

ABSTRACT
SARS-CoV-2 is the cause of the current global pandemic of COVID-19; this virus infects multiple organs, such as the lungs and gastrointestinal tract. The microbiome in these organs, including the bacteriome and virome, responds to infection and might also influence disease progression and treatment outcome. In a cohort of 13 COVID-19 patients in Beijing, China, we observed that the gut virome and bacteriome in the COVID-19 patients were notably different from those of five healthy controls. We identified a bacterial dysbiosis signature by observing reduced diversity and viral shifts in patients, and among the patients, the bacterial/viral compositions were different between patients of different severities, although these differences are not entirely distinguishable from the effect of antibiotics. Severe cases of COVID-19 exhibited a greater abundance of opportunistic pathogens but were depleted for butyrate-producing groups of bacteria compared with mild to moderate cases. We replicated our findings in a mouse COVID-19 model, confirmed virome differences and bacteriome dysbiosis due to SARS-CoV-2 infection, and observed that immune/infection-related genes were differentially expressed in gut epithelial cells during infection, possibly explaining the virome and bacteriome dynamics. Our results suggest that the components of the microbiome, including the bacteriome and virome, are affected by SARS-CoV-2 infections, while their compositional signatures could reflect or even contribute to disease severity and recovery processes.
KEYWORDS: COVID-19, genetic mutation, virome, bacteriome, dysbiosis
Introduction
The current pandemic of coronavirus disease 2019 (COVID-19), which is caused by the recently emerged severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has led to more than seventy million infections and one million fatalities as of December 2020. While a number of vaccines are in clinical trials,1–3 the number of infections and fatalities will continue to increase for a substantial length of time and place considerable health and economic burdens on the entire international community. Several medications have also been tested for their efficacy against COVID-19, including remdesivir4 and hydroxychloroquine,5 but none has shown promising results in completed clinical trials. It is therefore likely that clinical handling of COVID-19 will become routine in most countries, and addressing many complications of this disease, such as cytokine storm and organ failure in severe cases or long-term carriage and reactivation of the virus in treated patients,6–8 will require a substantial amount of research into the characteristics of patients under treatments.
While COVID-19 primarily manifests in the respiratory system by pneumonia, the virus can infect many other organs, especially the gastrointestinal (GI) system.9 Studies have reported that GI symptoms, usually diarrhea, can predate respiratory manifestations,10,11 and after viruses are cleared from respiratory systems in treated patients, they can still be detected in fecal samples or rectal swabs for several more days.12 Fecal-oral transmission is also indicated by recent epidemiological analysis13 and requires consideration in clinical treatment, as well as in efforts to prevent viral spread. The gut microbiome, which is an important component of the function of the GI system, is well recognized for its importance in participating in metabolism and immune modulations,14,15 and compositional and functional homeostasis of the gut microbiome is necessary to maintain proper immune functions and defend against infections.16 The antiviral effects of the gut microbiome can be long-reaching, as the gut microbiome not only affects viral infections in the GI system17 but can also produce metabolites or influence different lymphocytes to modulate influenza infections in the lung.18 For COVID-19, there is currently no evidence of a direct causal relationship between the bacteriome and COVID-19 susceptibility and severity, but studies have suggested that COVID-19 severity in patients is indeed highly correlated with the bacteriome, which can be utilized as a biomarker.19 Dysbiosis in the gut bacteriome has been reported in patients already infected with SARS-CoV-2, and shifts in the relative abundance of the patient’s intestinal bacteriome during treatment compared with healthy controls suggest potential recovery.20
Another frequently neglected aspect of the GI system that is attracting increasing attention is the gut virome, the collection of large numbers of phages and eukaryotic viruses that infects the host.21,22 On the one hand, phages prey on bacteria/archaea and compose a dynamic circle of interaction, and in a number of diseases in which microbial dysbiosis is prominent, virome shifts and potential “dysbiosis” can also be observed. On the other hand, eukaryotic viruses also establish a degree of symbiosis with the host and modulate host immune responses and may leverage the host response to infection and/or bacteriome shifts.23 SARS-CoV-2 infects host intestinal epithelial cells and may also place stress on the immune microenvironment in the gut24 which, in turn, may lead to alterations in the virome, and investigating virome changes may help to elucidate their connections to bacteriome alterations and potential recovery. With improvements in the enrichment method of virus-like particles (VLPs) combined with metagenomic sequencing, insights into the virome may serve to deepen our knowledge of the delicate interactions among host immune system, gut bacteriome and viruses.
In our research, we analyzed data from 13 patients who were admitted for COVID-19 and analyzed the genomic signature of viruses in the GI tract compared with that of the respiratory system; more importantly, we profiled the bacteriome/virome shifts that occurred in patients during treatment and demonstrated that they synchronize in alternations and respond distinctly to antibiotic treatments. Many of the critical clinical parameters were determined to be associated with bacteriome/virome composition, which might eventually contribute to the understanding of the disease and management of patients.
Results
Cohort description
In our study, 13 patients with COVID-19 were recruited, and 37 fecal samples were collected. The patients included seven females and six males, and their median age was 48 years old (range: 15 to 85). The mean body mass index (BMI, kg/m2) of these patients was 23.8, with five patients being overweight (> 25). Among the 13 COVID-19 patients, eight were antibiotic-naive and the remaining five were treated with antibiotics (Table 1). Based on the condition of the patients prior to their hospitalization, the disease severity of these subjects was classified into three categories, namely severe (3), moderate (7) and mild (3). Additionally, to compare the dynamics of the bacteriome/virome between healthy people and COVID-19 patients, samples of five healthy individuals from our previous study25 were selected as healthy controls in this work.
Table 1.
Cohort information of patients with COVID-19, included in our study
| ID | Gender | Age | BMI | Antibiotics | Antiviral drugs | Comorbidities | Disease severity | GI symptoms (duration) |
|---|---|---|---|---|---|---|---|---|
| BJ-1 | Male | 45 | 24.8 | Moxifloxacin | LPV/r | Hypertension | Moderate | Diarrhea (three days) |
| BJ-3 | Male | 15 | 28.69 | Not used | LPV/r | None | Mild | Nil |
| BJ-4 | Male | 67 | 24.69 | Not used | Nil | Hypertension | Moderate | Nil |
| BJ-5 | Male | 48 | 24.85 | Piperacillin/ tazobactam, Cefuroxime |
LPV/r | None | Severe | Abdominal distention (two days) |
| BJ-9 | Male | 47 | 18.93 | Not used | LPV/r | Hyperthyroidism | Mild | Nil |
| BJ-10 | Male | 67 | 26.29 | Moxifloxacin, Piperacillin/ tazobactam |
Arbidol | Gallstones | Severe | Diarrhea (three days) |
| BJ-2 | Female | 39 | 21.08 | Not used | LPV/r, Ribavirin | None | Moderate | Diarrhea (two day) |
| BJ-6 | Female | 54 | 27.47 | Not used | LPV/r, Arbidol | None | Moderate | Nil |
| BJ-7 | Female | 65 | 21.57 | Not used | Nil | Hypertension | Moderate | Nil |
| BJ-8 | Female | 85 | 25.8 | Levofloxacine | Arbidol | Arthritis | Severe | Nil |
| BJ-11 | Female | 69 | 19.47 | Not used | LPV/r | Hypertension | Moderate | Constipation (three days) |
| BJ-12 | Female | 44 | 20.56 | Moxifloxacin | LPV/r | None | Moderate | Diarrhea (three days) |
| BJ-13 | Female | 41 | 26.3 | Not used | LPV/r | None | Mild | Nil |
LPV/r: Lopinavir/Ritonavir
To investigate the evolution dynamics of the SARS-CoV-2 genome among patients in our cohort, we utilized next-generation sequencing (NGS) to sequenced the whole genome of the SARS-CoV-2 virus after amplification with designed primer pools using viral DNA obtained from throat swabs (three patients) and/or fecal samples (six patients) from 13 COVID19 patients. In the end, we obtained a total of 17 SARS-CoV-2 genomes for further analysis. Notably, for two patients, the whole genome of SARS-CoV-2 were obtained from both throat swabs and fecal samples (Table S1).
Viral genome in patients respiratory tracts vs GI tracts
Across the 17 genomes, the average coverage was 97.1% (ranging from 80.5% to 99.9%). The mean coverage depth was 21,628 X (ranging from 3,870 X to 41,890 X) across the whole genome per sample, which was large enough to meet the requirement for SNP calling26 (Table S1). Using Illumina reads and the first viral genome sequence (NC_045512) as a reference, we identified 68 highly confident SNPs in total, 31 of which were fixed in all genomes. Among the remaining 37 SNPs, 24 were nonsynonymous and led to amino acid changes in the ORF1ab (15), S (4), ORF3a (2), M (2) and N (1) gene products (Table S2).
Close inspection of genetic variants in individual patients demonstrated low diversity and mutation rates in SARS-CoV-2. Clustering analysis based on the SNPs identified in all 17 genomes within 7 patients demonstrated that the majority of the sequenced genomes belonged to the same clade, while only one sample with relatively few mutations clustered with the viral reference (Figure 1a). Similar results were observed in two individual patients for whom both respiratory and intestinal SARS-CoV-2 genomes were obtained, suggesting that the SARS-CoV-2 genomes in the respiratory and intestinal tracts of patients, if both detectable, originated from the same strain (Figure 1b).
Figure 1.
