

Abstract
Phagosomal compartments are critical in microbial defense as vesicles that degrade invading organisms. In a broader context, vesicular trafficking plays an important role in shuttling many different types of cargo that are critical for proper function of the cell. Endosomal and phagosomal vesicles are thus important locations for the assembly of intracellular signaling platforms that mediate host responses. Isolation of phagosomes from cells is an important technique that allows for a detailed study of phagosomal components and signaling complex assembly. However, purification of phagosomes had previously been challenging and it has been difficult to obtain sufficient purity of the phagosomal fractions. Here we modify a new magnetic isolation technique that greatly simplifies purification of phagosomes and isolates vesicles with sufficient purity for analysis.
Keywords: Phagocytosis, Phagosomes, Magnetic beads, Toll-like receptors, TLR2, Lyme disease, Borrelia burgdorferi
1. Introduction
Toll-like receptor (TLR) signaling requires the assembly of signaling platforms to transduce recognition of cognate ligands into cellular immune responses [1, 2]. Assembly of these signaling platforms may occur at different cellular locations dependent upon the specific TLR involved [3–7]. The location at which the TLR engages its ligand and the signaling platform is assembled may affect both the composition of the adaptor molecules recruited to the platform as well as the outcome of activation [5, 7]. Thus understanding the movement of ligands, innate immune receptors and adaptor molecules has become an area of great interest. Although live imaging microscopy and confocal microscopy have been invaluable tools these techniques are often performed using over-expressed fluorescently tagged proteins. This overexpression can lead to artifacts and the technique suffers from limitations in the number of molecules that can be tracked simultaneously. Isolation of phagosomes containing specific molecules allows for probing of the contents of these vesicles using western blotting or mass spectroscopy. Prior attempts at phagosomal purification had proved to be challenging [8–12]. A number of laboratories had purified phagosomes using latex beads, fractionation and gradient purification. However, these procedures required multiple purification steps and were complicated by the fragility of the phagosomes and the inability to obtain sufficiently pure fractions.
Recently, we adapted a magnetic purification technique for the isolation of phagosomes for the study of TLR2 ligand engagement of TLRs within phagosomes [6]. We utilized streptavidin coated 2.8μm magnetic Dynabeads which were then easily coated with TLR-biotin ligands or just with biotin alone. Bone marrow derived macrophage (BMDM) were allowed to phagocytose the beads and cells were then lysed. Using a neodymium magnet we were able to perform a one-step magnetic enrichment of the phagosomal fraction. We confirmed the purity of phagosomal enriched fraction with two methods: 1) by western blotting for molecules that are at the phagosome versus those that are cytoplasmic and 2) by fluorescent cell surface labeling of the plasma membrane.
We have successfully isolated phagosomal fractions using this technique and have been able to analyze by western blotting the presence of TLR signaling molecules at the phagosome. Our magnetic beads coated with TLR2 ligand mimic the internalization of the pathogen Borrelia burgdorferi. This process could easily be adopted to the study of other TLR ligands or organisms. We hope that the description of our procedure will help other investigators in answering similar questions about cellular trafficking and phagosomal signaling.
2. Materials
2.1. Bone marrow derived macrophage (BMDM) or other phagocytic cells or cell line
Dulbeco’s modified Eagle Medium (DMEM)
Fetal Bovine Serum (FBS)
Penicillin/Streptomycin/Amphothericin; tissue culture grade 100X
L929 cell supernatant
2.2. Phagosomal enrichment
TLR-biotin ligands (in this case Pam3CSK4-biotin)
Endotoxin free water
2.8 μm Dynabeads M-270
1 x Dulbecco’s Phosphate Buffered Saline (PBS): tissue culture grade, no calcium or magnesium
Complete Protease Inhibitor tablet
Neodymium block magnet 2”× 1” × 1”
Mammalian Cell Lysis Buffer: Pierce proprietary detergent in 25mM bicine buffer (pH 7.6)
NuPAGE LDS Sample Buffer (4X): Lithium dodecyl sulfate
2.3. Antibodies and Western Blot reagents
Total Jnk Antibodies
Vamp1/2/3 Antibodies
CD11b-FITC or CD11b coupled with anti-rabbit Alexa488
Tris Buffered Saline: 6.05 g Tris and 8.76 g NaCl in 800 mL of H2O. Adjust pH to 7.5
Bovine Serum Albumin
Tween 20
Immobilon-P PVDF membrane (0.2μm): Polyvinylidene Fluoride
Running Buffer/Transfer Buffer: Bio-Rad Tris Glycine 10X buffer
3. Methods
3.1. Preparation of Pam3CSK4-coated magnetic beads
TLR-biotin conjugates (Invivogen) are re-suspended in endotoxin free-water provided by the manufacturer at their suggested concentrations. Our studies were performed with the TLR2 ligand Pam3CSK4-biotin at a stock concentration of 1mg/ml.
