

Targeting α-synuclein to intact brainstem neurons drives gastrointestinal dysregulation seen in prodromal Parkinson’s disease.
Abstract
No disease-modifying therapy is currently available for Parkinson’s disease (PD), the second most common neurodegenerative disease. The long nonmotor prodromal phase of PD is a window of opportunity for early detection and intervention. However, we lack the pathophysiological understanding to develop selective biomarkers and interventions. By using a mutant α-synuclein selective-overexpression mouse model of prodromal PD, we identified a cell-autonomous selective Kv4 channelopathy in dorsal motor nucleus of the vagus (DMV) neurons. This functional remodeling of intact DMV neurons leads to impaired pacemaker function in vitro and in vivo, which, in turn, reduces gastrointestinal motility, a common early symptom of prodromal PD. We identify a chain of events from α-synuclein via a biophysical dysfunction of a specific neuronal population to a clinically relevant prodromal symptom. These findings will facilitate the rational design of clinical biomarkers to identify people at risk for developing PD.
INTRODUCTION
α-Synuclein is an established causal driver of Parkinson’s disease (PD): Multiplications or point mutations in the SNCA gene lead to familial forms of PD (1, 2), single-nucleotide polymorphisms near its locus increase the risk for idiopathic PD (3), and α-synuclein aggregates are a main constituent of the Lewy pathologies (LPs) that are a hallmark of PD and other synucleinopathies (4). These discoveries have motivated extensive research into developing therapies aimed at either silencing the SNCA gene, reducing α-synuclein production, preventing α-synuclein aggregation, promoting degradation of intracellular and extracellular α-synuclein, or preventing its propagation to other cells (5, 6). However, the lessons from Alzheimer’s disease clinical trials targeting amyloid-β with similar strategies (7) indicated that early detection before manifestation of clinical core symptoms might also be essential for PD (5, 6). Therefore, for future disease-modifying therapies to be successful, they will require early detection of the α-synucleinopathies, which in most (but not all) cases appear in the brainstem years before clinical diagnosis (8–10). During this prodromal period, future patients with PD already begin to suffer from a variety of nonmotor symptoms (NMS), which can precede clinical diagnosis by decades (8). It stands to reason that particular NMS are associated with the appearance of LPs in particular anatomical regions. For example, constipation and dysautonomia (11, 12) are likely related to the appearance (in Braak stage I) (13) of misfolded α-synuclein in the medullary dorsal motor nucleus of the vagus (DMV) that innervates the gastrointestinal (GI) tract (14, 15). While animal studies suggest that synucleinopathies in the enteric nervous system (ENS) itself may also contribute to GI dysmotility (16, 17), 90% of patients with PD exhibit synucleinopathies in the DMV (18), and therefore, it is important to elucidate whether and how DMV contributes to dysautonomia. Furthermore, is dysautonomia caused only by substantial DMV cell loss or established even earlier by LP-induced functional changes in surviving DMV neurons? This difference has important implications, as PD-selective DMV pathophysiology is likely to facilitate earlier and more selective detection compared to the more generic ensuing phenotypes caused by cell loss and neuroinflammation. In the present study, we test this hypothesis by using an adult-onset mouse model of a selective medullary α-synucleinopathy. We use this model to identify an α-synuclein–induced channelopathy in DMV neurons that drives prodromal dysautonomia.
RESULTS
Selective induction of α-synucleinopathy in vagal motoneurons of adult mice reduces vagal output and GI motility
To determine whether α-synucleinopathy restricted to the medulla reproduces dysautonomia characteristic of prodromal PD, we injected adeno-associated viruses (AAVs) harboring either the mutated human A53T-SNCA gene (AAV-A53T) or an empty vector (AAV-EV) into the right cervical vagus nerve of C57BL/6JRccHsd mice (Fig. 1A). Six weeks later, transgenic A53T α-synuclein protein was detected bilaterally within afferent sensory fibers of the medullary nucleus of the solitary tract (NST) and area postrema (Fig. 1B) as well as within ipsilateral cholinergic motoneurons of the DMV (Fig. 1, B and C) (19). Unbiased stereological analysis at this time point demonstrated that AAV injections per se—whether α-synuclein–expressing or empty vectors—induced only mild cell loss with a median of approximately 20%—presumably due to mechanical damage to the vagus nerve fibers (fig. S1). Moreover, while AAV injection into the cervical vagus perturbed the distribution of microglia in the DMV (20), there was no difference between AAV-EV– and AAV-A53T–inoculated mice in the total number iba-1–positive microglia or in the number of resting and activated microglia (fig. S2). Thus, neither mild DMV cell loss nor neuroinflammation alone can explain divergent phenotypes in this mouse model.
Fig. 1. Model of adult-onset α-synucleinopathy in the mouse dorsal medulla.
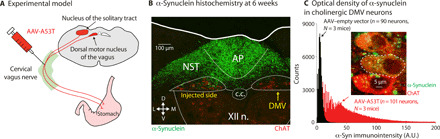
(A) Injection of AAVs harboring the mutated human A53T-SNCA gene into the cervical vagus causes the transfection of the DMV and other regions such as the nucleus of the solitary tract (NST). (B) Six weeks after injection, α-synuclein (green) is visible in the area postrema (AP), in the NST, and in the DMV, where immunohistochemical staining for choline acetyltransferase (ChAT) can be seen (red). XII n., hypoglossal nucleus; c.c., central canal. (C) Optical density measurements of the intensity of human α-synuclein fluorescent immunolabeling (green in inset) within regions of interest (dashed white line in inset) marked around the somata of ChAT-expressing neurons (red in inset) reveals a significant rightward shift (P = 3.5 × 10−3; t189 = 2.96, LMEM) in the distribution of immunointensity values from AAV-A53T–transfected relative to AAV-EV–transfected vagal motoneurons. A.U., arbitrary units.
A recent study has shown that generating synucleinopathy in the ENS by injecting preformed fibrils of α-synuclein into the upper GI tract can directly slow down GI motility (17). Because we corroborated that injection of AAV-A53T into the cervical vagus led to expression of α-synuclein in vagal terminals, as previously published (21), we wanted to determine whether this could directly affect the intrinsic motility of the upper GI tract. To this end, we used excised segments of the ileum in an organ bath, 6 weeks after bilateral injection of the cervical vagus (fig. S3A).We found that neither the frequency and amplitude of spontaneous contractions (fig. S3B) nor the responses of the ileal muscle to the muscarinic agonist pilocarpine (fig. S3C) were reduced by mutant α-synuclein expression. Similarly, responses of the ileal muscle to electric field stimulation (EFS) of the nerves were not altered.
Having determined that the upper GI smooth muscles, pacemakers, and nerves were intact in AAV-A53T–treated mice, we tested whether the AAV-A53T–injected mice nevertheless exhibited slowed GI motility in vivo. Six weeks after the injection, the AAV-A53T–injected mice produced larger stool than the AAV-EV–injected mice (fig. S4), which is suggestive of slowed GI motility (22). Direct measurements of time of passage confirmed a slowing of GI motility at 6, but not 3, weeks after inoculation (Fig. 2A). Thus, we hypothesized that the expression of mutant α-synuclein in the DMV reduced GI motility by inhibiting the electrical activity of DMV motoneurons, which normally maintain proximal gastric tone and augment GI motility. Selective, unilateral transfection of cholinergic DMV neurons with AAVs harboring the inhibitory designer receptor exclusively-activated by designer drugs (DREADDs), hM4Di, was sufficient to recapitulate the slowed motility induced by AAV-A53T. This was observed when the mice were intraperitoneally injected with the DREADD agonist clozapine-N-oxide (CNO) but not when intaperitonally injected with vehicle. Moreover, intraperitoneal injection of CNO into mice that did not express DREADDs had no effect on GI motility, either (Fig. 2B).
Fig. 2. GI motility is slowed in the mouse model of medullary α-synucleinopathy.
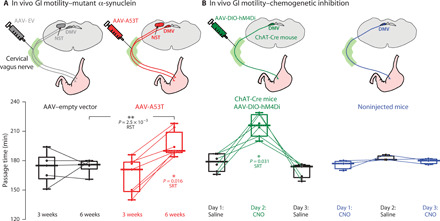
(A) Mice inoculated with AAV-A53T, but not with AAV-EV, exhibited slowed GI motility at 6, but not 3, weeks after inoculation [one-way analysis of variance (ANOVA), F3,22 = 5.66, P = 5 × 10−3]. (B) Injection of AAVs harboring Cre-dependent hM4Di DREADDs into the cervical vagus of ChAT-Cre mice leads to selective transfection of cholinergic motoneurons in the DMV. Intraperitoneal injection of the DREADD ligand, CNO, but not vehicle (saline), slowed GI motility (one-way ANOVA, F2,17 = 12.76, P = 4 × 10−4), indicating that silencing these neurons suffices to recapitulate the effect of AAV-A53T transfection. Injection of CNO had no effect on GI motility in noninjected mice (one-way ANOVA, F2,6 = 2.51, P = 0.16). RST, two-tailed Wilcoxon rank sum test; SRT, two-tailed Wilcoxon signed-rank test.
