

Abstract
Background
Idiopathic pulmonary fibrosis (IPF) is a complex lung disease, characterized by progressive lung scarring. Severe COVID-19 is associated with substantial pneumonitis and has a number of shared major risk factors with IPF. This study aimed to determine the genetic correlation between IPF and severe COVID-19 and assess a potential causal role of genetically increased risk of IPF on COVID-19 severity.
Methods
The genetic correlation between IPF and COVID-19 severity was estimated with linkage disequilibrium (LD) score regression. We performed a Mendelian randomization (MR) study for IPF causality in COVID-19. Genetic variants associated with IPF susceptibility (P<5 × 10−8) in previous genome-wide association studies (GWAS) were used as instrumental variables (IVs). Effect estimates of those IVs on COVID-19 severity were gathered from the GWAS meta-analysis by the COVID-19 Host Genetics Initiative (4,336 cases & 623,902 controls).
Findings
We detected a positive genetic correlation of IPF with COVID-19 severity (rg=0·31 [95% CI 0·04–0·57], P = 0·023). The MR estimates for severe COVID-19 did not reveal any genetic association (OR 1·05, [95% CI 0·92–1·20], P = 0·43). However, outlier analysis revealed that the IPF risk allele rs35705950 at MUC5B had a different effect compared with the other variants. When rs35705950 was excluded, MR results provided evidence that genetically increased risk of IPF has a causal effect on COVID-19 severity (OR 1·21, [95% CI 1·06–1·38], P = 4·24 × 10−3). Furthermore, the IPF risk-allele at MUC5B showed an apparent protective effect against COVID-19 hospitalization only in older adults (OR 0·86, [95% CI 0·73–1·00], P = 2·99 × 10−2) .
Interpretation
The strongest genetic determinant of IPF, rs35705950 at MUC5B, seems to confer protection against COVID-19, whereas the combined effect of all other IPF risk loci seem to confer risk of COVID-19 severity. The observed effect of rs35705950 could either be due to protective effects of mucin over-production on the airways or a consequence of selection bias due to (1) a patient group that is heavily enriched for the rs35705950 T undertaking strict self-isolation and/or (2) due to survival bias of the rs35705950 non-IPF risk allele carriers. Due to the diverse impact of IPF causal variants on SARS-CoV-2 infection, with a possible selection bias as an explanation, further investigation is needed to address this apparent paradox between variance at MUC5B and other IPF genetic risk factors.
Funding
Novo Nordisk Foundation and Oak Foundation.
Keywords: Mendelian randomization, Covid-19, Idiopathic pulmonary fibrosis, MUC5B, Mucin
Research in context.
Evidence before this study
Idiopathic pulmonary fibrosis (IPF), a complex lung disease characterized by progressive lung scarring, shares a number of major risk factors with COVID-19. Furthermore, severe COVID-19 is associated with substantial pneumonitis and IPF patients are at increased risk of severe COVID-19 and related mortality.
Added value of this study
By performing a Mendelian randomization analysis, this study identifies a causal relationship of IPF with severe COVID-19. Intriguingly, however, it demonstrates that the MUC5B allele has a different, protective, effect compared with all other IPF associated variants. This raises two important themes. One is the possibility that there are distinct endotypes of pulmonary fibrosis that reflect the underlying causal genetic variant and the other is that public health measures such as shielding or self-isolation policies introduce a bias that lead to an exaggeration of protective effects of genes that are over-represented in at risk populations.
Implications of all of the available evidence
Due to the diverse impact of IPF causal variants on SARS-CoV-2 infection, further investigation is needed to address this apparent paradox between variance at MUC5B and other IPF genetic risk factors.
Alt-text: Unlabelled box
1. Introduction
Since the emergence of a novel severe acute respiratory syndrome (SARS) coronavirus 2
(SARS-CoV-2) in Wuhan, China in December 2019, there have been more than 95 million confirmed cases and over 2 million deaths worldwide [1]. SARS-CoV-2 infection, which causes coronavirus disease 2019 (COVID-19), ranges from asymptomatic to severe disease needing ICU admission and mechanical ventilation2. Infection estimates may vary considerably within populations due to the frequency of asymptomatic disease and inherent risk factors for symptomatic disease as well as public health protection policies [3]. It is estimated that about 45% of those infected are asymptomatic, while up to 10% require hospitalization [4,5]. Severe disease, which occurs in up to 20% of hospitalized patients, is associated with a high mortality rate [6].
Idiopathic pulmonary fibrosis (IPF) is a complex lung disease characterized by progressive lung scarring caused by damage to the alveolar epithelium followed by an abnormal wound-healing response causing deposition of dense fibrotic tissue, which ultimately leads to loss of lung function and death through respiratory failure [7]. Moreover, despite the drugs pirfenidone and nintedanib being approved for IPF treatment, there is still no cure as these drugs only slow disease progression, and half of IPF patients die within 3 to 5 years after diagnosis [7,8]. IPF is influenced by both genetic and environmental factors. Previous genome-wide association studies (GWAS) have revealed common genetic variants associated with IPF [9,10], with the largest GWAS of IPF to date detecting 14 loci [9]. The most strongly associated IPF variant [9,11], rs35705950, has its risk allele (T) associated with a five-fold increase in disease risk9 and over expression of mucin 5B in small-airway epithelial cells [12,13].
