

Abstract
Noncanonical RNA motifs help define the vast complexity of RNA structure and function, and in many cases, these loops and junctions are on the order of only ten nucleotides in size. Unfortunately, despite their small size, there is no reliable method to determine the ensemble of lowest energy structures of junctions and loops at atomic accuracy. This chapter outlines straightforward protocols using a webserver for Rosetta Fragment Assembly of RNA with Full Atom Refinement (FARFAR) (http://rosie.rosettacommons.org/rna_denovo/submit) to model the 3D structure of small noncanonical RNA motifs for use in visualizing motifs and for further refinement or filtering with experimental data such as NMR chemical shifts.
Keywords: RNA 3D structure prediction, RNA Motifs
1. Introduction
RNA plays critical roles in all living systems through its ability to adopt complex 3D structures and perform chemical catalysis [1]. RNA structure appears modular in nature, defined through base pairing interactions. Nucleotides can either form structured helices composed of canonical Watson–Crick base pairs or small unpaired or noncanonical base paired regions in the form of junctions and loops (motifs) [2–4]. Helices are, for the most part, structurally similar to each other, leaving noncanonical motifs to define the vast complexity of RNA structure and function. These noncanonical elements define the topology of the 3D structure of RNA by orienting the helices to which they connect and by forming long-range tertiary contacts that can lock specific global RNA conformations in place. In addition to defining the overall 3D structure of RNA [5, 6], noncanonical motifs are the sites of small molecule binding and chemical catalysis [7–10]. Many noncanonical motifs are on the order of only ten nucleotides in size. Unfortunately, despite their small size, there is no reliable method to determine the ensemble of lowest energy structures of junctions and loops at near atomic accuracy. Nevertheless, to model RNA at high resolution, it is critical to achieve accurate solutions for these small motifs.
When their structures are solved experimentally, most motifs turn out to form complex arrangements of non Watson–Crick hydrogen bonds and a wide range of backbone conformations. Due to the large number of interactions possible and each nucleotide’s many degrees of internal freedom, it remains difficult to determine the lowest energy conformation [11]. Fragment assembly of RNA with full atom refinement (FARFAR) was an early attempt to help address this problem. FARFAR adapted the well-developed Rosetta framework for protein structure modeling to predict and design RNA noncanonical motifs [12]. Out of a 32-target test set, 14 cases gave at least one out of five models that were better than 2.0 Å all-heavy-atom RMSD to the experimentally observed structure. While not perfect, this level of accuracy can be combined with even sparse experimental data, such as 1H chemical shifts, to obtain high confidence structural models, as was demonstrated recently in blind predictions with the CS-ROSETTA -RNA method [13]. The motif models can also form building blocks for modeling more complex RNAs and has been tested in the RNA-Puzzles trials [14]. Application of FARFAR method for large RNAs with complex folds has been reviewed recently [15]. The current bottleneck for some of these motifs and for larger RNAs is the difficulty of complete conformational sampling [11]. On-going work with stepwise assembly (SWA) attempts to resolve this issue [16], but this more advanced procedure requires greater computational expense and a complex workflow that is not yet straightforward to implement on a public server, except in the special case of one-nucleotide-at-a-time crystallographic refinement [17]. Stepwise assembly is available in the main Rosetta codebase, but is not further discussed here.
This chapter outlines straightforward protocols that are enabling expert scientists and citizen scientists in the Eterna platform [18] to access FARFAR 3D RNA modeling through a simple web server. FARFAR (RNA De Novo) is part of the Rosetta Online Server that Includes Everyone (ROSIE) software, a push to give wide access to the algorithms found in the Rosetta 3.x framework [19]. The web server requires no initial setup for the user; all that is needed is to supply a sequence and an optional secondary structure to obtain all-atom models for an RNA motif of interest.
1.1. FARFAR Calculation
The FARFAR structure-modeling algorithm is based on two discrete steps. First, the RNA is assembled using 1–3 nucleotide fragments from existing RNA crystal structures whose sequences match subsequences of the target RNA. Fragment Assembly of RNA (FARNA) uses a Monte Carlo process guided by a low-resolution knowledge-based energy function [20]. Afterwards, these models can be further refined in an all-atom potential to yield structures with hydrogen bonds with realistic geometries and fewer clashes; the resulting energies are also better at discriminating native-like conformations from non-native conformations [12]. The two-stage protocol is called fragment assembly of RNA with full atom refinement (FARFAR).
2. Materials
FARFAR (RNA De Novo) is a webserver implementation of the Rosetta RNA fragment assembly algorithm server using the ROSIE framework. ROSIE is a web front-end for Rosetta 3 software suite, which provides experimentally tested and rapidly evolving tools for the high-resolution 3D modeling of nucleic acids, proteins, and other biopolymers. FARFAR (RNA De Novo) can be reached using any of the standard web browsers such as Apple Safari, Microsoft Internet Explorer, Mozilla Firefox, and Google Chrome here: http://rosie.rosettacommons.org/rna_denovo/submit.
3. Methods
This protocol outlines the steps to use the FARFAR (RNA De Novo) webserver located on the ROSIE website. Although it is possible to submit jobs without creating an account, having an account yields numerous benefits, such as email alerts when jobs are finished, as well as the ability to create private jobs that are not visible to other users. It is highly recommended to create an account when first visiting ROSIE. In addition to the FARFAR webserver, ROSIE also hosts many other Rosetta based applications with a continuous stream of novel applications in development.