To determine whether vagal motoneuron firing rates were indeed slower in the AAV-A53T–injected mice, we performed in vivo single-unit extracellular recording of DMV neurons under isoflurane anesthesia (23). Using juxtacellular labeling combined with immunohistochemistry (Fig. 3A), 65% (17 of 26) of the recorded neurons were positively identified as cholinergic DMV neurons (Fig. 3B), with the remainder being identified within the DMV via electrolytic lesions. The median firing rate of vagal motoneurons transfected with AAV-A53T was 57% slower than those transfected with AAV-EV. This reduction in firing rate was selective to vagal motoneurons, as neurons in the neighboring NST, where mutant α-synuclein was also expressed (Fig. 1B), discharged at control rates (Fig. 3C).
Fig. 3. In vivo extracellular recording and juxtacellular labeling of cholinergic DMV neurons in the mouse model of medullary α-synucleinopathy.
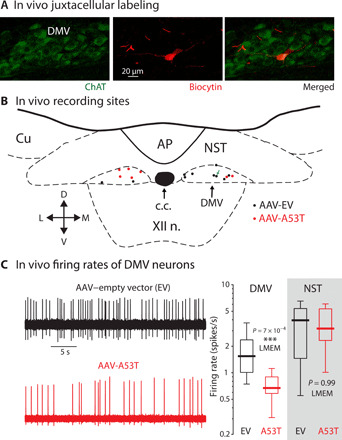
(A) Left: ChAT-expressing (green) DMV neurons. Middle: Juxtacellularly labeled DMV neuron (red) after in vivo extracellular recording from an anesthetized mouse injected with AAV-A53T. Right: Merge of both images. (B) Map of recording sites of juxtacellularly labeled neurons (black and red dots correspond to AAV-EV– and AAV-A53T–transfected neurons, respectively). Green arrow indicates location of neuron from (A). Cu, cuneate nucleus. (C) Left: Examples of extracellular recordings from mice injected with an AAV-EV (black) or with AAV-A53T (red). Right: Box plot of firing rates demonstrates that AAV-A53T transfection leads to a reduction in the firing rates of DMV neurons (EV: n = 15 neurons, N = 6 mice; A53T: n = 12 neurons, N = 7 mice, t25 = 3.87) but not NST neurons (EV: n = 8 neurons, N = 5 mice; A53T: n = 8 neurons, N = 5 mice, t14 = 0.016). LMEM, linear mixed-effects model.
α-Synucleinopathy in mouse vagal motoneurons slows their autonomous pacemaker frequency because of elevated surface density of functional Kv4 channels
DMV motoneurons are autonomous pacemakers (24), meaning that they generate repetitive activity due to their intrinsic pacemaker currents even in the absence of synaptic input. Thus, the most parsimonious explanation for the reduced in vivo firing rates in the AAV-A53T–injected mice is that their intrinsic pacemaker frequency is lowered. To test this, we first measured the frequency-current relationship (f-I curve) of DMV motoneurons in vitro, by recording them in the whole-cell configuration from acute medullary slices 6 weeks after AAV injection. The f-I curve characterizes neuronal excitability across the entire dynamic range of the neuron’s input-output relationship. We found that the f-I curve of DMV motoneurons from AAV-A53T–injected mice was shifted to the right, indicating a reduction in excitability in comparison to controls (Fig. 4A). In particular, the measurement in the zero-current condition demonstrated that the autonomous firing rate of DMV motoneurons from the AAV-A53T–injected mice was reduced. To quantitatively compare the reduction in the autonomous in vitro firing rate with that observed in vivo, we repeated the measurement of the autonomous firing rate (in the presence of synaptic blockers for glutamate and γ-aminobutyric acid receptors) in the extracellular cell-attached configuration (thereby maintaining the metabolic integrity of the neurons). We found that the autonomous firing rates were reduced in AAV-A53T–injected mice by 53%, relative to controls (Fig. 4B). This close agreement with the results from the in vivo recordings (Fig. 3C) suggests that slowing of vagal motoneuron discharge in the intact animal is due mainly to a cell-autonomous change in excitability. These findings demonstrate that the ability of either intrinsic or synaptic conductances to depolarize the membrane potential of DMV motoneurons is curtailed by an α-synuclein–induced modification of their membrane biophysics (i.e., ion channels that control the frequency of autonomous pacemaking).
Fig. 4. Reduction of pacemaker frequency and intrinsic excitability of DMV neurons in the mouse model of medullary α-synucleinopathy is associated with a dysregulation of Kv4 channels.

(A) Frequency-current (f-I) curves of DMV neurons transfected with AAV-A53T (n = 14 neurons, N = 5 mice) are right-shifted relative to control (n = 22 neurons, N = 7 mice), indicating reduced excitability (t249 = 2.43). (B) Left: Examples of cell-attached recordings of DMV neurons that were transfected with either AAV-EV (black) or AAV-A53T (red) in acute brain slices. Right: Box plot of firing rates demonstrates a slowing of the pacemaker frequency in DMV neurons transfected with AAV-A53T (EV: n = 19 neurons, N = 6 mice; A53T: n = 23 neurons, N = 9 mice; t40 = 3.32) that is similar to the reduction observed in vivo. Preincubation of the slices in either 1.3 μM phrixotoxin-2 (EV: n = 24 neurons, N = 3 mice; A53T: n = 33 neurons, N = 4 mice), a Kv4-selective antagonist or 400 μM 4-AP, which, in the submillimolar range, is a selective blocker of A-type Kv currents and which is also approved for clinical use (EV: n = 20 neurons, N = 3 mice; A53T: n = 26 neurons, N = 3 mice) (46, 47) occluded the reduction in firing rates (LMEM for initial six-group comparison identified a significant reduction only for the A53T-treated mice with no drugs, t139 = 4.12, P = 6.6 × 10−5) by significantly elevating the firing rates in A53T-injected mice (phrixotoxin-2: +1.3 spikes/s, t54 = 3.8; 4-AP: +1.6 spikes/s, t47 = 3.99; post hoc) relative to the no-drug condition.
One ionic current that curtails the rate of membrane depolarization and the firing rate of DMV neurons is the A-type, fast, inactivating, voltage-dependent K+ current carried by Kv4 channels (25, 26). Functional Kv4 channels act like a brake on spontaneous subthreshold depolarizations and slow down pacemaker frequencies. Conversely, when their Kv4 currents are reduced (e.g., because of rundown in whole-cell mode), DMV neurons speed up (25). To test whether Kv4 channels were responsible for the α-synuclein–induced reduction in firing rates, we measured the autonomous firing rate of DMV neurons in the presence of 1.3 μM phrixotoxin-2, a Kv4 selective neurotoxin (25), or in the presence of 400 μM 4-aminopyridine (4-AP, a nonselective antagonist of A-type Kv4 currents in the submillimolar range), which is a drug approved for various neuromuscular diseases (27). Both drugs occluded the reduction in firing rates in the AAV-A53T–injected mice relative to controls. This result supports the conclusion that the α-synuclein–induced firing rate reduction was due to a dysregulation of the Kv4 channels (Fig. 4B).
To investigate the source of the Kv4 current dysregulation, we compared the Kv4 current biophysics in DMV motoneurons treated with either AAV-A53T or AAV-EV (Fig. 5A). While the voltage dependencies and kinetics of the A-type current in DMV neurons did not differ between the AAV treatments (Fig. 5B), the surface density of the Kv4 current was significantly higher in mice injected with AAV-A53T (Fig. 5C). The constitutive Kv4 window current in the subthreshold range (Fig. 5B) prolongs the membrane depolarization and the lingering in the subthreshold range (25), which suffices to explain the reduced pacemaker frequency. Our data argue against an up-regulation of functional Kv4 channels, because the total whole-cell Kv4 current did not differ between the AAV treatments, pointing to an alternative mechanism.
Fig. 5. Kv4-channel density is elevated in the mouse model of medullary α-synucleinopathy.
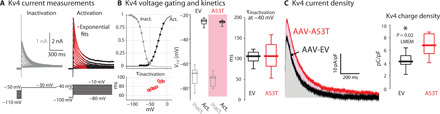
(A) Voltage-clamp measurements of A-type currents yield voltage activation and inactivation curves and time constants of inactivation. Note that a window current flows constitutively in the subthreshold voltage range where the voltage activation and inactivation curves overlap. (B) Comparison of voltage dependency of activation and inactivation (EV: n = 11 neurons; A53T: n = 8 neurons) and channel inactivation kinetics (EV: n = 9 neurons; A53T: n = 8 neurons) between AAV-EV– (N = 6) and AAV-A53T– (N = 4) transfected mice reveals no difference. (C) Left: Example of measurements of the inactivating A-type current density (depolarization to −40 mV), which is carried by Kv4 channels, from DMV neurons transfected with either AAV-EV (black, n = 11 neurons, N = 6 mice) or AAV-A53T (red, n = 8 neurons, N = 4 mice). Areas under the curves in gray and pink, respectively, indicate that the Kv4 charge densities are significantly higher (t17 = 2.57) in the DMV neurons transfected with AAV-A53T (box plots).