COVID-19 and IPF both begin with lung injury, and their most severe consequences are seen in elderly males, with male IPF patients showing a high risk of COVID-19 mortality [14,15]. Thus, it is plausible that there are shared pathogenic mechanisms between severe COVID-19 and IPF, which may relate to an underlying shared genetic etiology. Should there be shared genetic and pathological mechanisms, this would provide some rationale for investigating whether repurposing of anti-fibrotic therapy could be a treatment strategy for patients with COVID-19.
This study uses the genetic determinants of IPF, estimated from IPF GWAS summary statistics [9,10] to perform two-sample Mendelian randomization analysis [16] to assess whether a causal relationship of genetically mediated IPF risk with COVID-19 severity is plausible [17]. We also tested the association of the most strongly associated IPF variant [9,11], rs35705950 at the MUC5B gene locus, with different COVID-19 control groups and in an age-stratified analysis. In addition, we analyze common genetic variation across the entire genome to estimate the genetic correlation between IPF [9] and COVID-19 severity [17].
2. Methods
2.1. Study population
We extracted association summary statistics from the largest meta-analysis GWAS of IPF to date (4124 cases and 20,465 controls) [9] as well as the only exome-wide association study (ExWAS) of IPF using whole genome and whole exome sequenced samples (752 cases and 119,055 controls) [10]. All IPF cases and controls were of genetically determined European ancestries. The summary statistics for the outcome of COVID-19 severity was extracted from the COVID-19 Host Genetics Initiative (HGI) GWAS meta-analysis [17], available at https://www.covid19hg.org/results/. These COVID-19 HGI summary statistics are from the fourth round of GWAS meta-analysis, publicly available since October 20th, 2020, but without the 23 and Me cohort. Although the 23 and Me cohort was part of the COVID-19 HGI GWAS meta-analysis, downloadable results that included the 23andMe cohort were only publicly available for the top 10,000 variants due to 23 and Me data use restrictions. The COVID-19 HGI is an international collaborative effort that aims to study the genetic determinants of COVID-19 susceptibility, hospitalization and severity. The COVID-19 HGI has gathered clinical and genetic data and performed GWAS meta-analysis of multiple cohorts with a fixed effects inverse variance weighting. The analysis was adjusted for age, age [2], sex, age*sex, genetic ancestry principal components and other study-specific covariates. An allele frequency of 0.001 and an imputation info score of 0.6 was applied to each study before meta-analysis. For the primary analysis of very severe respiratory confirmed COVID-19 vs population (COVID-19 HGI analysis A2), severe cases were defined as hospitalized patients with confirmed COVID-19 by RNA PCR, serologic testing, or physician diagnosis that had very severe respiratory complications (N = 4336). Population controls were defined as individuals who tested negative for COVID-19, were never tested, or had an unknown testing status (N = 623,902). About 89·6% of COVID-19 severe cases and 99·9% of controls were of European ancestries. Additional COVID-19 association analyses of rs35705950 at the MUC5B gene locus were undertaken using a further four COVID-19 HGI GWAS datasets: i) COVID-19 hospitalization (COVID-19 HGI analysis B2: 6406 cases and 902,088 population controls) ii) COVID-19 susceptibility (COVID-19 HGI analysis C2; 14,134 cases and 1284,876 population controls), restricted to European ancestry individuals, iii) hospitalized COVID-19 cases vs non-hospitalized COVID-19 (COVID-19 HGI analysis B1: 2430 cases and 8478 controls) and iv) COVID-19 positive cases vs lab and/or self-reported negative COVID-19 controls (COVID-19 HGI analysis C1: 24,057 cases and 218,062 controls).
2.2. Genetic instrument variants
We used the 15 independent genetic variants associated with IPF at genome wide significance (P<5 × 10−8) as instrumental variables (Supplementary Table 1). The genetic instruments were independent, as they were located at least more than 45 Mb away from each other with LD r2<0.002. F-statistic, as implemented before [40], was used to detect how strong instrument for the IPF exposure were the genetic IVs tested. An F-statistic less than 10 indicates a weak instrument [41]. For the only genetic variant out of the 15 that was not represented in the COVID-19 severity outcome GWAS [17], we selected the next best available genetic variant based on posterior probability from IPF credible sets [9], while having an LD r2>0·9 with the index variant using the CEU European sub-cohort of the 1000 Genomes Phase 3 dataset (original SNP is rs2077551; proxy SNP is rs17652520) (Supplementary Table 1). Of the 15 genetic instruments, rs35705950 at the MUC5B gene locus explains 5·9–9·4% of IPF liability in the general population, while the remaining 14 loci collectively explain up to 3% of IPF liability in the general population [11]. The effect allele for the 15 independent genetic variants associated with IPF was aligned in each study to be the same as to allow correct effect estimates for IPF and COVID-19 outcomes. Palindromic SNPs were taken care since association results were always performed on the forward strand, whether from imputation data [9] or exome sequencing data [10].
