

B cell acute lymphoid leukemia uses the mitochondrial respiration as the main energy source that enhances drug resistance.
Abstract
How metabolic status controls the fates of different types of leukemia cells remains elusive. Using a SoNar-transgenic mouse line, we demonstrated that B cell acute lymphoblastic leukemia (B-ALL) cells had a preference in using oxidative phosphorylation. B-ALL cells with a low SoNar ratio (SoNar-low) had enhanced mitochondrial respiration capacity, mainly resided in the vascular niche, and were enriched with more functional leukemia-initiating cells than that of SoNar-high cells in a murine B-ALL model. The SoNar-low cells were more resistant to cytosine arabinoside (Ara-C) treatment. cyclic adenosine 3′,5′-monophosphate response element–binding protein transactivated pyruvate dehydrogenase complex component X and cytidine deaminase to maintain the oxidative phosphorylation level and Ara-C–induced resistance. SoNar-low human primary B-ALL cells also had a preference for oxidative phosphorylation. Suppressing oxidative phosphorylation with several drugs sufficiently attenuated Ara-C–induced resistance. Our study provides a unique angle for understanding the potential connections between metabolism and B-ALL cell fates.
INTRODUCTION
B cell acute lymphoblastic leukemia (B-ALL) is a severe malignant hematopoietic disorder that gives rise to the clonal expansion of hematopoietic stem/progenitor cells, which commonly occurs in children/youth. Several therapeutic regimens have been shown to achieve desirable effects in the treatment of B-ALL, including chemotherapy, bone marrow (BM) transplantation, chimeric antigen receptor T cell (CAR-T) therapy, or combinations of these treatments (1–4). However, the effectiveness of current strategies is hindered by drug resistance, lack of major histocompatibility complex–matched donor hematopoietic stem cell (HSC) sources, patient nonresponsiveness, or induced toxicity in the case of CAR-T therapy (5). Although chemotherapy has been considered one of the most effective methods of B-ALL treatment, 20% of patients relapse upon treatment with cytosine arabinoside (Ara-C), anthracycline antibiotics, or other chemotherapeutic drugs (6). Increasing evidence shows that a small population of leukemia cells, termed leukemia-initiating cells (LICs), may contribute to drug resistance or leukemia relapse (7). For example, immunophenotypic CD34+CD19+ LICs may be closely connected with leukemia progression and drug resistance in human B-ALL (8). However, specific markers showing B-ALL–LIC enrichment and the mechanisms by which chemotherapies lead to drug resistance remain largely unknown.
Recently, metabolic status has been found to play a pivotal role in the development of many different cancer types, including both hematological and solid cancers. For example, mutations in isocitrate dehydrogenase 1/2 (IDH1/2) have been reported to efficiently initiate several types of cancers, such as acute myeloid leukemia (AML), glioblastoma, and colon cancer (9–11). Acute/chronic myeloid LICs mainly use glycolysis as their energy source (12); fructose metabolism enhances the proliferation of Ph+ (Philadelphia chromosome positive)–B-ALL cells (13); fasting can effectively induce B-ALL differentiation at both the early and later stages of disease (14). However, the precise metabolic profiles of different nutrients in B-ALL are unclear, and whether metabolic status is closely connected with drug resistance is unknown.
Currently, drug resistance in B-ALL cells is thought to result from changes in many intrinsic factors (such as transcription factors or epigenetic modifications) and extrinsic factors (such as niche components). For example, mutations in the transcription factor IKZF1 (ikaros family zinc finger 1) lead to a notable reduction in sensitivity to the treatment of tyrosine inhibitors in B-ALL cells (15); the down-regulation of TWIST2 (twist family BHLH transcription factor 2) also results in resistance to the chemotherapeutic agents etoposide, daunorubicin, and dexamethasone (16). In addition, BM niches have been found to play key roles in leukemia development and chemotherapeutic resistance (7, 17). Remodeled leukemic niches may contain different cell types (such as endothelial cells, osteoblast cells, and mesenchymal stem cells), and their secreted proteins/cytokines/growth factors [such as stromal cell–derived factor 1, interleukin-3 (IL-3), IL-6, and hyaluronic acid] may differ from those secreted under physiological conditions to enhance leukemia cell expansion or chemotherapy resistance (17, 18). Although most of the recent studies have focused on niche components during AML development (19) and drug resistance (20), several lines of evidence indicate that niche factors also support the initiation and drug resistance of B-ALL. For example, growth and differentiation factor 15 secreted from Nestin+ cells remodels the BM niche upon Ara-C treatment and leads to chemotherapeutic resistance (21). However, how the unique metabolism of different nutrients contributes to the drug resistance of B-ALL cells awaits further investigation.
Although some chemical dyes including TMRM (tetramethylrhodamine methyl ester), MitoTracker, and H2DCFDA (2’,7’-dichlorodihydrofluorescein diacetate) have been used for the evaluation of mitochondrial activity and reactive oxygen species levels in different types of leukemia cells (22), they indirectly reflect the metabolic states of leukemia cells. The determination of the intermediate metabolites by commercial kits is another way to evaluate metabolic status. Nevertheless, currently, only limited metabolites can be routinely measured. Although metabolomic analysis offers a large advantage in the determination of all potential metabolites (23), it is still difficult to perform the necessary assays with the limited number of LICs available because of their rare frequency. Currently, sensitive and specific monitoring of the in vitro/in vivo dynamic changes in the metabolism of certain nutrients in a single leukemia cell or LIC in real time remains very challenging (24).
A highly responsive reduced form of nicotinamide adenine dinucleotide (NADH)/nicotinamide adenine dinucleotide (NAD+) sensor (SoNar) has been reported recently to be used in live cell and in vivo metabolic study (25, 26). SoNar is an intrinsically ratiometric sensor with two excitation wavelengths (420 and 485 nm), and it responds to NADH and NAD+ with opposing changes in fluorescence ratio. SoNar is strictly responsive to the NADH/NAD+ ratio rather than the absolute concentrations of either the two nucleotides. SoNar exhibits a 1500% dynamic range under different NAD+/NADH ratios, making it one of the most responsive genetically encoded sensors currently available. Thus, it enables better real-time tracking of the subtle changes of cellular metabolic states. Using SoNar, we recently showed that AML-LICs have a unique preference for glycolytic metabolism, which is closely connected with AML-LIC fate, including their capacity for self-renewal, homing, and localization to an the endosteal niche (27). The SoNar sensor can be used to sensitively and specifically monitor cytosolic NADH/NAD+ levels both in vitro and in vivo, which is intricately related to energy metabolism of the AML-LICs. Here, we report on the SoNar-transgenic mice that were established and the metabolic characteristics of B-ALL cells that were evaluated, including their potential connection to cell fate determinations and drug resistance.
RESULTS
SoNar can indicate the glycolysis or oxidative phosphorylation states of B-ALL cells at the single-cell level
To determine whether SoNar is able to precisely indicate the level of glycolysis or oxidative phosphorylation in lymphoid leukemia cells, we used a human B-ALL cell line, Nalm6, for ectopically expressed SoNar (hereafter termed SoNar-Nalm6), followed by the measurement of the ratio of fluorescence upon excitation at 405 and 488 nm. An approximate fourfold increase in the ratio of SoNar fluorescence was observed upon cell incubation with the lactate dehydrogenase A inhibitor oxamate (fig. S1, A and B). In contrast, pyruvate reduced the SoNar signal to 50% that of the control group (fig. S1, A and B). The pH-control SoNar sensor, iNapc, was also ectopically expressed in Nalm6 cells, and changes in the ratio of SoNar fluorescence were negligible after treatment with either pyruvate or oxamate (fig. S1, C and D). The SoNar-Nalm6 cells, but not the iNapc-Nalm6 cells, responded to stimulation by several inhibitors of the mitochondrial respiration (rotenone) or the malate-aspartate shuttle inhibitor [aminooxyacetate (AOA)], as indicated by slightly increased levels of SoNar fluorescence (especially upon rotenone treatment; fig. S1, A to D). The flow cytometric analysis also revealed that the ratio of SoNar fluorescence in the SoNar-Nalm6 cells was increased to 3.2-fold compared with that in the control cells upon oxamate treatment and was reduced upon pyruvate stimulation (fig. S1, E and F). In addition, SoNar fluorescence was also notably increased after treatment with rotenone (1.57-fold) but not AOA (fig. S1, E and F).
To further characterize the SoNar pattern in murine B-ALL cells, we generated a transgenic mouse strain expressing SoNar driven by the cytomegalovirus early enhancer/chicken β-actin promoter. Briefly, the pCAG vector containing SoNar DNA was linearized, purified, and microinjected into fertilized eggs of FVB mice. SoNar DNA was randomly incorporated into the genome, which may result in either single- or multiple-copy transgenes. In the current study, transgenic SoNar heterozygote mice were used for all the experiments. SoNar mice were further verified at the DNA and mRNA levels by polymerase chain reaction (PCR) and quantitative reverse transcription PCR (RT-PCR) (fig. S1, G and H), respectively. Fluorescence imaging also showed that SoNar was expressed in most tissues, such as BM, liver, spleen, skeletal muscle, myocardium, and kidney (fig. S1I). Furthermore, mononuclear cells isolated from peripheral blood or total BM cells expressed different ratios of SoNar fluorescence, as revealed by microscopy (fig. S1J). The SoNar-transgenic mice were then used for the subsequent establishment of the N-myc–induced leukemia model (28). N-myc overexpression in HSCs and committed progenitor B cells can lead to the initiation of the precursor B-ALL/lymphoma (pre–B-ALL/LBL). N-myc–induced B-ALL cells are positive to negative for the precursor B cell marker CD43 and negative for the mature B cell marker immunoglobulin M (IgM). Consistently, the murine B-ALL cells mainly expressed markers for B lymphoid cells (B220+) but not myeloid cells (Mac-1+/Gr-1+; fig. S1K), and these leukemia cells were mainly undifferentiated B220+CD43+ B progenitor cells, not differentiated B220+IgM+cells (fig. S1K).
We thus analyzed the SoNar fluorescence in the BM B-ALL cells (SoNar-ALL) and found that three cell fractions had distinct fluorescent ratios: SoNar-low (16.2%; SoNar ratio, 0 to 0.5), SoNar-middle/mid (72.1%; SoNar ratio, 0.5 to 1.5), and SoNar-high (11.7%; SoNar ratio, >1.5) (Fig. 1, A and B). Supplementation with exogenous oxamate and pyruvate in vitro sensitively and specifically enhanced and decreased the SoNar fluorescence ratio in the SoNar-ALL cells, respectively (Fig. 1, C and E). Moreover, treatment with the oxidative phosphorylation inhibitor rotenone and the malate-aspartate shuttle inhibitor AOA also led to a moderate increase in SoNar fluorescence (Fig. 1, C and E). Furthermore, the pH-control sensor, iNapc (29), was used to produce the retrovirus for the infection in murine primary BM B-ALL cells. The results showed that the iNapc-expressing cells had no response to any of the four metabolic inhibitors used (Fig. 1, D and F), excluding the possibility of interference by pH variations. Sequential treatment with pyruvate and oxamate, and vice versa, led to dynamic changes in SoNar ratios, indicating that SoNar sensitively and specifically reflects the NADH/NAD+ levels in a single live cell in the B-ALL model (Fig. 1, G to I, and movie S1). Consistently, the alteration of SoNar fluorescence in the primary B-ALL cells upon treatment with oxamate, pyruvate, rotenone, or AOA was sensitively detected by flow cytometry (Fig. 1, J to K). These results prompted us to evaluate the metabolic heterogeneity of B-ALL cells using the SoNar sensor.