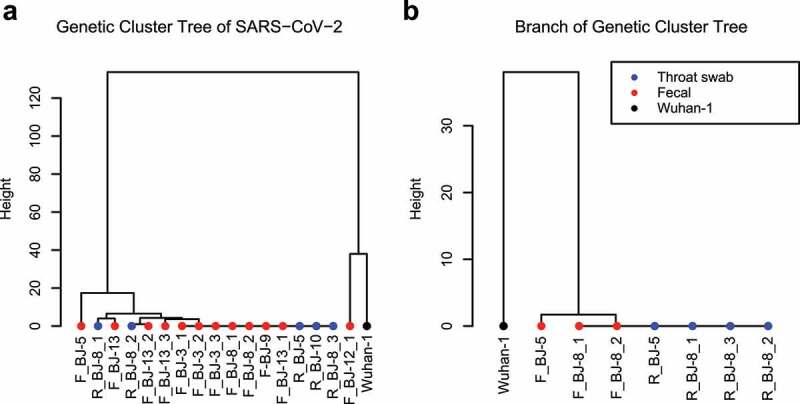
Overview of genomic SNP mutations in the SARS-CoV-2 genomes reconstructed in our study. (a) Clustering of SARS-CoV-2 genomes using Manhattan distances of total SNPs. Blue solid circles represent throat swab samples; red solid circles represent fecal samples; Wuhan-1 (NC_045512) was used as the outgroup. (b) A subbranch tree of (a), showing genomes of SARS-CoV-2 from throat swabs and fecal samples collected in two patients who had both
We further examined the genomic variations of SARS-CoV-2 and their correlation with different disease severities in our cohort. Among the patients, we observed three different categories regarding the severity of cases, namely, severe (3), moderate (1) and mild (3) cases. The patients did not exhibit significant differences in terms of anthropometric measures, including age (P = .42, Kruskal-Wallis test), and sex (P = .66, Kruskal-Wallis test), and when we determined the frequencies of SNPs identified in the genomic analysis, we observed that no SNPs were significantly enriched in any of the groups (P > .05, Fisher’s exact test). A recent human genetic study identified key host genetic variations and their correlations with disease severity,27 while in contrast, our study was not able to identify specific genetic mutations of SARS-CoV-2 that may contributing to disease severity in our cohort.
Virome composition and shifts along treatment
To investigate viral components of the GI tract and, more importantly, potential shifts along treatment with antibiotics and antiviral medications (details in Table 1) within COVID-19 patients, we specifically analyzed the virome in our cohort using an integrated viral genome database as a reference (methods). Viral DNA and RNA were purified from preparations of enriched virus-like particles (VLPs) obtained from stool samples and characterized by metagenomic sequencing based on methods developed in our lab.25 As expected, the viromes of COVID-19 patients include DNA/RNA viruses (Herelleviridae family, Virgaviridae family), bacteriophages (Caudovirales order, CrAss-like phage, Inoviridae family, Microviridae family, Myoviridae family, Podoviridae family, and Siphoviridae family) that infect bacterial cells, and some unclassified viruses in the gut (Figure 2a). Despite the significant individual differences in viral composition, shared viruses were observed to primarily belong to the Microviridae family. SARS-CoV-2 nucletides were determined to be notably low in relative abundance compared with VLP-originated nucleotides and could not be detected in the majority of patients (reaching coverage ≥ 30% and read number ≥ 10 in only one patient).
Figure 2.
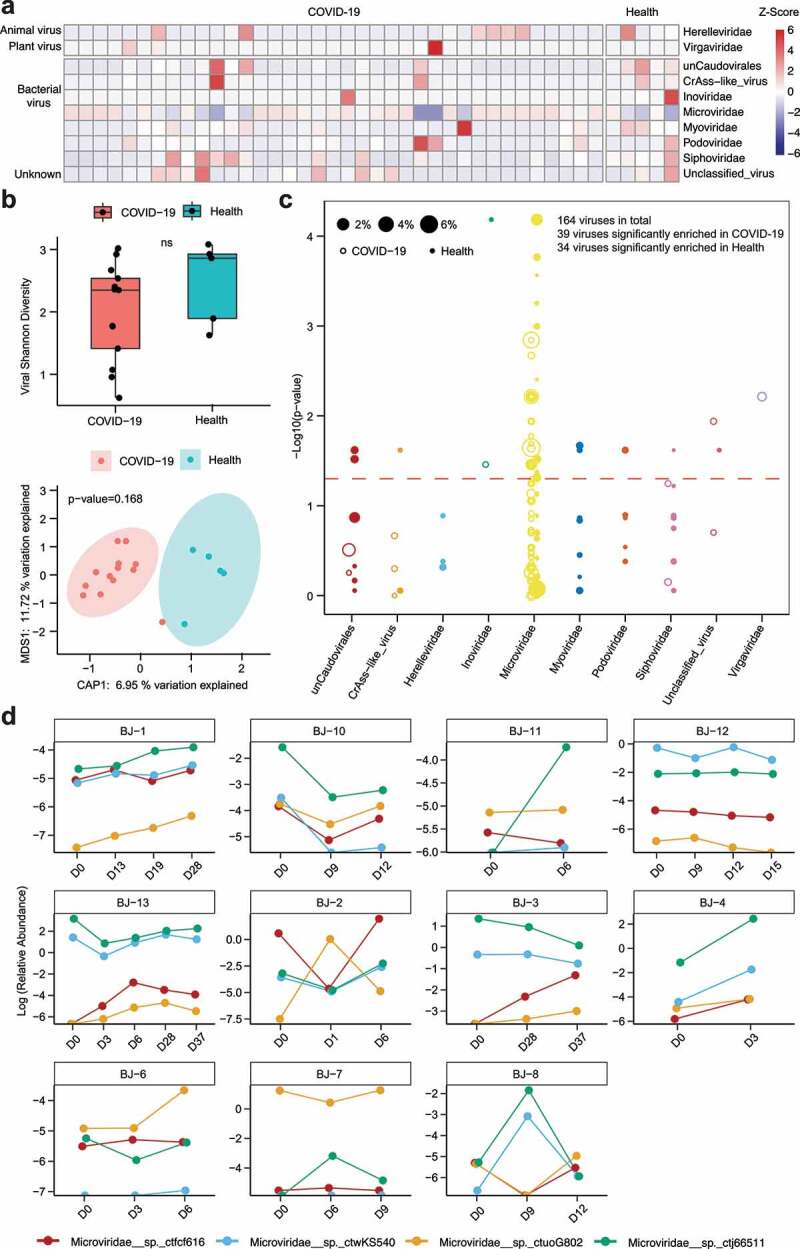
Composition and alterations of viral communities among COVID-19 patients and healthy controls. (a) Heatmap showing viral composition in COVID-19 patients’ fecal samples (n = 37) and healthy controls. The relative abundance of viral communities was normalized to Z-Scores. (b) Alpha – and beta-diversity of the virome between COVID-19 patients (n = 13) and healthy controls are shown with the Shannon index and constrained PCoA analysis based on Bray-Curtis dissimilarity, respectively. Red points represent COVID-19 patients, and blue solid circles represent healthy controls. (c) Comparison of viral abundance between COVID-19 patients and healthy controls. In this bubble plot, each dot represents one species of virus on the x-axis, clustered by their respective order, and the y-axis value denotes the inverse log10 p value, with those above the dashed line (p < .05) significantly different between healthy controls and patients. The size of each dot corresponds to the mean relative abundance in both COVID-19 patients and healthy controls. Hollow circles represent viruses enriched in patients with COVID-19, while solid circles represent viruses enriched in healthy individuals. (d) Dynamic changes of differential viral relative abundance along treatment within COVID-19 patients fecal samples
Alpha – and beta-diversity analyses based on the Shannon index and Bray-Curtis distance, respectively, showed no significant differences between COVID-19 patients and healthy controls regarding the composition of the baseline virome (Figure 2b, P = .168, PERMANOVA). Comparison of baseline viromes from COVID-19 patients with healthy individuals using the Wilcoxon rank-sum test showed that the abundance of some phages (including Inviridae and Microviridae) and a plant-RNA virus (Virgaviridae) identified by blastn as cucumber green mottle mosaic virus (CGMMV) as well as unclassified viruses were significantly higher in COVID-19 patients than in healthy subjects (Figure 2c). Notably, we found that the virome was relatively stable across multiple time points in patients (Fig. S1a). To study shifts in viral abundance over the course of treatment, we traced viral groups that differed significantly between COVID-19 patients and healthy controls and found that the alteration in viral abundance over time was inconsistent (Figure 2d). Nevertheless, we found a strong correlation between the composition of viral and bacterial communities in both COVID-19 patients and healthy controls, as determined by Procrustes analysis (Fig. S2a, P = .001). Additional network analysis demonstrated that bacterial species, including Bacteroides vulgatus, Faecalibacterium prausnitzii, and Ruminococcus gnavus, and three Microviridae bacteriophages, that is, Microviridae__sp._ctmin955, Microviridae__sp._ctvbz116, and Microviridae__sp.ctjA9876, compose central network nodes and potential keystone species that could play important roles in mediating the interactions between the viral and bacterial communities (Fig. S2b); none of the eukaryotic viruses, however, was determined to be central in terms of network structure.
Bacteriome shifts along treatment
To determine the impact of COVID-19 on the bacterial communities in the gut, we compared the compositions of gut bacterial communities in COVID-19 patients with those of healthy controls. In keeping with published studies,19,20 our initial finding was that the intestinal bacterial diversity of COVID-19 patients was significantly lower than that of healthy controls (Figure 3a, P < .05, Wilcoxon test), thereby exhibiting the signs of potential dysbiosis. Additionally, COVID-19 patients treated with antibiotics showed a trend of lower bacterial diversity than patients who did not receive antibiotics (Figure 3b), suggesting that antibiotic confounding effects be considered in future studies of gut bacteriome shifts in COVID-19 patients. Principal coordinate analysis (PCoA) based on Bray-Curtis distance showed a significant difference in the gut bacteriome between COVID-19 patients and healthy controls (Figure 3c, PERMANOVA, P = .004). Compared to healthy controls, bacterial abundance differential analysis showed that Ruminococcus gnavus, Eggerthella, Coprobacillus, Lachnospiraceae bacterium 2_1_58FAA, Clostridium ramosum, Eggerthella lenta and Lachnospiraceae bacterium 1_4_56FAA were significantly enriched in COVID-19 patients, while Alistipes_sp_AP11, Roseburia intestinalis, Burkholderiales bacterium 1_1_47, Eubacterium_hallii, Parasutterella_excrementihominis, Alistipes indistinctus, Coprobacter fastidiosus, Eubacterium eligens, Bacterioidales bacterium ph8, Bacterioides salyersiae, Odoribacter splanchnicus, Alistipes shahii, Ruminococcus bromii and Bacteroides massiliensis were significantly depleted (Figure 3d, P < .05). Notably, we also observed that the patients had a relatively stable bacteriome across multiple time points (Fig. S1b).
Figure 3.