2.3. Mendelian randomization analysis
To investigate causality of IPF with COVID-19 severity, two-sample Mendelian randomization (MR) analysis was performed using the random-effects inverse-variance weighted method (IVW) [18], implemented in the R (version 3.6.1) [19] package MendelianRandomization (version 0.5.0). MR relies on three main assumptions: (1) the instrumental variables must be strongly associated with the exposure, (2) must not be associated with factors that confound the relationship between exposure and outcome, and (3) can only be associated with the outcome through affecting the exposure. Sensitivity analysis was performed with the weighted median [20] and MR-Egger methods [21], which are less powerful than the IVW method if all MR assumptions hold, but are more robust to invalid instruments and horizontal pleiotropy [22]. The random-effects IVW method [18], which assumes that all genetic variants are valid instruments, regresses the effect sizes of variant COVID-19 severity associations against effect sizes of the variant IPF associations, assuming that the strength of the association of the genetic instruments with IPF is not correlated with the magnitude of the pleiotropic effects or that the pleiotropic effects have an average value of zero. On the contrary, since the weighted median method uses the median instrumental variable from all variants, it is robust to pleiotropy when >50% of the weight comes from valid instruments [20]. By using a weighted regression with an unconstrained intercept, MR-Egger [21] does also not assume that all variants are valid instruments. Therefore, MR-Egger is also more robust to specifically genetic pleiotropy. If the MR-Egger intercept term differs significantly from zero, then the genetic variants are not all valid.
We used three different approaches to detect potentially pleiotropic SNPs. First, we used an IVW leave-one-out analysis, implemented in the MendelianRandomization R package to determine whether any outliers could bias the overall causal estimate. Second, pleiotropy against other phenotypes was also determined through PheWAS lookups of each genetic instrumental variable (and their high correlated proxy variants at LD r2≥0·8) against Phenoscanner [23] and GeneATLAS [24], with association results deemed significant at P<1 × 10−5 (restricted to non-cancer diseases, non-lung and non-blood trait phenotypes). Finally, MR-PRESSO outlier test [25], implemented in the MRPRESSO (version 1.0) R package [19] was also performed to detect horizontal pleiotropic IVs, with SNPs being called pleiotropic at P<0·05.
2.4. Age-stratified COVID-19 severity GWAS
To detect possible age-related effects of the IPF genetic instrument rs35705950 at the MUC5B gene locus on COVID-19 severity, two GWAS of COVID-19 severity were performed for individuals greater and lower than 60 years of age, respectively.
2.5. Genetic correlation
Analysis of genetic correlation between IPF and COVID-19 severity was performed using the LD score regression method [26,27] applied to GWAS summary statistics of both diseases [9,17] using only variants with MAF>1% in both GWAS that were present in the HapMap3 recommended SNP list [26].
2.6. Ethics
Covid-19 HGI and the IPF GWAS consortia have ethical approvals from their respective cohorts [9,10,17]. Patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of our research.
2.7. Role of funding source
The Funders had no role in study design, data collection, data analyses, interpretation, or writing of this study.
3. Results
3.1. Genetic correlation of IPF with COVID-19 severity
