

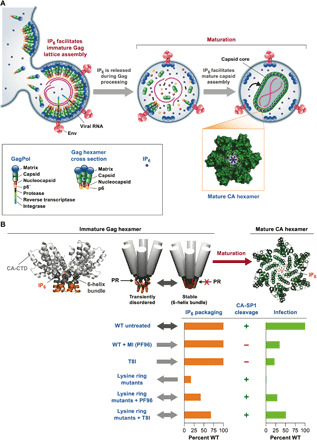
HIV immature lattice stability affects capsid maturation by altering IP6 packaging.
Abstract
HIV virion assembly begins with the construction of an immature lattice consisting of Gag hexamers. Upon virion release, protease-mediated Gag cleavage leads to a maturation event in which the immature lattice disassembles and the mature capsid assembles. The cellular metabolite inositiol hexakisphosphate (IP6) and maturation inhibitors (MIs) both bind and stabilize immature Gag hexamers, but whereas IP6 promotes virus maturation, MIs inhibit it. Here we show that HIV is evolutionarily constrained to maintain an immature lattice stability that ensures IP6 packaging without preventing maturation. Replication-deficient mutant viruses with reduced IP6 recruitment display increased infectivity upon treatment with the MI PF46396 (PF96) or the acquisition of second-site compensatory mutations. Both PF96 and second-site mutations stabilise the immature lattice and restore IP6 incorporation, suggesting that immature lattice stability and IP6 binding are interdependent. This IP6 dependence suggests that modifying MIs to compete with IP6 for Gag hexamer binding could substantially improve MI antiviral potency.
INTRODUCTION
The inositol phosphate IP6 is a newly described cofactor that has been proposed to be essential for HIV replication. It is an abundant polyanion found in all cells and is selectively packaged and enriched into HIV virions when they bud from the cell. Knockout (KO) of the biosynthetic kinases inositol polyphosphate multikinase (IPMK) or inositol pentakisphosphate 2-kinase (IPPK) decreases IP6 availability in the cell and reduces HIV production (1). Kinase KO does not alter HIV infectivity because the virions made in KO cells recruit IP6 molecules as efficiently as in wild-type (WT) cells. In the case of IPMK KO, loss of the kinase reduces but does not abolish IP5 (inositol 1,3,4,5,6-pentakisphosphate) and IP6 levels in the cell, due to the presence of alternative pathways for IP5 synthesis. In IPPK KOs, there is a decrease in IP6 but not IP5, and HIV packages IP5 instead (1).
HIV packages IP6 into virions using two rings of lysine residues, K158 and K227 [capsid (CA) numbering], that are found in immature Gag lattice hexamers (1). A crystal structure of the complex suggests that a single IP6 molecule is bound per hexamer and located immediately above a six-helix bundle (6HB) that forms at the junction between CA and adjoining spacer peptide 1 (SP1) domains of the Gag precursor (2). Mutation of either lysine leads to a profound loss of both virus production and infectivity. This reduction in infectivity occurs concomitantly with a reduction (but not complete loss) of IP6 packaging, at least for K227A or K227I mutants. Although both lysines bind IP6 in a hexamer structure, K158 preferentially engages the five equatorial phosphates, while K227 interacts with the axial phosphate. This binding hierarchy is reflected in the relative importance of the two lysine residues in HIV infectivity, with K158 mutants being significantly less infectious than K227 counterparts (3). Together, the data on kinase KOs and IP6-binding mutants suggest that inhibiting IP6 recruitment could be a highly effective approach for preventing HIV infection.
Before the discovery of IP6 as an HIV cofactor, a series of antiretroviral compounds that bind to the immature lattice were developed. These compounds, called maturation inhibitors (MIs), interfere with Gag processing by slowing the removal of SP1 from the C terminus of CA (4–6). The CA-SP1 cleavage site is buried within the 6HB in the immature Gag lattice, suggesting that some unfolding of the bundle is required for viral protease (PR) cleavage (7). Upon propagation in the presence of MI, HIV acquires resistance mutations that are thought to confer escape from MIs by altering the kinetics of CA-SP1 processing or decreasing the stability of the immature lattice. For instance, SP1-A1V has faster CA-SP1 cleavage kinetics, is replication competent, and is resistant to all MIs tested (8, 9). However, many MI resistance mutations are thought to work by counteracting the increased lattice stability induced by MI binding. Different MIs drive acquisition of different sets of escape mutations. The first-generation MI, bevirimat (BVM), selects for mutations around K227 (5, 8), while the structurally distinct MI, PF46396 (PF96), selects for mutations around K158 (10, 11). Meanwhile, the BVM analogs 7m and 7r (12) select for mutations around both the K158 and K227 lysine rings (9). Just as mutation of K158 causes a greater loss in infectivity than loss of K227, so MI escape mutants around K158 are generally less replication competent than their K227 counterparts. Several of the highly replication-defective, MI-resistant mutants are MI dependent, likely because the MI offsets the lattice-destabilizing effect of the resistance mutations. In the absence of MI, these lattice-destabilizing, MI-dependent viruses are rescued when they acquire mutations, such as SP1-T8I, that stabilize the CA-SP1 6HB (8, 11).
In this study, we investigated the relationship between immature Gag lattice stability, MI sensitivity, and IP6 incorporation. Our results suggest that immature lattice stability is finely balanced to promote efficient viral assembly but allow subsequent proteolytic processing and maturation upon particle release from the cell. While both the physiological cofactor IP6 and antiviral MIs increase lattice stability, only MIs do so in a manner that blocks CA-SP1 cleavage and virion infectivity. IP6 promotes virion infectivity by stabilizing the mature CA hexamers that form the building blocks of the HIV capsid. This role for IP6 during maturation highlights the importance of its initial packaging into virions through its binding to (and stabilization of) the immature lattice. We show that IP6 binding and immature lattice stability are interdependent; IP6 binding increases lattice stability while stabilizing the lattice increases IP6 binding. IP6 binding–deficient HIV-1 mutants can simultaneously restore IP6 incorporation and infectivity through the acquisition of compensatory mutations that stabilize the lattice. Moreover, treatment with the MI PF96 can also restore the IP6 incorporation and infectivity defects induced by these mutations. The IP6 binding–deficient mutants are completely insensitive to MIs in terms of CA-SP1 processing and remain so even in the presence of some compensatory, lattice-stabilizing mutations. These observations demonstrate a role for immature lattice stability in proper virion maturation and infectivity that is independent of CA-SP1 cleavage. We propose that IP6 incorporation is the key feature linking immature lattice stability to virion maturation and infectivity in this context. These findings suggest that designing MIs that not only inhibit CA-SP1 processing but also block IP6 incorporation may provide more potent antiretrovirals with dual inhibitory mechanisms and a high barrier to resistance.
RESULTS
MIs do not prevent virion incorporation of IP6
IP6 and MIs both bind the same central cavity in immature Gag hexamers (Fig. 1A). A recent structure of BVM complexed with the immature hexamer obtained by microelectron diffraction suggested that the IP6-coordinating residue K227 may be directly involved in BVM binding (13). This suggests that IP6 and MIs may bind competitively. To test this, we produced HIV-1 virions in cells supplemented with tritiated inositol and in the presence of three different MIs: PF96, BVM, and the BVM analog 7m. In agreement with previous reports (5, 9, 10), all three MIs significantly inhibited maturation, as visualized by the presence of unprocessed CA-SP1 (p25) in purified virions (Fig. 1, B and C, and fig. S1). As expected, the MIs also reduced virus infectivity (Fig. 1D). Having confirmed that virions had been produced with MIs at inhibitory concentrations, we extracted the inositol phosphates from the remaining samples and measured their levels of IP6 incorporation by strong anion exchange (SAX)–high-performance liquid chromatography (HPLC) fractionation and scintillation counting (Fig. 1E). Unexpectedly, none of the MIs reduced IP6 incorporation (Fig. 1F). This result shows that MIs do not prevent IP6 binding and that, contrary to structural predictions, MIs and IP6 may not be mutually competitive for binding to their targets. This finding is in contrast to the apparently competitive nature of BVM and PF96 binding (11). Although the data indicate that MIs do not compete with IP6 for 6HB binding, it is currently unclear whether all MI and IP6 binding sites are occupied in the immature Gag lattice.
Fig. 1. MIs do not prevent IP6 incorporation into HIV-1 particles.
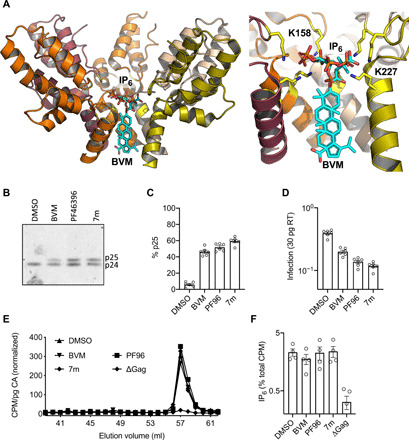
(A) Model of an immature hexamer showing the binding sites of IP6 and BVM, based on 6BHR (22) and 6N3U (13). (B) p24 Western blot of virions produced in HEK293T cells in the presence of different MIs. All three MIs interfere with CA-SP1 (p25) cleavage to CA (p24). DMSO, dimethyl sulfoxide. (C) Quantification of the processing defect observed in (B), expressed as the percentage of CA + CA-SP1 that remains as uncleaved p25. Error bars depict means ± SEM of band intensity on Western blots with samples from at least three independent experiments. (D) Infectivity of virions produced in the presence of different MIs (see Materials and Methods), normalized to quantity of input virus [per 30 pg of reverse transcriptase (RT)]. Error bars depict means ± SEM of at least three independent experiments carried out with at least two replicates. (E) Inositols extracted from purified virions produced in the presence of the indicate MIs and 3H inositol and fractioned by SAX-HPLC. The levels of different 3H inositol–containing species are quantified by scintillation counting. Fractions corresponding to the IP6 peak are displayed for a selection of viruses produced under different MI treatment conditions. The counts per minute (CPM) of tritiated IP6 are normalized to the amount of capsid (CA) in the sample. (F) IP6 incorporation expressed as the percentage of total CPM in virus samples from (E). P < 0.007 in all cases between MI treatment and ∆Gag, and P > 0.3 between different MI-treated samples. In all cases, the concentrations of MIs were PF96 (5 μM), BVM (2 μg/ml), and 7m (100 nM). Error bars depict mean CPM ± SEM from a minimum of three independent experiments. Statistical analysis was performed using Student’s t test.