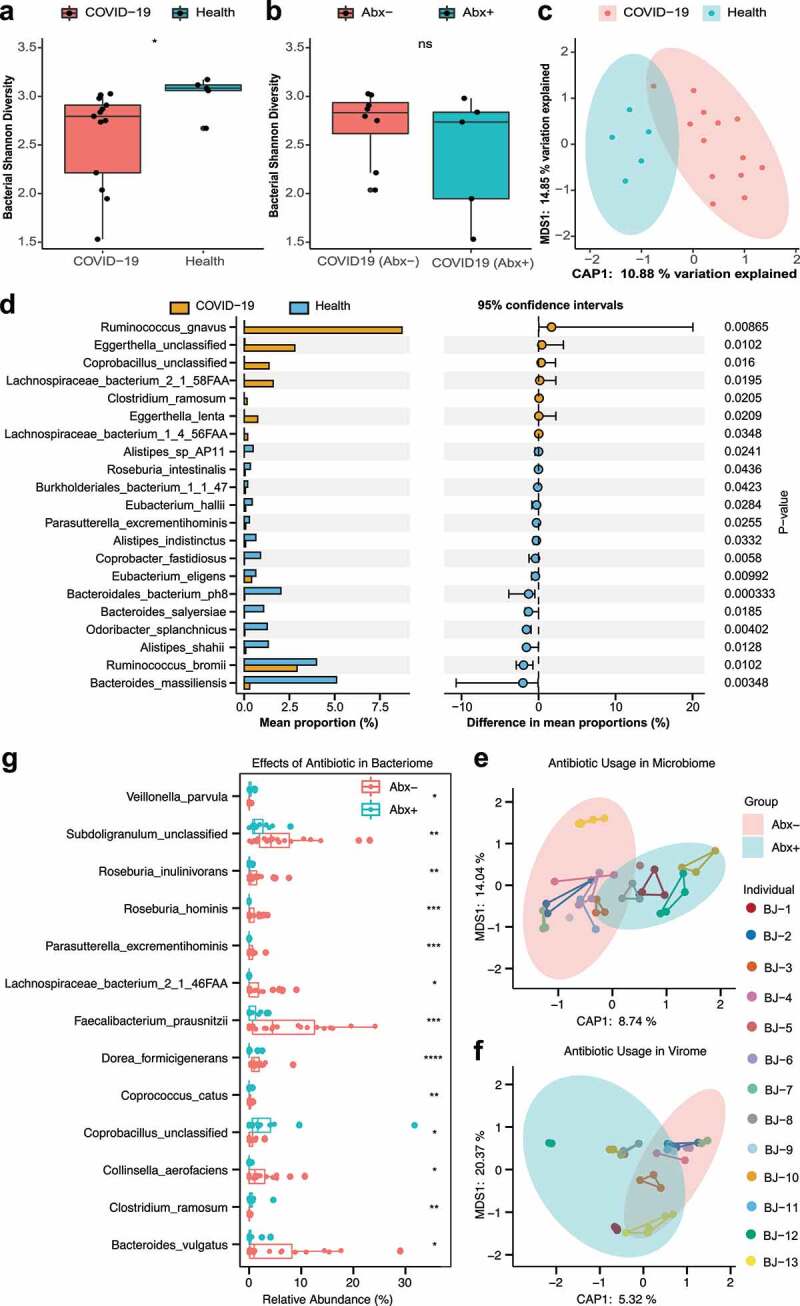
Alteration of bacterial communities and impact of antibiotics in COVID patients. (a) Box plot of bacterial Shannon diversity among COVID-19 patients and healthy controls. * represents P < .05 (Wilcoxon rank-sum test). (b) Box plot of bacterial Shannon diversity among COVID-19 patients without antibiotics (n = 8) and with antibiotics (n = 5). Abx– and Abx+ represent patients who were not treated with antibiotics and those who were treated with antibiotics during hospitalization, respectively (Wilcoxon rank-sum test). (c) Constrained PCoA plot based on Bray-Curtis dissimilarity between COVID-19 patients and healthy controls, P = .004, PERMANOVA. (d) Bar plot of differential bacterial communities among COVID-19 patients and healthy controls, which was performed using the Wilcoxon rank-sum test (P < .05). The orange bar represents fecal samples from patients with COVID-19, and blue bar represents healthy controls. (e) Constrained PCoA plot of the bacteriome in patient fecal samples treated with antibiotics and patient fecal samples without antibiotics using Bray-Curtis dissimilarity, P = .001, PERMANOVA. Different samples of the same individual are connected by the same colored line. (f) Constrained PCoA plot of viral communities in patients’ fecal samples treated with antibiotics and patients’ fecal samples without antibiotics using Bray-Curtis dissimilarity, P = .021, PERMANOVA. (g) Boxplot of differential bacterial communities identified by MaAsLin2 within COVID-19 patient fecal samples treated with/without antibiotics. (Wilcoxon rank-sum test). * P < .05; ** P < .01; *** P < .001; **** P < .0001; colors: red represents patients treated without antibiotics while blue represents patients treated with antibiotics
Subsequently, we traced bacterial groups that differed between healthy controls and COVID-19 patients and studied their dynamics during treatment with antibiotics and antiviral medications (details in Table 1). Among the enriched bacterial communities in COVID-19 patients, Lachnospiraceae bacterium 2_1_58FAA and Ruminococcus gnavus showed signs of decrease over the course of treatment, except in three individuals, while other bacteria fluctuated over time and exhibited no clear pattern (Fig. S3a). Similarly, among depleted bacterial groups in COVID-19 patients, there were no signs of recovery of bacteria (Fig. S3b). However, bacterial diversity did significantly increase over the course of treatment in three individuals out of 11 patients, indicating a potential recovery of bacterial composition and functionality; the effects of this recovery on the host remain to be explored, but previous studies did suggest that an increase in bacterial diversity signatures improved host health status28 (Fig. S4).
Responses of the bacteriome and virome to antibiotics
Previous studies have shown that the usage of antibiotics has a significant effect on bacterial abundance in the human gut,29 but little is known about the effect on viral abundance, particularly the effect of antibiotics in treating viral infections. Using PCoA analysis of bacterial abundance based on Bray-Curtis distance, we first observed that there was a significant difference between COVID-19 patients using antibiotics and those not using antibiotics (Figure 3e, P = .001, PERMANOVA). In addition, a similar analysis also identified significant differences in the composition of the virome within COVID-19 patients (Figure 3f, P = .021, PERMANOVA).
To investigate the specific effect of antibiotics on the abundance of bacterial and viral communities in COVID-19 patient fecal samples, we utilized MaAsLin2 (multivariate analysis by linear models) to identify differential bacterial and viral species. Using this method, ten species, including Subdoligranulum, Roseburia inulinivorans, Roseburia hominis, Parasutterella excrementihominis, Lachnospiraceae bacterium 2_1_46FAA, Faecalibacterium prausnitzii, Dorea formicigenerans, Coprococcus catus, Collinsella aerofaciens and Bacteroides vulgatus, showed a significant decrease correlated with antibiotic usage, while Veillonella parvula, Coprobacillus and Clostridium ramosum showed a significant increase, and no virus was identified as a differential species between with antibiotic-treated and antibiotic-untreated in COVID-19 patients (Figure 3g, P < .05, Wilcoxon test).
Clinical parameters associated with bacteriome/virome
Next, we systematically analyzed COVID-19 patients’ clinical information to further investigate the potential contributions of the gut bacteriome/virome to host health and recovery. The clinical data that we examined included disease severity (severe, moderate and mild), chemical characteristics of patient blood collected on the same day as fecal sampling, and profile of lymphocyte cells. In total, we collected clinical data from 18 samples from ten patients regarding nine routine blood parameters and five immune cell counts (Table S3).
PCoA based on Bray-Curtis distance showed significant differences in bacterial communities among disease severities (Figure 4a, P = .001, PERMANOVA), as did the viral communities (Figure 4b, P = .001, PERMANOVA). Next, we performed a species – and genus-level differential enrichment analysis of bacterial and viral communities within COVID-19 cases of varying disease severity. At the species level, a total of 19 bacterial taxa were observed to be enriched in various disease severities. Eighteen out of 19 showed significant enrichment in severe cases of COVID-19, including Corynebacterium durum, Rothia mucilaginosa, Enterococcus faecium, and Campylobacter gracilis, while only Eubacterium rectale showed significant enrichment in mild cases of COVID-19 (Table 2, FDR < 0.05, Wilcoxon test). At the genus level, such genera as Corynebacterium, Enterococcus, Rothia, Megasphaera, and Campylobacter were significantly enriched in severe cases, while Eubacterium was depleted in severe cases (Table S4). Additionally, 17 viruses, including 14 Microviridae phages, one Inoviridae phage, one Podoviridae phage and one unclassified virus, were enriched in severe cases, while no viral communities were determined to be enriched in mild cases (Table S5, FDR < 0.05, Wilcoxon test).
Figure 4.
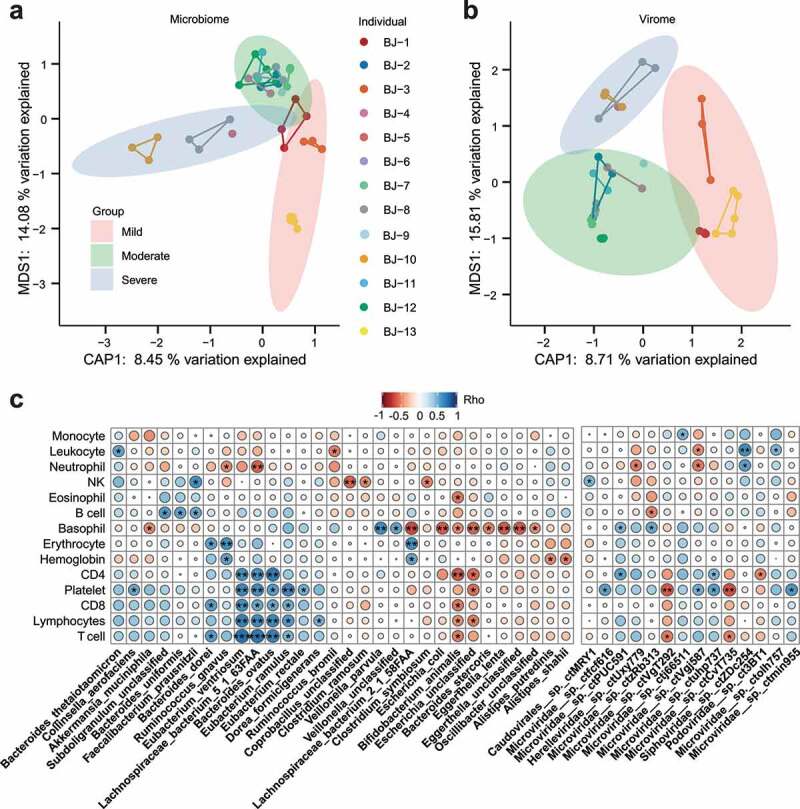
Correlation of clinical information with bacterial and viral communities. (a) Constrained PCoA analysis for bacterial communities of COVID-19 patients’ fecal samples with three categories of disease severity, P = .001, PERMANOVA. Different samples of the same individual are connected by the same colored line. (b) Constrained PCoA analysis for viral communities of COVID-19 patients’ fecal samples with three categories of disease severity, P = .001, PERMANOVA. (c) Correlation heatmap between clinical information of COVID-19 and bacteriome/virome relative abundance with Spearman’s rank correlation coefficient. Red circles represent Rho < 0, blue circles represent Rho > 0, and the circle radius reflects the correlation coefficient (Rho). * P < .05; ** P < .01
Table 2.
