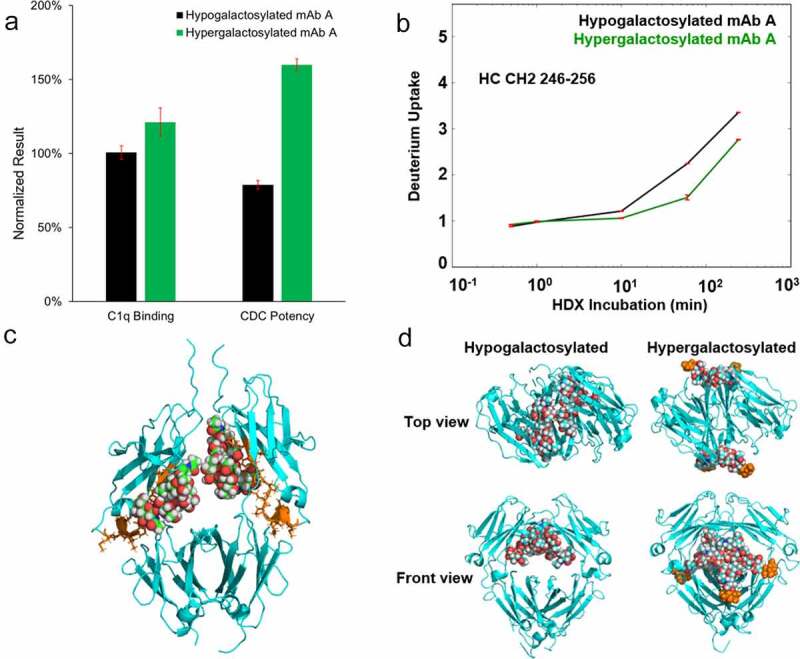
Characterization of hypogalactosylated and hypergalactosylated mAb A. (a) C1q binding and CDC potency of hypogalactosylated mAb A and hypergalactosylated mAb A; (b) Time-course of deuterium incorporation results (including ARDD) for HC CH2 Domain peptide segment 246–256; (c) The Fc region of a single hypogalactosylated mAb A joined by the disulfide bonds at the hinge region. The glycans and the proteins are shown using space-filling and cartoon representations, respectively. The peptide with different hydrogen/deuterium exchange rate between hypogalactosylated and hypergalactosylated mAb A is colored in orange; (d) Top view and front view of representative Fc structures of mAb A with and without terminal galactose obtained from 10 μs explicit solvent Molecular Dynamics simulations. The glycans and the proteins are shown using space-filling and cartoon representations, respectively. The terminal galactose in the hypergalactosylated structure are colored in orange
Since the hexamer of IgG1 forms through Fc-Fc interaction on the cell surface, Fc galactosylation may affect hexamer formation by altering the Fc conformation.24,26 To probe the Fc conformational change from galactosylation, we analyzed the hypogalactosylated and hypergalactosylated mAb A with hydrogen/deuterium exchange mass spectrometry (HDX-MS). No significant differences in hydrogen/deuterium (H/D) exchange uptake were found in the variable heavy chain, light chain, CH1 and CH3 domains between the two samples, including the C1q binding sites (D270, K322, P329 and P33124,31), meaning that the galactosylation does not directly alter the conformation of the C1q binding sites. The only peptide segment with a different H/D exchange uptake between hypogalactosylated and hypergalactosylated mAb (Figure 3b) is peptide 246–257 located in the CH2 domain, which agrees well with the previous study.32,33 This specific region was found to be a critical site for hexamer formation31,34 (orange cluster in Figure 3c).
To better understand the effect of galactosylation on the Fc conformation, we performed 10 explicit solvent Molecular Dynamics (MD) simulation of the Fc with and without the terminal galactose. The simulation results show that the conformations of the Fc domain with and without galactose are dramatically different. The representative conformational structures obtained from a clustering analysis of the combined MD trajectories show that the entire glycan structures are buried within the Fc for hypogalactosylated species whereas glycans extend outside for hypergalactosylated Fc (Figure 3d, Video S1 and S2). The glycans are further apart in the hypergalactosylated species than in the hypogalactosylated species, as observed by the histogram calculation of the distances between center of mass of the two glycans (Figure S4). The hypergalactosylated Fc dimer also has less backbone fluctuation than the hypogalactosylated Fc dimer (Figure S5).35 These MD results show when the glycans are buried inside the Fc core in the hypogalactosylated Fc dimer, the glycans tend to interact more strongly with each other and that results in a more compact structure. On the other hand, the glycans are extended outside the Fc core in the hypergalactosylated Fc dimer and solvates in water. Glycans are more flexible and spaced further apart.
Diebolder et al. discovered that combining E345R, E430G and S440Y (RGY) mutations stabilizes IgG1 hexamer in solution.21 The RGY mutant of mAb B, an IgG1 with the same Fc as mAb A was produced to study the terminal galactose impact on the IgG1 hexamer. Native MS shows the existence of monomer, dimer, and hexamer of the RGY mutant of mAb B in solution (Figure S6A). The pentamer was generated from in-source fragmentation of the hexamer during electrospray ionization, which is confirmed by isolation of the hexamer ions (Figure S6B). The RGY mutant was glycoengineered under 4⁰C to prevent thermally induced hexamer dissociation. The native MS spectra (Figure 4a) shows the shifted m/z spectrum resulting from the addition of terminal galactose hypergalactose hexamers are expected to have stronger Fc interaction, which could potentially mask solvent-exposed amino acids with high proton affinities. Compared to the hypo- species, the hypergalactose species was observed with significantly lower charge states, supporting this change in structure.