3.1. Main Page Form
This demonstration of FARFAR (RNA De Novo) uses the GCAA tetraloop; the whole structure was determined through NMR spectroscopy by Jucker et al. (PDB 1ZIH) [21]. This tetraloop has a sequence of gggcgcaagccu and secondary structure of ((((….)))) in dot parentheses notation (Fig. 1). Figure 2 shows the main submission form for the RNA De Novo server. The only required input is the sequence, from 5′ to 3′. This is typically in lowercase letters, but uppercase letters are acceptable and will be converted. Use a space, *, or + between strands (see below for a test case with-multiple strands). Note that this sequence is treated as RNA so that any T’s that appear in the sequence are automatically converted to U’s for the calculation. Next, enter the secondary structure, in dot-parentheses notation. This is optional for single-stranded motifs, but required for multi-strand motifs. Note that even if a location is “unpaired” in the input secondary structure (given by a dot, “ . ”), it is not forced to remain unpaired. Although this is optional for single stranded motifs, the results improve with the addition of the correct secondary structure. If uncertain about the secondary structure, consider utilizing the Vienna RNAfold webserver [22] (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) or other utilities described in this book. Alternatively, use chemical mapping techniques to estimate the secondary structure these methods have been recently tested in blind trials for their accuracy [23, 24]. In addition, note that there is currently a size submission limit of 32 nucleotides for FARFAR (RNA De Novo), as the amount of computation greatly increases as a function of number of residues.
Fig. 1.
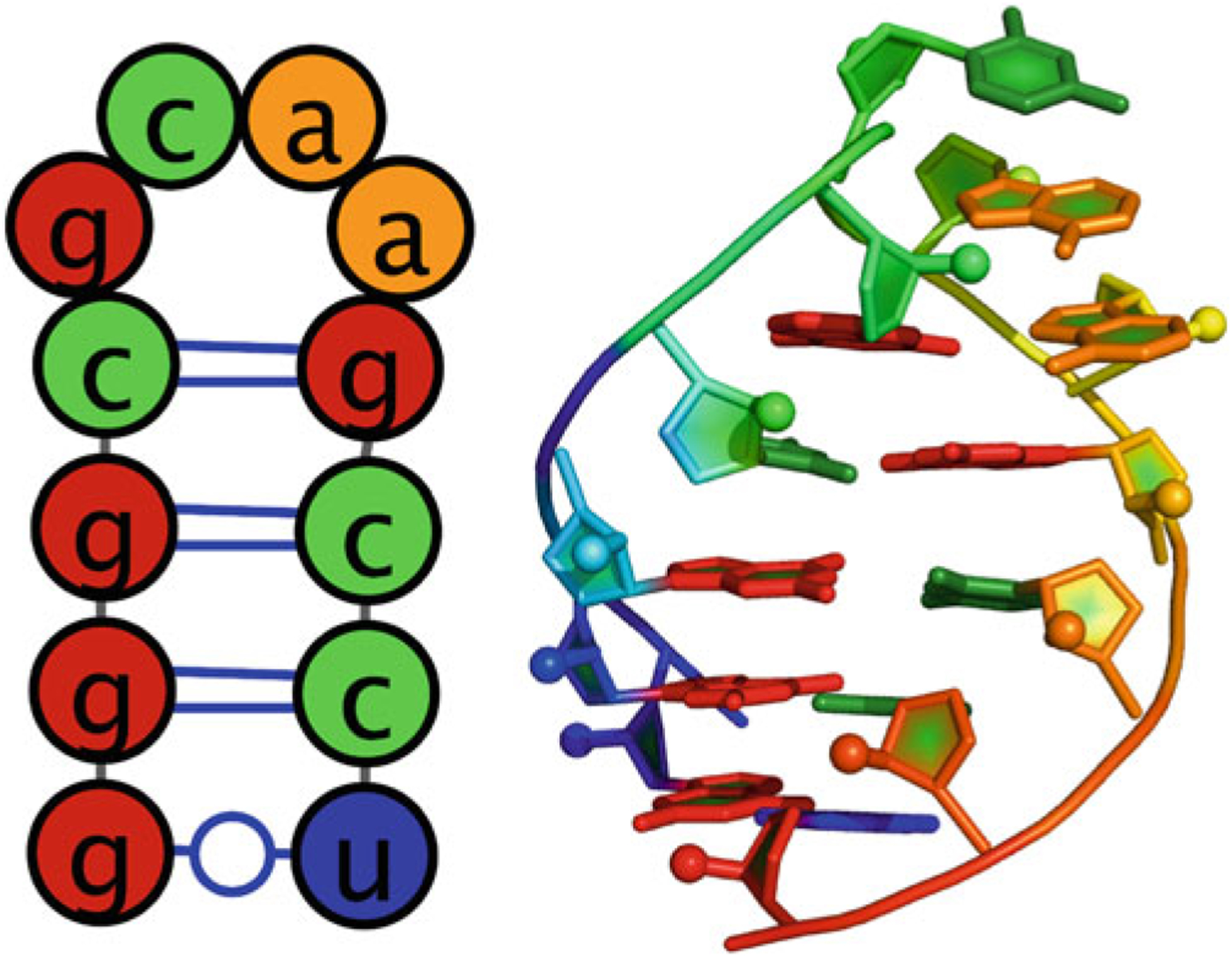
(Left) secondary structure of GCAA tetraloop. (Right) 3D structure of GCAA tetraloop (PDB: 1ZIH)
Fig. 2.