Bacterial groups with significant correlation (FDR < 0.05) with COVID-19 disease severity, identified with Wilcoxon rank-sum test
| Correlation | Species | p-value | FDR |
|---|---|---|---|
| Enriched in severe | s__Corynebacterium_durum | 9.7006E-05 | 0.01108106 |
| s__Rothia_mucilaginosa | 0.000105061 | 0.01108106 | |
| s__Enterococcus_faecium | 6.68909E-05 | 0.01108106 | |
| s__Campylobacter_gracilis | 0.000129225 | 0.01108106 | |
| s__Corynebacterium_glucuronolyticum | 0.001274125 | 0.030395375 | |
| s__Rothia_aeria | 0.001274125 | 0.030395375 | |
| s__Alloscardovia_omnicolens | 0.001118065 | 0.030395375 | |
| s__Enterococcus_avium | 0.001274125 | 0.030395375 | |
| s__Enterococcus_casseliflavus | 0.001118065 | 0.030395375 | |
| s__Leuconostoc_lactis | 0.001274125 | 0.030395375 | |
| s__Weissella_confusa | 0.001274125 | 0.030395375 | |
| s__Delftia_unclassified | 0.001393266 | 0.030395375 | |
| s__Eikenella_corrodens | 0.001274125 | 0.030395375 | |
| s__Campylobacter_concisus | 0.001274125 | 0.030395375 | |
| s__Citrobacter_freundii | 0.00141786 | 0.030395375 | |
| s__Enterobacter_aerogenes | 0.001274125 | 0.030395375 | |
| s__Streptococcus_infantis | 0.002441794 | 0.046529735 | |
| s__Megasphaera_micronuciformis | 0.002351595 | 0.046529735 | |
| Enriched in mild | s__Eubacterium_rectale | 9.04381E-05 | 0.031020272 |
Through Spearman’s correlation between the abundance of microbial communities and clinical data, three bacteria, Bacteroides ovatus, Lachnospiraceae bacterium 5_1_63FAA and Eubacterium ventriosum, were found to be positively correlated with inflammatory immune clinical parameters, such as CD4, CD8, lymphocytes and T cells, indicating that they may be potential proinflammatory agents (Figure 4c), while only one bacterial species, Faecalibacterium prausnizii, was positively correlated with NK cells. At the same time, we observed that Bifidobacterium animalis and Escherichia were negatively correlated with CD4, CD8, lymphocytes and T cells, while Corprobacillus, Clostridium ramosum and Clostridium symbiosum showed a significant negative association with NK cells. Notably, Bacteroides unifromis, Faecalibacterium prausnitzii and Subdoligranulum were determined to be positively associated with B cells. In the virome, we observed that one Caudovirales phage (Caudovirales__sp._ctMRY1) and two Microviridae phages (Microviridae__sp._ctPUC591, Microviridae__sp._ctuhp737) were significantly positively associated with NK cells and CD4, respectively, while another two Microviridae phages (Microviridae__sp.__ctVgT292, Microviridae__sp._ctCvT735) were significantly negatively associated with T cells (Figure 4c, P < .05).
Virome and bacteriome in SARS-CoV-2-infected mouse model
To further explore the potential mechanisms governing the bacteriome/virome in mice infected with SARS-CoV-2, we performed additional analysis on the intestinal contents of nine SARS-CoV-2-infected hACE2 transgenic mice, the most common model used currently for SARS-CoV-2 studies. Among these mice, four first were vaccinated using full-length spike protein from SARS-CoV-2 as an experimental vaccine (see Methods), and the remaining five were unvaccinated. After infection with SARS-CoV-2, the vaccinated group showed a decrease of more than 223-fold in viral load in the lung tissue compared to the unvaccinated group; these nine mice can be regarded as infected (vaccinated) vs. infected (unvaccinated) mice. Using second-generation sequencing, we obtained the metagenomic and metatranscriptomic data of the abovementioned nine mice and performed comparative analysis regarding the virome and bacteriome.
We explored the virome in mice through metagenomic and metatranscriptomic sequencing using an integrated viral genome database (see Methods); VLP enrichment was not performed, since handling of the materials was restricted to the facilities of the Biosafety Level III lab; instead, we used deep sequencing to mine the viruses contained in the intestinal content materials of the mice. Although SARS-CoV-2 infection was limited to lung tissues in our model, and it was not detected in the intestinal content materials, we identified various viruses in the mouse metagenomic and metatranscriptomic data, including Myoviridae, Siphoviridae and Podoviridae belonging to the order Caudoviridales, CrAss-like phage, Retroviridae and unclassified viruses (Figure 5a). Comparison of Shannon diversity index of viromes demonstrated higher diversity in infected (unvaccinated) mice than infected (vaccinated) mice in both metagenomic data and metatranscriptomic data, while the application of PCoA based on Bray-Curtis distance showed significantly differences between viromes in mouse metagenomic data but not in metatranscriptomic data (Figure 5b, PERMANOVA, P = .02). Compared to infected (vaccinated) mice, differential viral analysis showed that ten viruses belonging to Caudovirales order (three Siphoviridae, one Podoviridae, five Myoviridae and one phage of unknown family level classification) were significantly enriched, and three viruses (one unclassified virus, one Myoviridae and one phage of unknown family level classification) were depleted, in infected (unvaccinated) mice in metagenomic data, while only one phage (Podoviridae) in metatranscriptomic data was observed to be significantly enriched in infected (unvaccinated) mice (Figure 5c, P < .05, Wilcoxon test).
Figure 5.
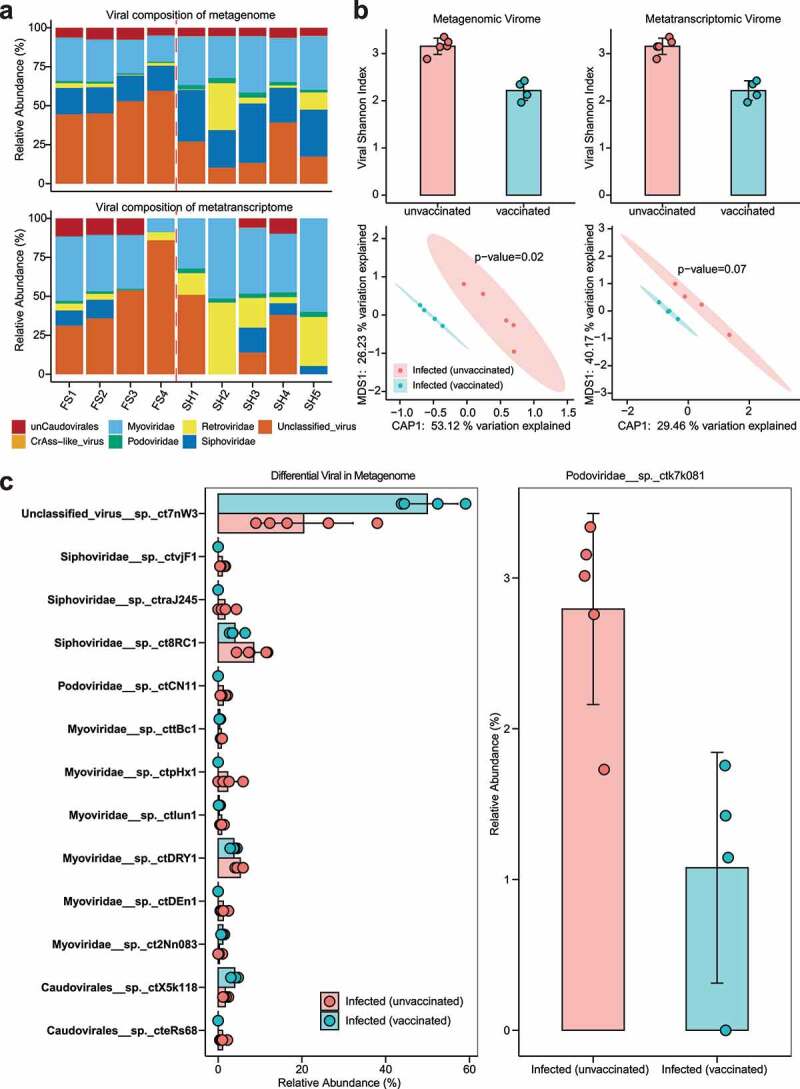
Composition and alteration of the virome between infected (unvaccinated) mice and infected (vaccinated) mice. (a) Composition of mouse viruses based on integrated viral genome databases in metagenomic and metatranscriptomic data. The left side of the dashed red line represents infected (vaccinated) mice, and the right side represents infected (unvaccinated) mice. “unCaudovirales” represents an unknown family under the order Caudovirales. (b) Alpha – and beta-diversity of the virome in infected (unvaccinated) and infected (vaccinated) mice based on metagenomic and metatranscriptomic data, respectively. P values were calculated using PERMANOVA. (c) Bar plot of differential viruses between infected (unvaccinated) and infected (vaccinated) mice based on the Wilcoxon test (P < .05). The graphs on the left represent differential viruses found in metagenomic data, and on the right, differential viruses found in metatranscriptomic data
Furthermore, metagenomic analysis showed higher flora diversity in infected (vaccinated) mice than in infected (unvaccinated) mice, consistent with bacterial results obtained with samples from human gut (Figure 6a). PCoA analysis based on Bray-Curtis distance showed a significant difference in the gut bacteriome among infected (vaccinated) and infected (unvaccinated) mice (Figure 6b, PERMANOVA, P = .011), which was in keeping with the bacterial beta diversity results observed for COVID-19 patients and healthy controls. Compared to infected (vaccinated) mice, the bacterial communities of infected (unvaccinated) mice showed a significant increase in Odoribacter and Akkermansia muciniphila, and a notable depletion of Lactobacillus reuteri and Bacteroides uniformis (Figure 6c, P < .05, Wilcoxon test). Similarly, the relative abundances of Akkermansia muciniphila and Odoribacter were higher in COVID-19 patients than in healthy controls (Figure 6d), suggesting that they may play important roles in the disease process. Furthermore, we found similar results in the metatranscriptome; the gut flora diversity was higher in infected (vaccinated) mice than in infected (unvaccinated) mice, and Akkermansia muciniphila was more transcriptionally active in infected (unvaccinated) mice (Fig. S5).