2.8 μm Dynabeads M-270 are provided in suspension from the manufacturer at 6×108 beads /ml. Beads are spun down at 500 x g for 5 minutes and re-suspended at a concentration of 1×106 beads/μl in endotoxin free water.
50μl of beads (5×107 beads) are incubated with 50μg of Pam3CSK4-biotin ligand (50μl) for 2 hours at room temperature or overnight at 4°C (see note 1).
Beads are spun down at 300 x g, washed twice in cell culture medium (DMEM 10%FBS) and re-suspended in 50μl of media for a final concentration of 1×106 Pam3CSK4-coated beads/μl (see note 2).
3. 2. Growth of Bone marrow derived macrophages
BMDM were generated according to standard protocols.
Bone marrow cells were flushed from mouse femurs and tibiae with sterile DMEM 10% FBS.
Cells are cultured in 100×15-mm plastic petri dishes (see note 3) for 5–7 days in DMEM 20% FBS supplemented with 30% L929 cell supernatant (see note 4) and 1% penicillin-streptomycin-amphotericin (see note 5).
After differentiation, aspirate media and undifferentiated progenitors and collect cells by pipetting with ice-cold media DMEM 10% FBS or ice-cold 1 x PBS. Collect the cells and count them.
3.3. Cell surface fluorescent labelling
BMDM are plated at 1×106 cells/well in 6 well tissue culture treated plates in DMEM 10% FBS and allowed to settle overnight (see note 6).
BMDM were cell surface labeled with CD11b-FITC antibody (Santa Cruz Biotechnology) at a 1:500 dilution for 1 hour in DMEM 10%FBS (see note 7). Alternatively you can use CD11b antibody (Abcam) at 1:500 for 1 hour followed by a secondary antibody conjugated to Alexa Fluor 488 at 1:1000 for 1 hour.
Cells were washed with media twice and fresh media was replaced.
3. 4. Phagocytosis and magnetic enrichment
-
1.
Cells are stimulated for 60 minutes (see note 8) with 10μl of 1×106 Pam3CSK4-coated beads/μl, for a final concentration of 1×107 beads per 1×106 cells, mimicking an MOI 10 (see note 9).
-
5.
Cells are collected by removing the media and then scraping in 200μl of cold 1x Tissue Culture Grade Phosphate Buffered Saline (PBS) containing Complete Protease Inhibitor Cocktail.
-
6.
The cells are then sonicated with 10 × 1 second pulses of 1–2 watts (see note 10).
-
7.
The cell lysate is then subjected to a neodymium magnet (see note 11) enrichment by applying the magnet to the side of the microfuge tube containing the lysate.
-
8.
The magnetic bead pellet is easily visible and the remaining lysate containing the membrane and cytosolic fractions is collected and saved for analysis by aspirating the supernatant with a pipet while the tube is next to the magnet.
-
9.
The magnetically enriched pellet is re-suspended in 70μl of cell lysis buffer. 10μl are set aside for measuring fluorescence. 20μl of 4x LDL SDS-PAGE running buffer is added to the remaining 60μl for western blot analysis.
3.5. Confirmation of phagosomal purity
Lysates are run on SDS-PAGE and transferred to a PVDF membrane for western blotting. We used total Jnk antibodies at 1:1000 to detect proteins specific to the cytoplasmic fraction or Vamp 1/2/3 antibodies at 1:1000 to detect proteins specific to the phagosomal membrane (Figure 1) (see note 12). However, others may wish to choose antibodies to other markers to suit their needs.
Membrane and cytosolic fractions versus phagosomal fractions are measured for FITC fluorescence using a standard fluorescent plate reader with a FITC filter. Data are plotted as mean fluorescent intensity (Figure 2).
Figure 1. Confirmation of phagosomal purity by western blot analysis of cellular fractions.
