

Abstract
The causative agent of severe acute respiratory syndrome (SARS) is the SARS-associated coronavirus, SARS-CoV. The nucleocapsid (N) protein plays an essential role in SARS-CoV genome packaging and virion assembly. We have previously shown that SARS-CoV N protein forms a dimer in solution through its C-terminal domain. In this study, the crystal structure of the dimerization domain, consisting of residues 270–370, is determined to 1.75Å resolution. The structure shows a dimer with extensive interactions between the two subunits, suggesting that the dimeric form of the N protein is the functional unit in vivo. Although lacking significant sequence similarity, the dimerization domain of SARS-CoV N protein has a fold similar to that of the nucleocapsid protein of the porcine reproductive and respiratory syndrome virus. This finding provides structural evidence of the evolutionary link between Coronaviridae and Arteriviridae, suggesting that the N proteins of both viruses have a common origin.
Coronaviruses are enveloped, single-stranded, positive-sense RNA viruses that infect a variety of mammals and birds. Although previously identified human coronaviruses cause only mild respiratory infections, in the 2003 outbreak of severe acute respiratory syndrome (SARS),3 a disease caused by a new type of coronavirus (SARS-CoV), there were more than 8,000 cases resulting in 774 deaths (∼10% mortality). Phylogenetic analysis suggests that SARS-CoV diverged early from group 2 coronaviruses and has evolved independently for a long period of time (1).
The coronavirus genome, containing ∼30,000 bases, is the largest among positive-sense RNA viruses (2, 3). It encodes non-structural proteins including the RNA polymerase and helicase, as well as the spike (S), envelope (E), membrane (M), and nucleocapsid (N) structural proteins. The coronavirus virion is about 120 nm in diameter and consists of a lipid envelope containing three or four anchored glycoproteins and a helical ribonucleoprotein core (4). The surface projections forming the crown-like structure observed via electron microscopy are made up of the S protein, which is responsible for receptor recognition and membrane fusion (5, 6, 7, 8, 9). The integral membrane proteins M and E are essential for virus budding. When co-expressed in animal cells, the M and E proteins are sufficient to form virus-like particles (10). The N protein interacts with the viral genome to form the ribonucleoprotein core and has been shown to be involved in viral RNA synthesis, transcriptional regulation of genomic RNA, translation of viral proteins, and budding (2, 11, 12).
Coronaviruses are related to arteriviruses by their similar genome organizations and viral replication mechanisms (3, 13). Recently, Coronaviridae and Arteriviridae were united to form the new order Nidovirales. The name of the order comes from a property common to both viruses, a nested set of subgenomic mRNAs for structural protein expression (Latin nidus, meaning nest). The replicase genes of arteriviruses and coronaviruses are thought to have a common origin since they both have conserved domains that are present at the same relative positions (2, 14, 15). The structural proteins, however, are thought to be unrelated due to differences in protein size and lack of sequence similarity (15).