An α-synuclein–induced, cell-autonomous shrinkage of vagal motoneurons drives a selective dysregulation of Kv4 current densities
α-Synuclein overexpression has been shown to cause vagal motoneurons to shrink (28). Measurement of the long axis of the somata of DMV neurons from animals transfected with AAV-A53T demonstrated that it was shorter in human α-synuclein–positive DMV neurons in comparison to neighboring human α-synuclein–negative DMV neurons (Fig. 6A). Note that human α-synuclein–negative neurons in AAV-A53T–transfected mice were not different in size compared to controls in the AAV-EV–transfected mice (Fig. 6A). This indicated that shrinkage of cell size was due to a cell-autonomous process and gave no evidence for the contribution of additional non–cell-autonomous conditions, such as neuroinflammation (fig. S2) (29, 30). To better gauge the degree of reduction in surface area, we filled DMV neurons from both AAV-EV– and AAV-A53T–transfected mice with biocytin. We then reconstructed their perisomatic region using fluorescent confocal microscopy to estimate their surface area. Using these morphological measurements, we found that the surface area was reduced by 42% from a median area of approximately 3800 μm2 in the AAV-EV–transfected mice to approximately 2200 μm2 in the AAV-A53T–transfected mice (Fig. 6B, left). These measurements were corroborated by an electrophysiological measurement of the membrane surface area. The whole-cell capacitance of a neuron scales with the somatic surface area because it is the product of the surface area and the specific capacitance of the lipid membrane (Cm). Measurements of the whole-cell membrane capacitance of the DMV neurons (Fig. 6B, right) demonstrated that it was reduced by 33% from approximately 32 picofarad (pF) in neurons transfected with AAV-EV to approximately 22 pF in DMV neurons transfected with AAV-A53T. Thus, there is a quantitative agreement between the A53T-SNCA–induced reductions of surface area estimated by either electrophysiological or morphological methods. In both AAV treatments, the ratio of the median whole-cell capacitance to the median surface area very closely reproduced the known value of Cm (1 μF/cm2) (31). In summary, the Kv4 channelopathy in human α-synuclein–positive DMV neurons is likely caused by a mismatch in the density of their Kv4 surface expression in shrinking DMV neurons.
Fig. 6. Selective shrinkage of human α-synuclein–positive DMV neurons.
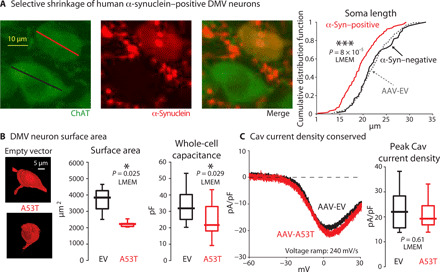
(A) Left: Examples of the measurement of the long axis of the soma of DMV neurons from mice injected with AAV-A53T. Staining for ChAT and human α-synuclein demonstrate the selective shrinkage of the human α-synuclein–positive (long axis marked in red) but not the neighboring human α-synuclein–negative neuron (long axis marked in black). Right: Cumulative distribution functions of the lengths of human α-synuclein–positive (red, n = 147) and α-synuclein–negative (black, n = 71) neurons from AAV-A53T–injected mice (N = 3) and of neurons from AAV-EV–injected mice (dashed, n = 252 neurons, N = 3 mice) indicate that the length of the α-synuclein–positive neurons is smaller (t464 = 3.67). (B) Left: Examples of biocytin-filled DMV neurons from AAV-A53T– and AAV-EV–injected mice that were reacted with streptavidin conjugated to Alexa Fluor 594. Middle: Box plots of the somatic surface areas of the biocytin-filled neurons as estimated with the FIJI software (EV: n = 4 neurons, N = 3 mice; A53T: n = 6 neurons, N = 4 mice; t8 = 2.75). Right: Whole-cell capacitance measurement of DMV neurons from mice transfected with either AAV-EV (n = 19 neurons, N = 8 mice) or AAV-A53T (n = 17 neurons, N = 6 mice). The morphological (t10 = 3.37) and physiological (t34 = 2.27) measurements demonstrate that the surface area of DMV neurons that express A53T α-synuclein shrinks. (C) Left: Examples of the Co2+-sensitive Cav current density in response to fast voltage ramps from DMV neurons transfected with either AAV-EV (black, n = 8 neurons, N = 5 mice) or AAV-A53T (red, n = 9 neurons, N = 6 mice). Right: Box plots of peak Cav current density, estimated by fitting activation curves to individual Co2+-sensitive current density measurements, indicate that they are equivalent (t15 = 0.52) under both AAV treatments despite the shrinkage.
Because Kv4 channel expression did not adapt to the A53T-driven reductions in surface area, we asked whether this maladaptation was selective to Kv4 channels. Therefore, we also measured voltage-activated Ca2+ (Cav) currents. We found that the total Cav currents in DMV neurons were reduced, similar as previously shown in a related mouse model, transgenic mice overexpressing human A53T α-synuclein (32). Intriguingly, the reduction in Cav currents of DMV neurons in the AAV-A53T–transfected mice matched the reduction in their surface area. Thus, in contrast to Kv4 channels, Cav channels may exhibit intact homeostatic control (Fig. 6C). Similarly, measures of other electrophysiological properties of the DMV pacemaking (e.g., action potential threshold, width, and afterhyperpolarization) were unaffected (fig. S5), ruling out changes to other voltage-activated channels involved in pacemaking (24, 25, 33). Together, these findings suggest that the A53T-induced Kv4 channelopathy in DMV neurons is both selective and maladaptive.
In summary, we have shown that expression of mutated α-synuclein in DMV motoneurons of adult mice leads to a selective elevation in the surface density of Kv4 channels in a cell-autonomous fashion that is associated with somatic shrinkage. The elevated Kv4 density slows the depolarization to action potential threshold and thereby reduces the DMV motoneurons’ pacemaker frequency. The ensuing reduction in vagal parasympathetic tone results in slowed GI motility (Fig. 7). This model provides a feasible pathophysiological mechanism for constipation in PD—one of its most common, early-onset prodromal symptoms. Elucidating the physiological underpinning of prodromal symptoms paves the way for a rational design of more selective clinical biomarkers for early diagnosis of the PD (e.g., testing of GI function with Kv4 modulators).
Fig. 7. Working model of how the medullary α-synucleinopathy reduces GI motility.
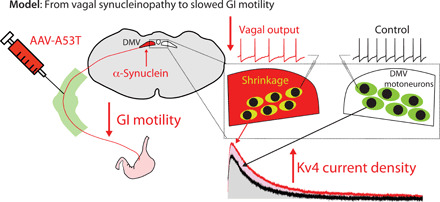
Expression of mutated α-synuclein in DMV motoneurons reduces vagal tone and GI motility due to a selective elevation in the surface density of Kv4 channels that is secondary to their shrinkage.
DISCUSSION
The notion that brain stem α-synucleinopathies are linked to prodromal NMS of PD, including dysautonomia (e.g., constipation) is not new (8, 34, 35). Earlier studies have even proposed that clinical autonomic measures may be sensitive enough to gauge the degree of brain stem LPs (36). However, it is unknown whether it is α-synuclein–induced cell loss in brain stem nuclei like the DMV (37, 38) that is responsible for the dysautonomia. Functional changes induced by α-synuclein might also contribute to vagal dysfunction and, importantly, are likely to precede cell loss–mediated impairments. These functional changes can be divided into cell-autonomous effects (e.g., altered physiology of DMV neurons that harbor α-synuclein pathology) (39) and nonautonomous effects (e.g., involvement of surrounding microglia leading to neuroinflammation) (30). In contrast to the strong evidence for neuroinflammation and cell loss in prodromal PD (30), we know nothing about functional changes in viable DMV neurons in humans and only very little in model systems (39). In a global A53T-SNCA transgenic mouse model, we previously reported in vitro reductions in activity-dependent calcium loading and oxidative enhancement of Kv4 channels in DMV neurons (32). In contrast, global transgenic expression of mutant α-synuclein led to in vivo and in vitro hyperexcitability with Kv4 dysfunction in dopamine substantia nigra neurons (23). These global transgenic models cannot capture prodromal PD states where α-synuclein pathology is initially restricted to a small number of sites. On the basis of immunohistochemical studies in human brains, Braak and colleagues have proposed a sequence of six stages of increasing expansion of LPs (13), which have been confirmed by others (16, 19) and may represent the most common spatiotemporal sequence of LPs. LPs in DMV are a hallmark of the earliest Braak stage I (40) and might be associated with various degrees of cell loss (30, 37, 38). The demonstration of α-synuclein spreading throughout neural circuits in rodent models (16, 17, 41–43) provided a plausible biological mechanism in support of the Braak hypothesis. Moreover, even processes that are currently believed to occur at the earliest stages, including the spread of α-synuclein from the periphery to DMV neurons were successfully modeled in rodents (16, 17, 19, 44). However, as potential functional implications are still unknown, we expanded these prodromal models to include functional physiological studies in vitro and in vivo.