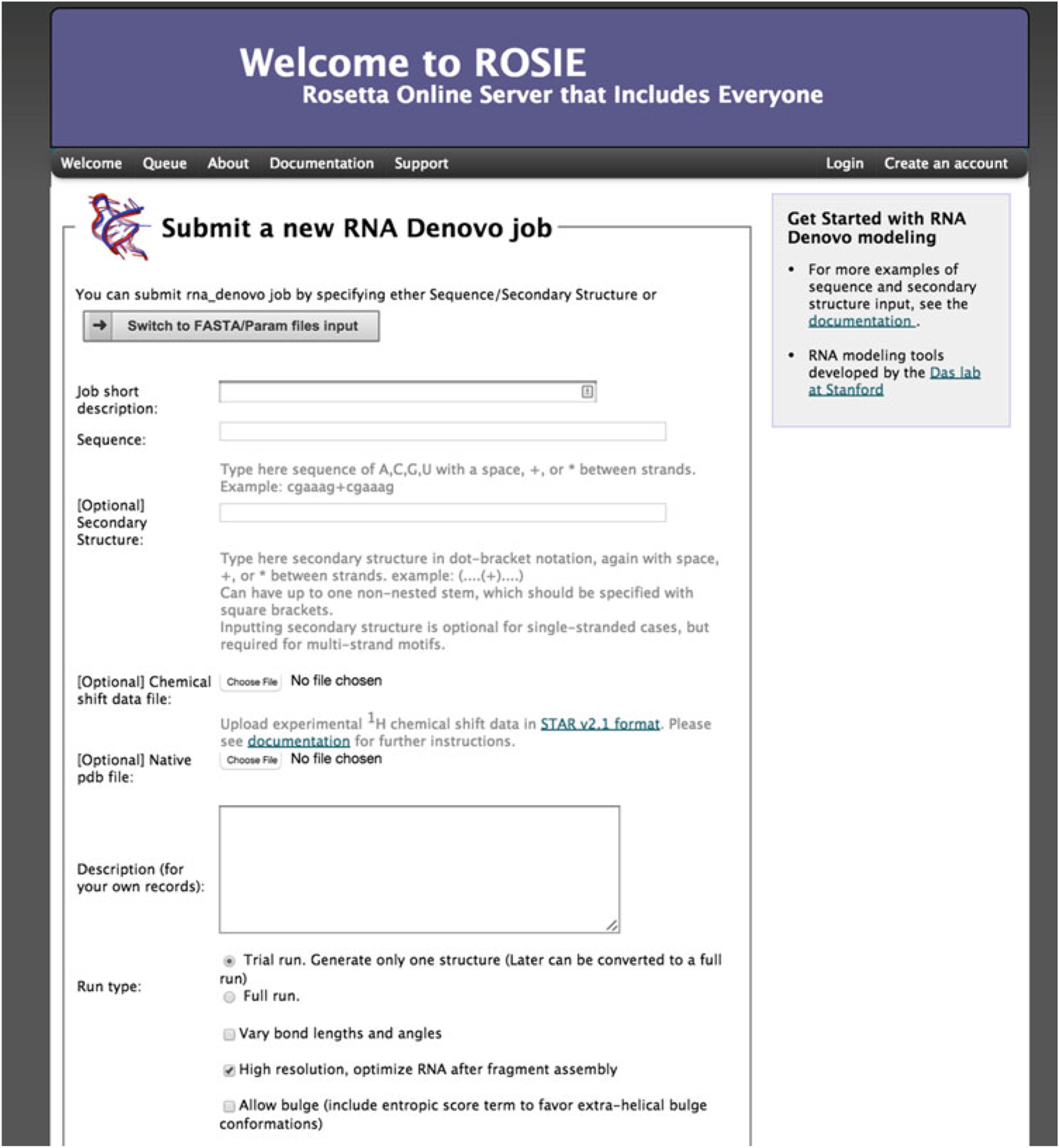
Main page of the FARFAR (RNA De Novo) webserver. Here the user can enter a sequence and secondary to submit a job to generation an all atom model of their construct
There are two more optional arguments. First is a file containing the 1H chemical shifts determined by NMR spectroscopy. The format of this file follows the STAR v2.1 format used by the Biological Magnetic Resonance Data Bank (BMRB) [25]. An example of the format is displayed in Fig. 3 with an explanation of each column. In addition, it is possible to supply a native structure for RMSD calculations. This file must be in PDB format, and for this case it is possible to download the structure from http://pdb.org/pdb/explore/explore.do?structureId=1zih. To supply a native structure, click the “Choose file” button next to native PDB-formatted file and select the appropriate file from your local hard drive.
Fig. 3.