Fig. 1. SoNar can indicate the glycolysis or oxidative phosphorylation states of B-ALL cells at the single-cell level.
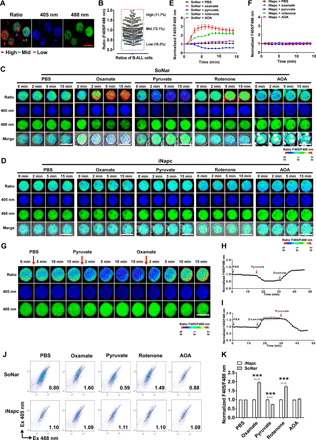
(A and B) Representative images of the ratio of SoNar fluorescence (F405/F488 nm) in murine SoNar B-ALL cells by confocal microscopy (A) and quantitative data are shown (B). A total of 204 SoNar B-ALL cells were calculated (n = 3). (C and D) Representative images of the ratio of SoNar fluorescence (C) and the control iNapc (D) fluorescence with excitation wavelengths at 405 and 488 nm in SoNar B-ALL cells at indicated time points upon treatments of phosphate-buffered saline (PBS), oxamate, pyruvate, rotenone, and AOA, respectively. (E and F) Quantification of the ratio of SoNar (E) and iNapc (F) in (C) and (D) is shown. A total of 25 to 30 SoNar B-ALL cells (E) and 25 to 30 iNapc-B-ALL cells (F) were measured (n = 3). (G and I) Shown are the representative images of the ratio of SoNar fluorescence in SoNar B-ALL cells upon the sequential treatments with pyruvate and oxamate (G) or vice versa (I). A total of 49 (H) or 58 (I) SoNar B-ALL cells were counted (n = 3). (J and K) Representative dot plots of the ratio of SoNar fluorescence in SoNar B-ALL cells upon treatments with PBS, oxamate, pyruvate, rotenone, and AOA (J). Quantification data in (J) are shown (K) (n = 3). This experiment was repeated independently three times. Scale bar, 10 μm. Data are represented as means ± SEM. Two-way analysis of variance (ANOVA) with Sidak’s multiple comparison test was used for the comparison of statistical significance (K). ***P < 0.001.
SoNar-low B-ALL cells prefer using oxidative phosphorylation as the main energy source
To characterize the metabolic profiles of different cell fractions in B-ALL cells with distinct SoNar fluorescence, we fluorescence-activated cell sorting (FACS)–purified SoNar-low and SoNar-high cells and evaluated the fluorescence ratios (Fig. 2A). The SoNar-high cells had an approximately 3.4-fold higher ratio of fluorescence than did SoNar-low cells, as indicated by either flow cytometric analysis (Fig. 2B) or confocal microscopy (Fig. 2C). As the control, the iNapc-low and iNapc-high cells had similar fluorescence ratios (fig. S2, A and B). The SoNar-low cells had a much lower fluorescence ratio changes than did SoNar-high cells upon oxamate or pyruvate stimulation (Fig. 2, D and E). In contrast, rotenone (but not AOA) treatment resulted in a greater fluorescence ratio changes in the SoNar-low cells than that in the SoNar-high cells (Fig. 2, F and G). Considering that the pyruvate treatment led to a minor response (Fig. 2E) and that the mitochondrial NADH oxidase inhibitor rotenone was much more efficient than the malate-aspartate shuttle inhibitor (AOA) in enhancing the SoNar ratio (Fig. 2, F and G), we speculated that the SoNar-low cells had a unique oxidative phosphorylation utilization profile while maintaining a glycolytic level similar to that of the SoNar-high cells. Flow cytometric analysis also showed a ~3.2-fold increase in the SoNar ratio upon rotenone treatment in the SoNar-low cells but showed minor or similar changes in these cells upon oxamate, pyruvate, or AOA stimulation (fig. S2, C and D).
Fig. 2. SoNar-low B-ALL cells prefer using oxidative phosphorylation as the main energy source.
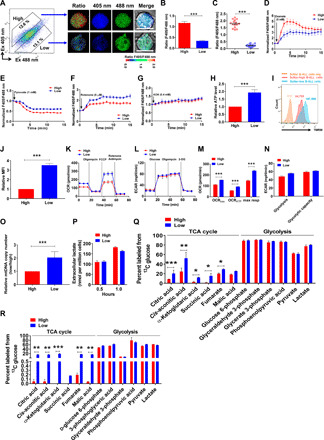
(A to C) FACS-purified SoNar-high and low B-ALL cells were evaluated for the ratio of SoNar fluorescence by confocal microscopy (A). Quantitative data in (A) as determined by either flow cytometric analysis (B) (n = 3) or confocal microscopy (C) (n = 3) are shown. Scale bar, 10 μm. (D to G) SoNar fluorescence ratios were measured in SoNar-high and low B-ALL cells upon treatment with oxamate, pyruvate, rotenone, and AOA. (H to J) ATP levels (H) and mitochondrial membrane potential (I) in SoNar-high and low B-ALL cells were measured by bioluminescence assay and TMRM staining (n = 3). MFI, mean fluorescence intensity. (K and L) OCRs (K) and extracellular acidification rates (ECAR) (L) were measured in SoNar-high and low B-ALL cells (n = 3). 2-DG, 2-deoxyglucose. mpH, mili potential of hydrogen. (M and N) basal respiration, ATP turnover, and maximum respiration in OCR (OCRbas, OCRATP, and max resp, respectively) (K) as well as glycolysis and glycolysis capacity in ECAR (L) were analyzed (n = 3). (O) Mitochondrial DNA (mtDNA) copies were detected in SoNar-high and low B-ALL cells (n = 3). (P) Extracellular lactate levels in SoNar-high and SoNar-low B-ALL cells (n = 3). (Q) The intermediate metabolites derived from glycolysis and TCA cycle were determined in SoNar-high and SoNar-low B-ALL cells after in vitro labeling with 13C-labeled glucose (n = 5). (R) 13C-labeled glucose was used for the in vivo labeling in SoNar B-ALL cells, followed by the measurement of intermediate metabolites derived from glycolysis and TCA cycle in SoNar-high and low leukemia cells (n = 3). Data are represented as means ± SEM. Student’s two-tailed unpaired t test (B, C, H, J, and O) and two-way ANOVA with Sidak’s multiple comparison test (M, N, P, Q, and R) were used for the comparison of statistical significance. *P < 0.05, **P < 0.01, and ***P < 0.001.
The SoNar-low (~10.9%) and SoNar-high (~11.1%) cells were then purified by flow cytometry according to SoNar ratio (fig. S2E) and subjected to several biochemical analyses of their respective metabolic state. Consistently, the SoNar-low cells had much higher levels of adenosine 5′-triphosphate (ATP) (Fig. 2H), greater mitochondrial potential (Fig. 2, I to J), and a higher oxygen consumption rate (OCR), as evidenced by the increase in basal respiration, ATP turnover, and maximum respiration (Fig. 2, K and M), but similar levels of extracellular acidification rate (ECAR) compared to these measures in the SoNar-high cells (Fig. 2, L and N). The SoNar-low cells also contained more mitochondrial DNA (mtDNA) copy numbers than did the SoNar-high cells (Fig. 2O). These results suggest that SoNar-low cells preferred oxidative phosphorylation while sustaining a glycolytic level similar to that of the SoNar-high cells. Consistently, the SoNar-low cells produced similar amounts of extracellular lactate (Fig. 2P).
Moreover, the in vitro metabolic flux analysis with 13C6 glucose (30, 31) showed that the SoNar-low cells had markedly enhanced trichloroacetic acid (TCA) cycle flux compared with that of the SoNar-high cells (Fig. 2Q). We also performed in vivo 13C6-labeling experiments (14) and demonstrated that many TCA intermediates were markedly increased in SoNar-low cells (Fig. 2R). The SoNar-low cells had a level of glycolysis similar to that of the SoNar-high cells as evaluated by both in vitro and in vivo metabolic flux assay (Fig. 2, Q to R), a finding that differs from the traditional notion that high oxidative phosphorylation is usually accompanied by a relatively low glycolytic level. In addition, SoNar-low cells had much higher level of both fatty acyl carnitines and fatty acid oxidation than that of SoNar-high cells, indicating that SoNar-low cells may also use fatty acid as an alternative resource of energy that depends on the TCA pathway (fig. S2, F and G) (32). It also seemed that glucose metabolism had the major contribution to the increase in oxidative phosphorylation level in SoNar-low cells as compared to the fatty acid or glutamine metabolism (fig. S2G). Consistently, SoNar-low cells had much higher levels of CD36, as a fatty acid translocase, than that of SoNar-high cells (fig. S2, H and I).
The SoNar-low B-ALL cell population may be enriched with more LICs with high leukemogenic activities than that of SoNar-high ones and resides in the vascular niche in the BM
To determine whether SoNar can serve as a functional metabolic indicator for LICs, we costained SoNar-low and SoNar-high cells with a potential LIC or leukemia precursor marker, CD43 (33), which showed much a higher percentage of CD43+ cells in the SoNar-low cells than that in the SoNar-high cells (25.4% versus 43.5%; Fig. 3, A and B). This finding is consistent with the observation of immunofluorescence staining showing an approximately threefold increase in the percentage of SoNar-low cells in the CD43+ B-ALL cells compared to that in their CD43− counterparts (30.1% versus 11.4%; Fig. 3, C and D). Notably, we demonstrated that the mice that received the SoNar-low cells underwent much faster leukemogenesis than did their counterparts, as displayed by much higher mCherry+ B-ALL cells in their peripheral blood (14.4% versus 2.2%; Fig. 3, E and F, and fig. S3A) and significantly reduced overall survival (26.0 days versus 43.5 days; Fig. 3G). Concomitantly, the percentage of B220+CD43+ immunophenotypic LICs in the BM of the recipients of the SoNar-low cells was much higher than that in the mice receiving SoNar-high cells (fig. S3, B and C). In contrast, the B220+IgM+ differentiated cells were much less in SoNar-low cells (fig. S3, B and C). More extensive infiltration was also observed in the recipients transplanted with SoNar-low cells, as evidenced by the increase in the relative weights of their spleens and livers (fig. S3, D and E) and the extent of hematoxylin and eosin staining (fig. S3F). However, the difference in engraftment may not have been due to changes in homing efficiency (fig. S3G).