IP6 binding–deficient mutants are resistant to MIs
Next, we asked whether mutants that have reduced IP6 incorporation respond differently to MIs. We compared the effect of MI treatment on the single-cycle infectivity of HIV-1 mutants K158A/I and K227A/I and found that none of these IP6 binding–deficient mutant viruses were inhibited by any of the MIs tested, in contrast to WT (Fig. 2, A and B). Most notably, the MI PF96 increased the infectivity of all mutants to levels similar to that of WT in the presence of the inhibitor (Fig. 2, A and B, and fig. S2). As MIs act by interfering with CA-SP1 proteolysis, we hypothesized that IP6 binding–deficient mutants may be resistant because they still undergo CA-SP1 processing in the presence of MI. Western blots of Gag proteins from purified virions revealed that while MIs reduced WT CA-SP1 processing, they did not alter CA-SP1 processing in any of the IP6 binding–deficient mutants (Fig. 2, C to F). The fact that PF96 does not alter mature CA levels in mutant viruses, together with the fact that it increases their infectivity more potently than it decreases WT infectivity, suggests that PF96 is somehow having an effect independent of altering CA-SP1 processing, in contrast to BVM or 7m that have no impact on these mutants.
Fig. 2. IP6 binding–deficient mutants escape maturation inhibition.
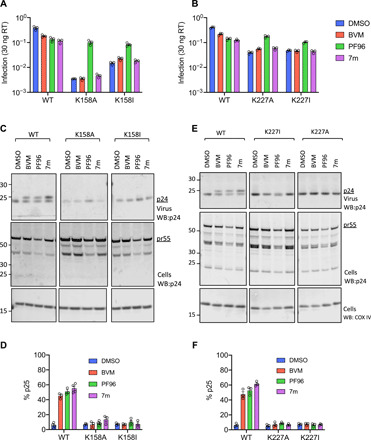
(A and B) Infectivity of WT and mutant viruses produced under different MI treatment conditions. Data are normalized to quantity of input virus (30 pg of RT). Error bars depict means ± SEM from three independent experiments, each containing at least two replicates. (C and E) p24 Western blots (WBs) of samples tested in (A) and (B). Western blots are shown for both virions and their corresponding producer cells alongside the corresponding loading control (COX IV). Note that the presence of MIs interferes with WT CA-SP1 processing but not that of K158 and K227 mutants. (D and F) Quantification of p25 levels from Western blots in (C) and (E) expressed as the percentage of CA + CA-SP1 that remains as uncleaved p25. Error bars depict means ± SEM of band intensity on Western blots with samples from three independent experiments.
IP6 binding–deficient mutants revert via acquisition of lattice-stabilizing mutations
The resistance of K158 and K227 mutants to inhibition by MIs reveals that they behave like MI escape mutants, which arise spontaneously upon propagation of HIV-1 in the presence of MIs. Previous work has described mutations within the major homology region (MHR) of CA (where K158 is located) that arose spontaneously upon passage of WT HIV-1 in the presence of the MI PF96 (11). MIs are proposed to decrease the efficiency of PR cleavage by increasing 6HB and immature hexamer stability (14). One mechanism by which some of these mutants are proposed to confer resistance is by decreasing immature hexamer stability, counteracting the stabilizing effect of MIs. The CA-SP1 processing data suggest that K158 and K227 mutants escape in the same way, as even in the presence of the compounds, there is complete cleavage of CA-SP1 to CA (Fig. 2, C to F). We reasoned that decreased 6HB stability of the lysine mutants is likely a result of their reduced IP6 occupancy (Fig. 3A). MI escape variants that are replication defective in the absence of MIs commonly acquire second-site mutations that increase their fitness, possibly by restoring some of their lost immature hexamer stability (8, 11). We therefore propagated K158A and K227A mutant viruses in a highly permissive T cell line (MT4) to determine whether they also gain mutations that improve fitness by increasing immature hexamer stability. Initially, we observed delayed replication kinetics of both mutants compared to WT HIV-1 (NL4-3) (Fig. 3B). K158A replicated with an ~8-day delay relative to WT, whereas K227A exhibited a less severe delay, although it produced lower levels of virus overall. Upon reinfection of naïve MT4 cells with reverse transcriptase (RT)–normalized supernatants, the passaged mutants replicated similarly, or with a small delay, relative to WT (Fig. 3C). We sequenced viral DNA isolated from the infected cells to determine what mutation(s) had been acquired in Gag by the virus to achieve this rescue. We identified three mutations—two in SP1 (in the 6HB) (T8I and M4L) and one in CA (N193H) (Fig. 3A). The T8I mutation arose independently in both K227A and K158A, while M4L and N193H arose only in K227A. Both T8I and N193H have previously been observed as two of the second-site suppressor mutations that rescue the replication of PF96-dependent MHR mutants in the absence of PF96 (11). The SP1-T8I mutation was also observed upon propagation of the assembly-deficient, BVM-resistant mutant SP1-A3V (8).
Fig. 3. IP6 binding–deficient mutants revert by acquiring second-site mutations that phenocopy MIs.
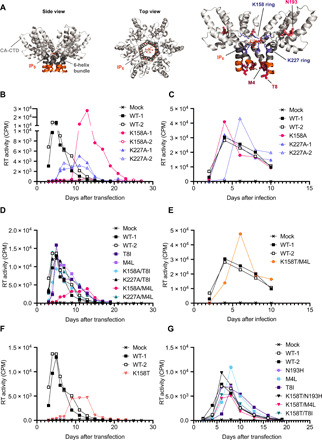
(A) Models of the CA C-terminal domain (CTD) in the immature Gag hexamer showing the location of IP6 and 6HB in side and top views. The position of the two lysine rings (K158 and K227) and sites of second-site compensatory mutations at M4, T8, and N193 are annotated in the final model. Model generated in PyMOL using Protein Data Bank 6BHR and 6N3U. (B) WT or mutant viruses were passaged in duplicate in the T cell line MT4. RT activity was measured in culture supernatant to determine the levels of viral production at the indicated time points. (C) Selected viruses were collected from culture supernatants from (B), RT-normalized, and used to infect fresh MT4 cells and RT activity measured as before. (D) Gag mutations arising during virus propagation were introduced into WT and lysine-mutant pNL4-3 plasmids and used to transfect fresh MT4 cells. (E) RT-normalized supernatant from (D) was used to infect fresh MT4s as in (C). (F) Passage of WT and K158T virus. (G) Gag mutations arising during virus propagation were introduced into the pNL4-3/K158T plasmid. The mutant molecular clones were used to transfect fresh MT4 cells.
To confirm that the SP1 mutations rescue, at least partially, the replication of K158A and K227A, we introduced the T8I or M4L mutations into proviral constructs containing the original K158A or K227A substitutions and transfected them into the MT4 T cell line. We found that each combination, excluding K158A/M4L, displayed similar replication kinetics to WT (Fig. 3D), confirming that these SP1 mutations are indeed capable of rescuing the replication of the lysine mutants in this cell line. As K158A/M4L was still substantially impaired in kinetics and viral yield, we passaged this virus further and obtained a threonine mutation at position 158. When transfected into MT4 cells, the double mutant K158T/M4L clone displayed substantially improved fitness relative to its K158A/M4L parent, exhibiting close to WT replication kinetics (Fig. 3, D and E). The ability of M4L to more fully rescue K158T relative to K158A suggests that the threonine mutation results in an immature Gag lattice that is more stable than that of K158A. To test this hypothesis, we passaged K158T and obtained an escape variant with the same N193H mutation that arose during rescue of K227A (Fig. 3F). Transfection of MT4 cells with K158T/N193H confirmed that this mutant replicated with similar kinetics to WT virus (Fig. 3G). While we observed almost complete rescue in MT4s, these cells are the most permissive of T cell line models. We therefore investigated whether the second-site compensating mutants were able to restore replication in the far less permissive SupT1 T cell line. Replication kinetics were significantly delayed in this T cell line relative to WT, particularly for those containing T8I. Mutants M4L and N193H conferred closer to WT kinetics when combined with less attenuated mutants K158T and K227A (fig. S3). These results demonstrate that the capacity to restore replication to IP6 binding–deficient mutants through the selection of second-site mutants varies in different T cell lines.
Second-site compensatory mutations rescue virus release and particle infectivity
Mutation of either K158 or K227 affects both viral production and infectivity. We therefore addressed whether the second-site compensatory mutations restore one or both of these properties. We first investigated virus production by carrying out virus release assays, which not only measure Gag expression but allow the efficiency of assembly and release to be compared. K158A, K158T, and K227A were all highly impaired for virus release (Fig. 4A and fig. S4A). Either SP1 mutation (M4L or T8I) or the CA mutation (N193H) was sufficient to restore the release of each lysine mutant to near WT levels. The exception was K158A/M4L, which remained substantially impaired (Fig. 4A). We directly compared the results of these virus release assays to changes in lysine mutant infectivity by comparing activity in single-round infectivity assays (Fig. 4B). There was a broad correlation between the infectivity data and the results of the virus release assay. For instance, K158A/M4L, which still has a virus release defect, is also the least infectious of the double mutants. The data also show that second-site mutants can partially restore lysine mutant infectivity independently of their restoration of virus release. For instance, K158T increases infectivity almost 10-fold without substantially altering virus release.