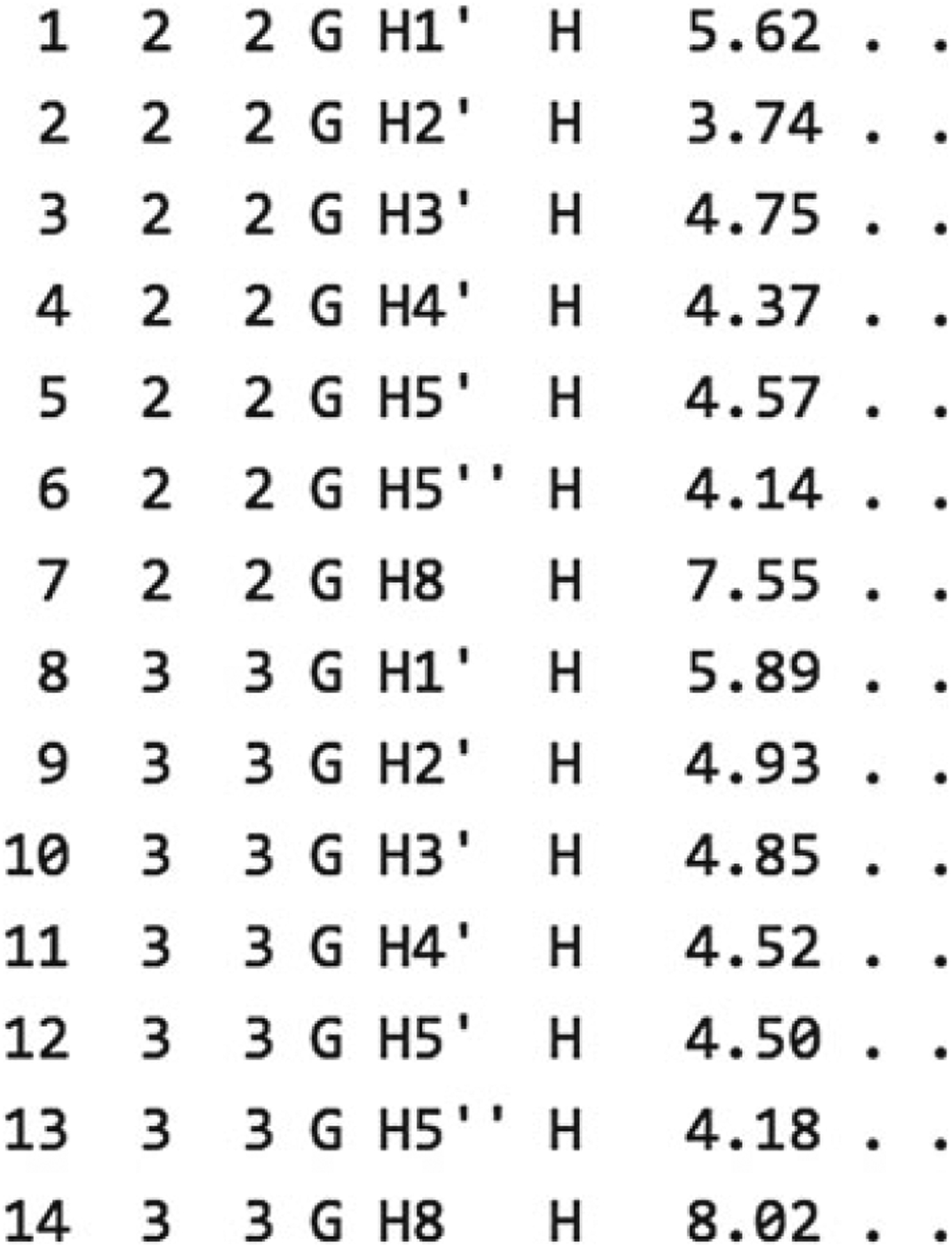
Example chemical shift data. Column description is as follows. (1) Atom entry number. (2) Residue author sequence code. (3) Residue sequence code. (4) Residue label. (5) Atom name. (6) Atom type. (7) Chemical shift value. (8) Chemical shift value error. (9) Chemical shift ambiguity code
There are two ways of running a FARFAR (RNA De Novo) job. The first is a trial run, which generates only one structure with a limited number of fragment assembly steps. This is for testing purposes only, and allows confirmation that the job is set up properly. The second is a full run that takes more computational time to complete and produces thousands of models. It is advised when setting up a job for a new sequence and secondary structure to always first run the job as a trial. Then, using www.pymol.org or your favorite viewer, open the PDB file; we use the PyMOL visualization script rr() available as part of the RiboVis package (https://ribokit.github.io/RiboVis/). This is particularly important if you have a multi-stranded motif—check that the strands are separated, and that any specified Watson–Crick pairs are reasonably paired. Once this is set up, go to the bottom of the page and click “Submit FARFAR (RNA De Novo) job”. Upon submission, a temporary status page will load (Fig. 4).
Fig. 4.