To quantify the shared genetic etiology of IPF with COVID-19 severity, we used the LD score regression method (LDSC) [26,27] and detected a positive genetic correlation of IPF susceptibility with COVID-19 severity (rg=0·31 [95% CI 0·04–0·57], P = 0.023 [LDSC]), which suggested a shared genetic etiology. A positive genetic correlation with COVID-19 hospitalization (rg=0·31 [95% CI 0·02–0·63], P = 0·035 [LDSC]) was also detected, but at low statistical significance and consequent wider confidence intervals. The genetic correlation between IPF risk and COVID-19 susceptibility was positive, though not statistically significant (rg=0·25 [95% CI −0·13–0·62], P = 0·193 [LDSC]).
3.2. Mendelian randomization analysis of COVID-19 severity
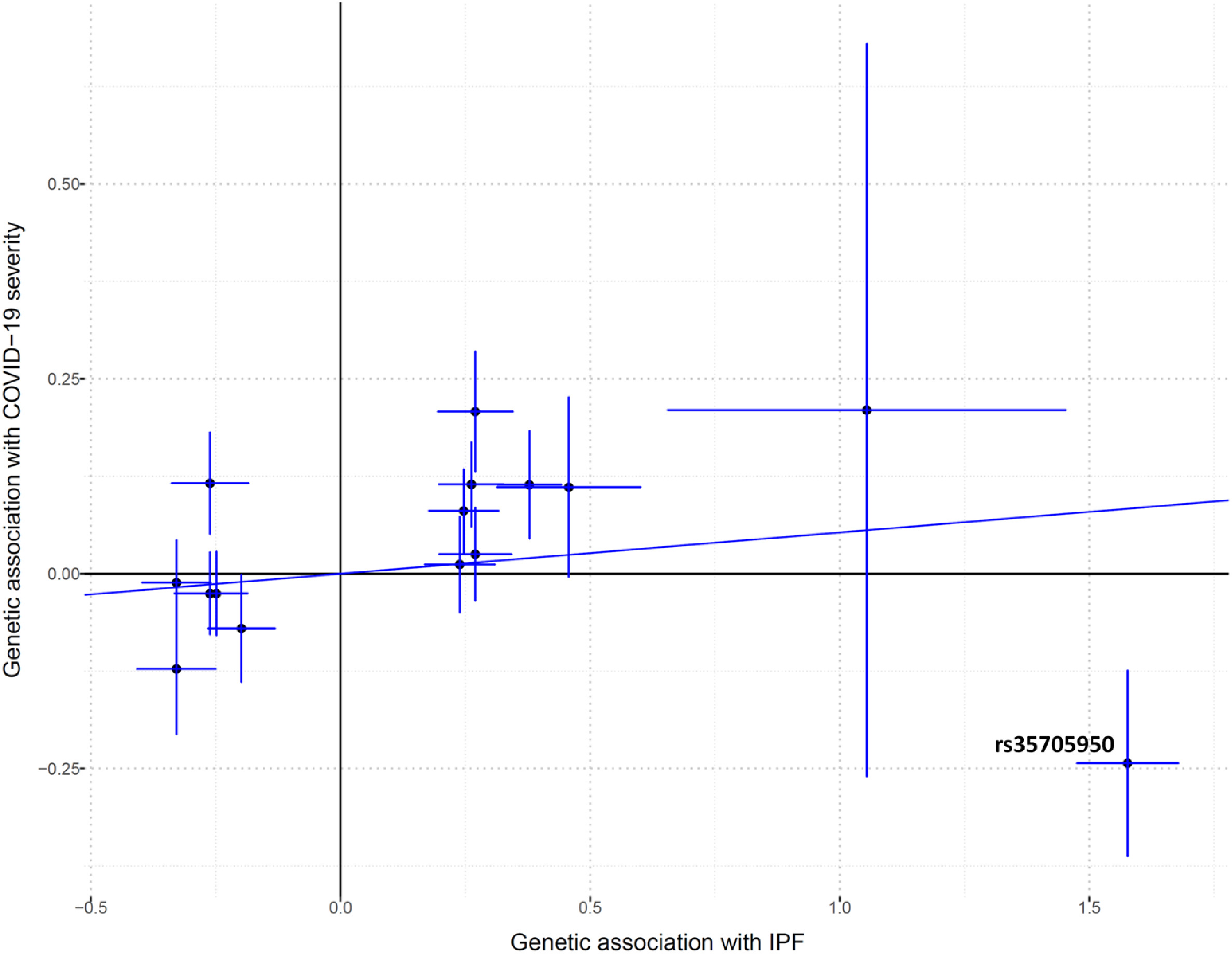
To determine whether the genetic correlation results have a known causal component, a two-sample MR analysis was performed to test the causal effect of IPF risk genes on COVID-19 severity. The F-statistic was 27, which show that the IVs tested were a strong instrument for the IPF exposure. Moreover, by using a known method for determining the statistical power of MR studies [42], we had 82% power to detect a true odds ratio of 1·125, or higher, for COVID-19 severity per standard deviation of IPF, assuming a 12·4% proportion of IPF variance explained from the IVs [11], with 4336 COVID-19 severe cases and 623,902 population controls. Genetically increased IPF risk was not associated with a higher risk of COVID-19 severity when compared to population controls using random-effects inverse variance weighted method (IVW) [18] on all 15 genetic instruments (OR 1·05, [95% CI 0·92–1·20], P = 0·43 [IVW]) (Table 1). However, a highly significant heterogeneity of effects (P = 3·30 × 10−16 [Cochran's Q]) prompted us to do IVW leave-one-out analysis to detect possible outlying genetic variants. As seen in Fig. 1, rs35705950 at the MUC5B gene locus appeared to be an outlier. Re-running the IVW MR analysis on the 14 non-MUC5B genetic instruments, we detected that a genetically increased IPF risk was associated with a higher risk of COVID-19 severity (OR 1·21, [95% CI 1·06–1·38], P = 4·24 × 10−3 [IVW]) (Table 1) (Fig. 2). Sensitivity analysis with the weighted median method [20] detected similar significant effect estimates (OR 1·19, [95% CI 1·07–1·32], P = 8·62 × 10−4 [weighted median]). Despite the reduced power of MR-Egger [21] compared to the IVW method [18], we detected consistent effect estimates for increased IPF risk and COVID-19 severity, albeit with broad confidence intervals (OR 1·30, [95% CI 0·67–2·51], P = 0·44 [MR-Egger]) (Table 1). Moreover, the MR-Egger intercept test indicated the absence of directional pleiotropy (P = 0·84 [MR-Egger intercept test]).
Table 1.
MR effect estimates of IVW, Weighted median and MR-Egger methods on the causal association of IPF with COVID-19 severity. Effect estimates are for all 15 IVs, 14 IVs without the rs35705950 at the MUC5B locus, and the 6 non-MUC5B IVs that do not associate with any other phenotype (Methods). IVs, genetic instrumental variables. OR, odds-ratio. CI Lower, lower confidence interval estimate. CI Upper, upper confidence interval estimate. P, p-value.
| Analysis | OR | 95% CI Lower | 95% CI Upper | P | Method |
|---|---|---|---|---|---|
| All 15 IVs | 1·054 | 0·924 | 1·203 | 4·31E-01 | IVW |
| 1·042 | 0·937 | 1·158 | 4·49E-01 | Weighted median | |
| 0.831 | 0.680 | 1·017 | 7·25E-02 | MR-Egger | |
| 14 IVs (no MUC5B) |
1·210 | 1·062 | 1·379 | 4·24E-03 | IVW |
| 1·188 | 1·074 | 1·315 | 8·62E-04 | Weighted median | |
| 1·296 | 0·669 | 2·510 | 4·43E-01 | MR-Egger | |
| 6 IVs (no other pheno) |
1·402 | 1·158 | 1·697 | 5·34E-04 | IVW |
| 1·349 | 1·164 | 1·565 | 7·30E-05 | Weighted median | |
| 1·233 | 0·442 | 3·439 | 6·89E-01 | MR-Egger | |
| 11 IVs (no MR-PRESSO outliers) |
1·194 | 1·106 | 1·290 | 5·48E-06 | IVW |
| 1·107 | 0·998 | 1·227 | 5·54E-02 | Weighted median | |
| 1·290 | 0·907 | 1·834 | 1·56E-01 | MR-Egger |
Fig. 1.