In our model, after injection of AAV-A53T into the cervical vagus, the AAVs inoculate both afferent fibers (anterogradely) and efferent fibers (retrogradely). Thus, in the NST and area postrema the α-synuclein immunoreactivity was restricted to afferent fibers, but not somata because their somata are located outside the cranium. Hence, the α-synuclein expressed in the afferent fibers did not cross trans-synaptically into their target neurons in the NST and area postrema. Similarly, α-synuclein taken up by efferent (including DMV motoneuron) fibers did not cross trans-synaptically into the GI tract (21), leaving, as we demonstrated here, the intrinsic physiological properties of the GI nerves and muscles intact. In addition, our stereological analysis demonstrated that α-synuclein expression in DMV neurons did not induce cell death or neuroinflammation per se. Thus, our model, well-suited to capture a very early time point in prodromal PD, enables the identification of phenotypes driven by the pathophysiology of DMV neurons. We have demonstrated that a selective physiological dysregulation of Kv4 channel density in DMV neurons is associated with a reduction in vagal output, which slows GI motility.
We have shown previously that DMV neurons in transgenic mice that globally overexpresses human A53T α-synuclein undergo cell-autonomous adaptations (compared with control mice) in the form of a transcriptional down-regulation of Cav currents, which leads to lower basal mitochondrial oxidative stress and improved oxidative function of Kv4 channels. The latter effect was only revealed through the dialysis of an antioxidant into the neuron via the patch pipette, because no difference was found in the amplitude, voltage dependence, or kinetics of the Kv4 currents (32). The current study was therefore motivated by the concern that these observed adaptations in expression and oxidative function of Cav and Kv4 channels reflected some developmental adaptation in the transgenic mouse model rather than an adaptive response that plausibly represents the disease process in adult patients with PD. Therefore, in the current, adult-onset brain stem–selective α-synucleinopathy model, we initially set out to replicate the Cav and Kv4 findings.
We were able to replicate the reduction in total Cav current, which suggests to us that mutant α-synuclein drives the down-regulation of Cav channels robustly regardless of whether it accumulates acutely or developmentally. Because basal mitochondrial oxidative stress in DMV neurons requires Ca2+ influx via Cav2 channels, the Cav2 current down-regulation reduces basal oxidative stress in DMV motoneurons (24, 32). Therefore, this response can be considered a protective response. When normalizing the total Cav current to a current of density, we found that in the adult-onset α-synucleinopathy model, current density was preserved despite the shrinkage. This scaling may represent a controlled homeostatic mechanism that titers the degree of channel down-regulation to match the shrunken cell size. Because high-voltage–activated Cav2 currents measured in DMV neurons flow primarily as a consequence of spiking, they do not strongly affect pacemaking frequency (25). We have observed in the A53T transgenic mice that the pacemaking frequency remained unaltered despite the Cav2 down-regulation (32).
In contrast to the Cav2 currents, the absolute Kv4 current amplitudes were unchanged in the adult-onset α-synucleinopathy model leading to an effective up-regulation of Kv4 current density, which was secondary to the cell shrinkage. Neither the shrinkage nor the Kv4 current density up-regulation was observed in the A53T transgenic mouse. Moreover (again, in contrast to the Cav2 currents) the pacemaker frequency of DMV neurons is highly sensitive to the amplitude of the Kv4 currents (Fig. 4B) (25). Accordingly, in transgenic A53T mice where there was no change in the size of the Kv4 currents, there is no change in pacemaker frequency (32), whereas in the current model, the up-regulation of the Kv4 current density reduced the autonomous firing rate. While it would have been nice to provide independent proof that selective up-regulation of Kv4 channel density in DMV motoneurons is sufficient to slow GI motility, this is not a manipulation that can be achieved easily in a controlled manner in vivo. Nevertheless, we believe that we provided compelling evidence, through a combination of complementary in vivo and in vitro experiments, for a chain of events leading from α-synucleinopathy, via the selective Kv4 channelopathy to the impaired GI motility (e.g., constipation) and dysautonomia that are prevalent in prodromal PD.
We made use of human mutated A53T α-synuclein, as it is an established cause for familial PD (2, 39). While corroboration of our finding in additional animals models of prodromal PD (e.g., that overexpress wild-type α-synuclein) (21) or in models of gut-to-brain transmission of synucleinopathies (16, 17, 44) is warranted, the aim of the present study was not to recapitulate a Braakian spread of pathology (which has already been firmly established in animal models) (16, 17, 21, 44) but rather to focus on the discovery of cellular pathophysiology. Our findings are orthogonal to the question of the spread of α-synuclein. Instead, we aimed to produce a selective synucleinopathy localized to the DMV, thereby replicating an early time point in PD progression. Although it has been established that the brainstem synucleinopathies are associated with prodromal symptoms (34), our study provides a direct link from α-synuclein via ion channel dysregulation and reduced neuronal activity in vivo to a prodromal NMS of PD. This establishes an additional perspective—cell-autonomous neuronal pathophysiology—in the etiology of PD to complement well-established pathological and molecular approaches. It also provides a set of questions for follow-up studies to understand which molecular and cell-biological mechanisms induce α-synuclein–mediated shrinkage and channel dysregulation (28, 45).
Clinical implications
In our model, the induction of an α-synucleinopathy in DMV motoneurons caused shrinkage and dysregulated surface densities of Kv4 channels, which, in turn, reduced firing rates. This reduction in vagal tone effectively slowed GI motility. If a similar mechanism is operative in prodromal PD patients with impaired GI motility, then the translational challenge would be to establish a selective functional test for prodromal patients or others at risk for PD. As we have identified Kv4 channels in DMV neurons as molecular targets, a selective pharmacological challenge would be conceptually most promising. While there is currently no selective Kv4 channel modulator in the clinic, the A-type Kv channel blocker 4-AP (46)—used to treat multiple sclerosis (47) and Lambert-Eaton myasthenic syndrome (27)—could potentially be useful. For readout of these tests, monitoring changes in GI motility is, in principle, possible. Even in the absence of selective Kv4 pharmacology, the detailed level of mechanistic understanding provided by our study could form the basis for an improved differential diagnosis of prodromal dysautonomia in PD.
MATERIALS AND METHODS
Experimental design
The prespecified objective of the study was to determine (i) the behavioral impact of selective expression of a mutated form of α-synuclein in DMV neurons (representing an early prodromal stage of PD) on GI motility, (ii) the impact of this selective α-synuclein expression on the biophysics and morphology of DMV neurons, and (iii) the molecular chain of events that link the pathophysiology of DMV neurons to the altered GI motility (the morphological hypothesis was suggested after the initiation of the study). To address these objectives, the study was composed of a behavioral arm, ex vivo (slice and organ bath physiology) and in vivo arms, and a histological arm. All arms revolved around the same backbone assay, which was the selective expression of a virally delivered mutated form of α-synuclein in the dorsal medulla of the adult C57BL/6J mouse, with the identical delivery of an empty vector serving as control. No part of the study was blinded.
The behavioral arm was composed of measuring the time of passage through the GI tract following gavage. The in vivo arm was composed of recording the spontaneous firing rate of DMV neurons (identified with juxtacellular labeling) in anesthetized mice. The organ bath arm was composed of measuring the spontaneous motility and responsiveness of excised ileal segments. The slice electrophysiology was composed of recording the autonomous firing rate, membrane currents, and other physiological measure from DMV neurons. The histology arm was composed of measuring morphological changes in DMV neurons as well as stereotaxic counts of cell loss and immunohistochemistry.
Individual mice were considered independent samples. Because of the nested design of most of the experiments, wherein we sampled multiple neurons (or ileal segments) from individual mice, we fit the data with linear mixed-effects models (LMEMs), where the response (e.g., firing rate, cell size, and capacitance) is modeled as a combination of a fixed effect, which is the treatment (e.g., the injection of either AAV-A53T or AAV-EV or drug versus control), and uncorrelated random effects due to each mouse. In LMEMs, repeated measures (e.g., individual neurons) per mouse are not considered statistically independent (48–51). Because every single mouse in the study had to undergo a viral injection into its cervical vagus, we required three mice per treatment per experiment. The only exception was the organ bath experiments (in the Supplementary Materials) where data from two mice yielded either a significant effect or the emerging control distribution was deemed to be so similar to the α-synuclein group that another mouse could not discriminate between the treatments.