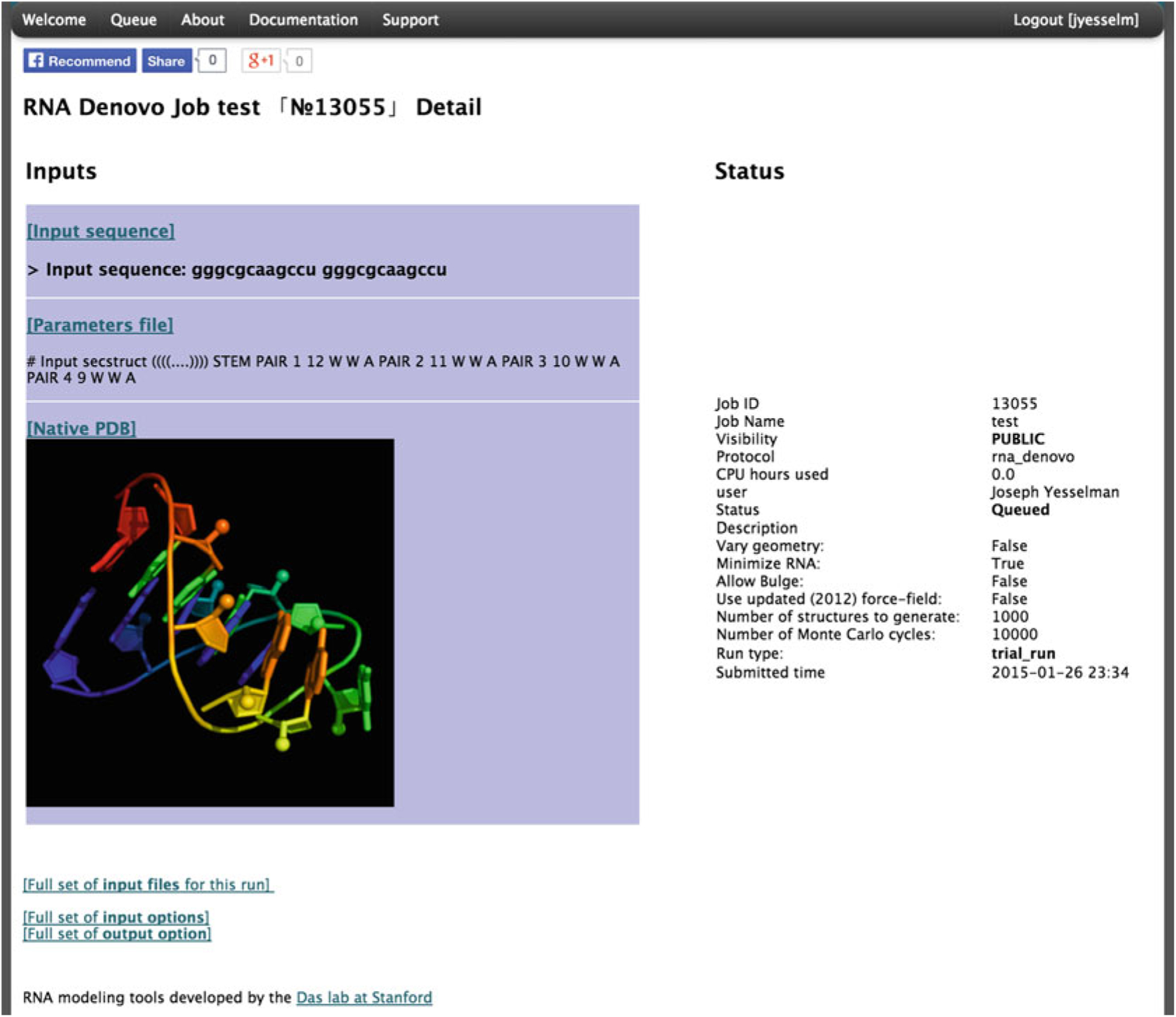
The status page for a submitted FARFAR (RNA De Novo) job
3.2. Advanced Options
In addition to the options discussed above, there are a few additional options that may be used occasionally. First is “Vary bond lengths and angles”; typically each residue has a set of bond lengths and angles between atoms that are based on idealized parameters. Checking this option will allow these parameters to vary slightly based on the Rosetta force field energy. This can increase conformational search space if you are interested in a specific interaction between residues and was used in previous benchmark studies, but requires more computational time [12].
When checked, “High resolution, optimize RNA after fragment assembly” will perform the all-atom refinement after fragment assembly; it is not recommended to uncheck this unless you are interested in quickly seeing the initial results or would like to perform your own high-resolution optimization. “Allow bulge (include entropic score term to favor extra-helical bulge conformations)”, will include conformations with residues bulged out and not interacting with other residues. If a residue is known to be extruded from the helix, this might be a good option to try to reduce the conformational space searched. When “Allow bulge (include entropic score term to favor extra-helical bulge conformations)” is checked, please note that residues that are bulged out will not be present in the final pdb model. “Number of structures to generate”, will change the number of final models, which will also greatly increase the time each run takes. “Number of Monte Carlo cycles”, controls the quality of each model; if models generated for a specific run have wildly different structures, then FARFAR has poor confidence in the accuracy (see next section). Increasing the number of Monte Carlo cycles can increase convergence, at the expense of greater computation.
3.3. Server Results
The server returns pictures of the best-scoring models from the five best-scoring clusters from the run in rank order by energy (Fig. 5). The clustering radius is 2.0 Å by default. Click on the [Model-N] link to download the PDB file. The server returns cluster centers (without pictures) for the next 95 clusters as, as well as the top 20 lowest-energy structures. These may be valuable if you are filtering models based on experimental data. The server also returns a “scatter plot” of the energies of all the models created. The x-axis is a distance measure from the native/reference model in RMSD (root mean-squared deviation) over all heavy atoms; if a reference model is not provided, then the RMSD is computed relative to the lowest energy model discovered by FARFAR. The y-axis is the score (energy) of the structure. In runs where a native structure is not supplied, the x-axis is a distance measure from the best scoring model found. As with nearly every Rosetta application, a hallmark of a successful run is convergence, visible as an energetic “funnel” of low-energy structures clustered around a single position. That is, near the lowest energy model there are additional models within ~2 Å RMSD. In such runs, the lowest energy cluster centers have a reasonable chance of covering native-like structures for the motif, based on our benchmarks. A hallmark of an unsuccessful run is a lack of convergence—few structures within 2 Å RMSD of the lowest energy model. Below the scatter plot, there is a detailed table of all the score terms used to calculate the final score as well as the RMSD to the native structure (if supplied). A description of the meaning of each term can be found in Table 1.
Fig. 5.