Fig. 4. Second-site compensatory mutations rescue virus release and restore infectivity of lysine mutants to close to WT levels.
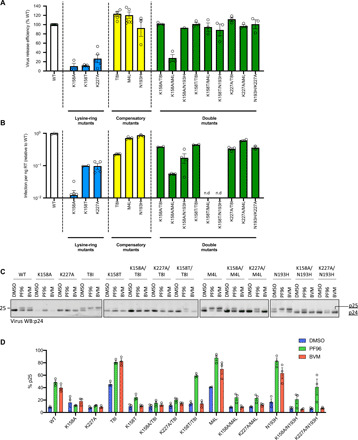
(A) Virus release assay. Virus release efficiency of the indicated Gag mutants was calculated on the basis of the level of p24 in purified virions quantified as a percentage of p24 and Pr55 expression in producer cells and p24 expression in purified virions. Data are normalized to give the efficiency relative to WT virus. Error bars depict the SEM from at least three independent experiments. (B) Infectivity of the same Gag mutants shown in (A). Infectivity is normalized to input virus (nanograms of RT) and presented as relative to WT levels. Error bars depict means ± SEM from two to four independent experiments. n.d, not determined. (C) p24 Western blots of virus obtained from transfection of K158, K227, and second-site mutants. Viruses were produced under control conditions (DMSO) or in the presence of the MIs BVM (2 μg/ml) or PF96 (5 μM). (D) Quantification of p25 levels from Western blots in (C) expressed as the percentage of CA + CA-SP1 that remains as uncleaved CA-SP1 (p25). Error bars depict means ± SEM of band intensity on Western blots with samples from three independent experiments.
The second-site mutations may increase viral production and infectivity by restoring lattice stability, analogous to their effects on MI escape mutants. To test this, we compared the CA-SP1 processing of second-site and IP6-binding mutants. The second-site mutations, in particular, T8I, increased the levels of CA-SP1 in virions relative to WT, consistent with an increase in stability of the 6HB within immature hexamers as previously suggested (Fig. 4, C and D, and fig. S4B) (11). In contrast, the CA-SP1 processing of all second-site mutants was increased when combined with either K158 or K227 mutations, consistent with the idea that loss of either lysine decreases hexamer stability. However, the processing assay is unable to directly measure the destabilizing effect of the IP6-binding mutants alone because WT virus is already almost completely processed. We therefore added MIs to the assay with the rationale that they would increase the stability of all viruses, allowing destabilizing effects to be observed. Addition of MIs to WT virus led to a substantial increase in unprocessed p25 and potentiated the processing defect of all second-site mutants, validating this approach (Fig. 4, C and D). MI addition revealed differences in the processing of IP6-binding mutant when combined with the second-site mutants. This was most notable for K158T, which was largely processed in the presence of PF96, whereas for K158T/T8I, over 50% of p25 remained unprocessed upon addition of the MI (Fig. 4D).
Second-site mutations maintain MI resistance
Second-site mutations improve the fitness of MI-passage mutants while maintaining resistance. We tested whether the same was true for IP6-binding mutants. Even with the addition of stabilizing second-site mutations, K158 and K227 variants remained largely resistant to MIs (Fig. 5, A to C, and fig. S5). This is despite the fact that, consistent with their increased hexamer stability, the three second-site mutants were individually more sensitive to MI treatment than WT virus. Next, we performed a series of replication experiments comparing the ability of viruses to replicate in the presence of MIs (Fig. 5, D to G, and fig. S6). In the case of WT virus, only 7r, a potent second-generation BVM derivative, had a substantial impact on replication kinetics, delaying replication by around 5 days (Fig. 5D). In contrast, K158A and K227A showed a profound fitness loss when compared to WT (Fig. 5E and fig. S6), and K158A only replicated in the presence of PF96. This is consistent with earlier data (11) showing the ability of PF96 to rescue both the production and infectivity defects imposed by K158A (Figs. 4D and 5, A to C). For T8I, replication in the presence of all MIs was severely delayed, consistent with exacerbation of its preexisting processing defect by MI treatment (Fig. 4C). The K158A/T8I double mutant replicated with WT kinetics in the presence or absence of MI (Fig. 5G), indicating that by combining T8I with K158A, as seen previously for virus release, CA-SP1 cleavage, and single-round infectivity, the replication of K158A and the MI inhibitor sensitivity of T8I were rescued. These data show that the K158 and K227 mutants behave like MI escape mutants that reduce immature lattice stability and respond in a similar way to the stabilizing effect of MI ligands and second-site mutations.
Fig. 5. Second-site revertants maintain MI resistance of IP6 binding–deficient mutants.
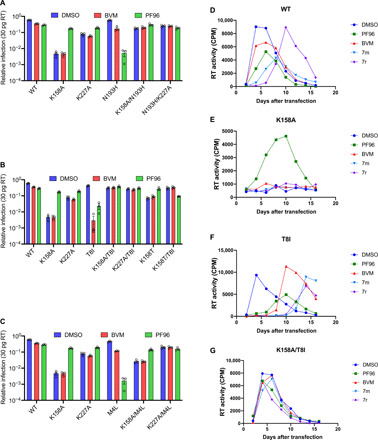
(A to C) The same selection of viruses and conditions that were tested for CA-SP1 processing in Fig. 4 was also tested for infectivity, after normalization for production (infection with 30 pg of RT). Error bars depict means ± SEM of at least three independent experiments, each containing two replicates. WT (D), K158A (E), T8I (F), and K158A/T8I (G) viruses were passaged in duplicate in the T cell line MT4 in the presence of DMSO, PF96 (5 μM), BVM (0.5 μM), 7m (0.5 μM), or 7r (0.5 μM). RT activity was measured in culture supernatant to determine the levels of viral production at the indicated time points.
Together, the virus release, CA-SP1 processing, infectivity, and replication data support the notion that losing K158 is more destabilizing than mutating K227 and that T8I is more stabilizing than the other compensatory mutations. T8I rescues K158A to a similar level of infection as it does the K227A mutant but the weaker stabilizing mutant M4L does not (Fig. 5, A to C). The addition of PF96 is needed to recover K158A/M4L to similar infection levels as the K158A/T8I double mutant. Conversely, the double mutant predicted to be most strongly stabilized on the basis of the CA-SP1 processing data (Fig. 4, C and D), K158T/T8I, displays reduced infectivity in the presence of PF96. This is consistent with the critical location within immature hexamers of K158 and T8. K158 is the primary coordinator of IP6, suggesting that its mutation will have the greatest impact on binding. Moreover, without IP6 binding, the remaining lysine ring, formed by K227, would be expected to become strongly destabilizing. Meanwhile, structural evidence suggests that T8 occupies a key position in the 6HB (15), which is predicted to become significantly more stable in the T8I mutant. To further explore the synergy between immature lattice stability, processing, and infectivity, we therefore focused on mutations of K158 and T8.
Second-site mutations and MI PF96 restore active IP6 packaging into HIV virions
A key question arising from these data is whether the combination of lysine ring and second-site mutations has made HIV-1 IP6 independent. To test this, we compared IP6 incorporation in K158A, T8I, and K158A/T8I viruses (Fig. 6A). Consistent with its critical role in coordinating IP6, we observed a significant loss of IP6 enrichment in K158A virions. However, the addition of T8I significantly increased IP6 incorporation to near WT levels. This suggests that rather than make HIV-1 IP6 independent, the second-site mutation restores the ability of the virus to actively package the metabolite. This result further suggests that a single lysine ring is sufficient for active IP6 packaging and that immature hexamer stability regulates IP6 binding. To test this, we measured how IP6 incorporation is altered by the MI PF96, which phenocopies T8I and is also proposed to stabilize immature hexamers (Fig. 6B) (7). Similar to T8I, we found that PF96 increased IP6 incorporation into K158A particles. This recovery of IP6 packaging may explain why PF96 markedly improves K158A infectivity.
Fig. 6. Rescue of IP6 binding–deficient mutant K158A restores the assembly of single capsids.
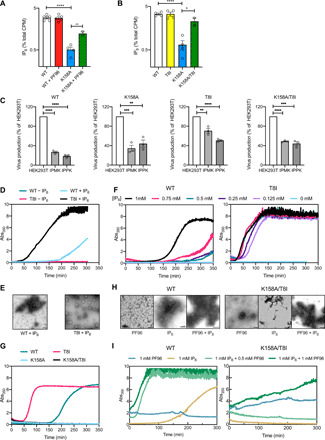
(A) IP6 in WT and K158A virions as a percentage of total CPM in the presence or absence of MI PF96. Error bars depict mean %CPM ± SEM from at least two independent experiments. Statistics were performed using Student’s t test (WT versus K158A, ****P < 0.0001; K158A versus K158A + PF96, **P = 0.0065). (B) IP6 incorporation (measured as the percentage of total CPM) in WT, T8I, K158A, or K158A/T8I viruses. Error bars depict IP6 as the percentage of total CPM ± SEM from at least two independent experiments. Statistical analysis was performed using Student’s t test (WT versus K158A, ****P < 0.0001; K158A versus K158A/T8I, *P = 0.011). (C) Relative virus production for WT, K158A, T8I, and K158A/T8I when produced in WT, IPMK KO, or IPPK KO HEK293T cells. Virus production in measured as RT activity compared to RT in WT virus produced in WT HEK293T cells. Measurements are the average of three independent experiments, and error bars depict this ± SEM. ****P < 0.0001 in all cases. K158A: ***P = 0.0004, **P = 0.0017. T8I: **P = 0.0041. T8I/K158A: ***P = 0.0001. (D to I) In vitro assembly reactions using 250 μM WT and mutant CA-SP1 in 50 mM tris-HCl (pH 8.0), 50 mM NaCl, 1 mM dithiothreitol (DTT) were monitored in real time by measuring the absorbance at 350 nm (Abs350). (D) Assembly of WT and T8I CA-SP1 recombinant protein into VLPs in the presence and absence of 1 mM IP6. (E) Negative stain images of material taken from D. Scale bar, 200 nm. (F) Assembly of WT and T8I CA-SP1 with indicated concentration of IP6. (G) Assembly of WT, K158A, T8I and T8I/K158A with 1mM IP6. (H) Negative stain EM images of material taken from I. (I) Assembly of WT and T8I/K158A CA-SP1 with 1 mM IP6 and/or 0.5 or 1 mM PF96 as indicated.