Coronavirus N proteins are composed of three distinct domains. High resolution structures of the N-terminal domain (∼130 residues) were determined by NMR (16) and crystallography (17). This region folds similarly to the U1A RNA-binding protein and is suggested to bind RNA (16, 17). The central region of the N protein has also been shown by several laboratories to be an RNA-binding domain (18, 19, 20, 21). No structural information is available for the central domain, possibly due to its high positive charge and flexible nature. Recently, secondary structure elements of residues 248–365 of SARS-CoV N protein have been reported from NMR studies; however, the tertiary structure was not resolved (22). We have previously shown that the full-length SARS N protein, consisting of 422 amino acids, forms a dimer in solution through its C-terminal domain. In this study, we report the crystal structure of the dimerization domain of SARS-CoV N protein consisting of residues 270–370. Consistent with biochemical studies showing that this domain mediates dimer formation of the N protein (17, 23), the structure consists of a dimer with extensive interactions between subunits, suggesting that the N protein is not stable in the monomeric form and that the dimeric structure represents the functional unit of the N protein. Furthermore, the dimerization domain of SARS-CoV N protein shows a similar fold to that of the N protein of porcine reproductive and respiratory syndrome virus (PRRSV), a member of Arteriviridae. The structural similarity between these two N proteins, in the absence of significant sequence similarity, adds further evidence that Coronaviridae and Arteriviridae have evolved from a common ancestor.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification—DNAs encoding various fragments of SARS-CoV (strain GD01) nucleocapsid protein, SARS N, were cloned into a pMCSG7 plasmid with an N-terminal His6 tag and a tobacco etch virus protease cleavage site. For selenomethionine (SeMet) derivatized protein, the methionine auxotroph strain B834(DE3) cells (Invitrogen) were transformed with the expression plasmid. The cells were grown to log phase at 37 °C in minimal medium supplemented with SeMet (Sigma) and cooled to 16 °C, and protein expression was induced by the addition of 0.1 mm isopropyl-1-thio-β-d-galactopyranoside. Cells were harvested by centrifugation at 4000 × g for 20 min. Cell pellets were resuspended in lysis buffer (20 mm Tris, pH 8.5, 200 mm NaCl) and lysed by sonication. Cell lysate was centrifuged at 90,000 × g for 45 min at 4 °C, and the supernatant was loaded onto a cobalt column (Clontech; TALON metal affinity resin). The column was then washed with lysis buffer followed by lysis buffer plus 5 mm imidazole. The protein was eluted with lysis buffer plus 100 mm imidazole. Fractions containing SARS N proteins were pooled and dialyzed against 20 mm Tris, pH 8.5, 150 mm NaCl to remove the imidazole. Tobacco etch virus protease was added to 10% (w/w), and the reaction mixture was incubated at 30 °C for 10 h. Complete removal of the affinity His tag was monitored by SDS-PAGE. Further purification was achieved by size exclusion chromatography (Amersham Biosciences; Superdex 75) in lysis buffer plus 10 mm dithiothreitol to keep all SeMet in the reduced form. Incorporation of SeMet was confirmed by mass spectrometry and fluorescence scans around the selenium potassium absorption edge. The native N protein fragments were expressed in BL21star(DE3) cells and purified similarly except that no dithiothreitol was used in the buffer.
Crystallization and Data Processing—Protein samples were dialyzed against 10 mm Tris at pH 8.5 and 70 mm NaCl and concentrated to 45 mg/ml. Crystals were grown by mixing protein solution with the reservoir solution containing 30–33% pentaerythritol ethoxylate, 15/4 ethoxylate/hydroxyl, 50 mm (NH4)2SO4, and 50 mm Bis-Tris, pH 6.5, at 1:1 ratio in sitting drops by vapor diffusion at 4 °C. Crystals were looped out of the drop and flash-frozen in liquid nitrogen. Data were collected at 100 K with a Quantum-Q315 CCD (ADSC) detector on beamline 19-ID at the Advanced Photon Source (Structural Biology Center, Argonne National Laboratory). X-ray diffraction data for the initial structure determination were collected to 2.2 Å resolution from crystals containing SeMet protein (see Table 1 , phasing set). Data from both lattices of the twin domains were processed separately with HKL2000 (24) and then merged together in Scalepack. Phases were obtained from the four selenium sites found by multiple wavelength anomalous diffraction using the program SOLVE (25) and improved by density modification using CNS (26). The initial model was built automatically using Arp/Warp in CCP4 (27). A higher resolution data set (1.75 Å) from a SeMet protein crystal (see Table 1, refinement set) was used for refinement. The resolution was extended over several cycles of model building in O (28) and refinement in REFMAC5 in CCP4 (27).
TABLE 1.