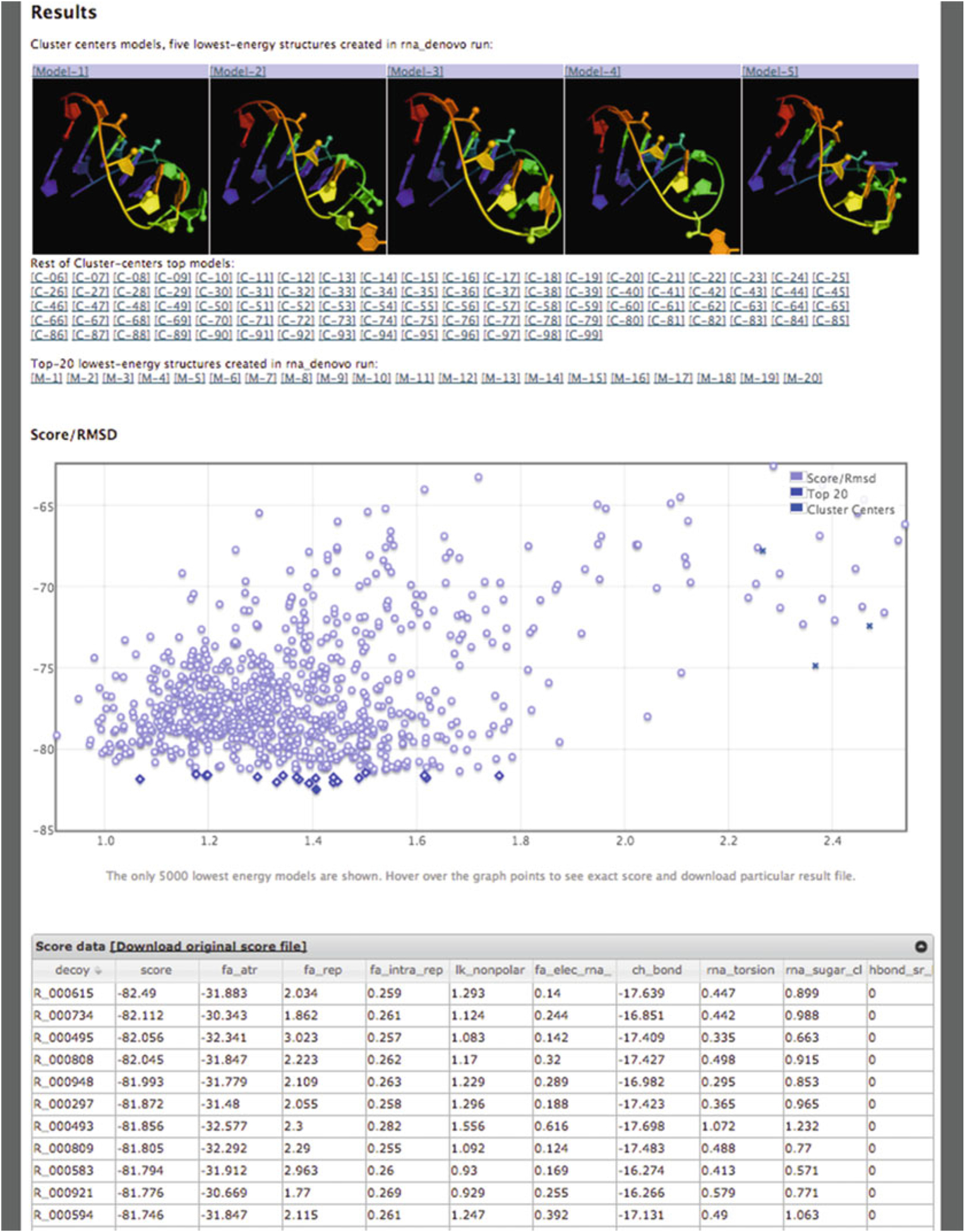
Results page for a RNA De Novo job
Table 1.
Score terms reported on RNA De Novo results page
| Term | Definition |
|---|---|
| Score | Final total score |
| fa_atr | Lennard-Jones attractive between atoms in different residues |
| fa_rep | Lennard-Jones repulsive between atoms in different residues |
| fa_intra_rep | Lennard-Jones repulsive between atoms in the same residue |
| lk_nonpolar | Lazaridis-Karplus solvation energy, over nonpolar atoms |
| fa_elec_rna_phos_phos | Simple electrostatic repulsion term between phosphates |
| ch_bond | Carbon hydrogen bonds |
| rna_torsion | RNA torsional potential |
| rna_sugar_close | Term that ensures that ribose rings stay closed during refinement |
| hbond_sr_bb_sc | Backbone-sidechain hbonds close in primary sequence |
| hbond_lr_bb_sc | Backbone-sidechain hbonds distant in primary sequence |
| hbond_sc | Sidechain-sidechain hydrogen bond energy |
| geom_sol | Geometric solvation energy for polar atoms |
| linear_chainbreak | For “temporary” chainbreaks, penalty term that keeps chainbreaks closed |
| N_WC | Number of Watson-Crick base pairs |
| N_NWC | Number of non-Watson-Crick base pairs |
| N_BS | Number of base stacks |
| Following are provided if the user gives a native structure | |
| rms | All-heavy-atom RMSD to the native structure |
| rms_stem | All-heavy-atom RMSD to helical segments in the native structure |
| f_natWC | Fraction of native Watson-Crick base pairs recovered |
| f_natNWC | Fraction of native non-Watson-Crick base pairs recovered |
| f_natBP | Fraction of native base pairs recovered |
Visual representation of convergence of the models generated by FARFAR (RNA De Novo) can be found in Fig. 6. As the figure demonstrates, there is high convergence in the top models found throughout the run. In addition, if one has 1H chemical shift data, those measurements can also be supplied, as described above; this can increase the convergence and accuracy of an FARFAR prediction run. Fig. 6 illustrates these improvements through a simple GA tandem motif; first generating models without 1H chemical shift data (Fig. 6d) yields the correct overall fold of the structure while incorrectly predicting the GA base pairs to be sheared instead of forming hydrogen bonds through their Watson-Crick edge [26]. The 1H chemical shift data adds sufficient restraints to resolve the base pairing discrepancy, with all top 20 models having the correct base pairing as the NMR solved structure. Both the native PDB and the chemical shift file can be downloaded from http://rosie.rosettacommons.org/documentation/rna_denovo.