We have previously shown that IP6 is required for both viral production and infectivity. Reducing cellular levels of IP5 or IP6, by knocking out key kinases required for its biosynthesis, decreases WT HIV production. We therefore tested how K158A, T8I, and K158A/T8I production is affected by changing cellular inositol phosphate levels. Production of K158A was reduced in both IPMK and IPPK KO cells, albeit not to the same extent as WT virus. This suggests that some degree of IP5 or IP6 binding still takes place via the remaining lysine residue, K227, and is consistent with previous observations (1). T8I was less affected by kinase KO than either WT or K158A viruses (Fig. 6C). This may be because T8I increases 6HB stability such that IP6 is no longer required or because this increases affinity of the 6HB for IP6 and compensates for the reduced cellular inositol phosphate concentration. The double-mutant K158A/T8I shows an intermediate sensitivity to kinase KO that is intermediate between the two single mutants.
T8I and PF96 restore immature lattice assembly
We previously proposed that IP6 is involved in formation of the immature lattice; IPPK and IPMK KO thus leads to a defect in virus production. Consistent with this, in vitro assembly data have shown that IP6 no longer promotes the immature lattice formation of lysine mutants such as K158A (2). To test our hypothesis that the stabilization of immature hexamers by second-site mutations such as T8I or the MI PF96 increases IP6 binding, we investigated their impact on the in vitro assembly of immature virus-like particles (VLPs). To this end, we produced recombinant CA-SP1 protein and measured its ability to assemble into VLPs in the presence or absence of IP6 (Fig. 6D). Comparing the kinetics of assembly by turbidity assay, we observed that T8I assembled more rapidly than WT CA-SP1 (Fig. 6D). Negative stain electron microscopy (EM) confirmed that the T8I assemblies in the turbidity assay were similar in morphology to WT immature VLPs (Fig. 6E). T8I-mutant CA-SP1 also assembled efficiently at reduced IP6 concentrations, with similar kinetics observed at 125 μM and 1 mM IP6. In contrast, the kinetics of WT CA-SP1 markedly decreased with IP6 concentration. Comparing the two proteins revealed that T8I assembles faster at 125 μM IP6 than WT at 1 mM (Fig. 6F). These data may explain why T8I virus is less sensitive than WT to reduced cellular IP6 levels upon IPMK or IPPK KO (Fig. 6C). Next, we investigated whether inclusion of T8I would restore CA-SP1 VLP assembly of K158A (Fig. 6G). However, we did not observe any assembly within the time scale of the experiment and at the IP6 concentrations tested (Fig. 6, G and I). This may be because the in vitro system used here does not recapitulate all components of immature assembly, for instance, the interaction between the nucleocapsid domain of Gag and the viral RNA. As we had previously observed that the effect of T8I and PF96 was additive in terms of inhibiting CA-SP1 processing (Fig. 4C), we tested whether inclusion of PF96 and IP6 together would stabilize K158A/T8I. We observed that the addition of both ligands allowed K158A/T8I to form immature VLPs, as evidenced both by turbidity assay and negative stain EM (Fig. 6, H and I). These data confirm that IP6 binding to IP6-binding mutant K158A can be restored indirectly by increasing the stability of the immature lattice. It also shows that PF96 and IP6 can act in concert to increase immature VLP assembly of WT CA-SP1, similar to the reported effect of BVM (16). This is consistent with the IP6 incorporation data suggesting that IP6 and PF96 binding is noncompetitive (Figs. 1F and 6A) and explains why the MI can concomitantly restore infectivity and IP6 incorporation in K158A virus.
Rescue of IP6 binding–deficient mutant K158A restores the assembly of single capsids
We previously suggested that IP6 is required not only for immature lattice assembly and particle release but also for building and stabilizing mature capsids. The above data suggest that second-site mutations such as T8I concomitantly restore IP6 recruitment and immature lattice stability. To test how this influences mature capsid formation, and thus infectivity, we carried out a series of cryo–electron tomography (cryo-ET) experiments. We purified WT, T8I, K158A, and K158A/T8I viruses, collected a series of tomograms from multiple grids per sample, and performed three-dimensional (3D) tomographic reconstructions on 60 to 130 virions per mutant. We assigned virions to one of four categories based on previous work (11, 17, 18) (Fig. 7A and fig. S7). We note, however, that in our data, as in all published cryo-ET results, the number of observations (i.e., tomograms) is small compared to the effectively infinite number of virions that exist. The majority of WT virions had undergone maturation, with <10% displaying a distinctive immature lattice. Of the mature WT cores, most had either a conical or tubular morphology, while a minor fraction (28%) were closed but irregular in shape. By contrast, T8I had fewer mature conical capsids (34% versus 52% for WT) and a larger fraction of immature virions (13% for T8I versus 5% for WT). This is in agreement with previous cryo-ET analysis (19) and is consistent with T8I causing stabilization of the 6HB and inhibiting CA-SP1 proteolytic processing (11, 17). Both K158A and the double mutant K158A/T8I also had fewer conical capsids than WT (43 and 34%, respectively, versus 52% for WT). It is therefore not clear from examining maturation of the lattice alone why K158A is substantially less infectious than WT and how T8I substantially increases infectivity. To attempt to understand this, we analyzed all mature virions (conical/tubular or irregular) in more detail. We assigned these virions to one of five categories based on previous work (18)—single cores, multiple cores, multilayered cores, and cores with additional structures that are either open or closed and attached to the core (Fig. 7B and fig. S7). Almost all WT viruses contained a single capsid core (88%), with a minor fraction containing an additional incomplete assembly. In contrast, there were few K158A virions with a single capsid (16%). Instead, substantial numbers of K158A virions contained multiple distinct cores or cores that were multilayered. This is in agreement with thin-section EM data, which showed that K158A virions frequently contained multiple capsids (20). There were also significant numbers of virions with additional open or closed structures (>70%). Notably, addition of T8I to K158A reversed these defects, and the majority of the double-mutant virions (64%) contained a single capsid core. These observations suggest that K158A may exhibit an over-assembly defect, in which it does not form a single capsid core but continues to assemble multiple lattice structures, and T8I somehow mitigates this phenotype. Given that the SP1 region is not present in mature capsids, the data support the notion that T8I rescues proper assembly indirectly by restoring active IP6 incorporation into the immature lattice. IP6 may be required to ensure that a single core is formed, as has been demonstrated in vitro (2), rather than other types of lattice structures. However, the differences in mature capsid formation between K158A and K158A/T8I do not fully account for the differences in infectivity between the mutants. Part of the explanation may be that not all the K158A mature cores that appear to be morphologically similar to WT are actually infectious. In many of the K158A virions, we noted the presence of density outside of what are otherwise correctly formed cores that may represent the unpackaged RNA genome (fig. S7). A similar phenomenon of condensed ribonucleoprotein located outside empty core structures has been reported for HIV virions that have undergone complete, but delayed, CA processing (18).
Fig. 7. Rescue of IP6 binding–deficient mutant K158A restores the assembly of single capsids.
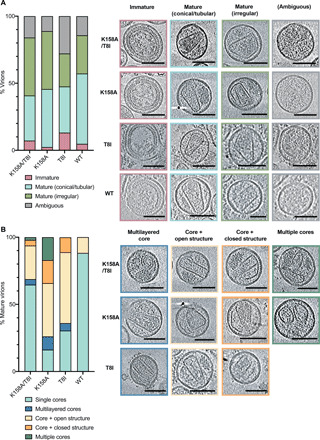
Cryo-ET was performed on the indicated HIV mutants. Tomograms were collected and reconstructions performed to assess capsid morphology. Capsids were classified into the indicated categories, and an example is shown for each mutant. A total of 64 WT, 69 T8I, 127 K158A, and 119 K158A/T8I tomograms were reconstructed, and the percentage of each category was calculated. (A) Virions were classified as immature (pink), mature with conical or tubular cores (cyan), mature with an irregular but closed structure (green), or ambiguous (partial or no lattices; gray), and their frequency was plotted as a percentage of all viruses. Representative tomograms of WT and mutant viruses for each category are shown as a gallery. (B) Virions with mature lattices were further subdivided into single cores (light blue), multilayered cores (dark blue), cores with additional open structures (light orange), cores with additional attached closed structures (dark orange), or multiple cores (dark green), and their frequency was plotted as a percentage of mature viruses. Representative tomograms of mutant viruses for each category are shown as a gallery. Scale bars, 100 nm.
