

Abstract
Aryl-substituted esters of a racemic diprotected 2-azido-1-alkanol were submitted to the Staudinger/aza-Wittig reaction in order to assess scope and establish conditions for their cyclization to the corresponding 2,4,5-trisubstituted oxazolines. Following the cyclization study, the (2R,3R)-antipode of the azidoalkanol was obtained in high ee by incubation of the corresponding racemic azidoacetate with pig liver esterase (PLE). The p-nitrobenzoate of the enantioenriched 2-azido-1-alcohol was cyclized by the Staudinger/aza-Wittig to give the corresponding (4R,5R)-disubstituted-2-(4-nitrophenyl) oxazoline. Selective reduction of the nitrophenyloxazoline to the corresponding aminophenyloxazoline using aluminum amalgam followed by direct azidation of the 2-(4-aminophenyl) moiety provided the corresponding (4R,5R)-2-(4-azidophenyl) oxazoline derivative. The azidophenyl oxazoline was reacted with a proven click partner 4-ethynylfluorobenzene under copper/sodium ascorbate mediation to provide the click triazole product in high yield.
Keywords: Aza-Wittig, Oxazolines, Azides, Click chemistry, Esterase
Graphical Abstract

1. Introduction
Oxazolines and their substituted derivatives occupy a unique niche in the realm of nitrogen-oxygen heterocycles. Oxazoline-derived structural cores are found in natural products, medicinals, experimental therapeutics and polymers and are also functional molecules as they have been employed as chiral directing groups, ligands and protecting groups in synthetic chemistry.1 Our research efforts have involved the design and development of oxazole-derived azides as reacting partners with arylacetylenes in the so called “click” reaction.2 The product triazoles of interest have evolved from the development of oxazole-based peptidomimetic inhibitors of Porphyromonas gingivalis adherence to Streptococcus gordonii using click chemistry.3 The adherence between P.gingivalis and S. gordonii is mediated by a protein-protein interaction which results in the generation of a dental biofilm. In turn the microbial community becomes resistant to many types of antibacterial therapy and ultimately leads to degradation of bone and dental structure. Consequently, any synthetic inhibitors of the adherence process will provide a unique therapy for the treatment of gingival disease propagated by P. gingivalis and the associated microbes. In the quest for adherence inhibitors with increased potency, we are presently exploring the replacement of the 2,4,5-trisubstituted oxazole motif with the corresponding 2,4,5-trisubstituted oxazolines (dihydrooxazoles). As evidenced by recent reports, oxazoline-based scaffolds are beginning to gain ground as functional peptidomimetics.4 In contrast to the more planar trisubstituted oxazoles, the corresponding oxazolines offer a more three-dimensional motif with two ring stereocenters. Therefore, our goals were to develop a synthesis of stereodefined trisubstituted oxazolines which will constitute the basic scaffold of new adherence inhibitors. The stereodefined oxazolines should have potential for further synthetic elaboration5 at the 4- and 5- positions, with the aid of suitable protecting groups,5 as well as suitably-positioned azide functionality for the ‘click’ cycloaddition. We carefully considered several of the more common routes to 2-substituted oxazolines which included the dehydration of N-acylaminoalcohols6a and the bimolecular cyclocondensation routes involving aminoalcohols and imidates6b or aminoalcohols and nitriles.6c Finally, we decided on the pursuit of an oxazoline synthesis based on the relatively mild Staudinger/aza-Wittig reaction of azido esters (Scheme 1).7 The advantages of the Staudinger/aza-Wittig scheme are the effective access to the azidoester intermediates as well as the straightforward accommodation of their stereocenters which will ultimately be expressed in the oxazoline product. Moreover, through enzyme-mediated enantioselective hydrolysis of azido acetate derivatives, access to either antiopodal intermediate en route to the target oxazoline can be realized.
Scheme 1.

Staudinger/aza-Wittig route to oxazolines.