Data collection and refinement statistics
Erroneous I/sigma values have been omitted, since overlapping reflections due to twinning render proper background estimates impossible.
|
Data Collection |
Refinement set |
Se-Met (phasing set) |
||
|---|---|---|---|---|
| Peak | Inflection | Remote | ||
| Space group C2 | ||||
| a, Å | 124.2 | 124.3 | ||
| b, Å | 50.5 | 50.6 | ||
| c, Å | 41.5 | 41.6 | ||
| β, o | 108.9 | 108.8 | ||
| Resolution range, Å | 30-1.75 | 50-1.9 | 50-2.2 | 50-2.2 |
| Wavelength, Å | 0.97929 | 0.97929 | 0.97940 | 0.94285 |
| Unique reflections | 24479 | 27506 | 24045 | 24131 |
| Completenessa | 99.2 (99.6) | 100.0 (100.0) | 99.2 (100.0) | 99.2 (100.0) |
| Redundancy | 2.9 (2.9) | 8.2 (7.8) | 9.4 (9.2) | 9.4 (9.2) |
| Rsymb | 7.4 (15.0) |
10.6 (16.5) |
12.8 (15.5) |
15.1 (21.2) |
| Refinement | ||||
| Rworkc, % | 18.7 | |||
| Rfree, % | 23.5 | |||
| Resolution range, Å | 30-1.75 | |||
| No. of reflections used | 23088 | |||
| No. of molecules in asymmetric unit | 2 | |||
| No. of non-hydrogen atoms in protein | 1511 | |||
| No of water molecules | 212 | |||
| No of sulfate ions | 1 | |||
| r.m.s.d.d from ideality | ||||
| Bond lengths, Å | 0.02 | |||
| Bond angles, o | 1.67 | |||
| Estimated coordinate error (Luzzati), Å | 0.16 | |||
| Average B-factor Å2 | 18.0 | |||
| Ramachandran statistics (non glycine) | ||||
| Most favored, % | 97.5 | |||
| Allowed, % | 2.5 | |||
| Generously allowed, % | 0.0 | |||
| Disallowed, % |
0.0 |
|||
Values in parentheses are for the highest resolution shell.
Rsym = Σ|Ii — 〈I 〉 |/ ΣIi, where Ii is the intensity of the ith observation and 〈I 〉 is the mean intensity of the reflection.
R = Σ| Fo | — | Fo |/Σ|Fo|, where Fo and Fc are the observed and calculated structure factors amplitudes. Rfree is calculated using 5% of the total reflections.
r.m.s.d., root mean square deviation.
RESULTS
Crystallization and Structure Determination—The full-length SARS-CoV N protein (SARS N) and 18 truncated constructs were expressed in bacteria and purified for crystallization. Although most constructs yielded either no crystals or merely clusters of small needles, a C-terminal construct containing residues 270–370 (cSARS-N) was readily crystallized to give well diffracting crystals. Although the crystals appear to be composed of a single lattice as viewed under a microscope fitted with a light polarizer, the diffraction pattern indicated that the crystals were unusually twinned, resulting in two overlapping monoclinic lattices of similar unit cell dimensions. The two lattices are related by a 180° rotation around the y axis followed by a 50° rotation around the z axis. As a result, nearly half of the observable reflections overlapped to some extent. Both native and SeMet derivatized proteins formed twinned crystals regardless of changes in temperature, precipitant, pH, and salt concentration. Because of the large number of partially overlapped reflections, only the central four pixels of each reflection were measured, these being proportional to the integrated intensity presuming the profile was fairly constant (details of the data processing procedure will be published elsewhere).
The structure of cSARS-N was determined by multiwavelength anomalous diffraction using a SeMet derivative to 1.75 Å resolution. The crystal belongs to space group C2, and the asymmetric unit contains a dimer of two subunits related by a 180° rotation. The final model, containing residues 270–366 of one subunit and residues 274–369 of the other, was refined to R work/R free of 18.7/23.5 (Table 1). A representative 2Fo – Fc electron density map is shown in Fig. 1 . The model, analyzed with PROCHECK (29), has 97.5% of non-glycine residues in the most favored regions of the Ramachandran plot, with no disallowed residues.