Rescue of IP6 binding–deficient mutant K158A restores capsid stability
We considered that the over-assembly defect of K158A may also be caused by lattices that rapidly form, disassemble, and reform because of a lack of IP6 to lock CA into a stable conical core. To investigate the stability of the K158A lattices that form inside HIV virions, we carried out complementary single-molecule capsid uncoating experiments. HIV virions were first immobilized for observation by total internal reflection fluorescence (TIRF) microscopy and then permeabilized using a pore-forming toxin. This perforation allows fluorescent cyclophilin A (CypA) molecules (“CypA paint”) present in the buffer to enter and bind to intact capsid lattices within each virion, whereby the CypA signal persists, while the capsid remains intact and then disappears upon capsid disassembly (Fig. 8, A and B). As reported previously (3, 21), survival analysis of all capsids in the field of view showed that WT capsid cores display distinct uncoating kinetics, with a majority of more stable capsids that disappear with a half-life of 10 to 15 min (Fig. 8, C and D) and a small subset of highly unstable capsids (half-life of <1 min; Fig. 8E). Cores present in T8I mutant virions behaved similarly to WT, but K158A mutant virions contained substantially fewer stable cores, with most lattices belonging to the unstable subset that are lost within 1 min (Fig. 8, C to E). Addition of T8I to K158A restored the fraction of longer-lived capsids to the same level as WT. We repeated these experiments in the presence of IP6 to address whether the mutant capsids differ in their ability to be stabilized. As before (3), we observed a remarkable increase in the stability of most WT cores in the presence of IP6. The small subset of highly unstable WT capsids could not be effectively stabilized by IP6 (Fig. 8E), consistent with these cores being incompletely assembled (21). Our observations were broadly similar for T8I, and most capsids were effectively stabilized by IP6. In contrast, far fewer K158A capsids were capable of being stabilized by IP6, and most cores underwent the same fast disassembly kinetics irrespective of the presence of IP6. This suggests that K158 cores are so unstable, due to decreased IP6 packaging, that they fall apart before they can recruit sufficient exogenous IP6 from the buffer, or that they are incompletely formed and cannot be stabilized. Last, we analyzed the effect of IP6 on K158A/T8I virions (Fig. 8, C to E). We found that the addition of T8I to K158A completely restored the subset of capsids that can be stabilized by IP6. A similar proportion of K158A/T8I capsids was stabilized by IP6 as WT and to a similar degree (i.e., increase in half-life to >1 hour). Overall, the effect of IP6 on the different CA variants is apparent when comparing the fraction of capsids that remained intact at 20 min (Fig. 8F). We observed a two- to threefold increase in intact capsids with IP6 for WT, T8I, and K158A/T8I but not for K158A due to the increase in unstable capsids for this mutant. Note that the CA proteins that comprise the capsids from K158A and K158A/T8I virions being measured in our experiments are actually identical in amino acid composition. Therefore, while T8I increases immature lattice stability, it cannot directly affect mature lattice stability. Thus, these findings support our hypothesis that it is the restoration of IP6 recruitment to K158A virions provided by T8I stabilization of the immature lattice that allows stable mature capsids to form.
Fig. 8. Rescue of IP6 binding–deficient mutant K158A restores capsid stability.
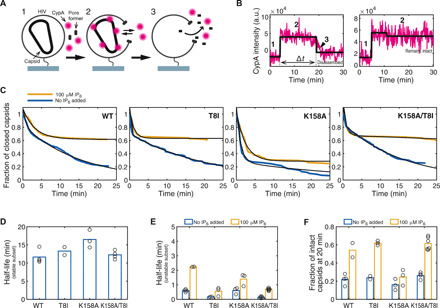
(A) Single-molecule TIRF uncoating assay by CypA paint. Capsids inside permeabilized virions are detected using Alexa Fluor 647–labeled CypA, which binds reversibly to the capsid exterior at a level that is proportional to the size of the CA lattice. (B) Representative CypA paint traces (magenta) with overlaid step fits (black) for a capsid that undergoes uncoating (left) and a capsid that remains intact (right). The CypA signal appears upon permeabilization (step 1) and remains constant, while the lattice is complete (step 2). Uncoating proceeds quickly once initiated, leading to rapid signal loss. The lifetime of the capsid is obtained from the duration (step length) of the CypA paint signal. a.u., arbitrary units. (C) Survival curves obtained from the lifetimes for all capsids in the field of view for WT, T8I, K158A, and K158A/T8I viruses in the absence (blue) and presence (yellow) of 100 μM IP6. Survival curves were fitted with a biexponential decay to extract the half-lives of the two stability types present under all conditions. (D) Half-lives of the stable subset of capsids in the absence of IP6. In the presence of IP6, this subset was so strongly stabilized that the half-life could not be reliably determined but was extended to several hours. (E) Half-lives of the unstable subset of capsids in the absence (blue) and presence (yellow) of 100 μM IP6. (F) Fraction of capsids that remain intact at 20 min in the absence (blue) and presence (yellow) of 100 μM IP6.
DISCUSSION
The data presented here reveal a cooperative relationship between IP6 binding and immature hexamer stability that ensures efficient HIV-1 infection. HIV-1 recruits IP6 into assembling virions via two lysine rings that are formed within immature Gag hexamers. IP6 promotes assembly of the immature lattice during virion formation at the plasma membrane, before budding (Fig. 9A). IP6 will not bind Gag monomers, as there are insufficient lysine side chains to coordinate the six phosphates. Mutations that decrease hexamer stability (i.e., favor monomers over hexamers) will therefore decrease IP6 affinity. Similarly, mutations that decrease IP6 affinity will decrease hexamer stability, because IP6 stabilizes the hexamer in the bound state. This explains why mutation of IP6-binding residues K158 or K227 reduces hexamer stability, in vitro assembly, and IP6 incorporation. It also explains why mutations or MIs that stabilize the hexamer increase IP6 incorporation. We have demonstrated this through the behavior of the MI PF96 and second-site mutations such as T8I, which stabilize immature hexamer mutants such as K158A, increase their incorporation of IP6 and the efficiency of virus release, alter CA-SP1 processing, restore particle infectivity, and rescue mature capsid formation (Fig. 9B). The cooperative effects of IP6, MIs, and immature hexamer mutations can influence both immature hexamer formation, which is a prerequisite for 6HB formation, and its localized melting to control protease accessibility. These data also make clear that MIs and IP6 are not competitive and are in agreement with in vitro assembly experiments in which BVM promoted the formation of immature VLPs (16). We propose that modification of MIs to compete with IP6 would add an additional mode of inhibition that is currently lacking and potentially significantly increase potency.
Fig. 9. IP6, MI, and CA residues influence the production of immature HIV virions and the subsequent correct processing and assembly of mature infectious virus.
(A) An overview of the role of IP6 in immature and mature viral production. IP6 is first recruited by Gag into virions that are assembling at the plasma membrane, where it promotes assembly of the immature lattice. Upon budding, Gag is cleaved by PR leading to release of IP6 from the lattice. Free IP6 present in the virion is then available to promote formation of the mature capsid by coordinating the R18 and K25 rings in mature hexamers. (B) The immature lattice and the 6HB must be sufficiently stable to promote virion production but not so stable that PR is unable to access the cleavage site in CA-SP1 and trigger maturation. MI PF96 or second-site mutations such as T8I increase 6HB stability without affecting IP6 packaging and reduce CA-SP1 processing and infectivity. Lysine ring mutations decrease 6HB stability, reducing IP6 packaging. These effects do not prevent processing but inhibit infectivity due to defects in mature capsid assembly. The addition of PF96 or a stabilizing mutation such as T8I to a lysine ring mutant rescues IP6 packaging by stabilizing the immature hexamer and thereby increasing affinity to the remaining ring residue.
Passage of the IP6 binding–deficient mutants K158A and K227A resulted in several second-site mutations that markedly increased viral replication. If IP6 were essential for virus replication, then one could anticipate two escape pathways: reversion of either mutation back to the WT sequence or the acquisition of second-site mutations that indirectly restore IP6 binding. In our experiments, we observed the latter pathway; mutations arose that increase IP6 binding indirectly by stabilizing the immature Gag hexamer and restoring binding of IP6 to the 6HB. This is in fact a more likely path to reversion because mutation of multiple residues in the hexamer can alter stability, providing the virus with several escape options. Moreover, reversion of alanine to lysine in either K158A or K227A requires two nucleotide changes (a transition and a transversion), whereas K158T or T8I mutations require only a single transition. The recovery of IP6-binding affinity through stabilization happens because a single lysine, either K158 or K227, is sufficient to retain some IP6 binding activity, as shown here and previously (1, 3). Packaged IP6 remains functionally important to viruses with a single lysine, as demonstrated by the fact that they remain sensitive to depletion of IP6 by KO of kinases IPPK or IPMK in virus-producer cells (1). Crucially, reducing the level of IP6 in producer cells reduces the number of virions produced but does not affect their specific infectivity (1). Thus, the susceptibility of K227A or K158A to IP6 depletion may not be obvious if infectivity data are normalized to production.
It is particularly notable that two of the second-site mutations (T8I and N193H) that arose upon passage of K158 and K227 mutant viruses were also acquired during selection experiments performed with replication-defective, MI-dependent mutants (8, 11). This is likely a consequence of the shared fitness requirement for an immature lattice that is stable enough to assemble and produce virions but not so stable as to prevent PR-mediated processing at the CA-SP1 junction. This notion of a “Goldilocks” zone of HIV-1 immature lattice stability is supported by the dual rescue observed with IP6 binding and second-site mutants. For instance, T8I rescues lysine mutant infectivity defects caused by lack of IP6 packaging, while the lysine mutants rescue the impaired CA-SP1 processing of T8I. The other 6HB mutation obtained in these experiments, M4L, shares the property of increased hexamer stability and would be predicted to increase the infectivity of previously described 6HB-destabilizing MI escape variants. The K158T mutation, while not a second-site mutation, can also be considered a stabilizing variant. While not as effective individually as the other mutants, K158T nevertheless increases viral production and decreases CA-SP1 processing, consistent with increased immature hexamer stability relative to K158A. K158T is reminiscent of K227I, which also rescues viral production relative to K227A, and likely stabilizes through a similar mechanism of hydrophobic packing that partially compensates for IP6 coordination. Together, our data suggest that because K158 and K227 mutants package less IP6 and are therefore less stable than WT, they are able to escape MIs, which can otherwise make immature hexamers hyperstable and defective for PR-mediated cleavage. The reduced hexamer stability of the lysine mutants also explains why PF96 actually promotes their infectivity; it restores hexamer stability and IP6 binding. The second-site mutations behave like MIs, stabilizing the immature hexamers and restoring infectivity and IP6 incorporation (Fig. 9B).