Fig. 3. The SoNar-low B-ALL cell population is enriched with functional LICs and resides in the vascular niche in the BM.
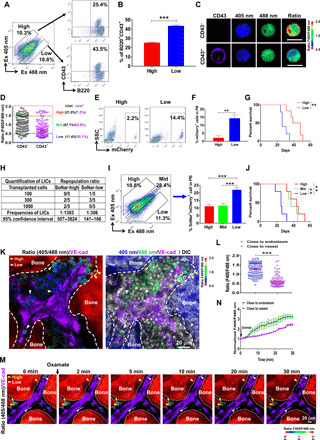
(A and B) The frequencies of B220+CD43+ cells evaluated by flow cytometry in SoNar-high and SoNar-low B-ALL cells are shown (B) (n = 3). This experiment was repeated independently three times. (C and D) The SoNar fluorescence ratios in CD43+/− B-ALL cells were determined by microscopy. A total of 94 to 95 SoNar B-ALL cells were counted (n = 3). Scale bars, 10 μm. (E to G) The mCherry+ B-ALL cell frequencies in the peripheral blood (PB) (E and F) (3 weeks) and overall survival (G) (log-rank test) of the recipient mice transplanted SoNar-high and SoNar-low cells are shown (n = 5). SSC, side scatter. (H) Functional LIC frequencies of primary B-ALL cells were determined by the limiting dilution assay using L-Calc software. (I and J) The frequencies of B-ALL cells in the peripheral blood (I) (3 weeks) and overall survival of the recipient mice (J) (log-rank test) receiving SoNar-high, SoNar-mid, and SoNar-low cells were determined (n = 5). (K and L) The B-ALL cell frequencies with indicated fluorescence ratios in the endosteal and vascular niches are shown. A total of 110 SoNar B-ALL cells were counted (n = 3). VE-cad, vascular endothelial cadherin; DIC, differential interference contrast. (M and N) The changes of the SoNar fluorescence ratio in B-ALL cells in the endosteal and vascular niche upon the oxamate stimulation are shown. A total of 46 to 47 SoNar B-ALL cells were counted (n = 3). Data are represented as means ± SEM. Student’s two-tailed unpaired t test (B, F, and L) and one-way ANOVA with Tukey’s multiple comparison test (I) were used for the comparison of statistical significance. **P < 0.01 and ***P < 0.001.
To determine the exact LIC frequencies in SoNar-low and SoNar-high cells, we transplanted 100, 300, and 1000 indicated leukemia cells into the recipients for the measurement of the functional LIC numbers according to the overall survival in each group (Fig. 3, E to G). As shown in Fig. 3H, the LIC frequency was ~1 in 306 SoNar-low cells, which was ~4.5-fold greater than that of the SoNar-high cells (1 in 1393). Furthermore, the SoNar-mid cells were also isolated and evaluated for the percentage of functional LICs, and the results showed that SoNar-mid cells had leukemogenic activity levels similar to those of the SoNar-high cells but lower than those of the SoNar-low cells, as evidenced by leukemia cell frequencies and overall survival (Fig. 3, I to J). These results demonstrated that SoNar-low cells may contain more LICs with high leukemogenic activities than that of SoNar-mid or SoNar-high cells and may use oxidative phosphorylation as their main energy source. Moreover, SoNar-low immunophenotypic B220+CD43+IgM−IgD− LICs also had much higher leukemogenic activities than that of the control ones as evidenced by the increased leukemia cell frequencies in the peripheral blood and reduced overall survival (fig. S3, H to I).
To unravel the potential connections between the oxidative phosphorylation status of B-ALL cells and their localization in the BM niche, we analyzed the ratios of SoNar fluorescence of the B-ALL cells in the BM niche. Most of the B-ALL cells with a low ratio of SoNar fluorescence were located close to the vascular niche, as indicated by anti–vascular endothelial cadherin antibodies labels, while the SoNar-high cells resided far from the vessels (Fig. 3, K and L). To directly monitor the dynamics of the metabolic status of B-ALL cells in the BM niche, we developed a surrogate technique to reflect the changes in SoNar fluorescence ratios: Scraping of the bony substance (see Materials and Methods) was followed by stimulation with oxamate in vitro. As shown in Fig. 3 (M and N) and movie S2, the SoNar-low cells were directly associated with vascular endothelial cells, and the increase in SoNar fluorescence ratios was only ~50% that of the SoNar-high cells over time. These findings indicate a potential and powerful tool for determining the metabolic profiles of B-ALL cells in the BM niche in vivo at the single-cell level with/without exogenous stimulation.
Because human B-ALL samples are usually accompanied with a BCR-ABL (Breakpoint Cluster Region-Tyrosine-Protein Kinase ABL1) mutant, we then examined the metabolic features of B-ALL cells in a BCR-ABL–induced murine B-ALL model, which showed that there also existed SoNar-low, SoNar-mid, and SoNar-high three populations (fig. S3, J and K). SoNar-low cells had much higher leukemogenic activities compared to that of the SoNar-high ones as evidenced by the increased leukemia cell frequencies in the peripheral blood, reduced overall survival (fig. S3, L and M), and the preference to residing in the vascular niche (fig. S3, N and O). Similarly, SoNar-low immunophenotypic B220+CD43+IgM−IgD− LICs from BCR-ABL–induced murine B-ALL model also had much higher leukemogenic activities than that of the SoNar-high ones (fig. S3, P and Q). Therefore, it is possible that some types of B-ALL driven by different gene mutants share similar metabolic features.
Pyruvate dehydrogenase complex component X promotes the oxidative phosphorylation of the SoNar-low B-ALL cells and enhances leukemogenesis
To gain better understanding of the molecular mechanisms that control the metabolic status and leukemogenic activity levels of the SoNar-low cells, we performed a microarray analysis using SoNar-low and SoNar-high cells and demonstrated that approximately 1497 and 3900 genes were up- and down-regulated, respectively (fig. S4A). A Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed that the genes of several metabolism-related pathways [calcium signaling pathway, Wnt signaling pathway, ErbB (Erythroblastic Leukemia Viral Oncogene Homolog) signaling pathway, and oxidative phosphorylation] were notably up-regulated in SoNar-low cells (Fig. 4A). Regarding the distinct features of glycolysis and oxidative phosphorylation between these two populations, we first analyzed the changes in several genes related to glucose, fatty acid, and glutamine metabolism (Fig. 4B). Quantitative RT-PCR revealed that several genes related to oxidative phosphorylation (Pdk2, Pdhe1a, Pdhx, Kgdh, Sdhb, Idh2, Mdh2, ND2, ND3, Cox6b1, Atp5l, and Atp6) and fatty acid metabolism (Cpt1a, Cpt2, Gcdh, Acat1, and Acadvl), but not glycolysis- or glutamine-related genes (Ldha, Pfkl, Pkm2, and Glut1 for glycolysis and Gls, Gss, and Glud1 for glutamine metabolism), were notably increased in the SoNar-low cells (Fig. 4B). Consistent with the enhanced protein level in the reported leukemia precursor marker CD43 (Fig. 3, A to C), the mRNA level of CD43 was also significantly increased in the SoNar-low cells than that of SoNar-high ones (Fig. 4B). One of the candidates among all the tested genes is important for oxidative phosphorylation, Pdhx, which was expressed at the highest level in the SoNar-low cells (Fig. 4B). PDHX has been reported to be the key component of the pyruvate dehydrogenase (PDH) complex to enhance PDH activity levels and facilitate the entry of pyruvate into the TCA cycle (34). These hints prompted us to ask whether PDHX is one of the main regulators that enhance oxidative phosphorylation utilization in SoNar-low cells.
Fig. 4. PDHX promotes the oxidative phosphorylation of the SoNar-low B-ALL cells and enhances leukemogenesis.
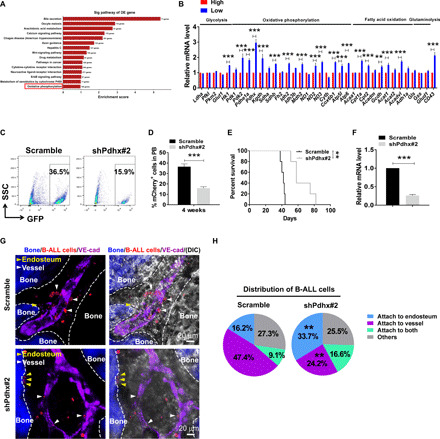
(A) KEGG analyses of pathways up-regulated in the SoNar-low B-ALL cells according to the microarray data. The candidate pathway of oxidative phosphorylation is highlighted in red. (B) Quantitative RT-PCR analysis of the expression levels of glycolysis, oxidative phosphorylation, fatty acid oxidation, and glutaminolysis related genes in SoNar-high and SoNar-low B-ALL cells (n = 3). (C to F) The GFP+ (green fluorescent protein–positive) B-ALL cell frequencies in the peripheral blood (C and D) (3 weeks after transplantation) and overall survival (E) of the recipients receiving the scrambled or Pdhx-knockdown (shPdhx#2) B-ALL cells (n = 5 mice per group, log-rank test). Relative mRNA expression of Pdhx in the scrambled and Pdhx-knockdown B-ALL cells is shown (F). (G and H) Representative images of the localization of the scrambled and Pdhx-knockdown B-ALL cells (mCherry+) in the endosteal (dashed lines) and vascular niche (vascular endothelial cadherin, purple) (G). Quantitative data of the leukemia cell frequencies attached to the endosteum and the vessel in (G) (H). Data are represented as means ± SEM. Student’s two-tailed unpaired t test (D and F) and two-way ANOVA with Sidak’s multiple comparison test (B and H) were used for the comparison of statistical significance. **P < 0.01 and ***P < 0.001.
To evaluate the PDHX function in leukemogenesis, we constructed several short hairpin RNAs (shRNAs) and evaluated their knockdown efficiency in L1210 cells, a mouse ALL cell line (fig. S4B). As shown in Fig. 4 (C to F), the recipient mice receiving N-myc+ Pdhx-knockdown B-ALL cells exhibited notably reduced leukemia development compared with that of the scrambled mice, as exhibited by the much lower frequency of leukemia cells in the peripheral blood (Fig. 4, C and D, and fig. S4C) and markedly extended survival upon transplantation (Fig. 4, E and F). Concomitantly, a much higher percentage of Pdhx-knockdown B-ALL cells than scrambled cells tended to reside in the endosteal niche (Fig. 4, G and H). Pdhx-knockdown B-ALL cells also had much lower levels of ATP and OCR but similar levels of ECAR (fig. S4, D to F). We also examined the knockdown efficiency in B-ALL cells and demonstrated that the PDHX protein level was reduced to ~18.9% of the wild-type (WT) one upon Pdhx knockdown (fig. S4G). These results indicate that PDHX may be indispensable for the maintenance of oxidative phosphorylation status, localization in the vascular niche, and leukemogenic activity of the SoNar-low cells.