Animals
All experiments were conducted on male C57BL/6JRccHsd mice except for the chemogenetic experiments (see below), which were conducted with one homozygous male ChAT-IRES-Cre transgenic mice (the Jackson Laboratory, stock no. 006410).
Cervical vagus injections
Six- to 7-week-old mice (>25 g) were anesthetized with an intraperitoneal mixture of ketamine (100 mg/kg) and medetomidine (83 μg/kg), and ointment was applied to prevent corneal dying. The mouse was held in place in a supine position with paper tape, and temperature was maintained at 37°C with a heating pad. Animals were hydrated with a bolus of injectable saline (5 ml/kg) mixed with an analgesic [carprofen (5 mg/kg)]. The hairs around the neck area were removed with an electric razor and hair-removing gel. Before cutting the skin, 70% alcohol and Betadyne were used to sterilize that area. The central line of the skin was vertically cut with surgical scissors from the sternum area to the lower jaw area (approximately 2 cm long), and the thoracic glands were gently teased apart. For unilateral injections, the right cervical vagus was exposed from beneath the carotid (after careful separation from fat and connective tissue) and held away from the pulsations with a paper pointer. The nerve was pierced with a glass pipette pulled to a very fine tip (three to four times thinner than the vagus nerve trunk), and AAV particles were injected with a Nanoject III injector (Drummond Scientific) at a speed of 1 nl/s for 30-s injection with 30-s pause for a total of 33 cycles (i.e., 990 nl in total). After the injection, the pipette was left for an extra 1 min before slowly being retracted. At this point, the injection step was terminated, and the paper pointer was gently removed so as not to damage the nerve. For bilateral injections, the process was repeated on the left side. Both glands were placed back above the neck region, and the skin was sutured. At the end, the Betadyne and alcohol sterilization was repeated. The animal was woken up with Antisedan (0.42 mg/kg, intraperitoneally) and kept warm until it was awake (approximately in 30 min). Animals were monitored and received carprofen for three additional days.
Adeno-associated viruses
The C57BL/6JRccHsd mice were injected with AAVs [5 × 1012 genome copies per ml (GC/ml)] that either harbor a human mutated A53T form of the α-synuclein gene (AAV1/2-CMV/CBA-human-A53T-alpha-synuclein-WPRE-BGH-polyA) or harbor an empty vector (AAV1/2-CMV/CBA-empty vector-WPRE-BGH-polyA) (Genedetect, New Zealand). The ChAT-IRES-Cre mice were injected with an AAV that harbors a Gi DREADD gene (AAV9-hSyn-DIO-hM4Di-mCherry, 8.8 × 1012 GC/ml, ELSC Vector Core Facility), which, unlike in the case of the C57BL/6JRccHsd mice, only expressed in the cholinergic neurons of the DMV and not in the NST.
In vivo GI motility assay
One week before AAV inoculation, the C57BL/6JRccHsd mice (>25 g) were lightly sedated with isoflurane until the breathing pattern reached to 1 cycle/s (observed from the chest movement). Mice were gavaged, between 10:00 a.m. and 11:00 a.m., with 0.3 ml of 6% carmine red (Sigma-Aldrich, Germany) mixed with 0.5% methyl cellulose (Sigma-Aldrich, Germany) using a 22-gauge feeding tube. After 2.5 hours, the animals were transferred into a clean cage with fresh beddings, to monitor the latency at which the first reddish feces pellets were observed. The gavage feeding was repeated twice per animal (once per day) to determine consistent and stable results. Mice that were inactive or asleep (approximately 15%) were excluded. The actual measurements of passage time were conducted 3 and 6 weeks after inoculation.
Stool mass measurement
Mice injected with either AAV-EV or AAV-A53T were housed individually between 10:00 a.m. and 11:00 a.m. in a clean cage with fresh beddings. Two and a half hours later, the stools were collected and dehydrated, and the total stool mass was measured.
Chemogenetics
ChAT-Cre transgenic mice that were inoculated with the AAV harboring the DREADDs were tested 5 to 6 weeks after inoculation. Each animal was subjected to three measurements on three different days. On the first day, the animals were gavaged and intraperitoneally injected with a vehicle injection of 0.9% saline (10 ml/kg). Two days later, to activate the Gi DREADDS overexpressed in the DMV, the mice received an identical gavage feeding with an intraperitoneal injection of water-soluble CNO (3 mg/kg) (Hello Bio, UK) dissolved in injectable 0.9% saline. Following an additional 2 days, the vehicle gavage was repeated. Noninoculated animals were used as controls and underwent the same measurements, except that the first and last measurements were with CNO injections, while the middle one was saline.
Ex vivo GI motility assay
Mice were perfused transcardially with ice-cold 0.9% phosphate-buffered saline (PBS) (for histological verification of DMV transfection, see below), and the entire GI tract from between the diaphragm and cecum was transferred into ice-cold Krebs solution carbogenated with 95% O2–5% CO2 and containing 118 mM NaCl, 4.7 mM KCl, 14.4 mM NaCHO3, 5 mM CaCl2, 1.2 mM MgSO4, 1.2 mM NaH2PO4, and 11.5 mM glucose. Sections of the ileum (approximately 1 cm long) were immersed in an organ bath (20-ml volume) filled with warm (37°C), carbogenated Krebs solution and tied with a silk suture to a force transducer. The transducer signal was logged onto a PC with a National Instruments analog-to-digital (A/D) board using custom-made code in LabVIEW (National Instruments). Sections were given 30 min to accommodate during which spontaneous contractions were recorded and were then subjected to increasing concentrations of pilocarpine (Sigma-Aldrich). EFS was conducted with a Grass S44 stimulator using 0.5-ms-long, 55-V pulses at 1, 2, 5, 10, 20, and 30 Hz for a duration of 15 s. The current was passed between two rings that surrounded (but did not touch) the ileal section inside the organ bath. We used spectral analysis to characterize spontaneous contractions and fit Hill equations to the dose-response curves of tension as a function of pilocarpine concentration or EFS frequency.
In vivo recordings and juxtacellular labeling
Mice were anesthetized in a nonrebreathing system with isoflurane (induction 2.5%, maintenance 0.8 to 1.4% in O2, 0.35 liters/min) and were placed in a stereotaxic frame (David Kopf), and eye lubricant (Vidisic, Bausch and Lomb, Berlin, Germany) was applied to prevent corneal drying. Lidocaine/prilocaine ointment (EMLA cream, AstraZeneca, Wedel, Germany) was used as a local analgesic at the incision site. Body temperature (33° to 36°C), heart rate (5 to 10 Hz), and respiration (1 to 2 Hz) were constantly monitored by the vital sign oximeter (MouseOX, STARR Life Sciences, USA). Bregma was set to be 1 mm lower than lambda so that the exposed brain stem will lie in a horizontal plane. All the neck muscles attached to the occipital bone were removed, and a small craniotomy (±0.5 mm from the midline) was bored at the caudal position −4.0 to −4.3 mm from lambda. A micromanipulator (SM-6, Luigs and Neumann) was used to lower the electrodes (1 to 2 μm/s) to the recording site [For DMV—anterior-posterior: −4.1 to −4.3 mm (from lambda), medial-lateral: ±0.1 to 0.3 mm (from lambda), dorsal-ventral: −2.8 to −3.0 mm (from the surface of cerebellum); for NST—same anterior-posterior and medial lateral, dorsal-ventral: −2.3 to −2.7 mm].
Glass electrodes (10 to 20 megohms; Harvard Apparatus) filled with 0.5 M NaCl, 10 mM Hepes, and 1.5% neurobiotin (Vector Laboratories) were used for recording. The extracellular single-unit activity was recorded for 1 to 3 min, and the signals were acquired with EPC-10 A/D converter (PatchMaster software, Heka; sampling rates of 12.5 kHz for spike train analyses and 20 kHz for AP waveform analyses). The extracellular signals were amplified 1000× (ELC-03 M, NPI Electronics), notch- and band-pass–filtered 0.3 to 5 kHz (single pole, 6 dB per octave, DPA-2FS, NPI Electronics). The signals were displayed both on an analog oscilloscope and via an audio monitor.
To identify the anatomical location and neurochemical identity of the recorded neuron, following extracellular single-unit recordings, the neurons were labeled with neurobiotin using a juxtacellular in vivo labeling technique (23, 52). Microionophoretic current was applied (1- to 7-nA positive current and 200-ms on/off pulse), via the recording electrode with continuous monitoring of the firing activity. The labeling was considered successful if the firing pattern of the neuron was modulated during current injection (i.e., to observe an increased activity during on-pulse and absence of activity in the off-pulse) and the process was stable for a minimum of 25 s followed by a rebound to spontaneous activity of the neuron after modulation. This procedure enabled us to map the recorded DMV neurons within the medulla. Thirty percent of recorded DMV neurons stopped firing either before or during the labeling procedure. In those cases, the labeling protocol was carried on until the current reached 5 to 7 nA, to create a microtrauma to verify the location within the DMV.