The fact that HIV-1 is capable of altering Gag lattice stability and CA-SP1 processing simply by changing residues within the immature hexamer poses the question why the virus needs IP6 in the first place. We have proposed (1, 3) that IP6 is required not only for immature lattice assembly but also for mature capsid formation. IP6 promotes assembly of mature capsids in vitro, while mutation of the IP6-coordinating residue, R18, in mature hexamers abolishes particle infectivity (1, 2). We suggest that this is a significant factor explaining why HIV-1 maintains IP6 binding. If IP6 were only required for immature lattice stabilization, then PF96 should be capable of functionally replacing it. Rather, the differing phenotypes of IP6 and MIs in a single round of infection suggest that while MIs only affect the immature lattice, IP6 also affects mature capsid assembly. MI potency is related to its ability to inhibit CA-SP1 processing, but processing alone is not predictive of the degree of infectivity loss in K227A and K158A. These mutants display similarly efficient CA-SP1 processing but differ in infectivity by two orders of magnitude. This discrepancy between CA-SP1 processing and changes in infectivity is also seen upon PF96 treatment; PF96 does not alter processing of either lysine mutant yet substantially increases their infectivity. What PF96 measurably changes are the levels of IP6 packaged into virions. There is a similar correspondence between infectivity and IP6 packaging with the addition of second-site compensatory mutations. The 6HB mutation T8I impairs CA-SP1 processing, although its infectivity is within one log of WT. When added to K158A, there is a small impact on processing, but the infectivity of the double mutant is increased by almost two orders of magnitude. Again, this is coincident with a marked restoration of IP6 packaging. These data are consistent with previous findings that the difference in infectivity between lysine mutants K158A and K227A cannot be explained by immature lattice assembly alone; similar levels of the two viruses are released from the cell (1), and in vitro data show that both mutants are similarly defective at in vitro assembly (22). It is notable that although the two structural classes of MI—BVM and its analogs and PF96—both have a similar mechanism of action (blocking CA-SP1 processing by stabilizing the 6HB), only PF96 is able to rescue the K158 and K227 mutants. This selective rescue by PF96 was also observed previously with the PF96-dependent MHR mutants (11). Structural studies of immature hexamers bound by BVM (and its analogs) and PF96 will be required to elucidate the basis for the selective ability of PF96 to rescue the lysine ring and MHR mutants.
Together, our findings support the model that IP6 is an important HIV cofactor that is required both for stabilizing the immature hexamer lattice and driving the formation of infectious mature capsids. Whether HIV can evolve to lose its dependence on IP6 as a cofactor in either immature or mature lattice assembly or indeed become independent of IP6 altogether, remains an open question. Of the two processes, we predict that it would be easier for immature lattice assembly to become IP6 independent, as K158 and K227 only appear to be involved in IP6 binding, whereas in the mature lattice, R18, or at least a charged pore, is required for nucleotide import (23). However, if IP6 were no longer actively recruited by the immature lattice, then HIV virions would likely have IP6 concentrations similar to that of the cytosol, and mature assembly would have to evolve to compensate. There is precedent among retroviruses to suggest that this is possible. A recent study showed that equine infectious anemia virus (EIAV) could form mature lattices at only 10 μM IP6 (24), which is lower than the concentration found in most cell types and suggests that it may not need to actively enrich IP6 similar to HIV. Ongoing studies will provide additional insights into the requirement for IP6, its roles throughout the HIV replication cycle, and whether this places any limitations on HIV evolution.
MATERIALS AND METHODS
Cells and plasmids
Human embryonic kidney (HEK) 293T CRL-3216 cells were purchased from American Type Culture Collection. All cells are regularly tested and are mycoplasma free. HEK293T and HeLa cell lines were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS), 2 mM l-glutamine, penicillin (100 U/ml), and streptomycin (100 mg/ml; Gibco) at 37°C with 5% CO2. The MT4 and SupT1 T cell lines were maintained in RPMI 1640 with l-glutamine (Corning) and supplemented with 10% FBS (GenClone), penicillin (100 U/ml), and streptomycin (100 mg/ml). Replication-deficient vesicular stomatitis virus glycoprotein (VSV-G)–pseudo-typed HIV-1 virions were produced in HEK293T cells using the packaging plasmid pMDG2, which encodes VSV-G envelope (Addgene plasmid no. 12259), pNL4-3–derived pCRV GagPol (HIV-1 clade B) (25), and pCSGW (26) as described previously (27). Mutagenesis of CA was performed using the QuickChange method (Stratagene) against pCRV GagPol, and primer sequences are given in table S1. The HIV-1 clade B infectious molecular clone pNL4-3 was used for all passage and virus release experiments. Mutant constructs were generated with the New England Biolabs (NEB) Q5 site directed mutagenesis kit (NEB, E0554), and primers (see table S1) were designed using the NEBaseChanger online tool.
Virus production
Viruses were produced from 2.5 × 106 cells in a 10-cm dish or 5 × 105 cells per well of a six-well plate, plated the day before. Transfection mixtures were made using 200 μl of Opti-MEM (Gibco), 1 μg of pMDG2, 1.5 μg of pCSGW, 1 μg of pCRV GagPol, and 12 μl of FuGENE6 (Promega). Mixtures were incubated at room temperature for 15 min and then added in entirety to 10-cm dishes or 60 μl added to a well of a six-well plate. Viral supernatants were harvested 48 hours after transfection, filtered through a 0.45-μm filter, and stored at −80°C. For MI treatment, cells were treated with 5 μM PF96, BVM (2 μg/ml), or 100 nM 7m as indicated at the time of transfection. Cells were media-changed to fresh media containing MIs 12 to 24 hours after transfection, and then viruses were harvested 5 hours later as above.
Infection experiments
For infection experiments with HEK293T, cells were seeded at 0.75 × 104 cells per well into 96-well plates and left to adhere overnight. Indicated amounts of virus were added, and the plates were scanned every 8 hours for up to 72 hours in an IncuCyte (Sartorius) to identify green fluorescent protein–expressing cells.
Virus quantification
The level of RT enzyme was quantified using quantitative reverse transcription polymerase chain reaction (PCR) as described previously with slight alterations (28). Briefly, 5 μl of viral supernatant was mixed with 5 μl of lysis buffer [0.25% Triton X-100, 50 mM KCl, 100 mM tris-HCl (pH 7.4), and 40% glycerol] and 0.1 μl of ribonuclease (RNase) inhibitor and incubated for 10 min at room temperature before diluting to 100 μl with nuclease-free water. Two microliters of lysate was added to 5 μl of TaqMan Fast Universal PCR Mix, 0.1 μl of MS2 RNA, 0.05 μl of RNase inhibitor, and 0.5 μl of MS2 primer mix, to a final volume of 10 μl. The reaction was run on an ABI StepOnePlus Real-Time PCR System (Life Technologies), with additional reverse transcription step (42°C for 20 min).
Purification and analysis of inositol phosphates
Virus particles were produced essentially as described above, but with cells precultured in inositol-free media supplemented with [3H] inositol (5 μCi/ml). The particles were concentrated by ultracentrifugation over a 20% (w/v) sucrose cushion (2 hours at 28,000 rpm), and resuspended in 1 ml PBS, followed by pelleting for 90 min at 15,000 rpm. Inositol phosphate extraction and analysis by HPLC were performed modifying a previously described protocol (29). Cells or pelleted virions labeled with [3H] inositol were resuspended in 200 μl of extraction solution (1 M perchloric acid and 5 mM EDTA) and incubated on ice or 100°C for 10 min. The samples were spun out at 13,000 rpm at 4°C for 5 min, and the supernatant was recovered. Supernatants from acid extractions were neutralized to pH 6 to pH 8 using 1 M potassium carbonate and 5 mM EDTA (approximately 100 μl) and incubated on ice with the lids open overnight at 4°C. Samples were spun at 13,000 rpm at 4°C for 5 min, and supernatant containing inositol phosphates was loaded onto HPLC or stored at 4°C. Inositol phosphates were resolved by SAX-HPLC on a PartiSphere SAX 4.6°—125-mm column (Hichrom). The column was eluted with a gradient generated by mixing buffer A (1 mM EDTA) and buffer B [1 mM EDTA and 1.3 M (NH4)2HPO4 (pH 4.0)] as follows: 0 to 5 min, 0% buffer B; 5 to 10 min, 0 to 30% buffer B; 10 to 85 min, 30 to 100% buffer B; 85 to 95 min, 100% buffer B. Fractions (1 ml) were collected and analyzed by scintillation counting after adding 4 ml of Ultima-Flo AP cocktail (PerkinElmer, 6013599).