Figure 4.
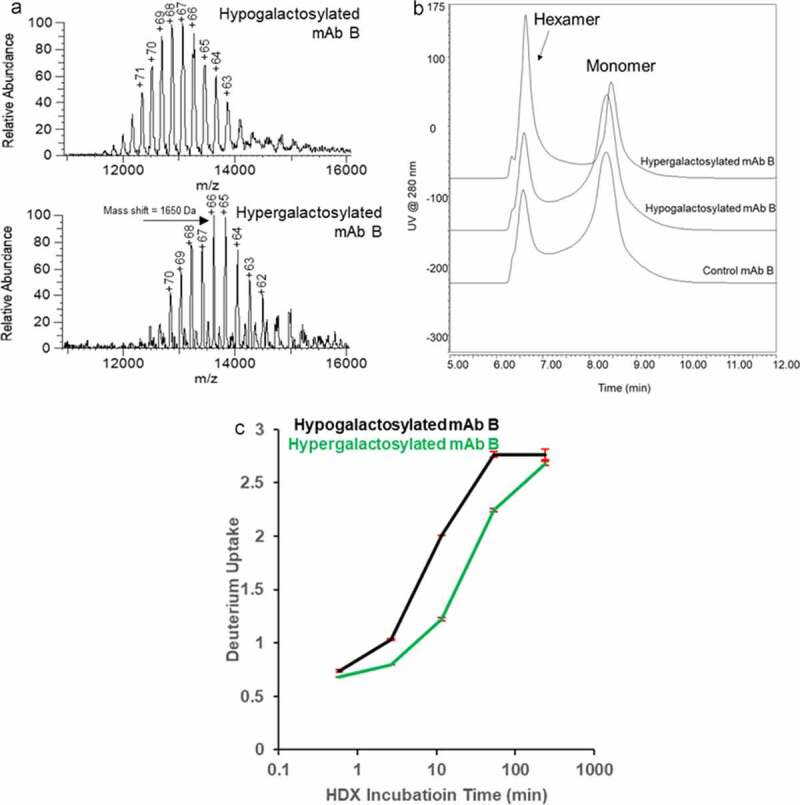
Characterization of hypogalactosylated and hypergalactosylated mAb B RGY mutants. (a) Reconstructed native mass spectra of hypogalactosylated and hypergalactosylated mAb B hexamer showing the mass to charge ratio (m/z) distribution shift from the galactosylation level difference; (b) Size exclusion chromatogram of hypogalactosylated and hypergalactosylated mAb B RGY mutants showing the Fc galactosylation enhance the hexamer formation; (c) Time-course of deuterium incorporation results for same HC CH2 Domain peptide in Figure 3
Size exclusion chromatography (SEC) was used to analyze the size distribution of both RGY mutants of mAb B and multi-angle light scattering (MALS) was used for peak attribution. All three mAb B mutants were separated into two peaks. (Figure 4b) The MALS characterization results (Figure S7, Table S1) shows the front peak has a molar mass around 900 kDa and the back peak has a molar mass around 150 kDa. The MALS results confirm the front peak is mAb B hexamer and the back peak is mAb B monomer. The overlaid chromatogram (Figure 4b) shows that the hypergalactosylated RGY mutant of mAb B has near twice as much hexamer in free solution as the hypogalactosylated RGY mutant of mAb B. The SEC result and glycan distribution is shown in Table S2. The glycan distribution and native MS data on hexamer show the hypergalactosylated variants are enriched in the hexamer. The HDX-MS results of hypergalactosylated and hypogalactosylated mAb B RGY mutants show the decreased deuterium uptake in the same segment on the CH2 domain in mAb A (Figure 4c), consistent with the mAb A HDX result. This result confirms conformational change induced by Fc galactosylation stabilizes the hexamer. H/D exchange results also demonstrate the hypergalactosylated mutant offered more solvent protection of the CH3 domain 432–446 region (Figure S8A), which is also a critical site in Fc dimer formation (Figure S8B). The characterization results of glycoengineered mAb B RGY mutants proved that the presence of a terminal galactose enhances the IgG hexamer formation for complement activation.
Discussion
We first proposed a consecutive model based on the glycosylation pathway shown in Figure 1a to describe the kinetics of galactosylation reaction of IgG1 in the Golgi apparatus of the CHO cell. The model was then validated by the good fit with large-scale manufacturing and small-scale R&D data. The data from small-scale R&D studies show slightly larger deviation, which might come from the higher heterogeneity between a small-scale bioreactors and different mixing mode, or even intentionally deviated cell culture conditions for R&D purposes. The IgG4s in this study are observed to deviate from the correlation by having less G1 and more G2 since the %G0 is below 70%. In this case, the second hypothesis is invalid. The galactose transfer reaction for IgG4 is relatively faster on G1 than on G0. The reason remains unclear, but might potentially link to the unique Fc conformation of IgG4.36 Although validating this correlation still requires more biological and physicochemical studies to reveal the entire process of galactose transfer in CHO cell culture under various conditions, the correlation derived from the consecutive reaction model could be used as tool to estimate the levels of each galactosylation species. The total galactose vs. %G0 plot from the model (Figure S9) is close to linear in the typical manufacturing region with the %G0 between 40% and 100%. Therefore, %G0 is a suitable surrogate for the inverse of total galactose to monitor for the purposes of quality control and process monitoring because %G0 is typically the most abundant glycoform and easier to measure precisely.