Fig. 6.
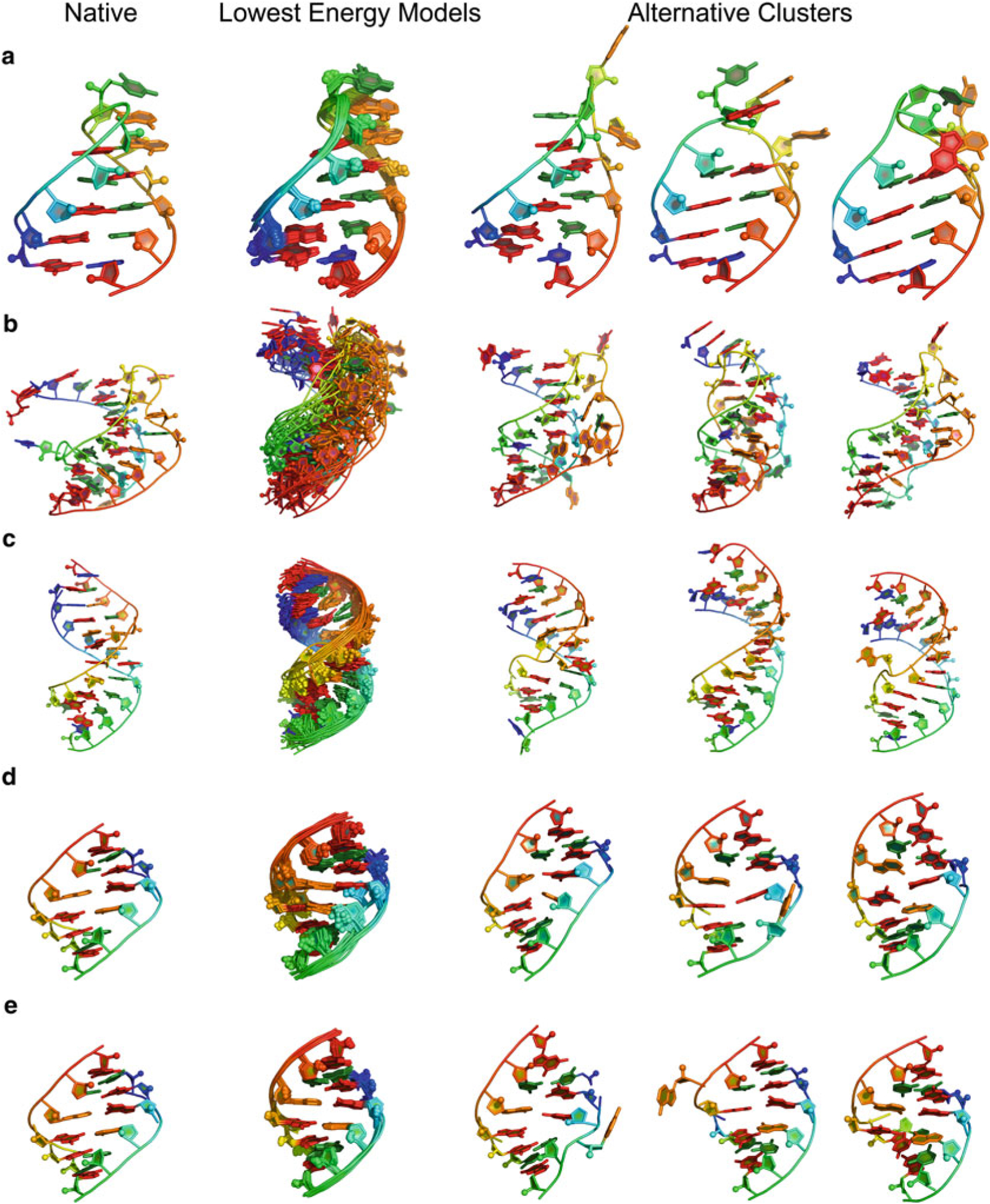
(a) GCAA tetraloop (1ZIH): RNA De Novo (through fragment assembly of RNA with full atom refinement, FARFAR) gives lowest energy models displaying structural convergence. (b) Pseudoknot (1L2X) [27], less converged then tetraloop–but also a larger RNA–gives models that are still within 3 Å heavy-atom RMSD for top model. (c) 4 × 4 internal loop solved by NMR at PDB ID 2L8F [28], converges despite presenting four noncanonical base pairs. (d) Tandem GA (1MIS) [26] without application of 1H chemical shifts. (e) Tandem GA with 1H chemical shifts, demonstrates the improved convergence with the addition of 1H chemical shift
Acknowledgments
We thank Sergey Lyskov for thoughtful discussions and expert assistance with the ROSIE platform development. We thank G. Kapral as well as elNando888 and Eterna players for testing and comments. Writing of this work was supported by a Burroughs-Wellcome Foundation Career Award and National Institutes of Health Grant R01GM100953.