Forest plot of IVW causal estimates, omitting each variant in turn. The estimate with the first labelled SNP includes all variants except the labelled variant, and so on. The IVW estimate including all variants ("IVW estimate") is also provided for reference. Estimates are in ln(OR).
Fig. 2.

Genetic association estimates of the 14 non-MUC5B instrumental variables not detected to be outliers. Horizontal error bars regards standard errors of IPF estimates, while vertical error bars regards standard errors of COVID-19 severity estimates. The line represents the IVW causal estimate of IPF on COVID-19 severity. IPF, idiopathic pulmonary fibrosis. Estimates are in ln(OR).
We also used Phenoscanner [23] and GeneATLAS [24] to detect if any of the genetic instrumental variables used in this MR study were associated with any other phenotypes. Eight out of the 14 non-MUC5B IVs were associated with at least one other non-cancer, non-lung and non-blood disease/trait. Those are rs12699415, rs28513081, rs12696304, rs7725218, rs9577395, rs59424629, rs62023891 and rs17652520 (Supplementary Table 1). Removing these 8 SNPs left us with 6 non-MUC5B SNPs (Supplementary Table 1). While the confidence intervals widened, the MR effect estimates restricting the analysis to these 6 SNPs remained consistent for IVW (OR 1·40, [95% CI 1·21–1·59], P = 5·34 × 10−4 [IVW]), weighted median (OR 1·35, [95% CI 1·20–1·50], P = 7·30 × 10−5 [weighted median]) and MR-Egger (OR 1·23, [95% CI 0·21–2·26], P = 0·69 [MR-Egger]) (Table 1). We also used MR-PRESSO [25] to further detect horizontal pleiotropic outlier IVs. Four out of the 14 IVs were detected as outliers (rs35705950 at MUC5B, rs2897075, rs9577395 and rs12610495) (Supplementary Table 1). Removing these 4 SNPs left us with 11 SNPs. MR effect estimates using these 11 IVs for IVW (OR 1·19, [95% CI 1·11–1·29], P = 5·48 × 10−6 [IVW]), weighted median (OR 1·11, [95% CI 1·00–1·23], P = 5·54 × 10−2 [weighted median]) and MR-Egger (OR 1·29, [95% CI 0·91–1·83], P = 0·16 [MR-Egger]) also remained consistent, i.e. increased IPF genetic risk associated with increased COVID-19 severity (Table 1).
3.3. MUC5B vs. COVID-19 with different control groups and in an age-stratified analysis
Since rs35705950 at the MUC5B gene locus was an outlier in the MR analysis, we assessed its association with COVID-19 susceptibility, hospitalization and severity. While the rs35705950 IPF risk allele T seems to be protective for COVID-19, its effect estimates and significance decrease as the case inclusion criteria expand to include less severe cases (Table 2). Using non-hospitalized COVID-19 cases as controls, rather than population controls, compared with hospitalized COVID-19 cases further decreased the protective effect size of the rs35705950 T allele (Fig. 3). Similarly, using lab/self-reported negative individuals as controls (instead of population controls) against COVID-19 positive cases also decreased the protective effect size of the rs35705950 T allele (Fig. 3). Furthermore, age stratified analysis for COVID-19 hospitalization and susceptibility showed that while the protective effect for hospitalization was similar for both age groups, albeit non-significant in under 60 s (over 60: hospitalization OR 0·86, [95% CI 0·73–1·00], P = 2·99 × 10−2 [Wald test]; under 60: hospitalization OR 0·85, [95% CI 0·64–1·06], P = 0·14 [Wald test]), the effect on susceptibility was reduced to the null in the under 60 s (over 60: susceptibility OR 0·87, [95% CI 0·77–0·98], P = 9·48 × 10−3 [Wald test]; under 60: susceptibility OR 0·99, [95% CI 0·90–1·09], P = 0·91 [Wald test]) (Table 2).
Table 2.