Virus release
Virus release assays were performed as described previously (30). Briefly, HEK293T cells were transfected with 2 μg of pNL4-3 WT or mutant plasmids in six-well plates. Polyethylenimine (1 mg/ml) was used as the transfection reagent. At 48 hours after transfection, viral supernatants were filtered and pelleted by ultracentrifugation at 4°C. Virus pellets and remaining cells were lysed and probed for Gag via Western blot. HIV–immunoglobulin (Ig) [National Institutes of Health (NIH) AIDS Reagent Program, catalog no. 3957] was used as the primary antibody for Gag detection, and an anti-human IgG horseradish peroxidase (HRP)–tagged antibody (Sigma-Aldrich catalog no. GENA933) was used as the secondary antibody. SuperSignal West Pico PLUS (Thermo Fisher Scientific, 34578) was used as the chemiluminescent substrate. Imaging and band quantification were performed using the Sapphire Biomolecular Imager and Azure Spot analysis software (Azure Biosystems). Virus release was calculated using the following formula
Virus passage
For transfection experiments, 3 × 106 cells were transfected with 3 μg of pNL4-3 WT or mutant plasmid using DEAE-dextran (0.7 mg/ml) as a transfection reagent. T cell transfections were carried out for 15 min at 37°C with 5% CO2. Cells were maintained in RPMI 1640 with l-glutamine (Corning) and supplemented with 10% FBS (GenClone), penicillin (100 U/ml), and streptomycin (100 mg/ml). Cells were split at a 1:2 ratio, and supernatant aliquots were collected every other day beginning at day 2 after transfection. Viral replication kinetics were measured via RT assay as described previously (31). For MT4 infections, selected viral supernatants were RT-normalized and used to infect cells at a 1 RT unit:10 cell ratio for 1 hour at 37°C with 5% CO2. Cell splitting schedules and replication kinetic measurements were implemented as described above. MIs were added to growth media at the following concentrations: PF96 (5 μM) and BVM, 7m, and 7r (0.5 μM); and these media were used for the entirety of the experiments. Second-site compensatory mutations were identified by isolating genomic DNA from cells at the peak of replication and sequencing PCR-amplified proviral sequences with Gag specific primers. Mutations of interest were introduced into the infectious molecular clone pNL4-3 via site-directed mutagenesis (Q5 mutagenesis kit, NEB) alone and in combination with other relevant mutations.
Western blotting
Samples were run on 4 to 12% bis-tris gels and transferred onto nitrocellulose membranes using iBlot (Life Technologies) and detected by enhanced chemiluminescence or by LI-COR for quantification. Anti-HIV p24 (183-H12-5C) was obtained from the NIH AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH; anti–HIV-1 p24 monoclonal (183-H12-5C) (catalog no. 3537) was obtained from B. Chesebro and K. Wehrly (32, 33), and loading control COX IV (P/N 926-42214) was obtained from LI-COR Biosciences. For virus release, samples were subjected to SDS–polyacrylamide gel electrophoresis (4 to 20%) and then transferred to a polyvinylidene fluoride membrane (Immobilon, Millipore) via semidry transfer (Bio-Rad Trans-Blot Turbo). The membrane was blocked for 1 hour with 5% nonfat milk in tris-buffered saline + 0.05% Tween 20 detergent (TBST) and incubated overnight at 4°C with anti-HIV IgG. The membrane was then washed with TBST, incubated for 2 hours with anti-human HRP-conjugated secondary antibody, and washed again. SuperSignal West Pico PLUS (Thermo Fisher Scientific) was used to reveal protein bands.
Virus particle production for tomography
Virus-like particles were produced in HEK293T as described above. Supernatants were harvested and passed through a 0.45-μm filter, followed by a 0.22-μm filter. The particles were concentrated by ultracentrifugation over a 20% (w/v) sucrose cushion (2 hours at 28,000 rpm in a Beckman SW32 rotor, Beckman Coulter Life Sciences). Resuspended particles were applied to a 6 to 18% iodixanol gradient (1.2% increment steps) and centrifuged for 1.5 hours at 250,000g in a Beckman SW40 rotor (Beckman Coulter Life Sciences) (34). The virus containing fraction was diluted in 1:10 phosphate-buffered saline (PBS) and concentrated by ultracentrifugation (45 min at 38,500 rpm in a Beckman SW40 rotor, Beckman Coulter Life Sciences). The pellet was resuspended in PBS and incubated at 4°C overnight to allow full resuspension.
Cryo-tomography
Ten-nanometer-diameter colloidal gold beads were added 1:1 to the purified HIV-1 mutants. Four-microliter sample-gold suspension was applied to a glow discharged C-Flat 2/2 3C (20 mA for 40 s). The grids were blotted and plunge-frozen in liquid ethane using an FEI Vitrobot Mark II at 15°C and 100% humidity. Tomography of WT virus was performed on a Tecnai F20 transmission electron microscope (FEI/Thermo Fisher Scientific), equipped with a Falcon III Direct Electron detector, operated at 200 kV, and controlled using Serial-EM (35). Tomographic tilt series were acquired under low-dose conditions with a tilt range between −40° and +40°, angular increments of 3°, defoci between −3 and −6 μm, and at a magnification of 50,000× giving a pixel size of 2.09 Å. Tomography of the mutants was performed on a FEI Titan Krios transmission electron microscope at 300 kV equipped with a Gatan K2 summit direct electron detector and a Gatan Quantum energy filter (Gatan imaging filter). Tilt series were acquired between −60° and +60° with increments of 3° using a dose symmetric scheme using Serial-EM (35). Images were collected at a magnification of 33,000× with 10 frames per tilt and a total dose of ∼120 e−/Å2 across all of the tilts. Frames were aligned in Serial-EM with a final pixel size of 3.667 Å per pixel in the unbinned image stacks. Tomograms were reconstructed using IMOD (4.9) (36). The alignment of 2D projection images of the tilt series was performed using gold beads as fiducial markers, and tomograms were reconstructed by back projection. The margin of error (MOE) for each virion category was calculated using the equation , based on a confidence level (CI) of 95% and where n is the total number of virions classified and p is the proportion of a particular classification. The number of virions per category was plotted in fig. S7, and the MOE was indicated with error bars.
CA-SP1 expression
CA-SP1 proteins were expressed in Escherichia coli C41 by inducing with 0.4 mM isopropyl-β-d-thiogalactopyranoside for 4 hours at 37°C. Bacterial pellets were resuspended in lysis buffer [100 mM tris (pH 8.0), 200 mM NaCl, 1 mM dithiothreitol (DTT), protease inhibitor (1 tablet/liter), bug buster, and 20 mM Imidazole] and lysed by sonication, and cellular debris was removed by centrifugation (1 hour at 20,000 rpm). The supernatant was filtered, affinity-purified with a Ni2+ resin, and eluted with imidazole. The eluted protein was dialyzed overnight in presence of ULP1 protease (ubiquitin-like-specific protease). Reverse Ni2+ chromatography was used to remove the SUMO tag and ULP1 protease. The protein was further purified by size exclusion chromatography [Superdex 75, 50 mM tris-HCl (pH 8), 50 mM NaCl, and 1 mM DTT].
Turbidity assays
A total of 1 mM IP6 and/or 0.5 mM or 1 mM PF96 was added to 250 μM CA-SP1 proteins. The increase in an absorbance at 350 nm was measured with a PHERAstar FSX Plate reader (BMG LABTECH) in 384-well plate for 100 min every 22 s with shaking after each measurement.
Negative stain
Samples from the turbidity assay were allowed to sediment overnight. Four microliters of sample was put onto a glow discharged carbon coated grid (Cu, 400 mesh, Electron Microscopy Services), washed, and stained with 2% uranyl acetate. Micrographs were taken at room temperature on a Tencai Spirit (FEI) operated at an accelerated voltage of 120 keV and Gatan 2k × 2k charge-coupled device (CCD) camera. Images were collected with a total dose of ~30 e−/Å2 and a defocus of 1 to 3 μm.
Virus production for TIRF microscopy
Replication-deficient HIV-1 virions without envelope protein were produced in HEK293T cells using pCRV-1 GagPol and CSGW, biotinylated using EZ-Link Sulfo-NHS-LC-LC-Biotin (Thermo Fisher Scientific, 21338), and purified as described (21, 37).
TIRF imaging of capsids in permeabilized viral particles
TIRF microscopy was carried out following the published method of Marquez et al. (21, 37). Briefly, biotinylated viral particles were captured onto coverslips attached to microfluidic flow cells and imaged using a custom built TIRF microscope with an ASI-RAMM frame (Applied Scientific Instrumentation), a Nikon 100× CFI Apochromat TIRF (1.49 numerical aperture) oil immersion objective and NicoLase laser system. Immobilized virions were treated with imaging buffer containing 200 nM perfringolysin O (PFO), to permeabilize the lipid envelope, and labeled CypA (0.5 to 1 μM), to detect the capsid. TIRF images were then acquired with a frequency of 1 frame/6 s using a 561-nm laser with a 20-ms exposure time for excitation and an Andor iXon 888 EMCCD camera for detection. Single-virion fluorescence traces were extracted from the TIRF image stacks using the JIM Immobilized Microscopy analysis package (https://github.com/lilbutsa/JIM-Immobilized-Microscopy-Suite) and further analyzed in MATLAB (The MathWorks Inc.) using the software adapted from previous work (38). Briefly, the duration of the CypA signal was extracted from fluorescence traces by step-fitting using change point analysis. Capsid stability was quantified as the time difference between acquisition of Alexa Fluor 568–CypA upon permeabilization and loss of fluorescence upon capsid uncoating.
Statistical analysis
Unless otherwise indicated, statistical analyses were Student’s t tests and performed using GraphPad Prism 9 software (GraphPad). Error bars depict the means ± SEM unless indicated otherwise.