PDHX was also down-regulated in a human B-ALL cell line, Nalm6, in which knocking down PDHX resulted in a significant decrease in proliferation in vitro (fig. S4, H and I). PDHX-knockdown Nalm6 cells contributed to the much lower leukemia cell frequencies in the peripheral blood and the markedly extended overall survival of the recipient mice (fig. S4, J to L). Consistently, the SoNar ratio was significantly increased in the PDHX-knockdown leukemia cells compared with that in the scrambled controls (fig. S4M). PDHX-knockdown Nalm6 cells responded more sensitively to oxamate treatment but less sensitively to rotenone stimulation (fig. S4, N to Q).
SoNar-low B-ALL cells are resistant to Ara-C treatment as regulated by cytidine deaminase pathways
To gain greater understanding of whether mitochondrial respiration is connected to the drug resistance of B-ALL cells, we treated SoNar-ALL cells with Ara-C in vivo for 2 days and found that the Ara-C–treated SoNar-ALL cells had a substantially decreased SoNar ratio (Fig. 5, A and B). The Ara-C–treated cells also had a ~3.6-fold increase in SoNar fluorescence ratio upon 5 min of rotenone stimulation, while there was only a 1.4-fold increase in SoNar fluorescence upon oxamate treatment. The slight or no changes were observed after pyruvate or AOA treatment (Fig. 5, C to F). Ara-C treatment also led to a significant increase in the mitochondrial potential in the B-ALL cells (Fig. 5G). These results indicate that mitochondrial respiration plays key roles in Ara-C–treated B-ALL cells. Ara-C–treated SoNar-expressing B-ALL cells mainly resided in the vascular niche (Fig. 5, H to J) and responded to a lesser extent to oxamate supplementation in vivo (Fig. 5, K and L, and movies S3 and S4). Treatment with Ara-C for 4 days also led to a marked reduction in the ratio of SoNar fluorescence (fig. S5A) and induced increased sensitivity in response to rotenone but not oxamate or pyruvate (fig. S5, B to E). In vivo Ara-C treatment for 4 days also led to a notable decrease in the SoNar ratio (fig. S5F) and an increase in the cell frequency of B-ALL cells in the vascular niche (fig. S5, G to H).
Fig. 5. SoNar-low B-ALL cells are resistant to Ara-C treatment as regulated by CDA pathways.
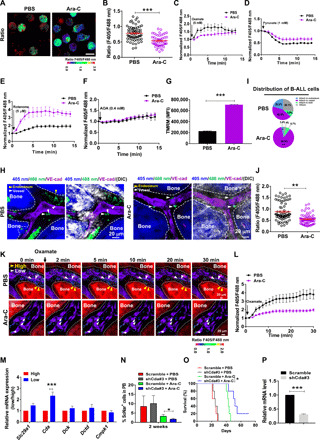
(A and B) The quantitation of fluorescence ratio in the SoNar B-ALL cells from the leukemia mice after 2-day Ara-C treatment. A total of 50 cells were counted (n = 3). Total BM leukemia cell numbers in each control or Ara-C–treated recipient mouse were 4.40 × 107 or 2.03 × 107, respectively. Scale bar, 10 μm. (C to F) The SoNar fluorescence ratio in the SoNar B-ALL cells in (A) was analyzed upon the stimulation with oxamate, pyruvate, rotenone, and AOA. (n = 3). (G) Mitochondrial membrane potential in Ara-C–treated SoNar B-ALL cells (n = 3). (H to J) The leukemia cell frequencies (H and I) and SoNar ratio (J) attached to the endosteum or vessel in the leukemic mice after 2-day Ara-C treatment. A total of 71 to 73 cells were counted (I and J) (n = 3). (K and L) The changes of SoNar ratio in SoNar B-ALL cells upon oxamate stimulation in (H) A total of 26 to 27 cells were counted (L) (n = 3). (M) The mRNA levels in genes related to the Ara-C transportation (Slc29a1) and catabolism (Cda, Dck, Dctd, and Cmpk1) in SoNar-high/SoNar-low cells (n = 3). (N to P) Percentages of the scrambled and Cda-knockdown TIB205 cells in the peripheral blood (N) and overall survival (O) of the recipient mice treated with/without Ara-C (n = 5 mice per group, log-rank test). Relative mRNA levels of Cda in (O) are shown (P) (n = 3). Data are represented as means ± SEM. Student’s two-tailed unpaired t test (B, G, J, and P), one-way ANOVA with Tukey’s multiple comparison test (N), and two-way ANOVA with Sidak’s multiple comparison test (I and M) were used for the comparison of statistical significance. *P < 0.05, **P < 0.01, and ***P < 0.001.
We then wondered about the underlying mechanisms that are required for the drug resistance to Ara-C. Several potential candidates related to the Ara-C transportation (Slc29a1) and catabolism (Cda, Dck, Dctd, and Cmpk1) were examined in SoNar-low and SoNar-high cells. One of these genes, Cda, which can convert Ara-C into uracil arabinoside (Ara-U) to decrease its leukemia cell killing and lead to drug resistance (35), was highly up-regulated in the SoNar-low cells (Fig. 5M). Moreover, SoNar-low cells had a much lower sensitivity in the SoNar fluorescence ratio changes upon Ara-C treatment in vitro than that in SoNar-high cells (fig. S5, I and J), indicating that SoNar-low cells may be more resistant to the Ara-C treatment. We had further determined the proliferation changes in SoNar-low and SoNar-high cells upon the Ara-C treatment in vitro at indicated time points and found that there were much less viable B-ALL cells in SoNar-high group compared to that of the SoNar-low ones (fig. S5K), indicating that Ara-C could kill more SoNar-high cells. Moreover, upon in vivo Ara-C treatment, the recipient mice transplanted with SoNar-low cells had approximately 14-day extended duration of survival normalized to that of the phosphate-buffered saline (PBS) treatment, which is shorter than that from the recipient mice transplanted with SoNar-high cells with 18-day extension in overall survival (fig. S5, L and M).
Consistently, the 2- or 4-day Ara-C treatment in vivo also led to a notable increase in Cda mRNA levels in the B-ALL cells, as determined by quantitative RT-PCR (fig. S5N). These data indicated that cytidine deaminase (CDA) may be a potential target for Ara-C–induced resistance in B-ALL cells. Cda was silenced in the TIB205 murine B-ALL cell line by shRNAs (fig. S5O), and then the cells were treated with Ara-C. Although knocking down Cda did not affect the proliferation of the TIB205 cells (fig. S5P), there was a significant increase in the ratio of SoNar fluorescence (fig. S5Q). After Ara-C treatment, in recipients receiving Cda-knockdown TIB205 cells, leukemia development was significantly slowed, as displayed by the reduced leukemia cell frequencies in the peripheral blood (Fig. 5N) and extended overall survival compared to their counterparts (Fig. 5O). The knockdown efficiency of Cda was further confirmed in the TIB205 cells from recipients (Fig. 5P). These findings support our notion that the sensitivities of Ara-C–induced drug resistance are mediated via CDA pathways.
Ara-C–resistant human B-ALL cell line has a higher level of oxidative phosphorylation
To test whether Ara-C–resistant human and murine B-ALL cell lines have similar metabolic properties, we analyzed the ratio change of SoNar fluorescence in the Nalm6 cells, which have been used for drug resistance in a previous study (36). As shown in fig. S6 (A to D), a 4- or 30-day treatment of Nalm6 cells with Ara-C led to a ~50% decrease in the SoNar ratio compared to that of the untreated controls. After 4 days of treatment with Ara-C in vivo, the SoNar-low Nalm6 cells resided more closely to the vascular endothelial cells in the BM niche compared to the control cells (fig. S6, E and F), which had a much lower ratio of SoNar fluorescence (fig. S6G), and were less sensitive to oxamate stimulation in the BM niche (fig. S6, H to I, and movies S5 and S6). Consistently, CDA expression was notably up-regulated in the Nalm6 cells after Ara-C treatment both in vitro and in vivo (fig. S6, J and K). Knocking down CDA (fig. S6, L and M) in the Nalm6 cells also resulted in a 1.3-fold increase in the SoNar ratio (fig. S6N), but had no effect on their survival (fig. S6O). Meanwhile, CDA was knocked down in two other B-ALL cell lines, Sup-B15 (with a BCR-ABL fusion gene) and REH (ETS Variant Transcription Factor 6-RUNX Family Transcription Factor 1) (with an ETV6-RUNX1 fusion gene). Consistently, the SoNar ratios were notably increased in both cell lines upon CDA knockdown but with no difference in overall survival (fig. S6, P and U). These results indicate that CDA also plays a role in the Ara-C resistance in human B-ALL cells.
Cyclic adenosine 3’,5’-monophosphate response element-binding protein transactivates the expression of PDHX and CDA to sustain leukemogenic activity and chemotherapeutic resistance
To gain a better understanding of PDHX and CDA fine-tuning in SoNar-low cells, we screened several signaling pathways that were up-regulated in SoNar-low cells according to the microarray data, such as the calcium signaling pathway and Wnt signaling pathway (Fig. 4A). We found that the phosphorylation level of cyclic adenosine 3’,5’-monophosphate response element-binding protein (CREB) was significantly up-regulated to ~1.7- to 3.8-fold (Fig. 6A). Consistently, calcium flux (both constitutive and ionomycin induced) was significantly increased in the SoNar-low cells compared with that in the SoNar-high cells (fig. S7, A and B). WT CREB efficiently transactivated Pdhx and Cda in a dose-dependent manner (Fig. 6, B and C), but the Creb mutant (S133A) did not enhance the transcription levels of Pdhx or Cda. Consistent with the luciferase assay results, using a chromatin immunoprecipitation (ChIP) assay, we confirmed that CREB could bind to the promoters of both Pdhx and Cda upon overexpression of the WT or mutant CREB in 293T cells (Fig. 6, D and E). We have further performed the ChIP assay on the endogenous CREB and found that it also bound to these two promoters (fig. S7, C and D). Knocking down Creb in B-ALL cells led to a marked delay in leukemia development, as evidenced by a decrease in leukemia cell frequencies in the peripheral blood and extended survival (Fig. 6, F and G). The knockdown efficiency in the primary B-ALL cells was evaluated by Western blotting (Fig. 6H). Consistently, the PDHX and CDA levels were also notably decreased in CREB knockdown (Fig. 6H). Creb-knockdown B-ALL cells tended to localize in the endosteal niche but not the vascular niche (Fig. 6, I and J), indicating that metabolic status is critical for the localization of B-ALL cells.
Fig. 6. CREB transactivates the expression of PDHX and CDA to sustain leukemogenic activity and chemotherapeutic resistance.