Although the Fc galactosylation could significantly enhance CDC potency, the mechanism is not very well understood. We first characterized the hypogalactosylated and hypergalactosylated IgG1 monomer with C1q binding in an SPR assay and a cell-based CDC potency assay. The difference in CDC potency between hypogalactosylated and hypergalactosylated variants is significantly higher than the increase of C1q binding affinity. A previous report showed that differences in C1q binding affinity for mAb galactosylation variants were similar to the difference of CDC potency,29 while the C1q binding was measured by immobilizing the mAb on the cell surface. Combining the results of both studies, one possible explanation of this unexpected mismatch in the results could be that Fc galactosylation elevates complement activation mainly through enhancing hexamerization on the cell surface because mAb only forms hexamers on the cell surface, but not on the gold surface because of differences in immobilization.21 The explanation is based on the hypothesis that CDC potency is enhanced by terminal galactose mainly because of increased mAb hexamerization.
To test this hypothesis, we investigated potential factors that could influence hexamerization in mAbs. The HDX-MS shows the hypergalactosylated mAb A has less deuterium uptake in the CH2 domain 246–257 region. This result demonstrates Fc galactosylation decreased the local solvent accessibility in this region and potentially disturbed the local hydrogen bonding; in other words, it increases the local hydrophobicity in the CH2 domain. Diebolder et al. showed IgG1-7D8 and its three mutants (I253A, I253Y and I253D) with descending order of CDC potency corresponding to the decreasing hydrophobicity of the mutated amino acid, as well as the increasing solvent accessibility in the same region in the CH2 domain (Table S3).21,35 In summary, both studies show the changes at the CH2 domain 246–257 region related to solvent-related interaction elevates the CDC potency of the mAb without directly affecting the C1q binding site to the mAb monomer. After we understood the galactosylation impact on the conformation of the mAb monomer, MD studies were used to understand the impact of galactosylation on Fc-Fc interaction. From the MD results, the Fc-Fc complexes are more stable with the terminal galactose with less steric effect and between solvation of the hydrophilic glycan groups. The MD data indicates that the galactosylation might affect the Fc-Fc interaction in the dimer, which is one important step of hexamer formation. Based on the HDX and MD results, we therefore hypothesize that Fc galactosylation affects Fc conformation and Fc-Fc interaction, which leads to the enhancement of hexamer formation and subsequently CDC potency. However, to test this hypothesis, more direct experimental evidence is required to demonstrate that terminal galactosylation enhances the IgG hexamer formation.
Typically, IgG hexamers only form on the cell surface after antigen binding and are not detectable in solution. We used the RGY-mutated mAb B to study the IgG1 hexamer structure because the hexamer of RGY mutant is stable in solution. Native MS has previously been used to analyze antibody hexamers,37 and was used in our study to confirm the existence of stable IgG hexamer in solution. Since Heck’s seminal work, other groups have applied this method to look at noncovalent lipids, stoichiometry questions, and the incorporation of backbone mutations.23,26,34,38 In this study, we observed that the MS conditions to release the monomer from the hexamer caused some backbone fragmentation of the glycans. The addition of a galactose to the hexamer at a charge of 66 would correspond to mass shift of 2.45 m/z units, requiring a resolution of 35000 @ 200 m/z, compared to a mass shift of ~8 Da if the released monomer was analyzed. Traditionally, lower resolution is used for large, unstable ions to limit signal decay, creating challenges for the MS tuning of this sample. Using post-signal processing and smoothing in Unidec 4.1, a mass shift of approximately 1650 Da was determined between the two species, which is equivalent to the mass of ~10 galactose. The HDX-MS on mAb B hexamer galactosylation variants shows the same results on the CH2 domain as monomer and increased protection in CH3 domain, which is the hexamer binding site. The SEC-MALS results on glycoengineering of mAb B RGY mutant provide clear proof that Fc galactosylation enhances IgG hexamerization.
In conclusion, we performed a systematic study on the formation and structure-function relationship of recombinant mAb Fc galactosylation and related biological activities. The galactosylation reaction of IgG1 in the Golgi apparatus of the CHO cell was described by a consecutive kinetic model and validated by hundreds of GMP manufacturing and R&D data points. The structural and functional studies reveal a molecular basis for Fc galactosylation-enhanced antibody hexamer formation, which is the primary source of increased complement activation through changes in the three-dimensional conformation of the mAb Fc region. These important new findings offer in-depth structural and functional insights into the Fc galactosylation and will help future mAb therapeutics development.
Materials and Methods
Materials
MAb A, a humanized monoclonal antibody IgG1 with CDC potency, was produced and purified at Genentech Inc. (South San Francisco, CA, USA). MAb B with E345R, E430G and S440Y (RGY) mutant was produced in Wuxi AppTec (Shanghai, China). The sialidase A, β-1,4-galactosidase and PNGase F were purchased from New England Biolabs (Ipswich, MA, USA). All other chemicals and reagents were purchased from Millipore-Sigma (St. Louis, MO, USA).