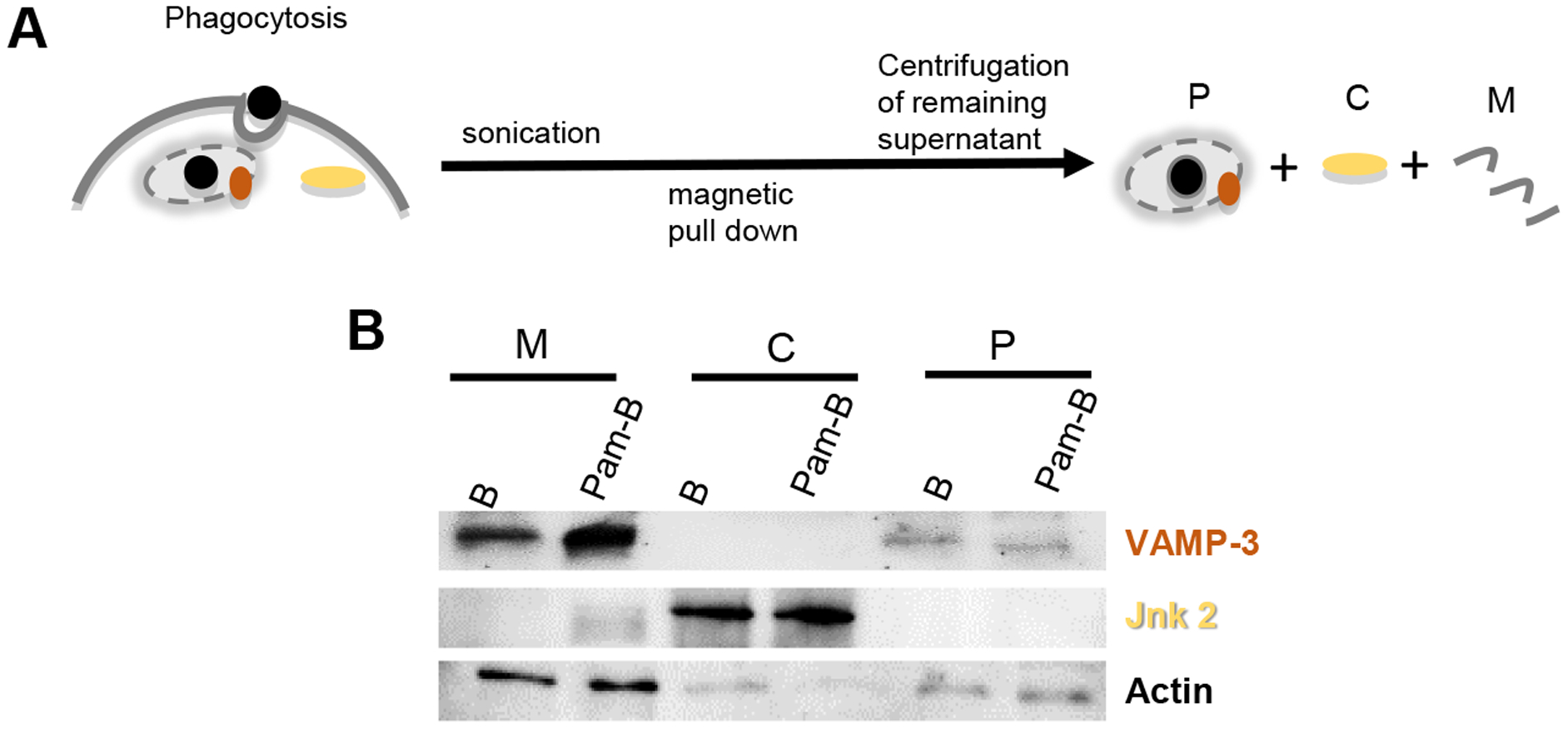
A) Streptavidin coated Magnetic Beads (B) or beads coated with Pam3CSK4-biotin (Pam-B) were allowed to be phagocytosed by bone marrow derived macrophage (BMDM) for 60 minutes. After phagocytosis cells were collected and sonicated. Lysates were subjected to magnetic pull down to collect the phagosomal fraction (P). The remaining supernatant was separated into cellular membrane (M) and cytosolic fractions (C) by centrifugation. B) Samples were loaded on SDS-PAGE as cell equivalents and blotted with the indicated antibodies. The purity of our phagosomal preparations was confirmed by detecting the presence of VAMP-3 at the phagosome but not cytosolic fractions, while the Jnk2 kinase was found in the cytosol but not the phagosomal fraction.
Figure 2. Confirmation of phagosomal purity by fluorescent labeling of the plasma membrane.