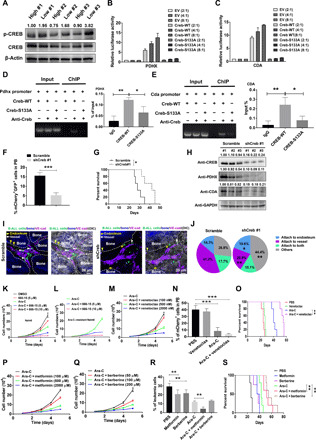
(A) CREB and p-CREB protein levels were measured in SoNar-high/SoNar-low B-ALL cells. (B and C) The luciferase activities were measured in 293T cells coexpressing with Pdhx (B) and Cda (C) luciferase reporter and WT or inactive form of Creb (Creb-S133A; n = 3). EV, empty vector. (D and E) ChIP assays were analyzed with 293T cells expressing WT or mutant Creb. The amplification of the CREB-binding sequence of Pdhx (D) and Cda (E) was presented as a percentage of input DNA (n = 3). (F and G) The mCherry+GFP+ cell frequencies in the peripheral blood (F) and overall survival (G) were determined in the recipient mice transplanted with the scrambled and Cda-knockdown B-ALL cells (n = 5 per group). (H) CREB protein levels of the scrambled and Creb-knockdown B-ALL cells in (G) were measured. Ratios of CREB/actin, PDHX/actin, and CDA/actin were normalized against the scrambled #1. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (I and J) The frequencies of the scrambled and Creb-knockdown B-ALL cells in the endosteal and vascular niches are shown. (K to M, P, and Q) The proliferation rates were evaluated with Nalm6 cells (K) or Ara-C–resistant Nalm6 cells (L, M, P, and Q) upon the Ara-C treatment with/without CREB inhibitor (666-15) (L), venetoclax (M), metformin (P), and berberine (Q) (n = 3). DMSO, dimethyl sulfoxide. (N, O, R, and S) The B-ALL cell frequencies (N and R) and overall survival (O and S) were determined in the leukemia mice upon the Ara-C treatment with/without venetoclax (O), metformin (S), and berberine (S) (n = 5 mice per group). Data are represented as means ± SEM. Student’s two-tailed unpaired t test (F), one-way ANOVA with Tukey’s multiple comparison test (B, C, D, E, N, and R), and two-way ANOVA with Sidak’s multiple comparison test (J) were used for the comparison of statistical significance. *P < 0.05, **P < 0.01, and ***P < 0.001.
To test the function of CREB in drug resistance, we used a potent and selective CREB inhibitor (666-15) to inhibit CREB-mediated gene transcription activity, which has been reported to be able to efficiently suppress the phosphorylation of CREB at S133 site (fig. S7E) (37, 38) and demonstrated that 666-15 could efficiently suppress the transcriptional activities of both Pdhx and Cda in a dose-dependent manner (fig. S7, F and G). Specifically, the 666-15 treatment efficiently inhibited both nondrug-resistant and drug-resistant Nalm6 cell growth in vitro and had a synergistic effect with Ara-C treatment (Fig. 6, K and L). In contrast, the 666-15 treatment had no effect on the cell proliferation of human cord blood CD34+ cells (fig. S7H). Ara-C treatment also resulted in an up-regulation of p-CREB in either Nalm6 cells or murine primary B-ALL cells (fig. S7, I and J), indicating that there is a positive feedback loop to enhance Ara-C–induced resistance.
To test whether directly targeting oxidative phosphorylation was efficient to overcome Ara-C–induced drug resistance, we treated the B-ALL cells with several mitochondrial metabolism inhibiting drugs, including venetoclax, metformin, and berberine, which have been shown to efficiently suppress mitochondrial respiratory function and have been used in clinical trials (venetoclax) or in the clinic (metformin and berberine) (39–41). We first treated Nalm6 cells with these three drugs and found that the ATP levels (fig. S7K), mitochondrial potential values (fig. S7L), and OCRs (fig. S7, M to O) were indeed significantly reduced upon treatment. Although venetoclax alone had some effect on the proliferation of the Ara-C–resistant Nalm6 cells in vitro at high doses (Fig. 6M), only the in vivo combination treatment of Ara-C with venetoclax (but not venetoclax alone) in the N-myc–induced murine B-ALL model led to a notable reduction in the proliferation of the Ara-C–resistant B-ALL cells (Fig. 6, N and O). Moreover, in vitro treatment with metformin or berberine alone also markedly suppressed the proliferation of the Ara-C–resistant Nalm6 cells (Fig. 6, P and Q). Notably, the combination treatment of Ara-C with metformin or berberine also efficiently inhibited the proliferation of Ara-C–resistant B-ALL cells and reduced the overall survival of the recipient mice (Fig. 6, R and S), which may open a unique avenue for targeting chemotherapeutic resistance.
Human primary B-ALL cells have higher levels of oxidative phosphorylation and reside in the vascular niche
To evaluate the metabolic status of human leukemia cells, we ectopically expressed SoNar in human primary B-ALL cells and transplanted the cells into recipient mice. About 10.4 and 17.7% of the human B-ALL cells were in the SoNar-low and high cell populations, respectively (Fig. 7, A and B). SoNar-low cells were less sensitive to oxamate treatment but highly responsive to rotenone treatment (Fig. 7, C to F). Similar to the results in mouse B-ALL cells, the slight or no ratio changes in SoNar fluorescence were observed in the SoNar-low cells upon pyruvate or AOA stimulation (Fig. 7, D and F). The SoNar-low cells had much higher ATP levels and OCRs but similar levels of ECARs compared to the SoNar-high cells (fig. S8, A and C). CD34+CD19+ leukemia precursor cells had much lower SoNar ratio than did the more differentiated CD34−CD19+ cells (Fig. 7, G and H). The SoNar-low cells had much greater leukemogenic activity than did the SoNar-high cells, as exhibited by higher leukemia cell frequencies in the peripheral blood and reduced overall survival (Fig. 7, I to M, and fig. S8, D and E). The SoNar-low cells tended to reside in the vascular niche and had low SoNar fluorescence ratio (Fig. 7, N-P). The treatment with Ara-C in vivo also resulted in a decrease in the SoNar ratio (Fig. 7Q) and less responsiveness to oxamate treatment (Fig. 7, R and S, and movies S7 and S8), findings consistent with the high expression level of CDA upon Ara-C treatment (fig. S8F). In vivo Ara-C treatment led to an increase in the frequency of the B-ALL cells residing in the vascular endothelial niche (fig. S8, G and H). In summary, a working model (Fig. 7T) was depicted and showed that the SoNar-low B-ALL cells had a much higher level of oxidative phosphorylation and tended to localize in the vascular niche, which may be enriched with LICs with high leukemogenic activities, and resistant to Ara-C treatment. CREB transactivated PDHX and CDA to maintain the metabolic status of the B-ALL cells, which was tightly connected with their proliferation capacity and chemotherapeutic resistance.
Fig. 7. Human primary B-ALL cells have higher levels of oxidative phosphorylation and reside in the vascular niche.
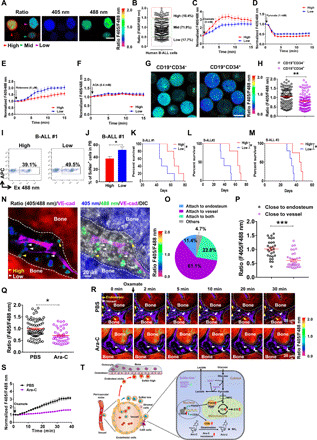
(A and B) The SoNar fluorescence ratio was determined in SoNar+ human primary B-ALL cells by confocal microscopy (A). A total of 222 cells were examined (B). Scale bar, 10 μm. (C to F) The SoNar ratio in human SoNar-high/SoNar-low B-ALL cells was evaluated upon stimulation with oxamate, pyruvate, rotenone, and AOA. A total of 25 SoNar-high and SoNar-low cells were measured (n = 3). (G and H) The fluorescence ratio was analyzed in SoNar+CD19+CD34−/SoNar+CD19+CD34+ human B-ALL cells (105 to 108 cells were examined; n = 3). Scale bar, 10 μm. (I to M) Shown are the frequencies in the peripheral blood (I and J) (AML#1) and overall survival (K to M) (AML#1 to AML#3) of nonobese diabetic–severe combined immunodeficient (NOD-SCID) mice receiving SoNar-high/SoNar-low human primary B-ALL cells (n = 5 mice per group). (N to P) Shown are the frequencies (N and O) and SoNar fluorescence ratio (P) of SoNar+ human B-ALL cells in the endosteal and vascular niches (28 to 36 cells were examined; n = 3). (Q) Shown is the SoNar ratio in human B-ALL cells after a 2-day Ara-C treatment (42 to 68 cells were examined; n = 3). Total BM leukemia cell numbers in each control or Ara-C–treated recipient mouse were 2.17 × 107 or 1.03 × 107, respectively. (R and S) Shown are the SoNar ratio changes in human B-ALL cells upon the oxamate stimulation in the BM of the NOD-SCID mice after a 2-day Ara-C treatment (26 cells were examined; n = 3). MPC, mitochondrial pyruvate carrier; ETC, electron transport chain. (T) Working model for the connections between B-ALL cell fates and metabolic status based on the SoNar sensor. Data are represented as means ± SEM. Student’s two-tailed unpaired t test (H, J, P, and Q) was used for the comparison of statistical significance. *P < 0.05, **P < 0.01, and ***P < 0.001.
DISCUSSION
Currently, the Warburg effect has been found in many types of solid cancers and some hematological disorders (such as AML) (27, 42, 43). However, increasing evidence also shows that mitochondrial respiration seems to be required or even critical for AML development (22, 44). These seemingly contradictory results suggest the possibility that leukemia cells may need both glycolysis and oxidative phosphorylation to sustain growth. We speculate that bulk leukemia cells are very heterogeneous and that they may be enriched with different cell subpopulations with different metabolic preferences. In addition, it is also possible that different leukemia cell types have different metabolic profiles (45). Consistently, here, we show that B-ALL cells heavily depended on oxidative phosphorylation to maintain their cell fate commitments, although the mechanisms by which the different leukemia cell types exhibit their unique metabolic features require further exploration. One potential way to solve this puzzle is to delineate the metabolic map by single-cell sequencing at various levels of mRNA, DNA, and/or epigenetic modification. It will be definitely helpful when proteomics or metabonomics analysis can be conducted at the single-cell level, although these technologies are still not fully available or are currently under development (46, 47). Although some studies have also reported that human B-ALL–LICs are enriched with CD34+CD38+CD19+ cells (48), it is still debatable whether B-ALL–LICs exist due to the lack of suitable immunophenotypic surface markers or other signatures that can be used to functionally determine leukemogenic activity (49). Here, our data reveal that functional B-ALL–LICs may be highly enriched in the SoNar-low cells (with 4.6-fold greater frequency than the SoNar-high cells). Moreover, we noticed that even in the same cell cycle phase, there still existed three distinct SoNar-low, SoNar-mid, and SoNar-high cell populations (fig. S9, A to C) and SoNar-low cells had much higher leukemogenic activities than that of the SoNar-high ones (fig. S9, D and E). These data support the notion that metabolic signatures may serve as unique markers for determining the level to which the LICs may be enriched in a cell population.