References
- 1.Cech TR, Steitz JA (2014) The noncoding RNA revolution—trashing old rules to forge new ones. Cell 157(1):77–94 [DOI] [PubMed] [Google Scholar]
- 2.Leontis NB, Westhof E (2003) Analysis of RNA motifs. Curr Opin Struct Biol 13(3):300–308 [DOI] [PubMed] [Google Scholar]
- 3.Hendrix DK, Brenner SE, Holbrook SR (2006) RNA structural motifs: building blocks of a modular biomolecule. Q Rev Biophys 38(03):221. [DOI] [PubMed] [Google Scholar]
- 4.Leontis NB, Lescoute A, Westhof E (2006) The building blocks and motifs of RNA architecture. Curr Opin Struct Biol 16(3):279–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore PB (1999) Structural motifs in RNA. Annu Rev Biochem 68(1):287–300 [DOI] [PubMed] [Google Scholar]
- 6.Brion P, Westhof E (1997) Hierarchy and dynamics of RNA folding. Annu Rev Biophys Biomol Struct 26(1):113–137 [DOI] [PubMed] [Google Scholar]
- 7.Lauhon CT, Szostak JW (1995) RNA aptamers that bind flavin and nicotinamide redox cofactors. J Am Chem Soc 117(4):1246–1257 [DOI] [PubMed] [Google Scholar]
- 8.Paige JS, Wu KY, Jaffrey SR (2011) RNA mimics of green fluorescent protein. Science 333(6042):642–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doudna JA, Lorsch JR (2005) Ribozyme catalysis: not different, just worse. Nat Struct Mol Biol 12(5):395–402 [DOI] [PubMed] [Google Scholar]
- 10.Lilley DM (2005) Structure, folding and mechanisms of ribozymes. Curr Opin Struct Biol 15(3):313–323 [DOI] [PubMed] [Google Scholar]
- 11.Sripakdeevong P, Beauchamp K, Das R (2012) Why Can’t We Predict RNA structure at atomic resolution? Nucleic Acids and Molecular Biology. Springer, Berlin, Heidelberg, pp 43–65 [Google Scholar]
- 12.Das R, Karanicolas J, Baker D (2010) Atomic accuracy in predicting and designing noncanonical RNA structure. Nat Methods 7(4): 291–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sripakdeevong P, Cevec M, Chang AT, Erat MC, Ziegeler M, Zhao Q et al. (2014) Structure determination of noncanonical RNA motifs guided by 1H NMR chemical shifts. Nat Methods 11(4):413–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cruz JA, Blanchet MF, Boniecki M, Bujnicki JM, Chen SJ, Cao S et al. (2012) RNA-Puzzles: A CASP-like evaluation of RNA three-dimensional structure prediction. RNA 18(4):610–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng CY, Chou FC, Das R (2015) Modeling complex RNA tertiary folds with Rosetta. Methods Enzymol 553:35–64 [DOI] [PubMed] [Google Scholar]
- 16.Sripakdeevong P, Kladwang W, Das R (2011) An enumerative stepwise ansatz enables atomic-accuracy RNA loop modeling. Proc Natl Acad Sci U S A 108(51):20573–20578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou F-C, Sripakdeevong P, Dibrov SM, Hermann T, Das R (2013) Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat Methods 10(1):74–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J, Kladwang W, Lee M, Cantu D, Azizyan M, Kim H, Limpaecher A, Yoon S, Treuille A, Das R, EteRNA Participants (2014) RNA design rules from a massive open laboratory. Proc Natl Acad Sci U S A 111(6): 2122–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lyskov S, Chou F-C, Conchúir SÓ, Der BS, Drew K, Kuroda D et al. (2013) Serverification of molecular modeling applications: the Rosetta Online Server That Includes Everyone (ROSIE). PLoS One 22;8(5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das R, Baker D (2007) Automated de novo prediction of native-like RNA tertiary structures. Proc Natl Acad Sci U S A 104(37): 14664–14669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jucker FM, Heus HA, Yip PF, Moors EH, Pardi A (1996) A network of heterogeneous hydrogen bonds in GNRA tetraloops. J Mol Biol 264(5):968–980 [DOI] [PubMed] [Google Scholar]
- 22.Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL (2008) The Vienna RNA websuite. Nucleic Acids Res 36(Web Server issue):W70–W74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kladwang W, Cordero P, Das R (2011) A mutate-and-map strategy accurately infers the base pairs of a 35-nucleotide model RNA. RNA 17(3):522–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miao Z, Adamiak RW, Blanchet M-F, Boniecki M, Bujnicki JM, Chen S-J et al. (2015) RNA-Puzzles Round II: assessment of RNA structure prediction programs applied to three large RNA structures. RNA 21:1066–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ulrich EL, Akutsu H, Doreleijers JF, Harano Y, Ioannidis YE, Lin J et al. (2008) BioMagResBank. Nucleic Acids Res 36(Database issue):D402–D408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu M, Turner DH (1996) Solution structure of (rGCGGACGC)2 by two-dimensional NMR and the iterative relaxation matrix approach. Biochemistry 35(30):9677–9689 [DOI] [PubMed] [Google Scholar]
- 27.Egli M, Minasov G, Su L, Rich A (2002) Metal ions and flexibility in a viral RNA pseudoknot at atomic resolution. Proc Natl Acad Sci U S A 99(7):4302–4307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lerman YV, Kennedy SD, Shankar N, Parisien M, Major F, Turner DH (2011) NMR structure of a 4 × 4 nucleotide RNA internal loop from an R2 retrotransposon: identification of a three purine-purine sheared pair motif and comparison to MC-SYM predictions. RNA 17(9):1664–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]