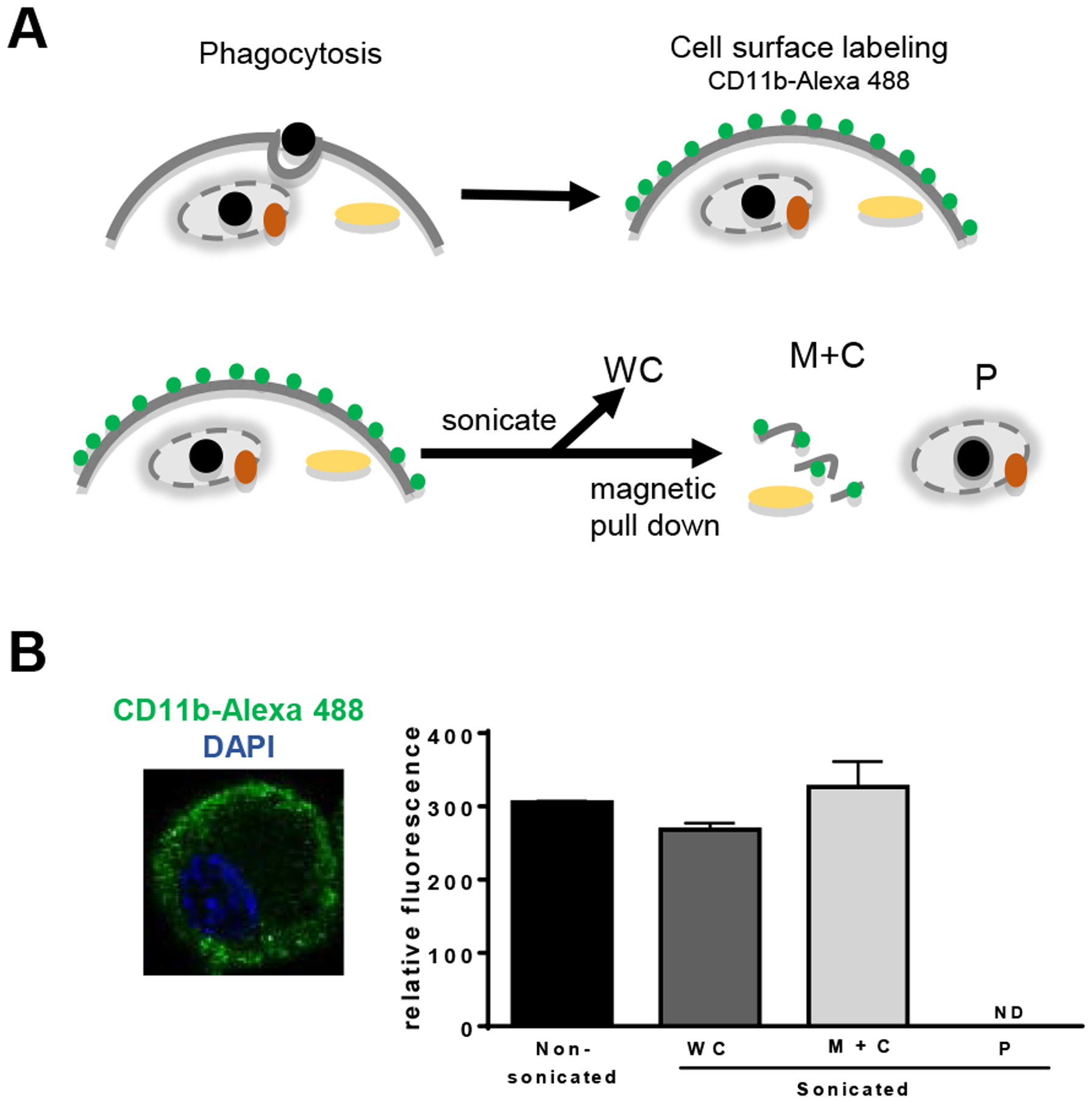
A) As a second method of confirmation of the purity of our phagosomal preparations, we stimulated BMDM with B or Pam-B for 60 min and subsequently surface stained the cells with CD11b antibodies. Cells were then sonicated and subjected to magnetic pull down to separate the phagosomal fraction from the membrane and cytosolic fractions. B) Immortalized macrophage were cell surface stained with antibodies to CD11b, followed by secondary antibodies conjugated to Alexa 488 (green). Slides were mounted using a DAPI containing mounting medium to stain the nuclei (blue) from Vectashield. Cell surface labeled BMDM post phagocytosis and fractionated as in part A were read on a fluorescent reader. Phagosomal fractions were void of any fluorescent signal. Shown is one representative experiment out of three independent experiments.
Notes
It will not be possible to shake the beads efficiently in such a small volume. If you prefer to shake overnight, increase the volume to at least 750μl in 1xPBS or endotoxin free water. Otherwise, make sure you occasionally flick the tube during the 2 hour incubation period.
It is best if beads coated with ligand or with biotin are prepared fresh before each experiment.