Increasing evidence has shown that abnormal metabolic signaling can also serve as the main triggers for leukemia initiation and progression. For example, IDH1/2 mutations can lead to the occurrence of AML or glioma (50, 51). However, how metabolic status determines a cancer cell fate remains controversial (52). Our studies may provide intriguing evidence showing that the oxidative phosphorylation level determines the fate of B-ALL cells. This supposition is consistent with previous studies from other groups, showing that intrinsic metabolic states are critical for leukemogenesis (51). In addition, although many reports demonstrate that different types of LICs reside in specific BM niches (53, 54), very rare studies have shown the precise niche of B-ALL–LICs (21, 55). Previous work has reported that B-ALL cells may reside in the vascular niche (56). Consistently, we, herein, also provide the data showing that SoNar-low cells are mainly located in the vascular niche, a finding that differs from that of AML-LICs and CML (chronic myeloid leukemia)–LICs, which preferentially reside in the endosteal niche.
In addition, we noticed that metabolic status was important for Ara-C resistance, as indicated by residual B-ALL cells having much higher levels of oxidative phosphorylation. The high levels of oxidative phosphorylation of B-ALL cells may fueled by both glucose metabolism and an alternative metabolic pathway of fatty acid oxidation upon chemotherapy as evidenced by increased level of fatty acyl carnitines (fig. S10A), enhanced OCR resulting from fatty acid oxidation (fig. S10B). It also seemed that glucose metabolism had the major contribution to the increase in oxidative phosphorylation level in Ara-C–resistant cells as compared to the fatty acid or glutamine metabolisms (fig. S10B). Consistently, several critical genes related to fatty acid oxidation, but not glutamine metabolism, were marked up-regulated in Ara-C–resistant cells (fig. S10C). Suppressing oxidative phosphorylation with several drugs (such as metformin, venetoclax, and berberine) enabled a markedly reversal of Ara-C–induced drug resistance both in vitro and in vivo.
Currently, the precise depiction of the metabolic dynamics of leukemia cells or LICs both in vitro and in vivo remains challenging. Most current studies mainly use certain types of dyes or chemical derivatives to evaluate the metabolic status of bulk leukemia cells or LICs (57), strategies with many drawbacks, such as insufficient permeabilization, limited retention time in cells, and severe cytotoxicity. In a previous study, we used a genetically encoded SoNar to show that AML-LICs mainly use glycolysis as the energy source (27). We herein further developed SoNar transgenic mice to determine the metabolic dynamics of B-ALL cells or LICs both in vitro and in vivo at the single-cell level using real-time imaging. Leukemia cells established with hematopoietic cells from transgenic mice had a much greater range of SoNar level than did that with plasmid-based SoNar. Moreover, it may be more physiologically relevant to use SoNar-transgenic mice than to use strategies based on artificial overexpression in certain types of cells, which may further promote the development of other metabolic sensors for the study of LIC metabolisms.
In summary, using a SoNar-transgenic mouse line, we revealed an unexpected metabolic feature of the B-ALL cells, which showed a preference for oxidative phosphorylation as the main energy source and localization in the BM vascular niche. B-ALL–LICs may be relatively more enriched in the SoNar-low cells, which were more resistant to Ara-C treatment. CREB-mediated PDHX and CDA pathways fine-tune the metabolic properties and drug resistance of B-ALL cells. Combination treatments of Ara-C with one of several oxidative phosphorylation inhibiting drugs sufficiently reversed drug resistance in both the mouse and human B-ALL models. Our studies open a unique avenue to study the metabolism of different types of leukemia cells or LICs. It also provides a unique angle for the development of potential strategies to overcome chemotherapeutic resistance.
MATERIALS AND METHODS
Generation, genotyping, mRNA level, and histology of SoNar transgenic mice
SoNar DNA consists of the sequence of cpYFP (circularly permuted yellow fluorescent protein), truncated T-Rex (78-211), and the linkers between them. To generate SoNar transgenic mice, SoNar DNA was constructed into pCAG vector with chicken β-actin promoter. The targeting construct was linearized, purified, and microinjected into FVB blastocysts. SoNar DNA was randomly incorporated into the genome and determined by the PCR assay. SoNar protein expression was measured by fluorescence microscopy or flow cytometry in the different tissues of SoNar mice. The FVB mice were eventually backcrossed with a C57BL/6 background. Heterozygote transgenic SoNar mice were used for all the experiments in the current study.
For the genotyping analysis of SoNar mice, the DNA of peripheral blood cells was extracted and determined by the PCR assay with specific primers for SoNar genomic DNA (table S1). For the mRNA analysis of SoNar mice, the RNAs of different tissues, including the skeletal muscle, myocardium, liver, spleen, lung, kidney, BM, brain, intestine, testis, and ovary, were extracted and verified by RT-PCR (table S1). For the histology analysis, SoNar mice were sequentially perfused with 4% paraformaldehyde. Different tissues were separated, fixed in 4% paraformaldehyde for 24 hours, and then dehydrated by incubation in 30% sucrose. The fixed tissues were embedded, frozen with liquid nitrogen, and then sliced into sections (50 μm) using a Leica CM1950 clinical cryostat. Fluorescence images were recorded using a Leica TCS SP8 SMD confocal microscope. C57BL/6 CD45.2 mice or nonobese diabetic–severe combined immunodeficient (NOD-SCID) mice at age of 6 to 8 weeks were ordered from the Shanghai Laboratory Animals Center (SLAC). All animal experiments were performed according to the Guidelines for Animal Care at Shanghai Jiao Tong University School of Medicine.
Establishment and analysis of the murine B-ALL model
To establish an N-myc–induced model (28) or BCR-ABL–p190–induced murine B-ALL, the MSCV–N-myc–Internal Ribosome Entry Site-mCherry or murine stem cell virus–BCR-ABL–p190-IRES-mCherry encoding plasmid and the packaging plasmid pCL-ECO (2:1) were used for the generation of retroviruses in 293T cells. Collected murine SoNar+ fetal liver Lin− cells were subsequently infected with collected MSCV–N-myc–IRES-mCherry or MSCV–BCR-ABL–p190-IRES-mCherry retrovirus. Two hundred thousand infected cells were injected into each lethally irradiated C57BL/6 recipient mouse by retroorbital injection. Leukemia development was evaluated by the frequency of mCherry+ cells in the peripheral blood of the recipient mice by flow cytometric analysis at indicated time points after transplantation. SoNar-low and SoNar-high B-ALL cells or immunophenotypic B220+CD43+IgM−IgD− LICs from primary leukemic mice were further purified for the metabolic analysis or transplantation experiments.
For the limiting dilution analysis, the indicated SoNar-low and SoNar-high B-ALL cells were collected from primary BM B-ALL cells and cotransplanted with 200,000 competitor cells into the lethally irradiated recipient mice by retroorbital injection. In some cases, SoNar B-ALL cells were treated with colchicine (30 nM) in vitro for 24 hours, followed by the isolation of SoNar-low and SoNar-high cells and transplantation into the recipient mice. In some drug resistance experiments, SoNar-expressing B-ALL cells were treated with 1 μM Ara-C for 24 hours, followed by the measurement of SoNar ratio changes. To determine LIC frequency of Ara-C–resistant B-ALL cells, SoNar-low and SoNar-high B-ALL cells were transplanted into the recipient mice after in vitro treatment with Ara-C. The overall survival of leukemic mice was recorded for the calculation of LIC frequency using L-Calc software from STEMCELL Technologies.
For the identification of the potential metabolic targets in SoNar-low or SoNar-high cells, lentiviral vector pLKO.1 was used to knock down the expression of murine Pdhx/Cda/Creb or human PDHX/CDA/CREB, followed by the evaluation of their functions in leukemogenesis. Briefly, the pLKO.1 and packaging plasmids of pSPAX2 and pMD2G were cotransfected into 293T cells to produce lentiviruses, which were further used for the subsequent infection in murine N-myc–mCherry+ BM leukemia cells, TIB205 cells [a murine B-ALL cell line, American Type Culture Collection (ATCC)], Nalm6 cells/Sup-B15 cells/REH cells (three human B-ALL cell lines, ATCC), or human primary B-ALL cells. A total of 1 × 104 infected N-myc+ BM leukemia cells, 5 × 106 TIB205 cells, 2 × 106 Nalm6 cells, or 5 × 106 human primary B-ALL cells were used for the transplantation into C57BL/6, BALB/C, or NOD-SCID mice, respectively, followed by the evaluation of the leukemia development as indicated by the leukemia cell frequencies in the peripheral blood at different time points and overall survival of the leukemic mice.
Live cell metabolic imaging
A 35-mm glass-bottom dish (Cellvis) was pretreated with poly-d-lysine hydrobromide (Sigma-Aldrich) and laminin (5 μg/ml) (Gibco) at 4°t for 2 days before imaging. SoNar-expressing leukemia cells were maintained in IMDM (Iscove’s Modified Dulbecco’s Media) (Gibco) supplemented with 2% fetal bovine serum and transferred to a pretreated dish. SoNar signaling was measured using a Nikon A1 confocal microscope with the excitation wavelengths at both 405 and 488 nm and the emission wavelength at 520 nm. To monitor the dynamic ratio changes of SoNar fluorescence upon the metabolic manipulation with different agonists or antagonists, 5 mM oxamate, 1 mM pyruvate, 5 μM rotenone, 0.4 mM AOA, and PBS control were individually added into the culture medium after the first image was acquired. In some cases, SoNar-expressing leukemia cells were sequentially treated with PBS, pyruvate, and oxamate, and vice versa, and SoNar signaling was recorded every minute for a total of 15 to 50 min. To determine the SoNar ratio changes within certain cell cycle phase, SoNar B-ALL cells were treated with colchicine (30 nM) in vitro for 24 hours, followed by the measurement of the ratios of SoNar fluorescence. The SoNar fluorescence ratios and pseudo-color images of SoNar signaling were evaluated by ImageJ software, and videos were exported as well. Usually, the dynamic changes of SoNar fluorescence ratios at indicated time points were normalized to that in the first image.
Study approval
Human primary B-ALL cells were collected from BM aspiration from the B-ALL patients in the Department of Hematology at the First People’s Hospital, Sixth People’s Hospital, or Tongren Hospital, Shanghai Jiao Tong University School of Medicine. Human sample information was listed in table S2. Written informed consent was notified and obtained from all the patients, and the standard procedures were approved by the Ethics Committee for Medical Research (Institutional Review Board) at Shanghai Jiao Tong University School of Medicine.