Glycoengineering of mAb A
The hypogalactosylated mAb A variant was prepared by incubating 100 μL 30 mg/mL mAb A drug substance with sialidase A (10 milliunits/mg protein) and β-1,4-galactosidase (10 milliunits/mg protein) in the buffer of 20 mM sodium acetate, 4% trehalose at pH 5.3 at 37⁰C for 24 hours. After 24 hours, another aliquot of each enzyme (10 milliunits/mg protein) was added and incubated at 37⁰C for 24 hours. The hypergalactosylated mAb A variant was prepared by incubating 100 μL mAb A 30 mg/mL with galactosyltransferase (90 milliunits/mg protein) at 37⁰C for 48 hours in the same sodium acetate buffer containing 20 mM uridine diphosphate galactose (UDP-Gal), and 25 mM MnCl2. Both variants were purified by Protein A chromatography using TSKgel Protein A-5PW column (Tosoh Bioscience LLC, King of Prussia, PA, USA) and buffer exchanged into the formulation buffer.
The rate controlled galactosylation of mAb A was performed by incubating 100 μL 30 mg/mL hypogalactosylated mAb A with galactosyltransferase (16 milliunits/mg protein) at 25⁰C in sodium acetate buffer at pH 5.3 containing 20 mM UDP-Gal and 1.25 mM MnCl2. The reaction was then quenched by 15 µL of 500 mM EDTA pH 8.0. The galactosylation variants were purified by Protein A chromatography using TSKgel Protein A-5PW column (Tosoh Bioscience LLC, King of Prussia, PA, USA) and buffer exchanged into the formulation buffer.
All the glycoengineering reactions described here could be scaled up and down by varying the amounts of the reagents proportionally.
RGY-mutated mAb B Galactosylation Variants Preparation
The hypogalactosylated RGY-mutated mAb B variant was prepared by incubating 5 mg/mL mAb B with sialidase A (10 milliunits/mg protein) and β-1,4-galactosidase (20 milliunits/mg protein) in sodium acetate buffer at pH 5.3 at 4⁰C for 7 days. The hypergalactosylated RGY-mutated mAb B variant was prepared by incubating mAb B with galactosyltransferase (90 milliunits/mg protein) at 4⁰C for 7 days in sodium acetate buffer at pH 5.3 containing 20 mM uridine diphosphate galactose (UDP-Gal), and 25 mM MnCl2. Both variants were purified by a 10 K Amicon Filter and reconstituted in the formulation buffer.
High Throughput Hydrophilic Interaction Chromatography For Relative Glycan Distribution Measurement
The protein samples were diluted to 1 mg/mL and treated with PNGase F in ammonium acetate buffer at pH 8.6 at 45⁰C for one hour. Released glycans were collected and labeled with 2-aminobenzamide (2-AB) using Signal™ 2-AB Labeling Kit (Agilent Technologies, Santa Clara, CA). Labeled glycans were analyzed on a UPLC ACQUITY H-Class system (Waters Corp., Milford, MA) using a ACQUITY UPLC Glycan BEH Amide 130 Å column (Waters Corp., Milford, MA). The mobile phase A was 100 mM ammonium formate at pH 4.5 and mobile phase B was acetonitrile. The gradient is 75% B to 63% B in 16.5 minutes. The 2-AB labeled glycans were detected by fluorescence at the excitation wavelength of 330 nm and the emission wavelength of 420 nm.
Hydrogen/Deuterium Exchange – Mass Spectrometry Analysis
H/DX MS was used to compare the conformational differences between the hypogalactosylated and hypergalactosylated mAbs. H/DX MS were performed with a fully automated LEAP RTC system (Leap Technologies, Carr, NC) coupled with an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, San Jose, CA), as shown in the two previous reports from our group.39,40 Samples were incubated in deuterium buffer at 20°C for labeling, followed by an in-line pepsin and protease XIII digestion and LC-MS analysis. Peptide identifications were based on accurate masses together with MS/MS fragmentation by Mascot search. Deuterium incorporation was determined by EXMS,41 followed by a python script. Averaged deuterium uptake difference (ARDD) was calculated by a modified python script.42,43
Native Mass Spectrometry
Sample was buffer exchanged, according to the manufacturer’s protocol, into 50 mM ammonium acetate using a Micro Bio-Spin 6 column (Bio-Rad Laboratories, Hercules, CA). Approximately 2 µL was loaded into a borosilicate glass tip (1.2 mm OD, 0.69 mm ID) that was pulled in-house on a P-1000 instrument (Sutter Instrument, Novato, CA) and sputter coated to 6 nm with Au-Pt (Leica Microsystems, DE). Static spray at a voltage of 1.2–1.3 kV was performed on a Q Exactive UHMR (Thermo Fisher Scientific, Waltham, MA). The mass spectrometry parameters were manually tuned per sample, with the most critical parameters including using the highest HCD pressure setting, operating the in-source trapping at −100 (reference) or −20 V (high galactose), a 100 ms injection time, and a resolving power of 8750. Data was processed in MetaUniDec v4.1.043 using a peak width of 90 m/z.