Effect estimates of rs35705950 at the MUC5B locus on COVID-19 severity (4336 cases & 623,902 controls; A2 analysis), hospitalization (6406 cases & 902,088 controls; B2 analysis) and susceptibility (14,134 cases & 1284,876 controls; C2 analysis). All, all the cohort. >60, sub-cohort of individuals older than 60 years. <60, sub-cohort of individuals younger than 60 years. The number of cases and controls for the age stratified analysis does not sum to the total number of cases and controls due to age missingnness in different cohorts. OR, odds-ratio of the IPF T risk allele. CI Lower, lower confidence interval estimate. CI Upper, upper confidence interval estimate. P, p-value. AF, allele frequency of rs35705950 T allele. N, sample size.
| Age | COVID-19 | OR | 95% CI Lower | 95% CI Upper | P | AF | N |
|---|---|---|---|---|---|---|---|
| All | Severity | 0·78 | 0·66 | 0·90 | 6·50E-05 | 0·108 | 628,238 |
| Hospitalization | 0·83 | 0·74 | 0·92 | 3·77E-05 | 0·112 | 908,494 | |
| Susceptibility | 0·93 | 0·88 | 0·99 | 9·18E-03 | 0·116 | 1299,010 | |
| >60 | Hospitalization | 0·86 | 0·73 | 1·00 | 2·99E-02 | 0·109 | 417,774 |
| Susceptibility | 0·87 | 0·77 | 0·98 | 9·48E-03 | 0·110 | 470,162 | |
| <60 | Hospitalization | 0·85 | 0·64 | 1·06 | 1·42E-01 | 0·112 | 83,265 |
| Susceptibility | 0·99 | 0·90 | 1·09 | 9·07E-01 | 0·107 | 314,152 |
Fig. 3.

Forest plot of the effect estimates of the association of rs35705950 IPF T risk allele at the MUC5B locus on various COVID-19 outcomes using different control population. A2 (Very severe respiratory confirmed covid vs. population) has 4336 cases and 623,902 controls. B1 (Hospitalized covid vs. not hospitalized covid) has 2430 cases and 8478 controls. B2 (Hospitalized covid vs. population) has 6406 cases and 902,088 controls. C1 (Covid vs. lab/self-reported negative) has 24,057 cases and 218,062 controls. C2 (Covid vs. population) has 14,134 cases and 1284,876 controls.
4. Discussion
Patients with Idiopathic pulmonary fibrosis (IPF) are at increased risk of COVID-19 mortality compared with the general population [14,15]. Whether the pathogenic mechanisms that lead to the development of IPF are causally related to the severity of COVID-19 is unknown and of paramount importance to inform preventive strategies and identify whether there is rationale for investigating the role of anti-fibrotic therapies in severe COVID-19 [28]. Using a two-sample Mendelian randomization approach and genome-wide genetic correlation analysis, we found that overall there was a genetic correlation between IPF and COVID-19 severity, but the genetic variants associated with IPF did not confer an increased risk of severe COVID-19. However, this was driven by a single outlier variant at the MUC5B locus, which had an apparently protective effect on the severity of COVID-19. Removal of this outlier demonstrated that, collectively, the remaining variants associated with increased IPF risk were associated with increased risk of severe COVID-19 (Table 1). This finding supports the epidemiological studies that have reported a strong association between IPF and COVID-19 severity [14,15,29].
It is intriguing that the most strongly associated IPF variant [9,11], rs35705950 at the MUC5B gene locus, which its risk allele (T) associated with a five-fold increase in IPF risk [9], is negatively associated with COVID-19 regardless of severity, suggesting that this IPF risk allele may protect against COVID-19. This association has been reported in a small study [30], and a protective role for MUC5B in airway defense has been described [31]. The IPF risk allele (T) of the rs35705950 is associated with increased MUC5B expression in lung tissue [13], which has been associated with muco-ciliary dysfunction and increased bleomycin-induced fibrosis in mice [32].
Alternatively, this apparent protective effect of the rs35705950 T allele against COVID-19 could be the consequence of selection bias in the COVID-19 GWAS. Whilst there is expected to be minimal misclassification in the COVID-19 case definitions, the general population control groups are likely to contain individuals who have never been exposed to the virus and thus whose severity of response to the virus is, as yet, unknown. The IPF patient population will be enriched for the rs35705950 T allele but less likely to be amongst the cases due to shielding behavior. The less protective effect of the rs35705950 T allele against COVID-19 when using COVID-19 negative individuals or non-hospitalized COVID-19 individuals as controls instead of using population controls further suggests that there might be a selection bias. The strong risk effect of this variant has previously been shown to introduce a bias in survival analyses [33]. This could account for the reduced protective effect of rs35705950 T allele in the younger age group strata of the COVID-19 GWAS as IPF is predominantly a disease affecting those over 60. However, the remaining 14 IPF variants are associated with an increased risk of severe COVID-19. Both IPF and severe COVID-19 are associated with increasing age and obesity and this may suggest shared a pathogenic role for cellular senescence [34,35] or metabolic syndrome [36]. The IPF genetic risk variant near the DPP9 gene on chromosome 19 has recently been reported as genome-wide significantly associated with COVID-19 severity [37]. In light of our findings of shared causal genetic etiology, it suggests that some of the molecular mechanisms that lead to IPF could also be important in the response to COVID-19. If that is the case, as already hypothesized in the literature [28,38], antifibrotic therapies used to treat IPF could have an important role in mitigating COVID-19 severity in IPF patients, and could potentially be evaluated in clinical trials to prevent the development of COVID-19 emergent pulmonary fibrosis.