FIGURE 1.

Electron density map. Stereo view of electron density map (2Fo – Fc, 1.5σ) around β-strand residues 333–337 of both subunits at the dimer interface.
Overall Structure—Sequence comparison shows that the dimerization domain of the nucleocapsid protein is conserved among the three groups of coronaviruses, suggesting a common structural and functional role of this domain (Fig. 2A ). The monomer of cSARS-N contains five short α-helices, one 310 helix, and two β-strands. The overall shape of the monomer resembles the letter C, with one edge formed by a β-hairpin extending away from the rest of the molecule (Fig. 2B). In contrast to many small proteins that are remarkably compact, the monomeric cSARS-N domain folds into an extended conformation with a large cavity in its center. It is therefore likely that the N protein is not stable in the monomeric form and that higher oligomerization of the polypeptide is necessary to produce a stable conformation. Consistent with previous biochemical studies suggesting that the full-length SARS N protein forms a dimer in solution through its C-terminal domain (17, 23), a compact dimeric form of cSARS-N was observed in the crystal (Fig. 2C). The dimer has a flat structure of approximate dimensions 48 × 42 × 25 Å. The two subunits in the dimer are almost identical, with a root mean square deviation of 0.36 Å over 92 Cα atoms out of 101 residues and the largest displacement of 0.56 Å. The dimer interface is largely formed by insertion of the β-hairpin of one subunit into the cavity of the opposite subunit (Fig. 2C). As a result, the four β-strands of the two subunits form an anti-parallel β-sheet, with 10 hydrogen bonds formed across the dimer interface by the main chain atoms of residues 330–340. Two large, pyramidal hydrophobic cores form a bow tie-shaped pocket that further stabilizes the dimer. Each core consists of base residues Phe-287, Leu-292, Trp-302, Ile-305, Phe-308, Tyr-334, Ile-338, Leu-340, Leu-354, Ile-358 from one subunit and apex residues Phe-315, Phe-316, Ile-321, and Leu-332 from the other subunit. The buried area in the dimer interface is 2093 Å2 per subunit, which accounts for 28% of the total solvent-accessible surface area of each subunit. The