Figure 6.
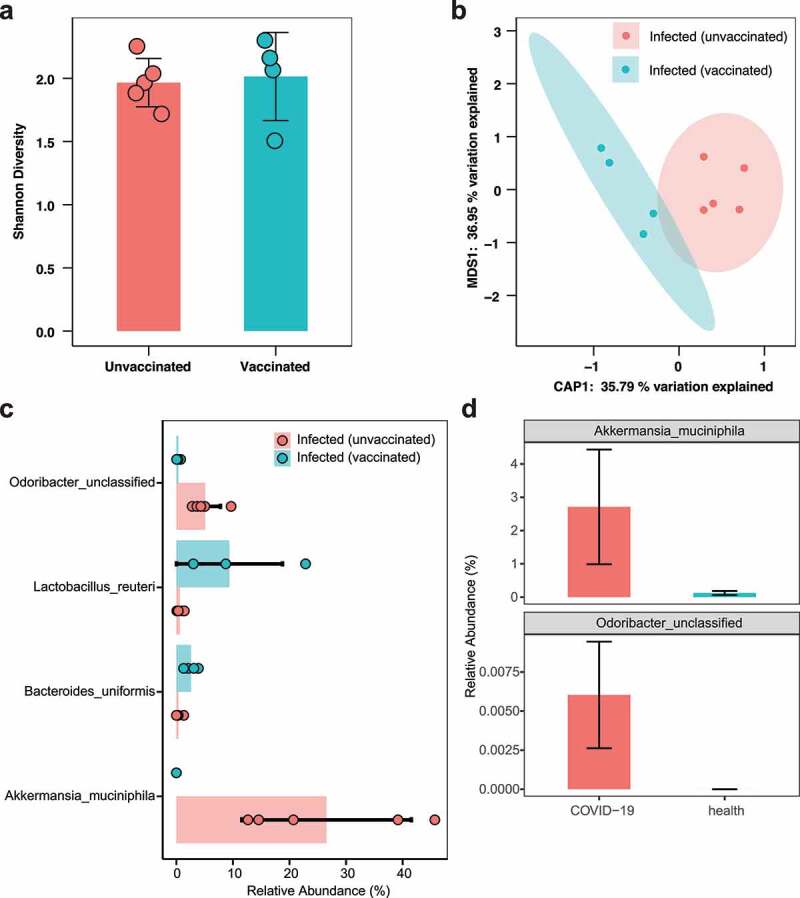
Alteration of the bacteriome between infected (unvaccinated) mice and infected (vaccinated) mice. (a) Comparison of bacterial Shannon diversity between infected (unvaccinated) and infected (vaccinated) mice. (b) Constrained PCoA analysis of bacteriome in mice using Bray-Curtis dissimilarity, P = .011, PERMANOVA. (c) Bar plot of differential bacterial communities among infected (unvaccinated) and infected (vaccinated) mice, which was performed using the Wilcoxon rank-sum test (P < .05). The red bar represents infected (unvaccinated) mice, and blue bar represents infected (vaccinated) mice. (d) Comparison of the relative abundances of Akkermansia muciniphila and Odoribacter between COVID-19 patients and healthy controls
Immune-related genes differentially expressed in control and infected mice
To elucidate that mechanisms underlying virome/bacteriome differences in mice, we performed transcriptomic analysis of mouse intestinal epithelial tissues, to investigate the host genetic expression differences in the gut after SARS-CoV-2 infection. Initial PCA showed significant differences in the expression of intestinal epithelial host genes in infected (unvaccinated) and infected (vaccinated) mice (Figure 7a). Analysis of differentially expressed genes showed 149 genes upregulated and 37 genes downregulated in infected (unvaccinated) mice compared to infected (vaccinated) mice (Figure 7b). Notably, 31 of these differentially expressed genes were involved in pathways associated with natural or adaptive immune responses, and enrichment analysis of immune-related genes showed that the vast majority of genes were highly expressed in infected (unvaccinated) mice (Figure 7c). Of these genes, a considerable proportion were involved in immune regulation as components of the immune system. For example, the polymeric immunoglobulin receptor (PIGR), which is upregulated in infected (unvaccinated) mice, was proven to play an important role in the maintenance of epithelial integrity and mucosal homeostasis in the colonic epithelium,30 and C3, a member of the complement system of serum proteins, is an import host defense mechanism for influenza A virus infection.31 Interleukin-15 (IL-15), as a cytokine, can perform antiviral functions via an interferon-dependent mechanism.32 Differential expression of other genes associated with SARS-CoV-2 virus infection was determined to be associated with SARS-CoV-2 virus infection. ACE2 expression was inhibited by upregulated zinc finger E-box-binding homeobox 1 (ZEB1) in SARS-CoV-2-infected lung cells,33 The apolipoprotein E (APOE) gene variant was determined to be correlated with the risk of Alzheimer’s disease and associated with COVID-19,34 and Mamoor demonstrated that Tribbles pseudokinase 1 (TRIB1) may be involved in the cellular response to COVID-19 infection.35 Other genes have anti-inflammatory or antiviral effects, such as inflammatory and metabolic pathways in mouse intestinal mucosa associated with MUC2 mucin.36 Hugh et al. demonstrated that multiple proteins involved in bacterial invasion can be modulated by Ras GTPase-activating-like protein IQGAP1 (IQGAP).37 Subsequently, we performed a gene ontology enrichment analysis, where many immune-related biological processes were significantly enriched, such as regulation of immune system processes, further suggesting that SARS-CoV-2 infection in mice induces the body’s immune system to resist viral invasion (Figure 7d).
Figure 7.
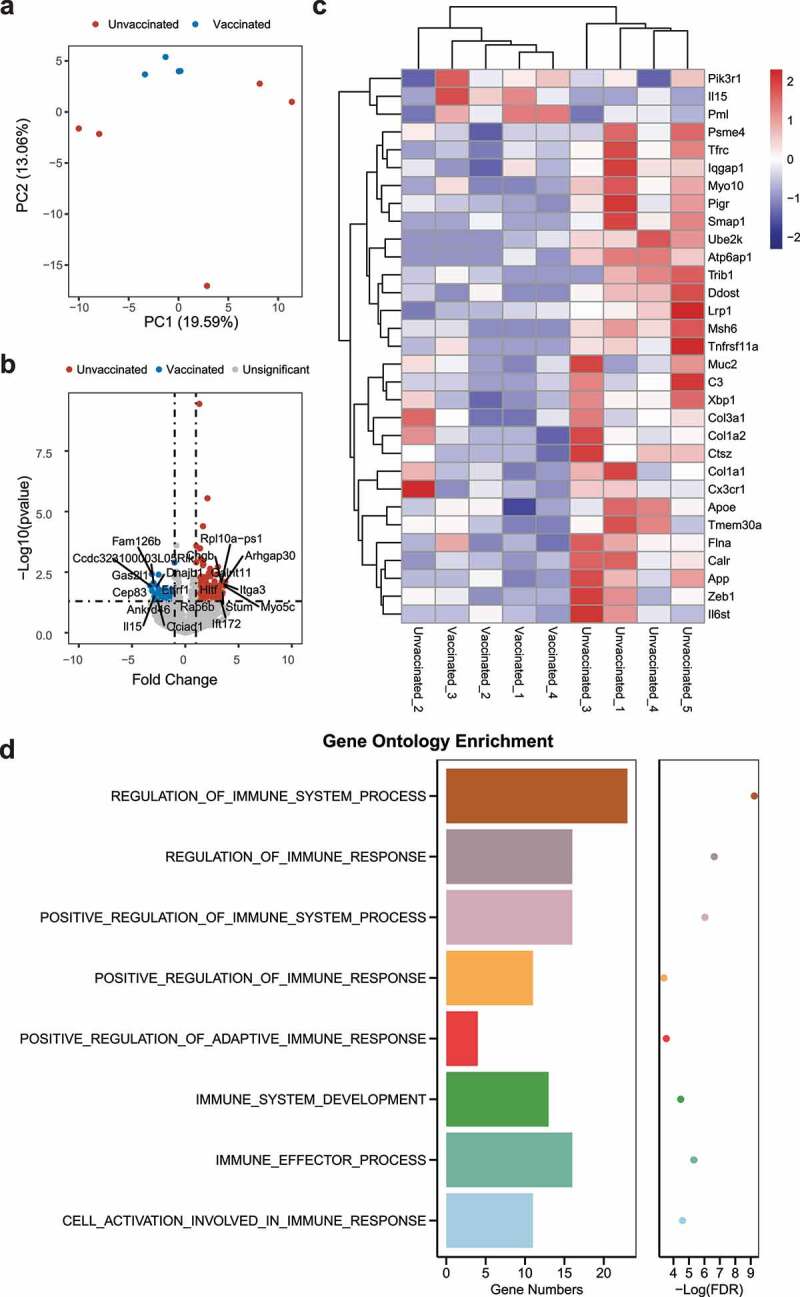
Differentially expressed genes between infected (unvaccinated) and infected (vaccinated) mice. (a) PCA plot of gene expression in mice. (b) Volcano plot of differential gene expression in infected (unvaccinated) versus infected (vaccinated) mice. Genes were considered significantly differentially expressed for a log 2 fold change and p value of 0.05. The top 10 (up – and/or downregulated) differentially expressed genes are shown. (c) Heatmap of immune-associated gene expression. (d) Gene ontology enrichment of differential genes in infected (unvaccinated) versus infected (vaccinated) mice
Discussion
COVID-19 is still rapidly spreading worldwide and exhibits nearly no sign of slowing, while multiple lines of vaccines are being developed. With the development of therapeutic measures, including antibodies and small molecules in various stages of clinical trials, one major concern is that rapid mutations of SARS-CoV-2,38 an RNA virus, could hinder the efficacy of vaccines or treatments. Thus, investigating genetic variations, particularly their potential correlation with the disease severity of COVID-19, is of considerable value for clinics. However, among the large number of reported genomes in the GISAID database, very little information is available regarding disease severity and other parameters, rendering it difficult to perform such analysis. In this study, we employed an amplification-based approach to profile the genomes of SARS-CoV-2 virus, and found that at least in our cohort, no SNP significantly contributed to disease severity or important clinical parameters. A recent study suggests that a particular D614G mutation in the spike protein increases its ability to infect host cells by increasing the efficiency of protease clearance,39 but this mutation was not found in our cohort, and the current findings did not mention the disease severity. Not all patients have a sufficient amount of viral RNA in their upper respiratory tracts (throat swabs) or gastrointestinal tracts (fecal samples) to permit viral genome sequencing, thereby limiting the number of genomes available; screening a larger number of patients could alleviate this problem.