After recordings, mice were euthanized (Na-pentobarbital, 1.6 g/kg) and transcardially perfused with 4% paraformaldehyde and 15% picric acid in PBS (pH 7.4). The perfused brain was stored in paraformaldehyde overnight and changed to 10% sucrose and 0.05% NaN3 solution for long-term storage.
Slice physiology
Six weeks after vagal transfection, mice were deeply anesthetized with intraperitoneal injections of ketamine (200 mg/kg) and xylazine (23.32 mg/kg) and perfused transcardially with ice-cold modified artificial cerebrospinal fluid (ACSF) oxygenated with 95% O2–5% CO2 and containing the following: 2.5 mM KCl, 26 mM NaHCO3, 1.25 mM Na2HPO4, 0.5 mM CaCl2, 10 mM MgSO4, 10 mM glucose, 0.4 mM ascorbic acid, and 210 mM sucrose. The cerebellum, pons, and medulla were rapidly removed, blocked in the coronal plane, and sectioned at a thickness of 240 μm in ice-cold modified ACSF. Slices were then submerged in ACSF, bubbled with 95% O2–5% CO2 and containing 2.5 mM KCl, 126 mM NaCl, 26 mM NaHCO3, mM 1.25 mM Na2HPO4, 2 mM CaCl2, 2 mM MgSO4, and 10 mM glucose. The slices were transferred to the recording chamber mounted on an upright Zeiss Axioskop fixed-stage microscope and perfused with oxygenated ACSF at 32°C. A 60×, 0.9–numerical aperture water immersion objective was used to examine the slice using standard infrared differential interference contrast video microscopy. Patch pipette resistance was typically 3 to 4.5 megohms. A junction potential of 7 to 8 mV was not corrected. For cell-attached current-clamp recordings of firing patterns, whole-cell current-clamp recordings, and voltage-clamp recordings of potassium currents, the pipette contained 135.5 mM KCH3SO3, 5 mM KCl, 2.5 mM NaCl, 5 mM Na-phosphocreatine, 10 mM Hepes, 0.2 mM EGTA, 0.21 mM Na2GTP, and mM 2 Mg1.5ATP (pH 7.3 with KOH, 280 to 290 mOsm/kg). For whole-cell voltage-clamp recordings of calcium currents, the pipette contained 111 mM CsCH3SO3, 12.5 mM CsCl, 1 mM MgCl2, 0.1 mM CaCl2, 10 mM Hepes, 1 mM EGTA, 0.21 mM Na2GTP, and 2 mM Mg1.5ATP (pH 7.3 with CsOH, 280 to 290 mOsm/kg). In some experiments, biocytin [0.2% (w/v)] was added to this internal solution. We used a mixture of synaptic receptor blockers containing the following: 50 μM d-APV, 5 μM NBQX, 10 μM SR 95531, 1 μM CGP 55845, 10 μM mecamylamine, and 10 μM atropine. For whole-cell calcium current recordings, Hepes-based bath solutions were used. The first solution contained 137 mM NaCl, 1.8 mM CaCl2, 1 mM MgCl2, 5.4 mM tetraethylammonium-Cl, 10 mM 4-AP, 0.001 mM tetrodotoxin, 5 mM Hepes, and 10 mM glucose (pH 7.3 with NaOH), and the second solution was identical except for an equimolar substitution of CaCl2 with CoCl2 (pH 7.3 with NaOH). Kv4 channels were blocked with preincubation of the slices in 1.3 μM phrixotoxin-2, a highly selective Kv4 channel antagonist (Alomone, Jerusalem, Israel).
Brain stem histology
The brain stem was sliced at 30-μm thickness with a cryostat (Leica CM1950), and the tissue was stored in an antifreeze solution. For the immunofluorescent staining, after 4× rinse/5 min in 0.1 M phosphate buffer (PB) solution, the slices were incubated in normal donkey serum for 1 hour. Then, the tissue was incubated overnight in the primary antibodies dissolved with 0.3% Triton X-100 and 0.1 M PB with 10% normal donkey serum (53). For iba-1 immunostaining, the protocol was slightly different: 3× rinse/5 min in 0.01 M PBS solution, and the slices were incubated in 5% goat serum for 30 min. The following primary antibodies were used: mouse anti-human α-synuclein [1:5000; Thermo Fisher Scientific; Research Resource Identifiers (RRID): AB_1954821], goat anti–choline acetyltransferase (ChAT) (1:100; Millipore; RRID: AB_262156), and rabbit anti–iba-1 (1:1000; Wako, 019-19741). On the second day, the tissue was incubated in the secondary antibodies at room temperature for 2 hours after washing steps. The following secondary antibodies were used: anti-mouse Alexa Fluor 488 (1:1000; Invitrogen), anti-goat Cy5 (1:1000), streptavidin conjugated to Cy3 (Abcam, UK), biotinylated donkey anti-rabbit (1:1000, Jackson ImmunoResearch), anti-rabbit Alexa Fluor 594 (1:000), and 4′,6-diamidino-2-phenylindole (DAPI) (1:6000; Invitrogen, D3571). The sections treated with biotinylated antibodies were further incubated for 1 hour in avidin-biotin-peroxidase solution (ABC Elite, Vector Laboratories, Burlingame, USA). The protein was lastly visualized by a 0.1 M PB solution containing 5% 3,3′-diaminobenzidine (DAB) (Sigma-Aldrich, Germany) and 0.02% H2O2. The sections were mounted on glass slides and dehydrated in a series concentration of ethanol, followed by an incubation of cresyl violet solution (Sigma-Aldrich, Germany). Afterward, the DAB/Nissl-stained sections were shortly incubated in xylene (Merck, Germany) and coverslipped using mounting gel (toluene solution, Thermo Fisher Scientific). The tissue of fluorescent staining was mounted (VECTASHIELD, Vector Laboratories) on glass slides after 4× rinse in 0.1 M PB.
The brain stem samples for the cohort of the juxtacellular recording were cut into 50-μm-thick slices (VT1000S microtome, Leica). Sections were rinsed in PBS and then incubated in a blocking solution (0.2 M PBS with 10% horse serum, 0.5% Triton X-100, and 0.2% bovine serum albumin). The following primary antibodies were used (in carrier solution, 22°C, overnight): monoclonal mouse anti-human α-synuclein (1:1000; 4B12, Thermo Fisher Scientific), the goat anti-ChAT (1:100; Millipore, RRID: AB_262156). The following secondary antibodies (in carrier solution, 22°C, overnight) Alexa Fluor 488 donkey anti-goat, Alexa Fluor 488 donkey anti-mouse, and streptavidin–Alexa Fluor 568 (1:1000; Molecular Probes) were used. Sections were rinsed in PBS and mounted on slides (VECTASHIELD, Vector Laboratories).
For semiquantitative optical density analysis of the human α-synuclein expression in DMV neurons, we used ImageJ software (http://rsbweb.nih.gov/ij/) to determine the mean immunosignal intensities of ChAT-positive regions of interest in the brain stem slices. We used three confocal slices through the DMV from each mouse with a total of three AAV-A53T– and three AAV-EV–injected mice for the analysis. Each confocal slice was a collapsed z-stack of 10 adjacent 1-μm-thick optical slices chosen so that they arose primarily from within individual DMV neurons. Thus, while it is not possible to rule out the possibility that some of the fluorescence arises from other processes (e.g., glia), most of the signal is confined to ChAT-immunopositive neuronal somatodendritic domains in the DMV.
For quantification of microglial activation, confocal images of the ipsilateral DMV at bregma −7.48 mm, bregma −7.32 mm, and bregma −7.20 mm were acquired from each animal with an Olympus confocal FV3000 inverted laser scanning microscope confocal microscope, under a 40× objective. Image analysis was performed in FIJI freeware (National Institutes of Health (NIH)]. The DMV was delineated on the basis of DAPI labeling of DMV neurons. Iba-1–immunoreactive microglia were counted using the cell counter plugin. The activation state of the microglia was determined according to morphologies outlined in a previous report (29). Microglia were considered activated (fig. S2) if their morphology was either hyperramified or hypertrophic.
Stomach and intestine histology
After 200 nl of AAV1/2-A53T-SNCA (the same virus used for vagal injection) intramuscularly injected in the stomach, 2 weeks after surgery, the mouse was euthanized for immunofluorescent staining. The staining procedure was processed as previously described (54).
Fluorescence microscopy and cell size analysis
Cell surface area
Confocal images of biocytin-filled neurons reacted with streptavidin conjugated to Cy3 were captured on a Nikon confocal 1AR microscope under a Plan Apochromat Lambda 60× oil objective. Images were oversampled in the z plane for maximum preservation of three-dimensional (3D) structure (pixel size xy = 0.138 μm; voxel depth z = 0.125 μm) and were processed and analyzed with FIJI software (ImageJ, NIH, version 2.0.0). The area encompassing the cell soma was cropped to discard neurites. A Gaussian blur was applied to the voxels (sigma radius = 1.0), and where necessary, cells were filled so that only voxels on the cell surface were used for calculations. A threshold was set for each cell, and the total surface area was analyzed using the 3D objects counter plugin (FIJI). Only complete cells were included in the analysis.