Our study also has some limitations, such as the modest variance explained by the non-MUC5B IPF genetic instruments, although within the range typical of complex traits. Nevertheless, the use of weak genetic instruments could only create biases estimates towards the null. Increased sample sizes, both from the IPF or COVID-19 GWAS could also have narrowed our confidence intervals around the true estimates, although we used the largest sample sizes for IPF and COVID-19 to date. Furthermore, MR-Egger results were not as compelling as the IVW or weighted median, suggesting that confounding factors could have biased the effect estimates. However, MR-Egger is usually considered as a sensitivity method which can also be biased in certain situations [39]. Moreover, the COVID-19 control groups, drawn from the general population, were of unknown virus exposure status, which might have further biased our causal estimates towards the null or create association with factors that increase likelihood of exposure to the virus, for example, demographic characteristics. In addition, survival bias could have attenuated or even reversed the rs35705950 estimates, due to missing potential cases among carriers of the rs35705950-T allele who already died due to IPF [43,44]. The strength of this potential bias would increase with greater average age of the COVID-19 case groups compared to the controls groups. However, we believe the bias would be negligible due to the rarity of IPF. Furthermore, although a large sample cohort was used in this study, we did not have access to further replication cohorts with COVID-19 severe cases, which would be important to confirm our findings.
In summary, our study provides genetic evidence that supports shared causal genetic etiology between IPF and COVID-19 severity that could inform the design of future preventive and therapeutic strategies to treat COVID-19.
Declaration of Competing Interest
JF works for Novo Nordisk A/S since January 2021. LVW receives funding from GSK and Orion, outside of the submitted work. RGJ Jenkins reports personal fees and other from Biogen, personal fees from Galapagos, other from Galecto, personal fees and other from GlaxoSmithKline, personal fees from Heptares, personal fees and other from MedImmune, personal fees from Boehringer Ingelheim, personal fees from Pliant, personal fees from Roche/InterMune, personal fees from MedImmune, personal fees from PharmAkea, personal fees from Bristol Myers Squibb, personal fees from Chiesi, personal fees from Roche/Promedior, other from RedX, other from NuMedii, other from Nordic Biosciences, outside the submitted work; and RGJ is supported by a National Institute of Health Research Professorship (NIHR ref: RP-2017-08-ST2-014). RGJ is a trustee for Action for Pulmonary Fibrosis.
Acknowledgments
Acknowledgments
We thank the patients, staff and investigators who contributed to the Covid-19 HGI and the IPF GWAS consortia. JF and BF received partial support from the Novo Nordisk Foundation (NNF18OC0053228) and the Oak Foundation (OCAY-18-598).LVW holds a GSK/British Lung Foundation Chair in Respiratory Research. The research was partially supported by the NIHR Leicester Biomedical Research centre and the NIHR Nottingham Biomedical Research centre; the views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. LMK holds a Medical Research Council IMPACT studentship (MR/N013913/1).
Consent for publication
All authors consent this study for publication.
Data Sharing: Supporting data is available in Supplementary Material, on https://www.covid19hg.org/results/ and https://github.com/genomicsITER/PFgenetics.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.ebiom.2021.103277.
Appendix. Supplementary materials
{kind=link}
References
- 1.WHO Coronavirus Disease (COVID-19) Dashboard. [cited January 22, 2021]; Available from: https://covid19.who.int/
- 2.Wiersinga W.J., Rhodes A., Cheng A.C., Peacock S.J., Prescott H.C. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (covid-19): a review. JAMA. 2020;324(8):782–793. doi: 10.1001/jama.2020.12839. [DOI] [PubMed] [Google Scholar]
- 3.Williamson E.J., Walker A.J., Bhaskaran K. Factors associated with COVID-19-related death using OpenSAFELY. Nature. 2020;584(7821):430–436. doi: 10.1038/s41586-020-2521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salje H., Tran Kiem C., Lefrancq N. Estimating the burden of SARS-CoV-2 in France. Science. 2020;369(6500):208–211. doi: 10.1126/science.abc3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lavezzo E., Franchin E., Ciavarella C. Suppression of a SARS-CoV-2 outbreak in the Italian municipality of Vo'. Nature. 2020;584(7821):425–429. doi: 10.1038/s41586-020-2488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berlin D.A., Gulick R.M., Martinez F.J. Severe Covid-19. N Engl J Med. 2020;383(25):2451–2460. doi: 10.1056/NEJMcp2009575. [DOI] [PubMed] [Google Scholar]
- 7.Lederer D.J., Martinez F.J. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378:1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 8.Ley B., Collard H.R., King T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431–440. doi: 10.1164/rccm.201006-0894CI. [DOI] [PubMed] [Google Scholar]
- 9.Allen R.J., Guillen-Guio B., Oldham J.M. Genome-Wide association study of susceptibility to idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2020;201(5):564–574. doi: 10.1164/rccm.201905-1017OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhindsa R.S., Mattsson J., Nag A. Identification of a novel missense variant in SPDL1 associated 317 with idiopathic pulmonary fibrosis. bioRxiv. 2020 doi: 10.1101/2020.06.29.178079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leavy O.C., Ma S.F., Molyneaux P.L. Proportion of Idiopathic pulmonary fibrosis risk explained by known common genetic loci in european populations. Am J Respir Crit Care Med. 2020 doi: 10.1164/rccm.202008-3211LE. Nov 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakano Y., Yang I.V., Walts A.D. MUC5B promoter variant rs35705950 affects MUC5B expression in the distal airways in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;193:464–466. doi: 10.1164/rccm.201509-1872LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seibold M.A., Wise A.L., Speer M.C. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drake T., Docherty A.B., Harrison E. Outcome of hospitalisation for COVID-19 in 330 patients with interstitial lung disease: an international multicentre study. medRxiv. 2020 doi: 10.1101/2020.07.15.20152967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esposito A.J., Menon A.A., Ghosh A.J. Increased odds of death for patients with interstitial lung disease and COVID-19: a case-control study. Am J Respir Crit Care Med. 2020;202(12):1710–1713. doi: 10.1164/rccm.202006-2441LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ebrahim S., Davey Smith G. Mendelian randomization: can genetic epidemiology help redress the failures of observational epidemiology? Hum Genet. 2008;123(1):15–33. doi: 10.1007/s00439-007-0448-6. [DOI] [PubMed] [Google Scholar]