Next, we analyzed the gut virome of COVID-19 patients by isolating VLPs and amplifying viral DNA and RNA with Oxford Nanopore sequencing. We found that while SARS-CoV-2 virus can be detected using a quantitative PCR approach in some patients’ fecal samples, overall, the presence of viral sequences in VLPs was low, and only four patients had detectable SARS-CoV-2 sequences in the gut virome data. It is likely that SARS-CoV-2 virus particles are relatively low in abundance and below the sequencing threshold, or in many cases, the virus nucleotides may be present but not enveloped. Overall, virome structure is closely associated with bacteriome structure and it is not entirely dominated by phages, as we detected a significant proportion of eukaryote-associated viruses, although due to a general lack of research and references, their taxonomy has not been fully elucidated. A few viruses related to plant RNA viruses and unclassified viruses were found to be enriched in multiple patients, suggesting that unestablished intestinal viruses could be involved in SARS-CoV-2 infections or, more importantly, could reflect or even affect the host immune status and response to viral infections. As the COVID-19 pandemic is still ongoing, and it is important to understand its pathology, as well as other important aspects, the virome we investigated may provide a different perspective for future efforts to study, characterize, and treat COVID-19, particularly regarding the early diagnosis and treatment of severe versus common cases, which may be confirmed as evidence accumulates and relevant research is determined to be useful.
Furthermore, the gut bacteriome affects an important proportion of host immune functions, and while previous studies of the gut-lung infection axis focused primarily on a mouse influenza model,40,41 several studies have also reported dysbiosis of the gut bacteriome in COVID-19 patients,19,20 denoted by a reduction in the diversity of bacterial communities. Our study of patients in Beijing adds to the findings obtained in Hong kong and Hangzhou and reaffirms that a decrease in the diversity of the gut bacteriome is common in patients infected with COVID-19. In addition, we found that Ruminococcus gnavus, which was significantly enriched in COVID-19, was associated with Crohn’s disease, producing an inflammatory polysaccharide that induces dendritic cells to produce cytokines, such as TNFα.42 Coprobacillus and Clostridium ramosum, which are enriched in the patients, were shown by Zuo et al. to be positively correlated with the severity of COVID-19.20 More critically, we observed a microbial signature of disease severity in which butyrate-producing bacterial groups, including Eubacterium, were depleted in severe cases of COVID-19, while opportunistic pathogens were enriched, including Corynebacterium, Enterococcus, Campylobacter, Cirtrobacter, and Enterobacter, which is in keeping with the findings of Zuo et al.20 regarding the general pattern of the gut microbiome in COVID-19 patients. Another study by Zuo et al.43 found an enrichment of Lachnospiraceae bacterium 1_1_57FAA in patients with low to nonexistent SARS-CoV-2 infectivity. However, COVID-19 patients in our cohort were observed to have higher levels of Lachnospiraceae bacterium 2_1_58FAA than healthy individuals. Since we are still in the early stages of studying and understanding microbiome differences and dynamics of COVID-19, the earliest studies may be expected to be limited in sample size and the universality of the results. These limitations have been observed for many biological and medical topics in which increasingly reliable results can be obtained by more independent studies, thereby overcoming potential cohort differences caused by cohort genetics, geographical factors and other environmental factors. We surmise that the differences we observed are due to 1) the relatively small number of cohorts used to date in microbiome-related research and 2) the cohort differences that do exist, which means that the marker taxa that we identified are cohort-specific instead of common or universal. Also, despite the necessity of utilizing antibiotics in treating secondary infections, we observed that they lead to additional shifts in the gut bacteriome of COVID-19 patients, as exemplified by the undesirable decrease in butyrate-producing bacteria (for instance Roseburia hominis and Faecalibacterium prausnitzii). Thus, it may be useful to control for this confounding effect while analyzing the bacteriome from patients, as it remains difficult to disentangle the effects of antibiotics vs. disease severity on the bacteriome.
Finally, deeper insights into the potential mechanisms underlying virome/bacteriome shifts were obtained using a mouse COVID-19 model, in which we investigated the gene expression in gut epithelial cells in tandem with the virome and bacteriome. In our mouse model, SARS-CoV-2 did not directly infect the GI tract, as demonstrated by sequencing efforts. Nonetheless, we found significant differences between infected (unvaccinated) mice and infected (vaccinated) mice in terms of host gene expression, where we found enrichment of immune – and anti-infection-related genes differentially expressed between groups. Among these genes, many were already reported to be key genes in host-microbiome or host-virus cross-talk, including IL15, C3 and MUC2, while genes related to SARS-CoV-2 infection or severity also exhibited changes in expression, including ZEB1 and pleiotropic APOE. This result, combined with the finding that SARS-CoV-2 was limited in the lungs of mice, suggests an overall modulation of immune functions in the host as a result of infection, rather than the changes simply being directly triggered by SARS-CoV-2. On the bacteriome side, the results in mice confirmed key characteristics of bacteriome changes observed in COVID-19 patients, particularly Odoribacter and Akkermansia muciniphila, both key bacteria implicated in a list of diseases,44,45 and their correlation to infection/disease severity warrants further investigation. Accompanying virome shifts are largely clustered in mouse bacteriophages and are tightly associated with shifts in the bacteriome. Importantly, the current investigations were primary and particularly limited by the biosafety requirements of experimenting with SARS-CoV-2, as well as the consequent limitations to sample size; in future research, more mechanistically oriented studies are warranted.
Methods and Materials
Study subject and design
In this study, a cohort of 18 subjects was recruited, including 13 COVID-19 patients from the Fifth Medical Center of PLA General Hospital (Fig. S6) and five healthy volunteers from our previous study.25 SARS-CoV-2-infected patients were confirmed using the Novel Coronavirus (SARS-CoV-2) Nucleic Acid Detection kit (BioGerm), as recommended by the CDC in China. According to the Seventh Revised Trial Version of the Novel Coronavirus Pneumonia Diagnosis and Treatment Guidance,46 the disease severity of patients infected with SARS-CoV-2 was classified into three categories: (1) severe, for adults, respiratory distress (≥ 30 breaths/min), oxygen saturation ≤ 93% at rest and arterial partial pressure of oxygen (PaO2)/fraction of inspired oxygen (FiO2) ≤ 300 mmHg (1 mmHg = 0.133 KPa). For children, tachypnea (RR ≥ 60 breaths/min for infants aged below 2 months; RR ≥ 50 BPM for infants aged 2–12 months; RR ≥ 40 BPM for children aged 1–5 years, and RR ≥ 30 BPM for children above 5 years old) was independent of fever and crying, oxygen saturation ≤ 92% on finger pulse oximeter taken at rest, labored breathing (moaning, nasal fluttering, and infrasternal, supraclavicular, and intercostal retraction), cyanosis, and intermittent apnea, lethargy and convulsion, difficulty feeding and signs of dehydration; (2) moderate, showing fever and respiratory symptoms with radiological findings of pneumonia; and (3) mild, the clinical symptoms were mild, and there was no sign of pneumonia on imaging. All subjects in our study provided written informed consent.
SARS-CoV-2 qPCR of COVID-19 patients
Throat swabs and fecal samples of COVID-19 patients were resuspended in sterile phosphate buffered saline (PBS) and treated by heating in a 56°C water bath for 30 min for inactivation. Viral RNAs of throat swabs and fecal samples with inactivating treatment were extracted based on the QIAamp Viral RNA Mini Kit Handbook (Qiagen). Viral RNAs were detected and quantified by real-time reverse-transcriptase-polymerase chain-reaction (RT-PCR) assay according to the novel coronavirus (2019-nCoV) N gene, ORF1ab gene, and S gene triple nucleic acid assay kit (MABSKY). The real-time RT-PCR included 17 μl of RT-PCR solution, 2 μl of primer probe mixture, 1 μl of mixed enzyme solution and 5 μl of RNAs in a final reaction volume of 25 μl. Reverse transcription at 50°C for 30 min, pre-denaturation at 95°C for 3 min, followed by 45 cycles of amplification reactions at 95°C for 5 s and 55°C for 30 s. The cycle threshold (Ct) values of real-time RT-PCR were converted into virus titers (TCID50) based on a standard curve prepared from 10-fold serial dilutions of SARS-CoV-2 provirus extracts (Table S6).
Amplification and sequencing of SARS-CoV-2 genome
According to the nCoV-2019 sequencing protocol reported by the ARTIC network,47 extracted RNAs were subjected to reverse transcription using the SuperScript III Reverse Transcriptase kit (Invitrogen) based on the manufacturer’s random hexamer primer protocol. Viral cDNAs were specifically amplified using a set of primers based on the nCoV-2019 sequencing protocol (V3). Library preparation was performed using an adjusted protocol for the Nextera XT DNA Library Preparation Kit from Illumina and sequencing was performed using NovaSeq (PE250).
Enrichment of virus-like particles and cDNA sequencing
Virus-like particles of inactivated fecal samples were enriched and purified according to the modified workflow we developed in a previous study.25 In brief, each fecal sample (approximately 500 mg) was resuspended in 15 ml PBS and centrifuged at 4,500 rpm for 10 min at the 4°C to remove large food residues (Beckman Coulter AllegraTM X-22 R). The supernatant was transferred to fresh tubes and centrifuged at 4,500 rpm for 10 min at the 4°C again. The supernatant was then filtered through a 0.45 μm PVDF membrane (Millipore) twice and centrifuged at 180,000 × g at 4°C for 3 h (Beckman Coulter XP-100). The pellets were resuspended in 400 µl PBS and treated with RNase A and DNase I at 37°C for 30 min. Viral nucleic acids, including DNA and RNA, were extracted by using a QIAamp MinElute Virus Spin Kit (Qiagen). The viral nucleic acids underwent reverse transcription, random amplification and agarose gel electrophoresis to obtain cDNA to prepare the sequencing library.