Cell diameter
Six weeks after injection with either AAV-EV or AAV-A53T, we took confocal images of ChAT/human α-synuclein staining from 30-μm-thick slides of DMV. In each slide, we collapsed 10 to 15 adjacent 1-μm-thick confocal images from the middle part of each slide in to a single image. We collected five such collapsed images that were approximately 150 μm apart from between AP: −7.32 to AP: −7.64 (from bregma). In each of the five images, we randomly selected ChAT-positive neurons with or without immunoactivity of anti-human α-synuclein from the medial-dorsal portion of DMV and measured the longest axis of their soma using the ImageJ software (NIH, USA).
Stereological quantification of DMV neuron number
The number of Nissl-stained DMV neurons was stereologically quantified, as previously described (28, 55). Briefly, the DMV was delineated from every fifth section of the DMV between bregma −7.32 and − 7.20 mm. Neuronal counts were performed on a Zeiss Axio Imager 2 microscope or an Olympus BX53, fitted with a Prior ProScan III motorized stage. The optical fractionator technique was used and was facilitated by either Stereo Investigator version 10, (MBF Biosciences) or new CAST software (Visiopharm). Total cell numbers were calculated according to the methods described previously (19). A guard zone of at least 1 μm was set. Coefficient of error was calculated with values <0.10 accepted.
Data and statistical analysis
Data were analyzed, and curve fitting was done using custom-made code on MATLAB (The MathWorks, Natick, MA) software. The two-tailed Wilcoxon rank sum test was used to test for changes of medians in two independent sample comparisons. The two-tailed Wilcoxon signed-rank test was used to test for changes of medians in matched-paired comparisons. In some cases, we first performed a two-tailed one-way analysis of variance (ANOVA) to compare among multiple groups. Throughout the main manuscript and the Supplementary Materials, uppercase Ns indicate the number of mice, and lowercase ns indicate the number of individual neurons (or ileal segment in the ex vivo GI motility assays) recorded. To fit LMEMs, to the data, we used the MATLAB (The MathWorks) FITLME command (48–51), and we reported N, n, the absolute value of Student’s tν statistic (where ν is the number of degrees of freedom), and the two-tailed P value of the fixed effect. Null hypotheses were rejected if the P values were below 0.05.
Acknowledgments
Funding: This work was supported by grants from the German-Israeli Foundation for Scientific Research and Development (no. I-1294-418.13/2015) to J.R. and J.A.G., the German Research Foundation Collaborative Research Center 815 “Redox Signaling” program to J.R., and the European Research Council (no. 646880) to J.A.G. We thank A. Troen for help with the stereological analyses, Y. Ritov for help with mixed-effects models, and E. Konyukhov and A. Shapochnikov for technical support. Ethics statement: All experimental procedures on mice adhered to and received prior written approval from the Hebrew University Institutional Animal Care and Use Committee and from the German Regierungspräsidium Darmstadt. Author contributions: Conceptualization: W.-H.C., J.R., and J.A.G. Methodology: W.-H.C., R.E.M., H.A.-Z., D.B.-Z., M.H., J.R., and J.A.G. Formal analysis: W.-H.C., R.E.M., and J.A.G. Investigation: W.-H.C., L.K., R.E.M., H.A-Z., D.B-Z., and J.A.G. Resources: J.B.K., J.M.B., R.Y., M.H., J.R., and J.A.G. Data curation: W.-H.C., R.E.M., J.R., and J.A.G. Writing—original draft: W.-H.C., R.E.M., J.R., and J.A.G. Writing—review and editing: W.-H.C., L.K., R.E.M., H.A.-Z., J.B.K., J.M.B., D.B.-Z., M.H., J.R., and J.A.G. Visualization: W.-H.C., R.E.M., J.R., and J.A.G. Supervision: J.R. and J.A.G. Project administration: J.R. and J.A.G. Funding acquisition: J.R. and J.A.G. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All the raw data are deposited in the Open Science Framework: https://osf.io/2c8rs/?view_only=89ee0c9bb8414aea8559819ddce5782d. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/11/eabd3994/DC1
REFERENCES AND NOTES
- 1.Farrer M., Wavrant-De Vrieze F., Crook R., Boles L., Perez-Tur J., Hardy J., Johnson W. G., Steele J., Maraganore D., Gwinn K., Lynch T., Low frequency of α-synuclein mutations in familial Parkinson’s disease. Ann. Neurol. 43, 394–397 (1998).9506559 [Google Scholar]
- 2.Polymeropoulos M. H., Lavedan C., Leroy E., Ide S. E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E. S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W. G., Lazzarini A. M., Duvoisin R. C., Iorio G. D., Golbe L. I., Nussbaum R. L., Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Simón-Sánchez J., Schulte C., Bras J. M., Sharma M., Gibbs J. R., Berg D., Paisan-Ruiz C., Lichtner P., Scholz S. W., Hernandez D. G., Krüger R., Federoff M., Klein C., Goate A., Perlmutter J., Bonin M., Nalls M. A., Illig T., Gieger C., Houlden H., Steffens M., Okun M. S., Racette B. A., Cookson M. R., Foote K. D., Fernandez H. H., Traynor B. J., Schreiber S., Arepalli S., Zonozi R., Gwinn K., van der Brug M., Lopez G., Chanock S. J., Schatzkin A., Park Y., Hollenbeck A., Gao J., Huang X., Wood N. W., Lorenz D., Deuschl G., Chen H., Riess O., Hardy J. A., Singleton A. B., Gasser T., Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 41, 1308–1312 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spillantini M. G., Crowther R. A., Jakes R., Hasegawa M., Goedert M., α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brundin P., Dave K. D., Kordower J. H., Therapeutic approaches to target alpha-synuclein pathology. Exp. Neurol. 298, 225–235 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lang A. E., Espay A. J., Disease modification in Parkinson’s disease: Current approaches, challenges, and future considerations. Mov. Disord. 33, 660–677 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Selkoe D. J., Alzheimer disease and aducanumab: Adjusting our approach. Nat. Rev. Neurol. 15, 365–366 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Postuma R. B., Berg D., Prodromal parkinson’s disease: The decade past, the decade to come. Mov. Disord. 34, 665–675 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Lim S.-Y., Fox S. H., Lang A. E., Overview of the extranigral aspects of Parkinson disease. Arch. Neurol. 66, 167–172 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Mahlknecht P., Pechlaner R., Boesveldt S., Volc D., Pinter B., Reiter E., Müller C., Krismer F., Berendse H. W., van Hilten J. J., Wuschitz A., Schimetta W., Högl B., Djamshidian A., Nocker M., Göbel G., Gasperi A., Kiechl S., Willeit J., Poewe W., Seppi K., Optimizing odor identification testing as quick and accurate diagnostic tool for Parkinson’s disease. Mov. Disord. 31, 1408–1413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldstein D. S., Dysautonomia in Parkinson’s disease: Neurocardiological abnormalities. Lancet Neurol. 2, 669–676 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Knudsen K., Szwebs M., Hansen A. K., Borghammer P., Gastric emptying in Parkinson’s disease – A mini-review. Park. Relat. Disord. 55, 18–25 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Braak H., Del Tredici K., Rüb U., de Vos R. A. I., Jansen Steur E. N. H., Braak E., Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Bonaz B., Sinniger V., Pellissier S., Vagal tone: Effects on sensitivity, motility, and inflammation. Neurogastroenterol. Motil. 28, 455–462 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Travagli R. A., Browning K. N., Camilleri M., Parkinson disease and the gut: New insights into pathogenesis and clinical relevance. Nat. Rev. Gastroenterol. Hepatol. 17, 673–685 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Kim S., Kwon S.-H., Kam T.-I., Panicker N., Karuppagounder S. S., Lee S., Lee J. H., Kim W. R., Kook M., Foss C. A., Shen C., Lee H., Kulkarni S., Pasricha P. J., Lee G., Pomper M. G., Dawson V. L., Dawson T. M., Ko H. S., Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103, 627–641.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Challis C., Hori A., Sampson T. R., Yoo B. B., Challis R. C., Hamilton A. M., Mazmanian S. K., Volpicelli-Daley L. A., Gradinaru V., Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat. Neurosci. 23, 327–336 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seidel K., Mahlke J., Siswanto S., Krüger R., Heinsen H., Auburger G., Bouzrou M., Grinberg L. T., Wicht H., Korf H. W., Den Dunnen W., Rüb U., The brainstem pathologies of Parkinson’s disease and dementia with Lewy bodies. Brain Pathol. 