- 17.The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur J Hum Genet. 2020;28(6):715–718. doi: 10.1038/s41431-020-0636-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burgess Stephen, Butterworth Adam S, Thompson Simon G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–665. doi: 10.1002/gepi.21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Core Team R. R Foundation for Statistical Computing; Vienna, Austria: 2020. R: a language and environment for statistical computing.https://www.R-project.org/ URL. [Google Scholar]
- 20.Bowden J., Davey Smith G., Haycock P.C., Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–314. doi: 10.1002/gepi.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowden J., Davey Smith G., Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burgess S., Foley C.N., Allara E., Staley J.R., Howson J.M.M. A robust and efficient method for Mendelian randomization with hundreds of genetic variants. Nat Commun. 2020;11(1):376. doi: 10.1038/s41467-019-14156-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamat M.A., Blackshaw J.A., Young R. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35(22):4851–4853. doi: 10.1093/bioinformatics/btz469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Canela-Xandri O., Rawlik K., Tenesa A. An atlas of genetic associations in UK Biobank. Nat Genet. 2018;50(11):1593–1599. doi: 10.1038/s41588-018-0248-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verbanck M., Chen C.Y., Neale B., Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–698. doi: 10.1038/s41588-018-0099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bulik-Sullivan B.K., Loh P.R., Finucane H.K. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47(3):291–295. doi: 10.1038/ng.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bulik-Sullivan B., Finucane H.K., Anttila V. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–1241. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.George P.M., Wells A.U., Jenkins R.G. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir Med. 2020;8(8):807–815. doi: 10.1016/S2213-2600(20)30225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gallay L., Uzunhan Y., Borie R. Risk Factors for mortality following COVID-19 in patients with pre-existing interstitial lung disease. Am J Respir Crit Care Med. 2020 doi: 10.1164/rccm.202007-2638LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Moorsel C.H.M., van der Vis J.J., Benschop C., Ruven H.J.T., Quanjel M., Grutters J.C. The MUC5B promotor polymorphism associates with severe COVID-19. medRxiv. 2020 doi: 10.1101/2020.05.12.20099333. 05.12.20099333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roy M.G., Livraghi-Butrico A., Fletcher A.A. Muc5b is required for airway defence. Nature. 2014;505(7483):412–416. doi: 10.1038/nature12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hancock L.A., Hennessy C.E., Solomon G.M. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun. 2018;9(1):5363. doi: 10.1038/s41467-018-07768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dudbridge F., Allen R.J., Sheehan N.A., Schmidt A.F., Lee J.C., Jenkins R.G., Wain L.V., Hingorani A.D., Patel R.S. Adjustment for index event bias in genome-wide association studies of subsequent events. Nat Commun. 2019;10(1):1561. doi: 10.1038/s41467-019-09381-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho S.J., Stout-Delgado H.W. Aging and lung disease. Annu Rev Physiol. 2020;82(1):433–459. doi: 10.1146/annurev-physiol-021119-034610. p. null. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nehme J., Borghesan M., Mackedenski S., Bird T.G., Demaria M. Cellular senescence as a potential mediator of COVID-19 severity in the elderly. Aging Cell. 2020;19(10):e13237. doi: 10.1111/acel.13237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jenkins G. Demystifying pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2020;319(3):L554–L559. doi: 10.1152/ajplung.00365.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pairo-Castineira E., Clohisey S., Klaric L. Genetic mechanisms of critical illness in Covid-19. Nature. 2020 doi: 10.1038/s41586-020-03065-y. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 38.Seifirad S. Pirfenidone: a novel hypothetical treatment for COVID-19. Med Hypotheses. 2020;144 doi: 10.1016/j.mehy.2020.110005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burgess S., Thompson S.G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32(5):377–389. doi: 10.1007/s10654-017-0255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rasooly D., Patel C.J. Conducting a reproducible mendelian randomization analysis using the R analytic statistical environment. Curr Protoc Hum Genet. 2019;101(1):e82. doi: 10.1002/cphg.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teumer A. Common methods for performing mendelian randomization. Front Cardiovasc Med. 2018;5:51. doi: 10.3389/fcvm.2018.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brion M.J., Shakhbazov K., Visscher P.M. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol. 2013;42(5):1497–1501. doi: 10.1093/ije/dyt179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delgado-Rodríguez M., Bias Llorca J. J Epidemiol Commun Health. 2004;58(8):635–641. doi: 10.1136/jech.2003.008466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schooling C.M., Lopez P.M., Yang Z. Use of multivariable Mendelian randomization to address biases due to competing risk before recruitment. Front Genet. 2021;11 doi: 10.3389/fgene.2020.610852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.