extended structure of the monomeric subunit and the extensive interactions between the subunits within the dimer suggest that the dimeric arrangement observed crystallographically represents the biological architecture of the coronavirus N protein. The structure of cSARS-N is consistent with biochemical studies showing that residues 1–284 of the SARS N protein are dispensable for dimer formation (23). The strong interactions between the two subunits explain the previous observation that a significant amount of denaturant (4 m urea) was necessary to disrupt the dimer in solution (23).
FIGURE 2.
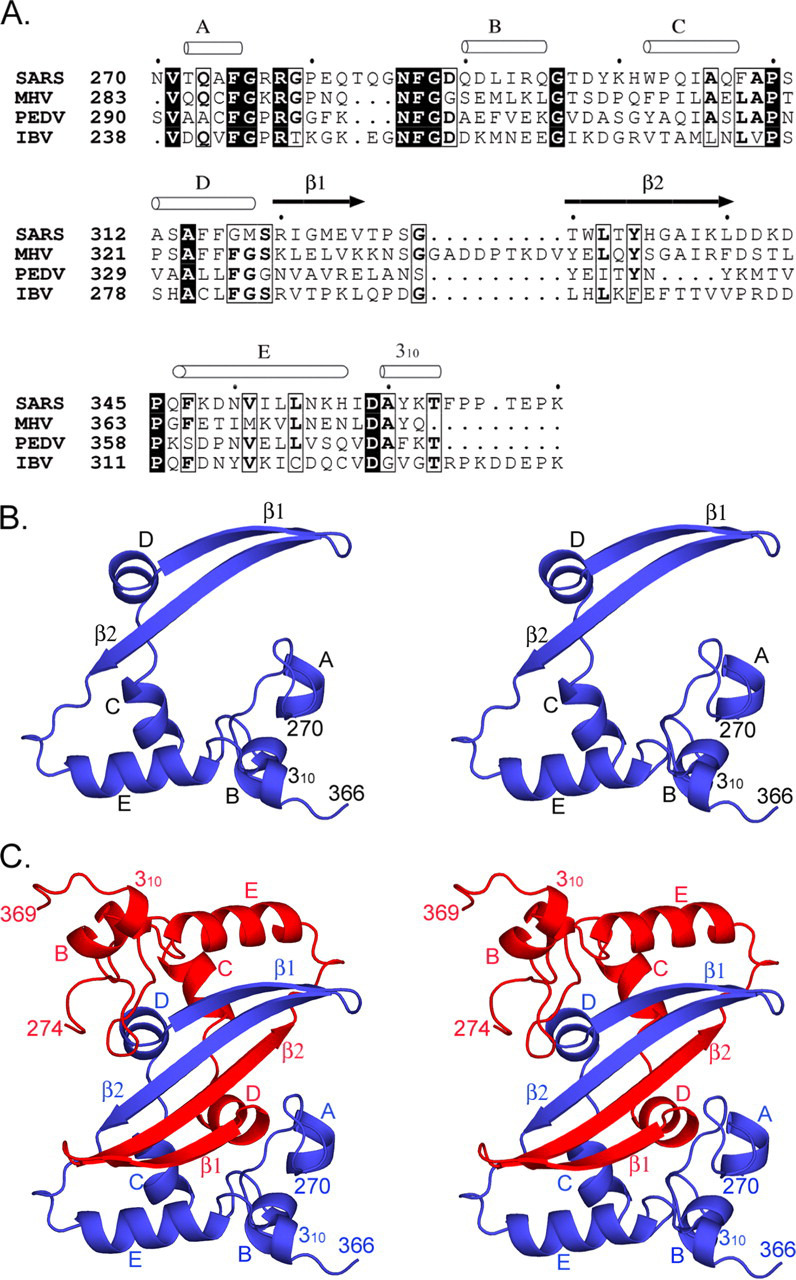
Structure of the SARS-CoV nucleocapsid dimerization domain.A, sequence alignment of the nucleocapsid protein dimerization domains of SARS-CoV (strain GD01) and representative coronaviruses from the three previously defined groups: mouse hepatitis virus (MHV, group 2), porcine epidemic diarrhea virus (PEDV, group 1), and avian infectious bronchitis virus (IBV, group 3). Solid boxes indicate strictly conserved residues, whereas open boxes indicate partially conserved residues (shown in bold letters). The secondary structure elements of SARS N protein are represented by cylinders (α-helices) and arrows (β-strands) above the sequences. B, cSARS-N monomer in stereo. C, stereo view of cSARS-N dimer with the two subunits colored in blue and red. Secondary structure elements and terminal residue numbers are labeled in the ribbon diagrams. The figures were prepared with ESPript (48) or PyMOL.
A recent NMR study reported secondary structural assignments of a SARS N protein construct that is similar to cSARS-N (22). Despite the difficulty in resolving the three-dimensional structure, a dimeric interface consisting of a four-stranded anti-parallel β-sheet and two α-helices was proposed (22). Comparing the crystal structure with the proposed NMR structure, one of the most notable differences is the location of the longest helix, helix E (residues 346–357). In the crystal structure, helices E of both subunits are located at the edge of the dimer molecule, away from each other (Fig. 2C), whereas in the structure proposed by NMR, they are placed at the interface to stabilize the dimer (22).