Statistics analysis
Statistical analysis was performed using GraphPad and SPSS software program, version 19.0. Data are represented as means ± SEM. All the experiments are performed independently at least three times. Data were analyzed with a Student’s t test (two-tailed), one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test, or two-way ANOVA with Sidak’s multiple comparison test according to the experimental design, and statistical significance was set at P < 0.05 (*P < 0.05, **P < 0.01, and ***P < 0.001). Other information related to Materials and Methods is included in the Supplementary Materials in the current version.
Acknowledgments
Funding: This work was supported by grants from National Basic Research Program of China (2019YFA0801800, 2018YFA0107000, and 2019YFA0904800), National Natural Science Foundation of China (NSFC) (81825001, 81570093, 31971052, 81900147, 81461138037, 31722033, 32030065, 31671484, 81873438, and 82000147), the innovative group of NSFC (81721004), Shanghai Science and Technology Commission (19XD1422100, 17ZR1415500, and 20JC1412000), The National Major Scientific and Technological Special Project for “Significant new drugs development” (2018ZX09201002-005), Research Unit of New Techniques for Live-Cell Metabolic Imaging (Chinese Academy of Medical Sciences, 2019RU01, 2019-I2M-5-013), Major Program of Development Fund for Shanghai Zhangjiang National Innovation Demonstration Zone (Stem Cell Strategic Biobank and Stem Cell Clinical Technology Transformation Platform, ZJ2018-ZD-004), innovative research team of high-level local universities in Shanghai. Author contributions: C.C., X.H., X.L., L.L, J. Zhu, J.X., Y.Z., Z.L., and J. Zheng designed the experiments, performed the experiments, analyzed data, and wrote the paper. H.S., D.H., H.G., T.Z., Z.Y., and L.X. performed the experiments. X.Z. and Y.Y. provided reagents and helped with the experiments. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The accession number for microarray data is GSE151106 (GEO datasets). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/11/eabd6280/DC1
REFERENCES AND NOTES
- 1.Park J. H., Riviere I., Gonen M., Wang X., Senechal B., Curran K. J., Sauter C., Wang Y., Santomasso B., Mead E., Roshal M., Maslak P., Davila M., Brentjens R. J., Sadelain M., Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan J., Niu Q., Deng B., Liu S., Wu T., Gao Z., Liu Z., Zhang Y., Qu X., Zhang Y., Liu S., Ling Z., Lin Y., Zhao Y., Song Y., Tan X., Zhang Y., Li Z., Yin Z., Chen B., Yu X., Yan J., Zheng Q., Zhou X., Gao J., Chang A. H., Feng X., Tong C., CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia 33, 2854–2866 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Li N., Wang S. A., Lin P., Jabbour E., Thompson P., Chen Z., Li S., Xu J., You M. J., Bueso-Ramos C. E., Medeiros L. J., Yin C. C., Relapsed B-acute lymphoblastic leukemia with aberrant myeloperoxidase expression following CAR T-cell therapy: A diagnostic challenge. Am. J. Hematol. 94, 1049–1051 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Pan J., Yang J. F., Deng B. P., Zhao X. J., Zhang X., Lin Y. H., Wu Y. N., Deng Z. L., Zhang Y. L., Liu S. H., Wu T., Lu P. H., Lu D. P., Chang A. H., Tong C. R., High efficacy and safety of low-dose CD19-directed CAR-T cell therapy in 51 refractory or relapsed B acute lymphoblastic leukemia patients. Leukemia 31, 2587–2593 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Santomasso B. D., Park J. H., Salloum D., Riviere I., Flynn J., Mead E., Halton E., Wang X., Senechal B., Purdon T., Cross J. R., Liu H., Vachha B., Chen X., DeAngelis L. M., Li D., Bernal Y., Gonen M., Wendel H. G., Sadelain M., Brentjens R. J., Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 8, 958–971 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faderl S., Thomas D. A., O’Brien S., Ravandi F., Garcia-Manero G., Borthakur G., Ferrajoli A., Verstovsek S., Ayoubi M., Rytting M., Feliu J., Kantarjian H. M., Augmented hyper-CVAD based on dose-intensified vincristine, dexamethasone, and asparaginase in adult acute lymphoblastic leukemia salvage therapy. Clin. Lymphoma Myeloma Leuk. 11, 54–59 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Ebinger S., Ozdemir E. Z., Ziegenhain C., Tiedt S., Castro Alves C., Grunert M., Dworzak M., Lutz C., Turati V. A., Enver T., Horny H. P., Sotlar K., Parekh S., Spiekermann K., Hiddemann W., Schepers A., Polzer B., Kirsch S., Hoffmann M., Knapp B., Hasenauer J., Pfeifer H., Panzer-Grumayer R., Enard W., Gires O., Jeremias I., Characterization of rare, dormant, and therapy-resistant cells in acute lymphoblastic leukemia. Cancer Cell 30, 849–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.le Viseur C., Hotfilder M., Bomken S., Wilson K., Rottgers S., Schrauder A., Rosemann A., Irving J., Stam R. W., Shultz L. D., Harbott J., Jurgens H., Schrappe M., Pieters R., Vormoor J., In childhood acute lymphoblastic leukemia, blasts at different stages of immunophenotypic maturation have stem cell properties. Cancer Cell 14, 47–58 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baer S. C., Casaubon L., Younes M., Expression of the human erythrocyte glucose transporter Glut1 in cutaneous neoplasia. J. Am. Acad. Dermatol. 37, 575–577 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Abdel-Wahab O., Levine R. L., Metabolism and the leukemic stem cell. J. Exp. Med. 207, 677–680 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mardis E. R., Ding L., Dooling D. J., Larson D. E., McLellan M. D., Chen K., Koboldt D. C., Fulton R. S., Delehaunty K. D., McGrath S. D., Fulton L. A., Locke D. P., Magrini V. J., Abbott R. M., Vickery T. L., Reed J. S., Robinson J. S., Wylie T., Smith S. M., Carmichael L., Eldred J. M., Harris C. C., Walker J., Peck J. B., Du F., Dukes A. F., Sanderson G. E., Brummett A. M., Clark E., McMichael J. F., Meyer R. J., Schindler J. K., Pohl C. S., Wallis J. W., Shi X., Lin L., Schmidt H., Tang Y., Haipek C., Wiechert M. E., Ivy J. V., Kalicki J., Elliott G., Ries R. E., Payton J. E., Westervelt P., Tomasson M. H., Watson M. A., Baty J., Heath S., Shannon W. D., Nagarajan R., Link D. C., Walter M. J., Graubert T. A., DiPersio J. F., Wilson R. K., Ley T. J., Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y.-H., Israelsen W. J., Lee D., Yu V. W. C., Jeanson N. T., Clish C. B., Cantley L. C., Vander Heiden M. G., Scadden D. T., Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 158, 1309–1323 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao P., Huang J., Zhang D., Zhang D., Wang F., Qu Y., Guo T., Qin Y., Wei J., Niu T., Zheng Y., SLC2A5 overexpression in childhood philadelphia chromosome-positive acute lymphoblastic leukaemia. Br. J. Haematol. 183, 242–250 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Lee W. C., Ji X., Nissim I., Long F., Malic enzyme couples mitochondria with aerobic glycolysis in osteoblasts. Cell Rep. 32, 108108 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Churchman M. L., Qian M., Te Kronnie G., Zhang R., Yang W., Zhang H., Lana T., Tedrick P., Baskin R., Verbist K., Peters J. L., Devidas M., Larsen E., Moore I. M., Gu Z., Qu C., Yoshihara H., Porter S. N., Pruett-Miller S. M., Wu G., Raetz E., Martin P. L., Bowman W. P., Winick N., Mardis E., Fulton R., Stanulla M., Evans W. E., Relling M. V., Pui C.-H., Hunger S. P., Loh M. L., Handgretinger R., Nichols K. E., Yang J. J., Mullighan C. G., Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell 33, 937–948.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thathia S. H., Ferguson S., Gautrey H. E., van Otterdijk S. D., Hili M., Rand V., Moorman A. V., Meyer S., Brown R., Strathdee G., Epigenetic inactivation of TWIST2 in acute lymphoblastic leukemia modulates proliferation, cell survival and chemosensitivity. Haematologica 97, 371–378 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L., O’Leary H., Fortney J., Gibson L. F., Ph+/VE-cadherin+ identifies a stem cell like population of acute lymphoblastic leukemia sustained by bone marrow niche cells. Blood 110, 3334–3344 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burt R., Dey A., Aref S., Aguiar M., Akarca A., Bailey K., Day W., Hooper S., Kirkwood A., Kirschner K., Lee S. W., Lo Celso C., Manji J., Mansour M. R., Marafioti T., Mitchell R. J., Muirhead R. C., Cheuk Yan Ng K., Pospori C., Puccio I., Zuborne-Alapi K., Sahai E., Fielding A. K., Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 134, 1415–1429 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saito Y., Chapple R. H., Lin A., Kitano A., Nakada D., AMPK protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell 17, 585–596 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karjalainen R., Pemovska T., Popa M., Liu M., Javarappa K. K., Majumder M. M., Yadav B., Tamborero D., Tang J., Bychkov D., Kontro M., Parsons A., Suvela M., Mayoral Safont M., Porkka K., Aittokallio T., Kallioniemi O., McCormack E., Gjertsen B. T., Wennerberg K., Knowles J., Heckman C. A., JAK1/2 and BCL2 inhibitors synergize to counteract bone marrow stromal cell-induced protection of AML. Blood 130, 789–802 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Duan C. W., Shi J., Chen J., Wang B., Yu Y. H., Qin X., Zhou X. C., Cai Y. J., Li Z. Q., Zhang F., Yin M. Z., Tao Y., Mi J. Q., Li L. H., Enver T., Chen G. Q., Hong D. L., Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell 25, 778–793 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Lagadinou E. D., Sach A., Callahan K., Rossi R. M., Neering S. J., Minhajuddin M., Ashton J. M., Pei S., Grose V., O’Dwyer K. M., Liesveld J. L., Brookes P. S., Becker M. W., Jordan C. T., BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12, 329–341 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng Y., Lin T.-Y., Lee G., Paddock M. N., Momb J., Cheng Z., Li Q., Fei D. L., Stein B. D., Ramsamooj S., Zhang G., Blenis J., Cantley L. C., Mitochondrial one-carbon pathway supports cytosolic folate integrity in cancer cells. Cell 175, 1546–1560.