Complement-Dependent Cytotoxicity Assay
The method is performed in 96-well tissue culture microtiter plates. In this assay, varying concentrations of mAb A reference material, control, or samples are incubated with WIL2-S cells (50,000 cells/well) in the presence of a fixed amount of human complement. The plates are then incubated at 37°C/5% CO2 in a humidified incubator for 1–2 hours, during which time the Fab region of the antibody binds to the CD20 receptor on the WIL2-S cells, and the Fc region binds to the complement, leading to cell lysis. At the end of the incubation, the relative number of viable cells is quantitated indirectly by adding 50 WL of the redox dye, alamarBlueTM, to each well and the plate is incubated at 37°C/5% CO2 in a humidified incubator for 15 to 26 hours. The fluorescence is read using a 96-well fluorometer with excitation at 530 nm and emission at 590 nm. AlamarBlueTM is blue and nonfluorescent in its oxidized state, but is reduced by the intracellular environment to a pink form that is highly fluorescent. The changes in color and fluorescence are proportional to the number of viable cells. The results, expressed in relative fluorescence units (RFU), are plotted against the mAb A concentrations, and a parallel line analysis program was used to estimate the relative potency of the antibody samples.
Surface Plasmon Resonance-based C1q binding assay
A Biacore T200 instrument (GE healthcare, Chicago, IL, USA) was used for SPR analysis. Biacore sensor S series chip SA (BR-1005-32, GE healthcare, Chicago, IL, USA) was washed with 1 M NaCl in 50 mM NaOH. Biotinylated anti-LC-kappa antibody (Catalog # 7103272100 Thermo scientific, San Jose, CA, USA) was diluted to 50 µg/mL and immobilized at the saturation level. Samples prepared at 10 µg/mL antibody concentration in assay running buffer (phosphate-buffered saline with 0.05% Tween 20) were captured for 100 seconds at 10 µL/min. Subsequently, C1q protein (Catalog # A400, Quidel, San Diego, CA, USA) were injected for 4 min at 30 µL/min. The dissociation phase was achieved by passing the same assay running buffer through the chamber for 100 seconds. The regeneration was performed with a single injection of 10 mM Glycine-HCl, pH 2.0 for 30 seconds at 30 µL/min. All experiments were performed at 25°C. Duplicate injections of each sample and a buffer blank were flowed over the two surfaces (a reference flow cell and a testing flow cell). Data were collected at a rate of 1 Hz. Readout was the maximum binding response during association phase, five seconds before the end of the C1q injection. A reference flow cell was run in conjunction to the testing flow cell to negate the effects of nonspecific binding. In addition, injections of blank running buffer were included on experimental flow cells. Signals from the reference flow cell and blank buffer injections were subtracted from the absolute response of sample injections on experimental flow cells (double subtraction method). Data were analyzed using Biacore T200 evaluation software and JMP software.
Size Exclusion Chromatography coupled with Multi-angle Light Scattering Analysis
The size variants distribution was analyzed using a UPLC ACQUITY H-Class system (Waters, Milford, MA) using a Tosoh TSKgel G3000SWXL column (7.8 mm X 300 mm, 5 μm). Samples (20 μg) were injected and isocratic eluted at 0.5 mL/min using 40 mM sodium phosphate, 60 mM sodium dibasic, 100 mM sodium sulfate at pH 6.8. The UV detection was at 280 nm. The molecular weights of the peaks were measured by a Wyatt μDAWN MALS detector and a Wyatt Optilab UT-rEX differential refractive index (dRI) detector.
Molecular Dynamics Simulation Protocol and Computational Analysis
The Fc structure of mAb A was obtained from the crystal structure. G0 and G2 glycans were built and attached to the Fc using the Glycan Reader & Modeler module in CHARMM-GUI44,45 (http://www.charmm-gui.org/input/glycan). CHARMM-GUI44,45 was used to prepare Molecular Dynamics input files. We used CHARMM36m force field for protein46 and the CHARMM36 force field for carbohydrates,47 and TIP3P water model48 for explicit solvation. Counter ions of Na+ and Cl− with the concentration of 0.15 M were added to neutralize the system.
The GPU implementation of Amber 2016 MD software package with the SPFP precision model49 was used for the MD simulation using the following protocol. First, the structure was relaxed with 2000 steps of conjugate-gradient energy minimization, using harmonic restraining potential with the force constant of 10 kcal/mol/Å2 to restrain the solute to the initial structure. Then, the solvent molecules were allowed to move using NPT ensemble with a temperature of 300 K. Another step of conjugate-gradient energy minimization was performed with 2000 steps while removing all the restrains. Next, the pressure was maintained at 1 atm and the thermostat temperature increased to 300 K over the course of 500 ps, while Harmonic positional restraints of strength 10 kcal/mol/Å2 was applied to the solute. The system was then equilibrated for 1 ns with a restraint force constant of 1 kcal/mol/Å2 All restraints were removed for the production stage. The hydrogen mass repartition option of Amber was used allowing a time step of 4 fs.50 The simulation time step was 4 fs. A 10 Å cutoff radius was used for range limited interactions, with Particle Mesh Ewald electrostatics for long-range interactions. The production simulation was carried out using NPT conditions. Langevin dynamics51 was used to maintain the temperature at 300 K with a collision frequency of 3 ps-1. The production stage of the MD simulation was performed for 500 ns. During dynamics the SHAKE algorithm52,53 was applied to constrain all bonds involving hydrogen atoms. Default values were used for all other simulation parameters.