Do not use tissue culture treated plates. The macrophage are strongly adherent cells and will be difficult to remove from tissue culture treated plates without trypsin. Grown on non-tissue culture treated plates they can be removed by pipetting with ice-cold media or ice cold tissue culture grade 1x PBS. If necessary, cells can be gently scraped off with a cell scraper.
Murine L929 fibroblasts (L-1 at ATCC) secrete Macrophage Colony Stimulating Factor (MCSF) necessary for macrophage differentiation. The cells are grown in 225 cm2 tissue culture treated flasks till confluency in 100 ml of DMEM 10% FBS. Cells are allowed to grow past confluency for 3–4 days. Longer is better but having too many dead cells is not desirable either, so 3–4 days is ideal. Supernatants are then collected and filter sterilized.
Although standard protocols call for a change in media every two days, we have found that addition of new media, rather than aspiration and change of media, generates larger yields of macrophage. Aspiration of the media results in significant loss of yet undifferentiated progenitors and lower macrophage yields. Also feeding the cells daily will help them differentiate faster. We recommend a daily media supplement of 5ml of DMEM, 20% FBS, 1% Pen/Strep/Amph., 30% L929 supernatant.
Being primary cells, BMDM will not grow overnight like immortalized cell lines. The number of cells plated the day before should remain constant.
This can also be done in 1 x PBS (tissue culture grade) at 37°C.
You may choose to synchronize your beads. The magnetic beads are relatively heavy and will sink to the bottom where they will come in contact with the cells (approximately within 10 minutes depending upon volume). If you are performing a 60 minute time point, it may not be necessary to synchronize the beads. For shorter phagocytosis times, you may choose to let the beads settle for 10 minutes, then remove all the media and wash once. Add new media and set that as your starting point for phagocytosis. Note that you will collect significantly less beads for shorter time points making your protein of interest harder to detect on a western blot. You may want to increase the numbers of cells and beads utilized if you are aiming for shorter time points.
60 minutes is a long time point for phagocytosis. BMDM are efficient phagocytes and will internalize beads within minutes. Within an hour the beads will have proceeded through most of the phagosomal maturation stages and will most likely be at the lysosomal stage. If you are interested in earlier phagosomal stages you will have to do a time course to optimize the correct time point.
Do not use any type of lysis buffer at this step as it will destroy your phagosomes. You may use a Dounce Homogenizer instead of sonication. Although more gentle on phagosomes, in our hands Dounce Homogenizing was also less effective at cell lysis and ultimately yielded a lower phagosomal enrichment fraction as many cells remained intact.
These magnets are quite powerful. Keep them away from any electronics. We recommend at minimum the size block magnet we recommended, bigger block magnets exist and may work even better.
We suggest that you pour a 1.5 mm 10% Tris-glycine polyacrylamide gel or use a gradient 4–20% Tris Glycine Biorad gel. Primary antibody incubation is recommended overnight in 5% BSA TBST.
References
- 1.Kawai T and Akira S, Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol Med, 2011. 3(9): p. 513–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kawai T and Akira S, The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol, 2009. 21(4): p. 317–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barton GM and Kagan JC, A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol, 2009. 9(8): p. 535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonham KS, et al. , A promiscuous lipid-binding protein diversifies the subcellular sites of toll-like receptor signal transduction. Cell, 2014. 156(4): p. 705–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kagan JC, et al. , Tram couples endocytosis of Toll-like receptor 4 to the induction of interferon beta. Nature Immunology, 2008. 9: p. 343–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mantegazza AR, et al. , Adaptor protein-3 in dendritic cells facilitates phagosomal toll-like receptor signaling and antigen presentation to CD4(+) T cells. Immunity, 2012. 36(5): p. 782–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sasai M, Linehan MM, and Iwasaki A, Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science, 2010. 329(5998): p. 1530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desjardins M and Griffiths G, Phagocytosis: latex leads the way. Curr Opin Cell Biol, 2003. 15(4): p. 498–503. [DOI] [PubMed] [Google Scholar]
- 9.Desjardins M, et al. , Molecular characterization of phagosomes. J Biol Chem, 1994. 269(51): p. 32194–200. [PubMed] [Google Scholar]
- 10.Desjardins M, ER-mediated phagocytosis: a new membrane for new functions. Nat Rev Immunol, 2003. 3(4): p. 280–91. [DOI] [PubMed] [Google Scholar]
- 11.Gagnon E, et al. , Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell, 2002. 110(1): p. 119–31. [DOI] [PubMed] [Google Scholar]
- 12.Stuart LM and Ezekowitz RA, Phagocytosis and comparative innate immunity: learning on the fly. Nat Rev Immunol, 2008. 8(2): p. 131–41. [DOI] [PubMed] [Google Scholar]