25, 121–135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ulusoy A., Rusconi R., Pérez-Revuelta B. I., Musgrove R. E., Helwig M., Winzen-Reichert B., Di Monte D. A., Caudo-rostral brain spreading of α-synuclein through vagal connections. EMBO Mol. Med. 5, 1119–1127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallaher Z. R., Ryu V., Herzog T., Ritter R. C., Czaja K., Changes in microglial activation within the hindbrain, nodose ganglia, and the spinal cord following subdiaphragmatic vagotomy. Neurosci. Lett. 513, 31–36 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ulusoy A., Phillips R. J., Helwig M., Klinkenberg M., Powley T. L., Di Monte D. A., Brain-to-stomach transfer of α-synuclein via vagal preganglionic projections. Acta Neuropathol. 133, 381–393 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janezic S., Threlfell S., Dodson P. D., Dowie M. J., Taylor T. N., Potgieter D., Parkkinen L., Senior S. L., Anwar S., Ryan B., Deltheil T., Kosillo P., Cioroch M., Wagner K., Ansorge O., Bannerman D. M., Bolam J. P., Magill P. J., Cragg S. J., Wade-Martins R., Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl. Acad. Sci. U.S.A. 110, E4016–E4025 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramaniam M., Althof D., Gispert S., Schwenk J., Auburger G., Kulik A., Fakler B., Roeper J., Mutant α-Synuclein enhances firing frequencies in dopamine substantia nigra neurons by oxidative impairment of A-type potassium channels. J. Neurosci. 34, 13586–13599 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldberg J. A., Guzman J. N., Estep C. M., Ilijic E., Kondapalli J., Sanchez-Padilla J., Surmeier D. J., Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson’s disease. Nat. Neurosci. 15, 1414–1421 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooper G., Lasser-Katz E., Simchovitz A., Sharon R., Soreq H., Surmeier D. J., Goldberg J. A., Functional segregation of voltage-activated calcium channels in motoneurons of the dorsal motor nucleus of the vagus. J. Neurophysiol. 114, 1513–1520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sah P., McLachlan E. M., Potassium currents contributing to action potential repolarization and the afterhyperpolarization in rat vagal motoneurons. J. Neurophysiol. 68, 1834–1841 (1992). [DOI] [PubMed] [Google Scholar]
- 27.Oh S. J., Shcherbakova N., Kostera-Pruszczyk A., Alsharabati M., Dimachkie M., Blanco J. M., Brannagan T., Lavrnić D., Shieh P. B., Vial C., Meisel A., Komoly S., Schoser B., Sivakumar K., So Y.; LEMS Study Group , Amifampridine phosphate (Firdapse®) is effective and safe in a phase 3 clinical trial in LEMS. Muscle Nerve. 53, 717–725 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Ulusoy A., Musgrove R. E., Rusconi R., Klinkenberg M., Helwig M., Schneider A., Di Monte D. A., Neuron-to-neuron α-synuclein propagation in vivo is independent of neuronal injury. Acta Neuropathol. Commun. 3, 13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rusconi R., Ulusoy A., Aboutalebi H., Di Monte D. A., Long-lasting pathological consequences of overexpression-induced α-synuclein spreading in the rat brain. Aging Cell 17, e12727 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Surmeier D. J., Obeso J. A., Halliday G. M., Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 18, 101–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perrins R., Biophysics of computation: Information processing in single neurons. Christof Koch. Q. Rev. Biol. 74, 494–494 (1999). [Google Scholar]
- 32.Lasser-Katz E., Simchovitz A., Chiu W.-H., Oertel W. H., Sharon R., Soreq H., Roeper J., Goldberg J. A., Mutant α-synuclein overexpression induces stressless pacemaking in vagal motoneurons at risk in Parkinson’s Disease. J. Neurosci. 37, 47–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bove C., Coleman F. H., Travagli R. A., Characterization of the basic membrane properties of neurons of the rat dorsal motor nucleus of the vagus in paraquat-induced models of parkinsonism. Neuroscience 418, 122–132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noorian A. R., Rha J., Annerino D. M., Bernhard D., Taylor G. M., Greene J. G., Alpha-synuclein transgenic mice display age-related slowing of gastrointestinal motility associated with transgene expression in the vagal system. Neurobiol. Dis. 48, 9–19 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Kaufmann H., Goldstein D. S., Autonomic dysfunction in Parkinson disease. Handb. Clin. Neurol. 117, 259–278 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Valappil R. A., Black J. E., Broderick M. J., Carrillo O., Frenette E., Sullivan S. S., Goldman S. M., Tanner C. M., Langston J. W., Exploring the electrocardiogram as a potential tool to screen for premotor Parkinson’s disease. Mov. Disord. 25, 2296–2303 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Eadie M. J., The pathology of certain medullary nuclei in parkinsonism. Brain 86, 781–792 (1963). [DOI] [PubMed] [Google Scholar]
- 38.Halliday G. M., Blumbergs P. C., Cotton R. G. H., Blessing W. W., Geffen L. B., Loss of brainstem serotonin- and substance P-containing neurons in Parkinson’s disease. Brain Res. 510, 104–107 (1990). [DOI] [PubMed] [Google Scholar]
- 39.Thakur P., Chiu W. H., Roeper J., Goldberg J. A., α-Synuclein 2.0 — Moving towards cell type specific pathophysiology. Neuroscience 412, 248–256 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Braak H., Ghebremedhin E., Rüb U., Bratzke H., Del Tredici K., Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 318, 121–134 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Luk K. C., Kehm V., Carroll J., Zhang B., O’Brien P., Trojanowski J. Q., Lee V. M.-Y., Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rey N. L., George S., Steiner J. A., Madaj Z., Luk K. C., Trojanowski J. Q., Lee V. M.-Y., Brundin P., Spread of aggregates after olfactory bulb injection of α-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol. 135, 65–83 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henrich M. T., Geibl F. F., Lakshminarasimhan H., Stegmann A., Giasson B. I., Mao X., Dawson V. L., Dawson T. M., Oertel W. H., Surmeier D. J., Determinants of seeding and spreading of α-synuclein pathology in the brain. Sci. Adv. 6, eabc2487 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holmqvist S., Chutna O., Bousset L., Aldrin-Kirk P., Li W., Björklund T., Wang Z.-Y., Roybon L., Melki R., Li J.-Y., Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 128, 805–820 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Fang F., Yang W., Florio J. B., Rockenstein E., Spencer B., Orain X. M., Dong S. X., Li H., Chen X., Sung K., Rissman R. A., Masliah E., Ding J., Wu C., Synuclein impairs trafficking and signaling of BDNF in a mouse model of Parkinson’s disease. Sci. Rep. 7, 3868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luca C. C., Nadayil G., Dong C., Nahab F. B., Field-Fote E., Singer C., Dalfampridine in Parkinson’s disease related gait dysfunction: A randomized double blind trial. J. Neurol. Sci. 15, 7–11 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Lecat M., Decavel P., Magnin E., Lucas B., Gremeaux V., Sagawa Y., Multiple sclerosis and clinical gait analysis before and after fampridine: A systematic review. Eur. Neurol. 78, 272–286 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Boisgontier M. P., Cheval B., The anova to mixed model transition. Neurosci. Biobehav. Rev. 68, 1004–1005 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Aarts E., Verhage M., Veenvliet J. V., Dolan C. V., Van Der Sluis S., A solution to dependency: Using multilevel analysis to accommodate nested data. Nat. Neurosci. 17, 491–496 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Forstmeier W., Wagenmakers E.-J., Parker T. H., Detecting and avoiding likely false-positive findings – A practical guide. Biol. Rev. 92, 1941–1968 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Lazic S. E., The problem of pseudoreplication in neuroscientific studies: Is it affecting your analysis? BMC Neurosci. 11, 5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schiemann J., Schlaudraff F., Klose V., Bingmer M., Seino S., Magill P. J., Zaghloul K. A., Schneider G., Liss B., Roeper J., K-ATP channels in dopamine substantia nigra neurons control bursting and novelty-induced exploration. Nat. Neurosci. 15, 1272–1280 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chiu W.-H., Carlsson T., Depboylu C., Höglinger G. U., Oertel W. H., Ries V., Selegiline normalizes, while l-DOPA sustains the increased number of dopamine neurons in the olfactory bulb in a 6-OHDA mouse model of Parkinson’s disease. Neuropharmacology 79, 212–221 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Abu-Gazala S., Horwitz E., Schyr R. B. H., Bardugo A., Israeli H., Hija A., Schug J., Shin S., Dor Y., Kaestner K. H., Ben-Zvi D., Sleeve gastrectomy improves glycemia independent of weight loss by restoring hepatic insulin sensitivity. Diabetes 67, 1079–1085 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Musgrove R. E., Helwig M., Bae E. J., Aboutalebi H., Lee S.-J., Ulusoy A., Di Monte D. A., Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular α-synuclein transfer. J. Clin. Invest. 130, 3738–3753 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/11/eabd3994/DC1