The protocol described above was repeated to generate 20 independent replicates of 500 ns trajectories, adding up to 10-microsecond trajectories for each structure. We used the CPPTRAJ tool available in AmberTools 201553 to calculate the center of mass distances and root mean square fluctuations. To obtained the representative structures shown in Figure 3d, clustering of trajectories was performed with CPPTRAJ using the DBSCAN clustering algorithm with the minimum number of points set to 15 and epsilon set to 7. The distance metric was the root mean square deviation using atoms in the glycans, as well as the atoms involved in the five disulfide bonds in the Fc region, representative of the overall of conformation of the Fc.
Supplementary Material
Acknowledgments
The authors will like to thank Feng Yang, John Stults, Tilman Schlothauer and David Michels for their critical review and helpful suggestions. The authors will also like to thank John Stults, David Michels, Matthew Kalo, Vikas Sharma, and John Joly for their support on the project.
Abbreviations:
ADCC: Antibody-dependent cell-mediated cytotoxicity; C1q: Complement component 1q; CDC: complement-dependent cytotoxicity; CHO:Chinese Hamster Ovary; CQA: critical quality attribute, GMP: Good Manufacturing Practice; HDX: hydrogen/deuterium exchange; HILIC: hydrophilic interaction chromatography; IgG: immunoglobulin G; mAb: monoclonal antibodies; MALS: multi-angle light scattering; MD: Molecular Dynamics; MOA: mechanism of action; MS: mass spectrometry; RGY: E345R, E430G and S440Y; R&D: research and development; SEC: Size exclusion chromatography; SPR: surface plasma resonance; TOF-MS: time-of-flight mass spectrometry; UDP-Gal: uridine diphosphate galactose
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Grilo AL, Mantalaris A. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2019;37(1):9–10. doi: 10.1016/j.tibtech.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 2.De La Torre BG, Albericio F.. The pharmaceutical industry in 2018. an analysis of fda drug approvals from the perspective of molecules. Molecules. 2019;24(4):4. doi: 10.3390/molecules24040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh G. Biopharmaceutical benchmarks 2014. Nat Biotechnol. 2014;32(10):992–1000. doi: 10.1038/nbt.3040. [DOI] [PubMed] [Google Scholar]
- 4.Reusch D, Tejada ML. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology. 2015;25(12):1325–34. doi: 10.1093/glycob/cwv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alt N, Zhang TY, Motchnik P, Taticek R, Quarmby V, Schlothauer T, Beck H, Emrich T, Harris RJ. Determination of critical quality attributes for monoclonal antibodies using quality by design principles. Biologicals. 2016;44(5):291–305. doi: 10.1016/j.biologicals.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 6.Sumit M, Dolatshahi S, Chu AA, Cote K, Scarcelli JJ, Marshall JK, Cornell RJ, Weiss R, Lauffenburger DA, Mulukutla BC, et al. Glycosylation dynamics in chinese hamster ovary cells fed-batch cultures using time course omics analyses. iScience. 2019;12:102–20. doi: 10.1016/j.isci.2019.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Del Val IJ, Kontoravdi C, Nagy JM. Towards the Implementation of Quality by Design to the Production of Therapeutic Monoclonal Antibodies with Desired Glycosylation Patterns. Biotechnol Progr. 2010;26(6):1505–27. doi: 10.1002/btpr.470. [DOI] [PubMed] [Google Scholar]
- 8.Eon-Duval A, Broly H, Gleixner R. Quality attributes of recombinant therapeutic proteins: An assessment of impact on safety and efficacy as part of a quality by design development approach. Biotechnol Progr. 2012;28(3):608–22. doi: 10.1002/btpr.1548. [DOI] [PubMed] [Google Scholar]
- 9.Hossler P. Protein glycosylation control in mammalian cell culture: past precedents and contemporary prospects. Adv Biochem Eng Biot. 2012;127:187–219. [DOI] [PubMed] [Google Scholar]
- 10.Gawlitzek M, Estacio M, Furch T, Kiss R. Identification of cell culture conditions to control n-glycosylation site-occupancy of recombinant glycoproteins expressed in cho cells. Biotechnol Bioeng. 2009;103(6):1164–75. doi: 10.1002/bit.22348. [DOI] [PubMed] [Google Scholar]
- 11.Del Val IJ, Nagy JM, Kontoravdi C. A dynamic mathematical model for monoclonal antibody N-linked glycosylation and nucleotide sugar donor transport within a maturing Golgi apparatus. Biotechnol Progr. 1730-1743;2011:6. [DOI] [PubMed] [Google Scholar]
- 12.Grainger RK, James DC, Cell Line CHO. Specific Prediction and Control of Recombinant Monoclonal Antibody N-Glycosylation. Biotechnol Bioeng. 2013;110(11):2970–83. doi: 10.1002/bit.24959. [DOI] [PubMed] [Google Scholar]