PromethION library preparation was performed according to the manufacturer’s instructions for barcoding cDNA/DNA and native DNA (SQK-LSK109, EXP-NBD104 and EXP-NBD114). When multiplexing, all the samples were pooled together. Oxford Nanopore Technologies (ONT) MinKNOW software (v.19.10.1) was used to collect raw sequencing data, and Guppy (v.3.2.4) was used for real-time base calling of the raw data.
Bacteriome sequencing
The PVDF membranes left over from the VLP enrichment and purification process were employed to isolate bacterial communities. The nucleic acids of bacterial communities in membranes were extracted by using the AllPrep PowerFecal DNA/RNA Kit (Qiagen). Similarly, a modified protocol for the Nextera XT DNA Library Preparation kit from Illumina was used for library construction and sequencing was performed using HiSeq XTen (PE250) and NovaSeq (PE150).
Transcriptome, metagenome and metatranscriptome sequencing in a mouse model
Nine hACE2 transgenic C57BL/6 mice at 6 to 8 weeks of age were randomly allocated to two groups. Four mice were immunized with 1 × 1011 vp of recombinant type 7 chimpanzee adenovirus vaccine expressing the spike protein of COVID-19 virus and five mice were inoculated with PBS. All mice received a second identical dose of vaccine or PBS on day 28. Fourteen days later, mice were infected with 5 × 105 TCID50 SARS-CoV-2 virus through the intranasal route. On day three postinfection, mice were sacrificed and necropsied to collect the small intestine and fecal contents. Mouse small intestines were homogenized and mixed with TRIzol for RNA extraction. An AllPrep Powerfecal DNA/RNA Kit (Qiagen) kit was used to extract DNA and RNA from the mouse fecal contents according to the manufacturer’s protocol. Viral particles in the lung were quantified using RT-PCR, and the infected groups averaged 8.72 in log(10) copies/g, which was significantly higher than that of the control group (average 6.37, P = .035, Wilcoxon test).
The ribosomal RNAs of mouse small intestine tissue were removed using the KAPA RiboErase Kit (HMR) Human/Mouse/Rat. Library preparation was performed using the KAPA RNA Hyperprep Kit, and sequencing was performed using HiSeq XTen (PE150). The metagenome and metatranscriptome were sequenced using NovaSeq 6000 (PE150) and HiSeq XTen (PE150), respectively. DNA libraries were prepared using the Rapid DNA Library Prep Kit for Illumina, and RNA libraries were prepared using the KAPA RNA Hyperprep Kit. The RibominusTM Transcriptome Isolation Kit – Bacteria (Invitrogen) was used to remove ribosomal RNAs.
SNP calling of SARS-CoV-2 genome
SNP calling was performed based on the modified Variant Calling Pipeline using GATK448 published by Mohammed Khalfan (https://gencore.bio.nyu.edu/variant-calling-pipeline-gatk4/). Briefly, qualified raw data were mapped to the SARS-CoV-2 reference genome (NC_045512) using BWA-MEM,49 and then bam files were marked in duplicate and sorted using Picard. Next, variant calling was performed using GATK (v4.1.6) followed by extracting SNPs and filtering SNPs with parameters ‘QUAL < 30 MQRankSum < −12.5 FS > 60.0 ReadPosRankSum < −8.0 MQ < 40.0 QD < 2.0 DP < 8.0ʹ. Finally, SnpEff (v4.3) was used to annotate and predict the effects of SNPs on genes.
Bioinformatics analysis of the virome and bacteriome in humans
To obtain a comprehensive viral genome, we integrated two viral genome databases using Mash50 and Nucmer51 with 95% nucleotide identity and 70% sequence coverage, including the NCBI Viral genome database and Cenote Human Virus Database,52 the latter of which includes 52,750 nonredundant virus OTUs extracted from nearly 6,000 human metagenomic samples, resulting in 61,843 integrated viral genomes. Adaptor and barcode sequences of ONT sequencing raw data were trimmed by using Porechop (v0.2.3)53 with default parameters. Trimmed reads were subjected to viral taxonomic classification by aligning them with the integrated viral genome database using minimap2 (v2.1.7).54 Only those viral genomes that met a coverage greater than 30% and had a mapped read number greater than 10 were retained for further analysis. The relative abundance of the virome was calculated using the average coverage depth of the viral genome, which was enumerated by samtools (v1.3.1 with ‘samtools coverage’).
For bacteriome analysis, bacterial raw reads were deduplicated using Kneaddata (v0.7.4 with parameters ‘–trimmomatic-options “SLIDINGWINDOW:4:20 MINLEN:50”’)55 to trim and filter low-quality sequences, as well as contaminating human reads with the GRCh38 human genome reference. Profiling of the composition of bacterial communities was performed using MetaPhlAn2 (v2.7.5)56 by mapping reads to clade-specific markers.
Virome/bacteriome and transcriptome analysis in a mouse model
Metagenomic and metatranscriptomic sequences were deduplicated using Kneaddata55 to trim and filter low-quality sequences, as well as contaminating mouse reads with the mm10 mouse genome reference. Viral composition analysis was performed using bowtie2 to align clean reads to the integrated viral genome database. Profiling the composition of bacterial communities was performed using MetaPhlAn2 (v2.7.5)56 by mapping reads to clade-specific markers.
To analyze the transcriptome of mouse intestinal epithelial tissues, adapter and low-quality sequences of raw data were removed and filtered using Fastp.57 Clean data were aligned to the mouse reference mm10 using HISAT258 with default parameters, and read count-matched host genes were calculated with HTseq.59 Differential expression gene analysis was performed used DESeq2.60 The FUMA website61 was used to conduct gene functional mapping and annotation.
Statistical analysis
All statistical analyses were performed in R(v3.6.1). Shannon diversity and principal coordinates analysis based on Bray-Curtis dissimilarities were performed by using vegan packages (v2.5–6). Two groups were compared using the Mann-Whitney-Wilcoxon rank-sum test, while three groups were compared using the Kruskal-Wallis test. Multivariate analysis was performed using PERMANOVA in vegan.
Analysis of differences in bacterial and/or viral communities between COVID-19 patients and healthy controls was performed using the Mann-Whitney-Wilcoxon rank-sum test. Differential bacterial and viral taxa among COVID-19 patients with/without antibiotic treatment were identified using Multivariate Association with Linear Models (MaAsLin2),62 and box plots were employed to show variances of the bacteriome and virome (Wilcoxon rank-sum test). The WilcoxTest function in the GSALightning package (v1.14) was used for differential enrichment analysis within different COVID-19 severities. Procrustes analysis is a statistical method that analyses the consistency of two-dimensional graphs produced by overlaying principal component analysis of two datasets. Procrustes analysis was performed based on the Bray-Curtis distances of eigenvalues for both bacteriome and virome using the Procrustes function in vegan packages. Correlations between the bacteriome and virome were calculated using SparCC,63 and an association network graph was generated using igraph packages (v1.2.5) in R (v3.6.1).
Supplementary Material
Acknowledgments
We are grateful to all participants in this study.
Funding Statement
This work was supported by the the National Natural Science Foundation of China (grant number 32041009 and 31771481) Strategic Priority Research Program of the Chinese Academy of Sciences (grant number XDB29020000), and Ministry of Science and Technology Special National Project on Investigation of Basic Resources of China program (grant number 2019FY101500).
Disclosure statement
None declared.
Author contributions
Jiabao Cao and Cheng Wang performed data analysis and draft manuscript. Yuqing Zhang and Guanlin Lei conducted the experiments. Kun Xu, Na Zhao, Jingjing Lu, Ning Zhang and Yuhuan Gong assisted in experiments and were responsible for metagenomic sequencing. Fanping Meng, Linxiang Yu, Jin Yan, Changqing Bai, Shaogeng Zhang and Zhu Chen collected samples and edited the manuscript. Yuhai Bi, Yi shi and Lianpeng Dai provided critical comments. Penghui Yang and Jun Wang designed and supervised the study.