2. Results and Discussion
Our synthesis begins with the readily-available unsaturated dibenzyl ether 1 in which each benzyloxymethyl group is positioned to become the substituent groups at positions 4 and 5 of the oxazoline target (Scheme 2).8 Epoxidation of 1 (mCPBA/CH2Cl2)9 gave the (dibenzyloxy)epoxide 2 (80%) which upon treatment with sodium azide (DMF/100 °C/24 h) gave the (dibenzyloxy) azido alcohols 3 (97%). In order to establish conditions and explore the scope of the Staudinger/aza-Wittig reaction the dibenzyloxyazidoester substrates 4, 5, 6, 7 were prepared from 3. Hence, (dibenzyloxy) azido alcohol 3 was esterified with the appropriate acid chlorides (RCOCl) in the presence of base to give azido esters 4–7 which were purified by column chromatography on silica gel. Treatment of the azidoesters 4–7 with triphenylphosphine (1.5 eq.) in tetrahydrofuran (rt to 40 °C/16 h) gave the corresponding oxazolines 8–11 in 50–94% isolated yield. Thus, the reagents/conditions of the Staudinger/aza-Wittig reaction for conversions of azidoesters 4–7 to oxazolines 8–11 were found to be of general applicability.
Scheme 2.

Preparation of oxazolines 8-11 from azidoesters 4-7. Reagents/Conditions: (a) mCPBA/DCM/0 °C to rt/24 h, 80%. (b) NaN3/DMF/100 °C, 24 h, 97%. (c) ROCl/Et3N or pyridine/DMAP/DCM/rt/16–48 h. (d) PPh3/THF/rt to 40 °C/16 h.
With the conditions of the Staudinger/aza-Wittig cyclization now established, we then prepared the (dibenzyloxy)azidoalcohol (2R,3R)-3a generated from enantioselective esterase hydrolysis of the racemic acetate derivative 12 (Scheme 3).10 Hence, the (dibenzyloxy)azidoalcohol 3 was then acetylated (acetic anhydride/pyridine/1.5 h/rt) to provide the (dibenzyloxy)-azidoacetate 12 (89%). Addition of the (dibenzyloxy)azidoacetate 12 to a suspension of pig liver esterase (PLE) in aqueous phosphate buffer (pH=7.4, 35 °C/7 days) gave the chromatographically-separable mixture of (2R,3R)-azidoalcohol 3a [α]D20 −14.2 (c=0.4, CHCl3) (42% based on racemic 12) and unhydrolyzed azidoacetates 12a/12b. We were satisfied that the aqueous conditions offered by the pH=7.4 buffer ensured the optimum performance of the esterase as was the case in our previous work with esterase enzyme systems.11 For comparison, we explored the enzymatic resolution of the azidoalcohol mixture using various lipases and vinyl acetate in organic solvents and found that the “reverse” enzymatic esterification mode was unsuccessful with these substrates. The absolute configuration of azidoalcohol (2R,3R)-3a was confirmed by correlation with products prepared in two separate previously reported syntheses from tartaric acid derivatives.12a–d Interestingly, after several enzymatic runs, our (2R,3R)-3a consistently gave a somewhat higher rotation than the azidoalcohol product obtained in one reported synthesis (Lit. [α]D20 −10.5, c=0.4, CHCl3).12a The enantiomeric purity of (2R,3R)-3a was determined by preparation of its Mosher ester 13a13 (See Supplementary Data) from (R)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetic acid [(DCC/DMAP/CH2Cl2), [α]D20 +19.0 (c=0.18, CHCl3)] and the ee was found to be >99% by 19F NMR analysis. After chromatographic separation of azidoalcohol 3a from the unhydrolyzed azidoesters, the presence of unhydrolyzed azidoester 12a was confirmed by saponification (LiOH/H2O) of the recovered azidoacetate mixture and revealed material of lower rotation, presumably due to the presence of the antipodal azidoalcohol derived from 12b. Therefore, the 12a/12b mixture could be recycled with fresh esterase to provide additional azidoalcohol (2R,3R)-3a. Starting with the esterase product azidoalcohol (2R,3R)-3a, treatment with 4-nitrobenzoyl chloride (pyridine/DMAP/CH2Cl2) afforded the corresponding 4-nitrobenzoyl ester (2R,3R)-7a [97%, [α]D20 −21.8 (c=0.57, CHCl3)]. Staudinger/aza-Wittig cyclization of 4-nitrobenzoyl ester (2R,3R)-7a (PPh3/THF) gave the (4R,5R)-2-(nitrophenyl)oxazoline 11a [78%, [α]D20 +12.1 (c=0.34, CHCl3)] after purification by flash-column chromatography. The reduction-azidation of the (nitrophenyl)oxazoline (4R,5R)-11a involved a two-step sequence whereby the arylnitro group was reduced with aluminum amalgam (THF/H2O)14,15 followed by direct treatment with sodium nitrite/sodium azide (AcOH/H2O) to give azidophenyl-oxazoline (4R,5R)-14 [69% over two steps, [α]D20 +8.0 (c=0.07, CHCl3)]. The (4R,5R)-2-(azidophenyl)-oxazoline 14 responded well to the click reaction with 1-ethynyl-4-fluorobenzene, a common co-reactant with several of our azidophenyloxazoles from previous studies.3a–c Thus, admixture of copper sulfate pentahydrate and sodium ascorbate to a solution of azidophenyloxazoline (4R,5R)-14 and 1-ethynyl-4-fluorobenzene in THF/H2O followed by stirring (16 h) gave the (4R,5R)-(oxazolinylphenyl)triazole click product (4R,5R)-15 [94%, [α]D20 +4.5 (c=0.16, CH2Cl2)] as a white solid after flash column chromatography on silica gel. The click product (4R,5R)-15 was characterized by 1H NMR as the 1,4-’anti’-disubstituted triazole whereby the regiochemistry of the cycloaddition is consistent with the copper (I)-catalyzed dipolar cycloaddition mechanism.16
Scheme 3.

Pig liver esterase-mediated hydrolysis of azidoacetate 12 giving (2R,3R)-3a followed by Mosher ester derivatization and acylation with 4-nitrobenzoyl chloride. Reagents/Conditions: (a) Ac2O/pyridine/rt/1.5 h, 89%. (b) PLE/phosphate buffer/35 °C/7 days, 42%. (c) R-(+)-α-methoxy-α-(trifluoromethyl)phenylacetic acid/DCC/DCM/rt/48 h, 61% (d) 4-nitrobenzoyl chloride/pyridine/DMAP/DCM/rt/16 h, 97%.
3. Conclusions
The employment of a stereoselective enzymatic hydrolysis using pig liver esterase (PLE) combined with the Stauginger/aza-Wittig cyclization proves to be a mild and effective route to stereodefined 2-aryl-4,5-disubstituted oxazolines. Under the ideal conditions using aqueous phosphate buffer, the key intermediary β-azidoacetate is a substrate which yields the corresponding non-racemic azido alcohol in good yield and high ee. The use of aqueous conditions in the enzymatic step is a noteworthy example in of “green chemical” transformation and an otherwise eco-friendly step. The reduction-azidization sequence involving the aluminum amalgam reduction of the nitrophenyloxazoline was noteworthy and is an effective application to click chemistry as a result of our previous studies. The application of the esterase hydrolysis scheme will be applied to the complete stereochemical array of 4,5-disubstituted oxazolinyl intermediates followed by their conversion to adherence inhibitors and will be reported in due course.
Supplementary Material
Scheme 4.

Conversion of 4-nitrobenzoyl ester (2R,3R)-7a to oxazoline (4R,5R)-11 followed by azidation to give click partner (4R,5R)-14 and subsequent click reaction to afford (4R,5R)-15. Reagents/Conditions: (a) PPh3/THF/rt to 40 °C/16 h, 78%. (b) Al(Hg)/THF/H2O/rt/30 min. (c) NaNO2/ NaN3/AcOH/H2O/0 °C to rt/1.5 h, 69% (two steps). (d) 4-ethynylfluorobenzene/CuSO4·5H2O/sodium ascorbate/THF/H2O/rt/16 h, 94%.
Highlights.
Cyclization under Mild Conditions
Enzyme Hydrolysis under Aqueous Conditions
High ee’s
Acknowledgments
Funding in part was provided by a University of Louisville Dissertation Completion Fellowship and NIH/NIDCR 1R01DE023206. Acknowledgement is made to the NSF (CHE1726633) for support of the Indiana University Mass Spectrometry Facility.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Data
Supplementary data features detailed experimental procedures, characterization data (1H, 13C and 19F NMR, FTIR, and HRMS data), and copies of spectra is provided.