- 13.Jedrzejewski PM, Del Val IJ, Constantinou A, Dell A, Haslam SM, Polizzi KM, Kontoravdi C. Towards controlling the glycoform: a model framework linking extracellular metabolites to antibody glycosylation. Int J Mol Sci. 2014;15(3):4492–522. doi: 10.3390/ijms15034492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naik HM, Majewska NI, Betenbaugh MJ. Impact of nucleotide sugar metabolism on protein N-glycosylation in Chinese Hamster Ovary (CHO) cell culture. Curr Opin Chem Eng. 2018;22:167–76. doi: 10.1016/j.coche.2018.10.002. [DOI] [Google Scholar]
- 15.Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29(4):310–12. doi: 10.1038/nbt.1839. [DOI] [PubMed] [Google Scholar]
- 16.Hodoniczky J, Zheng YZ, James DC. Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol Progr. 1644-1652;2005:6. [DOI] [PubMed] [Google Scholar]
- 17.Raju TS, Jordan RE. Galactosylation variations in marketed therapeutic antibodies. Mabs. 2012;4(3):385–91. doi: 10.4161/mabs.19868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer S, Leusen JH, Boross P. Regulation of complement and modulation of its activity in monoclonal antibody therapy of cancer. MAbs. 2014;6(5):1133–44. doi: 10.4161/mabs.29670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogers LM, Veeramani S, Weiner GJ. Complement in monoclonal antibody therapy of cancer. Immunol Res. 2014;59(1–3):203–10. doi: 10.1007/s12026-014-8542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu XY, Wang XY, Li RY, Jia SC, Sun P, Zhao M, Fang C. Recent progress in the understanding of complement activation and its role in tumor growth and anti-tumor therapy. Biomed Pharmacother. 2017;91:446–56. doi: 10.1016/j.biopha.2017.04.101. [DOI] [PubMed] [Google Scholar]
- 21.Diebolder CA, Beurskens FJ, De Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJ, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343(6176):1260–63. doi: 10.1126/science.1248943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cook EM, Lindorfer MA, Van Der Horst H, Oostindie S, Beurskens FJ, Schuurman J, Zent CS, Burack R, Parren PWHI, Taylor RP. Antibodies That Efficiently Form Hexamers upon Antigen Binding Can Induce Complement-Dependent Cytotoxicity under Complement-Limiting Conditions. J Immunol. 1762-1775;2016:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Jong RN, Beurskens FJ, Verploegen S, Strumane K, Van Kampen MD, Voorhorst M, Horstman W, Engelberts PJ, Oostindie SC, Wang GB, et al. Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. Plos Biol. 2016;14(1):1. doi: 10.1371/journal.pbio.1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ugurlar D, Howes SC, De Kreuk BJ, Koning RI, De Jong RN, Beurskens FJ, Schuurman J, Koster AJ, Sharp TH, Parren PWHI, et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science. 2018;359(6377):794–97. doi: 10.1126/science.aao4988. [DOI] [PubMed] [Google Scholar]
- 25.Van Den Bremer ET, Beurskens FJ, Voorhorst M, Engelberts PJ, De Jong RN, Van Der Boom BG, Cook EM, Lindorfer MA, Taylor RP, Van Berkel PH, et al. Human IgG is produced in a pro-form that requires clipping of C-terminal lysines for maximal complement activation. MAbs. 2015;7(4):672–80. doi: 10.1080/19420862.2015.1046665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang G, De Jong RN, Van Den Bremer ET, Beurskens FJ, Labrijn AF, Ugurlar D, Gros P, Schuurman J, Parren PW, Heck AJ. Molecular Basis of Assembly and Activation of Complement Component C1 in Complex with Immunoglobulin G1 and Antigen. Mol Cell. 2016;63(1):135–45. doi: 10.1016/j.molcel.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 27.Houde D, Peng YC, Berkowitz SA, Engen JR. Post-translational Modifications Differentially Affect IgG1 Conformation and Receptor Binding. Mol Cell Proteomics. 1716-1728;2010:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol. 2008;20(4):471–78. doi: 10.1016/j.coi.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 29.Peschke B, Keller CW, Weber P, Quast I, Lunemann JD. Fc-Galactosylation of Human Immunoglobulin Gamma Isotypes Improves C1q Binding and Enhances Complement-Dependent Cytotoxicity. Front Immunol. 2017;8:646. doi: 10.3389/fimmu.2017.00646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomann M, Schlothauer T, Dashivets T, Malik S, Avenal C, Bulau P, Ruger P, Reusch D. In vitro glycoengineering of IgG1 and its effect on Fc receptor binding and ADCC activity. PLoS One. 2015;10(8):e0134949. doi: 10.1371/journal.pone.0134949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Idusogie EE, Presta LG, Gazzano-Santoro H, Totpal K, Wong PY, Ultsch M, Meng YG, Mulkerrin MG. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J Immunol. 2000;164(8):4178–84. doi: 10.4049/jimmunol.164.8.4178. [DOI] [PubMed] [Google Scholar]
- 32.Aoyama M, Hashii N, Tsukimura W, Osumi K, Harazono A, Tada M, Kiyoshi M, Matsuda A, Ishii-Watabe A. Effects of terminal galactose residues in mannose alpha1-6 arm of Fc-glycan on the effector functions of therapeutic monoclonal antibodies. MAbs. 2019;11(5):826–36. doi: 10.1080/19420862.2019.1608143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hooker AD, James DC. Analysis of glycoprotein heterogeneity by capillary electrophoresis and mass spectrometry. Mol Biotechnol. 2000;14(3):241–49. doi: 10.1385/MB:14:3:241 [DOI] [PubMed] [Google Scholar]