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zacharias N. M., Baran N., Shanmugavelandy S. S., Lee J., Lujan J. V., Dutta P., Millward S. W., Cai T., Wood C. G., Piwnica-Worms D., Konopleva M., Bhattacharya P. K., Assessing metabolic intervention with a glutaminase inhibitor in real-time by hyperpolarized magnetic resonance in acute myeloid leukemia. Mol. Cancer Ther. 18, 1937–1946 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y., Hu Q., Cheng F., Su N., Wang A., Zou Y., Hu H., Chen X., Zhou H. M., Huang X., Yang K., Zhu Q., Wang X., Yi J., Zhu L., Qian X., Chen L., Tang Y., Loscalzo J., Yang Y., SoNar, a highly responsive NAD+/NADH sensor, allows high-throughput metabolic screening of anti-tumor agents. Cell Metab. 21, 777–789 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Y., Wang A., Zou Y., Su N., Loscalzo J., Yang Y., In vivo monitoring of cellular energy metabolism using SoNar, a highly responsive sensor for NAD+/NADH redox state. Nat. Protoc. 11, 1345–1359 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Hao X., Gu H., Chen C., Huang D., Zhao Y., Xie L., Zou Y., Shu H. S., Zhang Y., He X., Lai X., Zhang X., Zhou B. O., Zhang C. C., Chen G.-Q., Yu Z., Yang Y., Zheng J., Metabolic imaging reveals a unique preference of symmetric cell division and homing of leukemia-initiating cells in an endosteal niche. Cell Metab. 29, 950–965.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Sugihara E., Shimizu T., Kojima K., Onishi N., Kai K., Ishizawa J., Nagata K., Hashimoto N., Honda H., Kanno M., Miwa M., Okada S., Andreeff M., Saya H., Ink4a and Arf are crucial factors in the determination of the cell of origin and the therapeutic sensitivity of Myc-induced mouse lymphoid tumor. Oncogene 31, 2849–2861 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tao R., Zhao Y., Chu H., Wang A., Zhu J., Chen X., Zou Y., Shi M., Liu R., Su N., Du J., Zhou H. M., Zhu L., Qian X., Liu H., Loscalzo J., Yang Y., Genetically encoded fluorescent sensors reveal dynamic regulation of NADPH metabolism. Nat. Methods 14, 720–728 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X., Romero I. L., Litchfield L. M., Lengyel E., Locasale J. W., Metformin targets central carbon metabolism and reveals mitochondrial requirements in human cancers. Cell Metab. 24, 728–739 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nemkov T., D’Alessandro A., Hansen K. C., Three-minute method for amino acid analysis by UHPLC and high-resolution quadrupole orbitrap mass spectrometry. Amino Acids 47, 2345–2357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamaidi I., Zhang L., Kim N., Wang M.-H., Iclozan C., Fang B., Liu M., Koomen J. M., Berglund A. E., Yoder S. J., Yao J., Engelman R. W., Creelan B. C., Conejo-Garcia J. R., Antonia S. J., Mule J. J., Kim S., Sirt2 inhibition enhances metabolic fitness and effector functions of tumor-reactive T cells. Cell Metab. 32, 420–436.e12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeon Y. K., Min H. S., Lee Y. J., Kang B. H., Kim E. J., Park H. J., Bae Y., Lee H. G., Park W. S., Song H. G., Jung K. C., Park S. H., Targeting of a developmentally regulated epitope of CD43 for the treatment of acute leukemia. Cancer Immunol. Immunother. 60, 1697–1706 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eastlack S. C., Dong S., Ivan C., Alahari S. K., Suppression of PDHX by microRNA-27b deregulates cell metabolism and promotes growth in breast cancer. Mol. Cancer 17, 100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahlknecht U., Dransfeld C. L., Bulut N., Kramer M., Thiede C., Ehninger G., Schaich M., SNP analyses in cytarabine metabolizing enzymes in AML patients and their impact on treatment response and patient survival: Identification of CDA SNP C-451T as an independent prognostic parameter for survival. Leukemia 23, 1929–1932 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Consolaro F., Basso G., Ghaem-Magami S., Lam E. W.-F., Viola G., FOXM1 is overexpressed in B-acute lymphoblastic leukemia (B-ALL) and its inhibition sensitizes B-ALL cells to chemotherapeutic drugs. Int. J. Oncol. 47, 1230–1240 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phua T., Sng M. K., Tan E. H. P., Chee D. S. L., Li Y., Wee J. W. K., Teo Z., Chan J. S. K., Lim M. M. K., Tan C. K., Zhu P., Arulampalam V., Tan N. S., Angiopoietin-like 4 mediates colonic inflammation by regulating chemokine transcript stability via tristetraprolin. Sci. Rep. 7, 44351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu J., Dong J., Verzola D., Hruska K., Garibotto G., Hu Z., Mitch W. E., Thomas S. S., Signal regulatory protein alpha initiates cachexia through muscle to adipose tissue crosstalk. J. Cachexia. Sarcopenia Muscle 10, 1210–1227 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diaz-Flores E., Comeaux E. Q., Kim K. L., Melnik E., Beckman K., Davis K. L., Wu K., Akutagawa J., Bridges O., Marino R., Wohlfeil M., Braun B. S., Mullighan C. G., Loh M. L., Bcl-2 is a therapeutic target for hypodiploid B-lineage acute lymphoblastic leukemia. Cancer Res. 79, 2339–2351 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu J., Chen Z., Cui Y., Wei H., Zhu Z., Mao F., Wang Y., Liu Y., Berberine promotes XIAP-mediated cells apoptosis by upregulation of miR-24-3p in acute lymphoblastic leukemia. Aging (Albany NY) 12, 3298–3311 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramos-Penafiel C., Olarte-Carrillo I., Ceron-Maldonado R., Rozen-Fuller E., Kassack-Ipina J. J., Melendez-Mier G., Collazo-Jaloma J., Martinez-Tovar A., Effect of metformin on the survival of patients with ALL who express high levels of the ABCB1 drug resistance gene. J. Transl. Med. 16, 245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiao G., Chan L. N., Klemm L., Braas D., Chen Z., Geng H., Zhang Q. C., Aghajanirefah A., Cosgun K. N., Sadras T., Lee J., Mirzapoiazova T., Salgia R., Ernst T., Hochhaus A., Jumaa H., Jiang X., Weinstock D. M., Graeber T. G., Muschen M., B-cell-specific diversion of glucose carbon utilization reveals a unique vulnerability in B cell malignancies. Cell 173, 470–484.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen W.-L., Wang Y.-Y., Zhao A., Xia L., Xie G., Su M., Zhao L., Liu J., Qu C., Wei R., Rajani C., Ni Y., Cheng Z., Chen Z., Chen S.-J., Jia W., Enhanced fructose utilization mediated by SLC2A5 is a unique metabolic feature of acute myeloid leukemia with therapeutic potential. Cancer Cell 30, 779–791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pollyea D. A., Stevens B. M., Jones C. L., Winters A., Pei S., Minhajuddin M., D’Alessandro A., Culp-Hill R., Riemondy K. A., Gillen A. E., Hesselberth J. R., Abbott D., Schatz D., Gutman J. A., Purev E., Smith C., Jordan C. T., Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 24, 1859–1866 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kreitz J., Schönfeld C., Seibert M., Stolp V., Alshamleh I., Oellerich T., Steffen B., Schwalbe H., Schnütgen F., Kurrle N., Serve H., Metabolic plasticity of acute myeloid leukemia. Cell 8, 805 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toby T. K., Fornelli L., Srzentic K., DeHart C. J., Levitsky J., Friedewald J., Kelleher N. L., A comprehensive pipeline for translational top-down proteomics from a single blood draw. Nat. Protoc. 14, 119–152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doerr A., Single-cell proteomics. Nat. Methods 16, 20 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Kong Y., Yoshida S., Saito Y., Doi T., Nagatoshi Y., Fukata M., Saito N., Yang S. M., Iwamoto C., Okamura J., Liu K. Y., Huang X. J., Lu D. P., Shultz L. D., Harada M., Ishikawa F., CD34+CD38+CD19+ as well as CD34+CD38-CD19+ cells are leukemia-initiating cells with self-renewal capacity in human B-precursor ALL. Leukemia 22, 1207–1213 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Jiang Z., Deng M., Wei X., Ye W., Xiao Y., Lin S., Wang S., Li B., Liu X., Zhang G., Lai P., Weng J., Wu D., Chen H., Wei W., Ma Y., Li Y., Liu P., Du X., Pei D., Yao Y., Xu B., Li P., Heterogeneity of CD34 and CD38 expression in acute B lymphoblastic leukemia cells is reversible and not hierarchically organized. J. Hematol. Oncol. 9, 94 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inoue S., Li W. Y., Tseng A., Beerman I., Elia A. J., Bendall S. C., Lemonnier F., Kron K. J., Cescon D. W., Hao Z., Lind E. F., Takayama N., Planello A. C., Shen S. Y., Shih A. H., Larsen D. M., Li Q., Snow B. E., Wakeham A., Haight J., Gorrini C., Bassi C., Thu K. L., Murakami K., Elford A. R., Ueda T., Straley K., Yen K. E., Melino G., Cimmino L., Aifantis I., Levine R. L., De Carvalho D. D., Lupien M., Rossi D. J., Nolan G. P., Cairns R. A., Mak T. W., Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell 30, 337–348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paz J. L., Levy D., Oliveira B. A., de Melo T. C., de Freitas F. A., Reichert C. O., Rodrigues A., Pereira J., Bydlowski S. P., 7-ketocholesterol promotes oxiapoptophagy in bone marrow mesenchymal stem cell from patients with acute myeloid leukemia. Cell 8, 482 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agathocleous M., Meacham C. E., Burgess R. J., Piskounova E., Zhao Z., Crane G. M., Cowin B. L., Bruner E., Murphy M. M., Chen W., Spangrude G. J., Hu Z., DeBerardinis R. J., Morrison S. J., Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye H., Adane B., Khan N., Sullivan T., Minhajuddin M., Gasparetto M., Stevens B., Pei S., Balys M., Ashton J. M., Klemm D. J., Woolthuis C. M., Stranahan A. W., Park C. Y., Jordan C. T., Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell 19, 23–37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verma D., Zanetti C., Godavarthy P. S., Kumar R., Minciacchi V. R., Pfeiffer J., Metzler M., Lefort S., Maguer-Satta V., Nicolini F. E., Burroni B., Fontenay M., Krause D. S., Bone marrow niche-derived extracellular matrix-degrading enzymes influence the progression of B-cell acute lymphoblastic leukemia. Leukemia 34, 1540–1552 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Polak R., de Rooij B., Pieters R., den Boer M. L., B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 126, 2404–2414 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Sipkins D. A., Wei X., Wu J. W., Runnels J. M., Cote D., Means T. K., Luster A. D., Scadden D. T., Lin C. P., In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 435, 969–973 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Almeida M. J., Luchsinger L. L., Corrigan D. J., Williams L. J., Snoeck H. W., Dye-independent methods reveal elevated mitochondrial mass in hematopoietic stem cells. Cell Stem Cell 21, 725–729.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/11/eabd6280/DC1