Acknowledgments
Funding: D.L.M., N.R., and L.C.J. are supported by the Medical Research Council (MRC) (UK; U105181010) and a Wellcome Trust Investigator Award (200594/Z/16/Z). L.C.J. and T.B. are supported by a Wellcome Trust Collaborator Award (214344/A/18/Z). T.B. and K.M.R.F. are supported by NHMRC (Australia; APP1182212 and APP1158338). L.K. is supported by a PhD Fellowship of the Boehringer Ingelheim Fonds. J.A.G.B. is supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (ERC-CoG-648-432 MEMBRANEFUSION) and by the MRC (MC_UP_1201/16). A.S. is supported by the MRC (grant MR/T028904/1). Research in the Freed laboratory is supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH. A.B.K. is supported in part by an Intramural AIDS Research Fellowship. Author contributions: Study was conceived by D.L.M., A.B.K., E.O.F., and L.C.J. Manuscript was written by D.L.M., A.B.K., E.O.F., and L.C.J., with contributions from all authors. Experiments were performed by D.L.M., A.B.K., N.R., K.M.R.F, M.N., L.K., M.S.C.W., B.A., Z.K., and A.S. Analysis was carried out by all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/11/eabe4716/DC1
REFERENCES AND NOTES
- 1.Mallery D. L., Faysal K. M. R., Kleinpeter A., Wilson M. S. C., Vaysburd M., Fletcher A. J., Novikova M., Böcking T., Freed E. O., Saiardi A., James L. C., Cellular IP6 levels limit HIV production while viruses that cannot efficiently package IP6 are attenuated for infection and replication. Cell Rep. 29, 3983–3996.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dick R. A., Zadrozny K. K., Xu C., Schur F. K. M., Lyddon T. D., Ricana C. L., Wagner J. M., Perilla J. R., Ganser-Pornillos B. K., Johnson M. C., Pornillos O., Vogt V. M., Inositol phosphates are assembly co-factors for HIV-1. Nature 560, 509–512 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mallery D. L., Márquez C. L., McEwan W. A., Dickson C. F., Jacques D. A., Anandapadamanaban M., Bichel K., Towers G. J., Saiardi A., Böcking T., James L. C., IP6 is an HIV pocket factor that prevents capsid collapse and promotes DNA synthesis. eLife 7, e35335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kleinpeter A. B., Freed E. O., HIV-1 maturation: Lessons learned from inhibitors. Viruses 12, 940 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li F., Goila-Gaur R., Salzwedel K., Kilgore N. R., Reddick M., Matallana C., Castillo A., Zoumplis D., Martin D. E., Orenstein J. M., Allaway G. P., Freed E. O., Wild C. T., PA-457: A potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl. Acad. Sci. U.S.A. 100, 13555–13560 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou J., Yuan X., Dismuke D., Forshey B. M., Lundquist C., Lee K.-H., Aiken C., Chen C. H., Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J. Virol. 78, 922–929 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagner J. M., Zadrozny K. K., Chrustowicz J., Purdy M. D., Yeager M., Ganser-Pornillos B. K., Pornillos O., Crystal structure of an HIV assembly and maturation switch. eLife 5, e17063 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adamson C. S., Ablan S. D., Boeras I., Goila-Gaur R., Soheilian F., Nagashima K., Li F., Salzwedel K., Sakalian M., Wild C. T., Freed E. O., In vitro resistance to the human immunodeficiency virus type 1 maturation inhibitor PA-457 (Bevirimat). J. Virol. 80, 10957–10971 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urano E., Timilsina U., Kaplan J. A., Ablan S., Ghimire D., Pham P., Kuruppu N., Mandt R., Durell S. R., Nitz T. J., Martin D. E., Wild C. T., Gaur R., Freed E. O., Resistance to second-generation HIV-1 maturation inhibitors. J. Virol. 93, e02017-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blair W. S., Cao J., Fok-Seang J., Griffin P., Isaacson J., Jackson R. L., Murray E., Patick A. K., Peng Q., Perros M., Pickford C., Wu H., Butler S. L., New small-molecule inhibitor class targeting human immunodeficiency virus type 1 virion maturation. Antimicrob. Agents Chemother. 53, 5080–5087 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waki K., Durell S. R., Soheilian F., Nagashima K., Butler S. L., Freed E. O., Structural and functional insights into the HIV-1 maturation inhibitor binding pocket. PLOS Pathog. 8, e1002997 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urano E., Miyauchi K., Kojima Y., Hamatake M., Ablan S. D., Fudo S., Freed E. O., Hoshino T., Komano J., A triazinone derivative inhibits HIV-1 replication by interfering with reverse transcriptase activity. ChemMedChem 11, 2320–2326 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Purdy M. D., Shi D., Chrustowicz J., Hattne J., Gonen T., Yeager M., MicroED structures of HIV-1 Gag CTD-SP1 reveal binding interactions with the maturation inhibitor bevirimat. Proc. Natl. Acad. Sci. U.S.A. 115, 13258–13263 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keller P. W., Adamson C. S., Heymann J. B., Freed E. O., Steven A. C., HIV-1 maturation inhibitor bevirimat stabilizes the immature Gag lattice. J. Virol. 85, 1420–1428 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.L. Mendonça, D. Sun, J. Ning, J. Liu, A. Kotecha, M. Olek, T. Frosio, X. Fu, B. A. Himes, A. B. Kleinpeter, E. O. Freed, J. Zhou, C. Aiken, P. Zhang, CryoET structures of immature HIV Gag reveal a complete six-helix bundle and stabilizing small molecules distinct from IP6. bioRxiv 2020.10.31.363382 [Preprint]. 1 November 2020. 10.1101/2020.10.31.363382. [DOI]
- 16.Kucharska I., Ding P., Zadrozny K. K., Dick R. A., Summers M. F., Ganser-Pornillos B. K., Pornillos O., Biochemical reconstitution of HIV-1 assembly and maturation. J. Virol. 94, e01844-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fontana J., Keller P. W., Urano E., Ablan S. D., Steven A. C., Freed E. O., Identification of an HIV-1 mutation in spacer peptide 1 that stabilizes the immature CA-SP1 lattice. J. Virol. 90, 972–978 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mattei S., Anders M., Konvalinka J., Krausslich H. G., Briggs J. A. G., Muller B., Induced maturation of human immunodeficiency virus. J. Virol. 88, 13722–13731 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fontana J., Jurado K. A., Cheng N., Ly N. L., Fuchs J. R., Gorelick R. J., Engelman A. N., Steven A. C., Distribution and redistribution of HIV-1 nucleocapsid protein in immature, mature, and integrase-inhibited virions: A role for integrase in maturation. J. Virol. 89, 9765–9780 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Schwedler U. K., Stray K. M., Garrus J. E., Sundquist W. I., Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 77, 5439–5450 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marquez C. L., Lau D., Walsh J., Shah V., Guinness C. M., Wong A., Aggarwal A., Parker M. W., Jacques D. A., Turville S., Böcking T., Kinetics of HIV-1 capsid uncoating revealed by single-molecule analysis. eLife 7, e34772 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith W. B. II, McCaslin I. R., Gokce A., Mandava S. H., Trost L., Hellstrom W. J., PDE5 inhibitors: Considerations for preference and long-term adherence. Int. J. Clin. Pract. 67, 768–780 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Jacques D. A., McEwan W. A., Hilditch L., Price A. J., Towers G. J., James L. C., HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature 536, 349–353 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dick R. A., Xu C., Morado D. R., Kravchuk V., Ricana C. L., Lyddon T. D., Broad A. M., Feathers J. R., Johnson M. C., Vogt V. M., Perilla J. R., Briggs J. A. G., Schur F. K. M., Structures of immature EIAV Gag lattices reveal a conserved role for IP6 in lentivirus assembly. PLOS Pathog. 16, e1008277 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zennou V., Perez-Caballero D., Göttlinger H., Bieniasz P. D., APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J. Virol. 78, 12058–12061 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naldini L., Blomer U., Gallay P., Ory D., Mulligan R., Gage F. H., Verma I. M., Trono D., In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272, 263–267 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Price A. J., Jacques D. A., McEwan W. A., Fletcher A. J., Essig S., Chin J. W., Halambage U. D., Aiken C., James L. C., Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLOS Pathog. 10, e1004459 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vermeire J., Naessens E., Vanderstraeten H., Landi A., Iannucci V., van Nuffel A., Taghon T., Pizzato M., Verhasselt B., Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLOS ONE 7, e50859 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azevedo C., Saiardi A., Extraction and analysis of soluble inositol polyphosphates from yeast. Nat. Protoc. 1, 2416–2422 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Waheed A. A., Ono A., Freed E. O., Methods for the study of HIV-1 assembly. Methods Mol. Biol. 485, 163–184 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Willey R. L., Smith D. H., Lasky L. A., Theodore T. S., Earl P. L., Moss B., Capon D. J., Martin M. A., In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J. Virol. 62, 139–147 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Toohey K., Wehrly K., Nishio J., Perryman S., Chesebro B., Human immunodeficiency virus envelope V1 and V2 regions influence replication efficiency in macrophages by affecting virus spread. Virology 213, 70–79 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Wehrly K., Chesebro B., p24 antigen capture assay for quantification of human immunodeficiency virus using readily available inexpensive reagents. Methods 12, 288–293 (1997). [DOI] [PubMed] [Google Scholar]
- 34.Dettenhofer M., Yu X.-F., Highly purified human immunodeficiency virus type 1 reveals a virtual absence of Vif in virions. J. Virol. 73, 1460–1467 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mastronarde D. N., Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Kremer J. R., Mastronarde D. N., McIntosh J. R., Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Marquez C. L., Lau D., Walsh J., Faysal K. M. R., Parker M. W., Turville S. G., Böcking T., Fluorescence microscopy assay to measure HIV-1 capsid uncoating kinetics in vitro. Bio-protocol. 9, e3297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Böcking T., Aguet F., Harrison S. C., Kirchhausen T., Single-molecule analysis of a molecular disassemblase reveals the mechanism of Hsc70-driven clathrin uncoating. Nat. Struct. Mol. Biol. 18, 295–301 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/11/eabe4716/DC1