References and notes
- 1.(a) Chaudhry P Schoenan F Neuenswander B Lushington GH Aubé JJ Comb. Chem 9 (2007) 473–476. doi: 10.1021/cc060159t [DOI] [PMC free article] [PubMed] [Google Scholar]; For a recent review, See:; (b) Yoshimura A Saito A Yusubov MS Zhdankin VV Synthesis 52 (2020) 2299–2310. doi: 10.1055/s-0040-1707122 [DOI] [Google Scholar]
- 2.Gehringer M Laufer SA Angew. Chem. Int. Ed 56 (2017) 2–4. doi: 10.1002/anie.201710195 [DOI] [PubMed] [Google Scholar]
-
3.(a) Patil PC
Tan J
Demuth DR
Luzzio FA
MedChemComm
10 (2019) 268–279.doi: 10.1039/c8md00405f [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Patil PC
Tan J
Luzzio FA
Demuth DR
Antimicrob. Agents. Chemother
62 (2018) e00400–18. doi: 10.1128/AAC.00400-18 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Patil PC
Tan J
Demuth DR
Luzzio FA
Bioorg. Med. Chem
24 (2016) 5410–5417. doi: 10.1016/j.bmc.2016.08.059 [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Loner CM
Luzzio FA
Demuth DR
Tetrahedron Lett. 53 (2012) 5641–5644. doi: 10.1016/j.tetlet.2012.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Demuth DR
Luzzio FA
Antibiofilm Compounds US
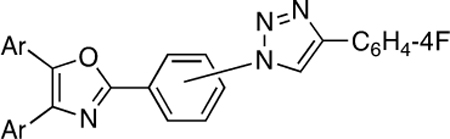
9,167,820, 2015. [Google Scholar]; A typical oxazole-based adherence inhibitor motif is shown below whereby the acetylenic click partner is 4-ethynylfluorobenzene (Ref 3c).
- 4.(a) Zhou M Qian Y Xie J Zhang W Jiang W Xiao X Chen S Dai C Cong Z Ji Z Shao N Liu L Wu Y Liu R Angew. Chem. Int. Ed 59 (2020) 7412–6419. doi: 10.1002/anie.202000505 [DOI] [PubMed] [Google Scholar]; (b) Taechalertpaisarn J Lyu RL Arancillo M Lin C-M Jiang Z Perez LM Ioerger TR Burgess K Org. Biomol. Chem 17 (2019) 908–915. doi: 10.1039/c8ob02901f [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li X Taechalertpaisarn J, Xin D, Burgess K, Org. Lett 17 (2015) 632–635. doi: 10.1021/ol5036547 [DOI] [PubMed] [Google Scholar]
- 5.(a) Liu G-Q, Yang C-H, Li Y-MJ Org. Chem 80 (2015) 11339–11350. doi: 10.1021/acs.joc.5b01832 [DOI] [PubMed] [Google Scholar]; (b) Castellano S, Kuck D, Sala M, Novellino E, Lyko F, Sbardella G, J. Med. Chem 51 (2008) 2321–2325. doi: 10.1021/jm7015705 [DOI] [PubMed] [Google Scholar]
- 6.(a) Phillips AJ, Uto Y, Wipf P, Reno MJ, Williams DR, Org. Lett 2 (2000) 1165–1168. doi: 10.1021/ol005777b [DOI] [PubMed] [Google Scholar]; (b) Meyers AI, Slade J, J. Org. Chem 45 (1980) 2785–2791. Doi: 10.1021/jo01302a008 [DOI] [Google Scholar]; (c) Trose M, Lazreg F, Lesieur M, Cazin CSJ, J. Org. Chem 80 (2015) 9910–9914. doi: 10.1021/acs.joc.5b01382 [DOI] [PubMed] [Google Scholar]