Availability of data and material
Raw reads of metagenomic sequencing of Virome and Bacteriome were deposited at GSA (Genome Sequence Archive) under Bioproject accession PRJCA003532.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Dai L, Zheng T, Xu K, Han Y, Xu L, Huang E, An Y, Cheng Y, Li S, Liu M, et al. A universal design of betacoronavirus vaccines against COVID-19, MERS, and SARS. Cell. 2020;182(3):722–33.e11. doi: 10.1016/j.cell.2020.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang N, Shang J, Jiang S, Du L.. Subunit vaccines against emerging pathogenic human coronaviruses. Front Microbiol. 2020;11:298. doi: 10.3389/fmicb.2020.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu F-C, Li Y-H, Guan X-H, Hou L-H, Wang W-J, Li J-X, Wu S-P, Wang B-S, Wang Z, Wang L, et al. Safety, tolerability, and immunogenicity of a recombinant adenovirus type-5 vectored COVID-19 vaccine: a dose-escalation, open-label, non-randomised, first-in-human trial. Lancet. 2020;395(10240):1845–21. doi: 10.1016/S0140-6736(20)31208-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y, Fu S, Gao L, Cheng Z, Lu Q, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395(10236):1569–1578. doi: 10.1016/S0140-6736(20)31022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geleris J, Sun Y, Platt J, Zucker J, Baldwin M, Hripcsak G, Labella A, Manson DK, Kubin C, Barr RG, et al. Observational study of hydroxychloroquine in hospitalized patients with covid-19. N Engl J Med. 2020;382(25):2411–2418. doi: 10.1056/NEJMoa2012410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ye Q, Wang B, Mao J. The pathogenesis and treatment of the `Cytokine Storm‘ in COVID-19. J Infect. 2020;80(6):607–613. doi: 10.1016/j.jinf.2020.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bi Q, Wu Y, Mei S, Ye C, Zou X, Zhang Z, Liu X, Wei L, Truelove SA, Zhang T, et al. Epidemiology and transmission of COVID-19 in 391 cases and 1286 of their close contacts in Shenzhen, China: a retrospective cohort study. Lancet Infect Dis. 2020;20(8):911–919. doi: 10.1016/S1473-3099(20)30287-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ye G, Pan Z, Pan Y, Deng Q, Chen L, Li J, Li Y, Wang X. Clinical characteristics of severe acute respiratory syndrome coronavirus 2 reactivation. J Infect. 2020;80(5):e7–e14. doi: 10.1016/j.jinf.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao F, Tang M, Zheng X, Liu Y, Li X, Shan H. Evidence for gastrointestinal infection of SARS-CoV-2. Gastroenterology. 2020;158(6):1831–1833.e3. doi: 10.1053/j.gastro.2020.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin L, Jiang X, Zhang Z, Huang S, Zhang Z, Fang Z, Gu Z, Gao L, Shi H, Mai L, et al. Gastrointestinal symptoms of 95 cases with SARS-CoV-2 infection. Gut. 2020;69(6):997–1001. doi: 10.1136/gutjnl-2020-321013. [DOI] [PubMed] [Google Scholar]
- 11.Luo S, Zhang X, Xu H. Don’t overlook digestive symptoms in patients with 2019 novel coronavirus disease (COVID-19). Clin Gastroenterol Hepatol. 2020;18:1636–1637. doi: 10.1016/j.cgh.2020.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Y, Guo C, Tang L, Hong Z, Zhou J, Dong X, Yin H, Xiao Q, Tang Y, Qu X, et al. Prolonged presence of SARS-CoV-2 viral RNA in faecal samples. Lancet Gastroenterol Hepatol. 2020;5(5):434–435. doi: 10.1016/S2468-1253(20)30083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Y, Li X, Zhu B, Liang H, Fang C, Gong Y, Guo Q, Sun X, Zhao D, Shen J, et al. Characteristics of pediatric SARS-CoV-2 infection and potential evidence for persistent fecal viral shedding. Nat Med. 2020;26(4):502–505. doi: 10.1038/s41591-020-0817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt TSB, Raes J, Bork P. The human gut microbiome: from association to modulation. Cell. 2018;172(6):1198–1215. doi: 10.1016/j.cell.2018.02.044. [DOI] [PubMed] [Google Scholar]
- 15.Kelly D, Conway S, Aminov R. Commensal gut bacteria: mechanisms of immune modulation. Trends Immunol. 2005;26(6):326–333. doi: 10.1016/j.it.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 16.Belkaid Y, Hand Timothy W. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karst SM. The influence of commensal bacteria on infection with enteric viruses. Nat Rev Microbiol. 2016;14(4):197–204. doi: 10.1038/nrmicro.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A. 2011;108(13):5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu S, Chen Y, Wu Z, Chen Y, Gao H, Lv L, et al. Alterations of the gut microbiota in patients with COVID-19 or H1N1 influenza. Clin Infect Dis. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuo T, Zhang F, Lui GCY, Yeoh YK, Li AYL, Zhan H, Wan Y, Chung ACK, Cheung CP, Chen N, et al. Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterology. 2020;159(3):944–55.e8. doi: 10.1053/j.gastro.2020.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shkoporov AN, Clooney AG, Sutton TDS, Ryan FJ, Daly KM, Nolan JA, McDonnell SA, Khokhlova EV, Draper LA, Forde A, et al. The human gut virome is highly diverse, stable, and individual specific. Cell Host Microbe. 2019;26(4):527–41.e5. doi: 10.1016/j.chom.2019.09.009. [DOI] [PubMed] [Google Scholar]
- 22.Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD, Lewis JD, Bushman FD. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res. 2011;21(10):1616–1625. doi: 10.1101/gr.122705.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alcami A, Koszinowski UH. Viral mechanisms of immune evasion. Immunol Today. 2000;21(9):447–455. doi: 10.1016/S0167-5699(00)01699-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dhar D, Mohanty A. Gut microbiota and Covid-19- possible link and implications. Virus Res. 2020;285:198018. doi: 10.1016/j.virusres.2020.198018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao J, Zhang Y, Dai M, Xu J, Chen L, Zhang F, Zhao N, Wang J. Profiling of human gut virome with oxford nanopore technology. Med Microecol. 2020;4:100012. doi: 10.1016/j.medmic.2020.100012. [DOI] [Google Scholar]
- 26.Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet. 2011;12(6):443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McLaren PJ, Carrington M. The impact of host genetic variation on infection with HIV-1. Nat Immunol. 2015;16(6):577–583. doi: 10.1038/ni.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med. 2015;21(8):895–905. doi: 10.1038/nm.3914. [DOI] [PubMed] [Google Scholar]
- 29.Langdon A, Crook N, Dantas G. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med. 2016;8(1):39. doi: 10.1186/s13073-016-0294-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murthy AK, Dubose CN, Banas JA, Coalson JJ, Arulanandam BP. Contribution of polymeric immunoglobulin receptor to regulation of intestinal inflammation in dextran sulfate sodium-induced colitis. J Gastroenterol Hepatol. 2006;21(9):1372–1380. doi: 10.1111/j.1440-1746.2006.04312.x. [DOI] [PubMed] [Google Scholar]
- 31.O’Brien KB, Morrison TE, Dundore DY, Heise MT, Schultz-Cherry S, Vartanian J-P. A protective role for complement C3 protein during pandemic 2009 H1N1 and H5N1 influenza A virus infection. PLoS One. 2011;6(3):e17377. doi: 10.1371/journal.pone.0017377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foong YY, Jans DA, Rolph MS, Gahan ME, Mahalingam S. Interleukin-15 mediates potent antiviral responses via an interferon-dependent mechanism. Virology. 2009;393(2):228–237. doi: 10.1016/j.virol.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 33.Stewart CA, Gay CM, Ramkumar K, Cargill KR, Cardnell RJ, Nilsson MB, Heeke S, Park EM, Kundu ST, Diao L, et al. SARS-CoV-2 infection induces EMT-like molecular changes, including ZEB1-mediated repression of the viral receptor ACE2, in lung cancer models. bioRxiv. 2020. doi: 10.1101/2020.05.28.122291. [DOI] [Google Scholar]
- 34.Kuo CL, Pilling LC, Atkins JL, Masoli JAH, Delgado J, Kuchel GA, et al. APOE e4 genotype predicts severe COVID-19 in the UK biobank community cohort. J Gerontol A. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mamoor S. TRIB1 is differentially expressed and transcriptionally induced in models of coronavirus infection. OSF Preprints. 2020. [Google Scholar]
- 36.Tadesse S, Corner G, Dhima E, Houston M, Guha C, Augenlicht L, Velcich A. MUC2 mucin deficiency alters inflammatory and metabolic pathways in the mouse intestinal mucosa. Oncotarget. 2017;8(42):71456–71470. doi: 10.18632/oncotarget.16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim H, White CD, Sacks DB. IQGAP1 in microbial pathogenesis: targeting the actin cytoskeleton. FEBS Lett. 2011;585(5):723–729. doi: 10.1016/j.febslet.2011.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, Masciovecchio C, Angeletti S, Ciccozzi M, Gallo RC, et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med. 2020;18(1):179. doi: 10.1186/s12967-020-02344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu J, He C-L, Gao Q-Z, Zhang G-J, Cao -X-X, Long Q-X, et al. The D614G mutation of SARS-CoV-2 spike protein enhances viral infectivity and decreases neutralization sensitivity to individual convalescent sera. bioRxiv. 2020. [Google Scholar]
- 40.Brown RL, Sequeira RP, Clarke TB. The microbiota protects against respiratory infection via GM-CSF signaling. Nat Commun. 2017;8(1):1512. doi: 10.1038/s41467-017-01803-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steed AL, Christophi GP, Kaiko GE, Sun L, Goodwin VM, Jain U, Esaulova E, Artyomov MN, Morales DJ, Holtzman MJ, et al. The microbial metabolite desaminotyrosine protects from influenza through type I interferon. Science. 2017;357(6350):498. doi: 10.1126/science.aam5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henke MT, Kenny DJ, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc Natl Acad Sci. 2019;116(26):12672. doi: 10.1073/pnas.1904099116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuo T, Liu Q, Zhang F, Lui GC-Y, Tso EYK, Yeoh YK, et al. Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut. 2020:gutjnl-2020-322294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13(9):R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geerlings SY, Kostopoulos I, de Vos WM, Belzer C. Akkermansia muciniphila in the human gastrointestinal tract: when, where, and how? Microorganisms. 2018;6(3):75. doi: 10.3390/microorganisms6030075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.NATCM . Diagnosis and treatment protocol for novel coronavirus pneumonia (Trial Version 7). Chin Med J (Engl). 2020;133(9):1087–1095. doi: 10.1097/CM9.0000000000000819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Itokawa K, Sekizuka T, Hashino M, Tanaka R, Kuroda M. A proposal of alternative primers for the ARTIC Network’s multiplex PCR to improve coverage of SARS-CoV-2 genome sequencing. bioRxiv. 2020. [Google Scholar]
- 48.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Current Protocols Bioinformatics. 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016;17(1):132. doi: 10.1186/s13059-016-0997-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delcher AL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30(11):2478–2483. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tisza MJ, Buck CB, New Comprehensive A. Catalog of the human virome reveals hidden associations with chronic diseases. bioRxiv. 2020. [Google Scholar]
- 53.Wick RR, Judd LM, Gorrie CL, Holt KE. Completing bacterial genome assemblies with multiplex MinION sequencing. Microbial Genomics. 2017;3(10). doi: 10.1099/mgen.0.000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H, Birol I. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McIver LJ, Abu-Ali G, Franzosa EA, Schwager R, Morgan XC, Waldron L, Segata N, Huttenhower C. bioBakery: a meta’omic analysis environment. Bioinformatics (Oxford, England). 2018;34(7):1235–1237. doi: 10.1093/bioinformatics/btx754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12(10):902–903. doi: 10.1038/nmeth.3589. [DOI] [PubMed] [Google Scholar]
- 57.Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i90. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England). 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. doi: 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mallick H, Tickle TL, McIver LJ, Rahnavard G, Nguyen LH, Weingart G, Ma S, Ren B, Schwager E, Subramanian A, et al. Multivariable association in population-scale meta’omic surveys. 2020. [DOI] [PMC free article] [PubMed]
- 63.Friedman J, Alm EJ, von Mering C. Inferring correlation networks from genomic survey data. PLoS Comput Biol. 2012;8(9):e1002687. doi: 10.1371/journal.pcbi.1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw reads of metagenomic sequencing of Virome and Bacteriome were deposited at GSA (Genome Sequence Archive) under Bioproject accession PRJCA003532.