- 34.Strasser J, De Jong RN, Beurskens FJ, Wang G, Heck AJR, Schuurman J, Parren P, Hinterdorfer P, Preiner J. Unraveling the macromolecular pathways of igg oligomerization and complement activation on antigenic surfaces. Nano Lett. 2019;19(7):4787–96. doi: 10.1021/acs.nanolett.9b02220. [DOI] [PubMed] [Google Scholar]
- 35. Walters BT, Jensen PF, Larraillet V, Lin K, Patapoff T, Schlothauer T, Rand KD, Zhang J. Conformational Destabilization of Immunoglobulin G Increases the Low pH Binding Affinity with the Neonatal Fc Receptor. J Biol Chem. 1817-25;2016:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davies AM, Rispens T, Ooijevaar-de Heer P, Gould HJ, Jefferis R, Aalberse RC, Sutton BJ. Structural determinants of unique properties of human IgG4-Fc. J Mol Biol. 2014;426(3):630–44. doi: 10.1016/j.jmb.2013.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dyachenko A, Wang G, Belov M, Makarov A, De Jong RN, Van Den Bremer ET, Parren PW, Heck AJ. Tandem native mass-spectrometry on antibody-drug conjugates and submillion da antibody-antigen protein assemblies on an orbitrap emr equipped with a high-mass quadrupole mass selector. Anal Chem. 2015;87(12):6095–102. doi: 10.1021/acs.analchem.5b00788. [DOI] [PubMed] [Google Scholar]
- 38.Heidemann J, Kolbel K, Konijnenberg A, Van Dyck J, Garcia-Alai M, Meijers R, Sobott F, Uetrecht C. Further insights from structural mass spectrometry into endocytosis adaptor protein assemblies. Int J Mass Spectrom. 2020;447. doi: 10.1016/j.ijms.2019.116240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang HM, Li C, Lei M, Lundin V, Lee HY, Ninonuevo M, Lin K, Han GH, Sandoval W, Lei DS, et al. Structural and functional characterization of a hole-hole homodimer variant in a “Knob-Into-Hole” bispecific antibody. Anal Chem. 2017;89(24):13494–501. doi: 10.1021/acs.analchem.7b03830. [DOI] [PubMed] [Google Scholar]
- 40.Liu PL, Gao X, Lundin V, Shi C, Adem Y, Lin K, Jiang GY, Kao YH, Yang F, Michels D, et al. Probing the impact of the knob-into-hole mutations on the structure and function of a therapeutic antibody. Anal Chem. 2020;92(1):1582–88. doi: 10.1021/acs.analchem.9b04855. [DOI] [PubMed] [Google Scholar]
- 41.Kan ZY, Mayne L, Chetty PS, Englander SW. ExMS: Data analysis for HX-MS experiments. J Am Soc Mass Spectr. 1906-1915;2011:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Q, Willison LN, Tripathi P, Sathe SK, Roux KH, Emmett MR, Blakney GT, Zhang HM, Marshall AG. Epitope mapping of a 95 kda antigen in complex with antibody by solution-phase amide backbone hydrogen/deuterium exchange monitored by fourier transform ion cyclotron resonance mass spectrometry. Anal Chem. 2011;83(18):7129–36. doi: 10.1021/ac201501z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marty MT, Baldwin AJ, Marklund EG, Hochberg GKA, Benesch JLP, Robinson CV. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal Chem. 2015;87(8):4370–76. doi: 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 1859-65;2008:11. [DOI] [PubMed] [Google Scholar]
- 45.Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, et al. Generator for NAMD, GROMACS, AMBER, openMM, and CHARMM/openMM simulations using the CHARMM36 additive force field. J Chem Theory Comput. 2016;12(1):405–13. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, De Groot BL, Grubmuller H, MacKerell AD Jr.. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods. 2017;14(1):71–73. doi: 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guvench O, Greene SN, Kamath G, Brady JW, Venable RM, Pastor RW, Mackerell AD Jr.. Additive empirical force field for hexopyranose monosaccharides. J Comput Chem. 2008;29(15):2543–64. doi: 10.1002/jcc.21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79(2):926–35. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 49.Le Grand S, Gotz AW, Walker RC. SPFP: speed without compromise-A mixed precision model for GPU accelerated molecular dynamics simulations. Comput Phys Commun. 2013;184(2):374–80. doi: 10.1016/j.cpc.2012.09.022. [DOI] [Google Scholar]
- 50.Hopkins CW, Le Grand S, Walker RC, Roitberg AE. Long-time-step molecular dynamics through hydrogen mass repartitioning. J Chem Theory Comput. 1864-1874;2015:4. [DOI] [PubMed] [Google Scholar]
- 51.Pastor RW, Brooks BR, Szabo A. An analysis of the accuracy of langevin and molecular-dynamics algorithms. Mol Phys. 1988;65(6):1409–19. doi: 10.1080/00268978800101881. [DOI] [Google Scholar]
- 52.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-integration of cartesian equations of motion of a system with constraints - molecular-dynamics of n-alkanes. J Comput Phys. 1977;23(3):327–41. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- 53.Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9(7):3084–95. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.