- 7.(a) Wang L, Ren ZL, Chen M, Ding MW, Synlett 25 (2014) 721–723 doi: 10.1055/s-0033-1340596 [DOI] [Google Scholar]; (b) Loos P, Ronco C, Riedrich M, Arndt HD, Eur. J. Org. Chem (2013) 3290–3315. doi: 10.1002/ejoc.201300160 [DOI] [Google Scholar]; (c) Nebot J, Romea P, Urpi F, Org. Biomol. Chem 10 (2012) 6395–6303. doi: 10.1039/c2ob25793a [DOI] [PubMed] [Google Scholar]; (d) Wu J, Liu JC, Wang L, Ding MW, Synlett 19 (2011) 2880–2882. doi: 10.1055/s-0031-1289865 [DOI] [Google Scholar]; (e) De Moliner F, Crosignani S, Banfi L, Riva R, Basso A, J. Comb. Chem 12 (2010) 613–616. doi: 10.1021/cc100122n [DOI] [PubMed] [Google Scholar]; (f) Takeuchi H, Yanagida S, Ozaki T, Hagiwara S, Eguchi S, J. Org. Chem 54 (1989) 431–434. doi: 10.1021/jo00263a033 [DOI] [Google Scholar]
- 8.(a) Schill H, DeMeijere A, Yufit DS, Org. Lett 9 (2007) 2617–2620. doi: 10.1021/ol070707r [DOI] [PubMed] [Google Scholar]; (b) Charette AB, Gagnon A, Fournier JF, J. Am. Chem. Soc 124 (2002) 386–387. doi: 10.1021/ja017230d [DOI] [PubMed] [Google Scholar]; (c) Wu FL, Ross BP, McGeary RP, Eur. J. Org. Chem 10 (2010) 1989–1998. doi: 10.1002/ejoc.200901264 [DOI] [Google Scholar]
- 9.(a) Schmidt Y, Breit B, Chem. Eur. J 17 (2011) 11780–11788. doi: 10.1002/chem.201100843 [DOI] [PubMed] [Google Scholar]; (b) Hachiya I, Matsumoto T, Inagaki T, Takahashi A, Shimizu M, Heterocycles 82 (2010) 449–460. doi: 10.3987/COM-10-S(E)16 [DOI] [Google Scholar]; (c) Schomaker JM, Bhattacharjee S, Yan J, Borhan B, J. Am. Chem. Soc 129 (2007) 1996–2003. doi: 10.1021/ja065833p [DOI] [PubMed] [Google Scholar]
- 10.For a recent review see:; Musa MM, Hollmann F, Mutti FG, Catal. Sci. Technol 9 (2019) 5487–5503. doi: 10.1039/c9cy01539f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Luzzio FA, Fitch RW, J Org. Chem 64 (1999) 5485–5493. doi: 10.1021/jo990293i [DOI] [PubMed] [Google Scholar]; (b) Luzzio FA, Duveau DY Tetrahedron Asymm. 13 (2002) 1173–1180. doi: 10.1016/S0957-4166(02)00313-0 [DOI] [Google Scholar]
- 12.(a) Aboulaala K, Goux-Henry C, Sinou D, Safi M, Soufiaoui M, J. Mol. Catal. A 237 (2005) 259–266. doi: 10.1016/j.molcata.2005.05.006 [DOI] [Google Scholar]; (b) Tanner D, Birgersson C, Gogoll A, Tetrahedron 50 (1994) 9797–9824. doi: 10.1016/S0040-4020(01)85546-0 [DOI] [Google Scholar]; (c) Kinugasa M, Harada T, Oku A, Tetrahedron Lett. 39 (1998) 4529–4532. doi: 10.1016/S0040-4039(98)00823-5 [DOI] [Google Scholar]; (d) The second previously reported synthesis of (2R,3R)-3a (rotation not reported) culminated in the corresponding (2R,3R) amino alcohol ([α]D20 +11.8, c=1.1, CHCl3), and accordingly, we reduced our (2R,3R)-3a to the corresponding amino alcohol which gave a comparable rotation ([α]D20 +13.6, c=0.08, CHCl3) [Google Scholar]
- 13.Eliel EL; Wilen SH; Mander LN Stereochemistry of Carbon Compounds (1994) John Wiley &Sons: New York, pp 222–223. ISBN: 0-471-01670-5 [Google Scholar]
- 14.Keck GE, Fleming S, Nickell D, Weider P, Synthetic Commun. 9 (1979) 281–286. doi: 10.1080/00397917908064153 [DOI] [Google Scholar]
- 15.Monsen PJ, Luzzio FA, Tetrahedron Lett. 61 (2020) 152575–152579. doi: 10.1016/j.tetlet.2020.152575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meldal M, Tornøe CW, Chem. Rev 108 (2008) 2952–3015. doi: 10.1021/cr0783479 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.