Structural Similarity to the Nucleocapsid Protein of Arterivirus—A DALI search of the Protein Data Bank did not identify any other proteins with similar folds to that of cSARS-N. However, common structural features can be found between cSARS-N and the nucleocapsid protein of PRRSV (Fig. 2). PRRSV belongs to the recently recognized family Arteriviridae, which is in the same order as Coronaviridae. The crystal structure of the C-terminal 65 residues of PRRSV N protein (30) is a homodimer. Each subunit consists of two central anti-parallel β-strands flanked by three α-helices. Although the sequences of PRRSV N and cSARS-N have no significant similarity (Fig. 3A ), superposition of the monomeric N proteins of PRRSV and SARS-CoV shows that the two have similar folds in a core region of ∼50 residues (Fig. 3, B and C). The central β-strands are superimposable, and the helices following the β-strands are of similar length and position (Fig. 3B). In addition, as proposed in the recent NMR study (22), the dimer interface of the two N proteins are very similar, noted by the intertwining of the central β-strands to form a four-stranded sheet (Fig. 3C). This is evident by a root mean square deviation of 2.24 Å for the Cα positions among 38 residues at the dimer interface.
FIGURE 3.
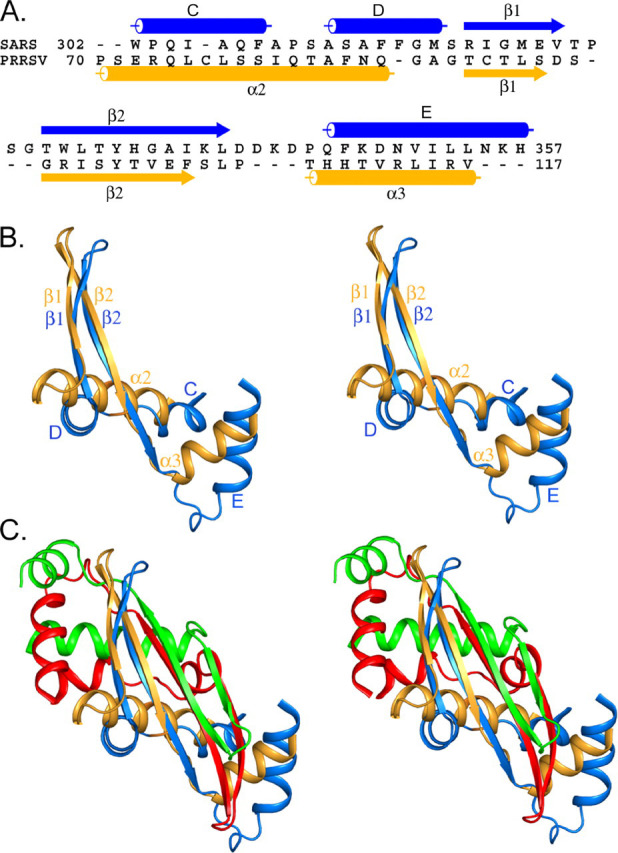
Structural similarity between Coronavirus and Arterivirus nucleocapsid proteins.A, structure-based alignment of the dimerization domains of SARS N and PRRSV N. The secondary structure elements of SARS N (blue) and PRRSV N (gold) are shown as cylinders (α-helices) and arrows (β-strands). B, superposition of SARS N residues 302–357 (blue) with PRRSV N residues 70–117 (gold) monomers in stereo. This view is obtained by a 90° counterclockwise rotation of the structure shown in Fig. 2B about a vertical axis. C, superposition of the dimeric SARS N (blue and red) and PRRSV N (gold and green) in stereo. The figures were prepared using Alscript (49) or PyMOL.
DISCUSSION
Specific packaging of the viral genome into the virion is a critical step in the life cycle of an infectious virus. The N protein plays an essential role in this process through self-association and interactions with viral RNA and other viral proteins. In an effort to understand the mechanism of how SARS N protein functions, we determined the crystal structure of its C-terminal dimerization domain. Extensive hydrogen-bonding and hydrophobic interactions are observed between the two subunits within the dimer found in the crystal, suggesting that the functional unit of the N protein is dimeric. Strong protein-protein interactions at the dimerization region may be essential to hold the putatively monomeric, highly charged RNA-binding domains in close proximity (16), thereby facilitating the formation of a large helical nucleocapsid core. Association of the N protein dimers is necessary for further assembly of the core. The full-length dimeric N protein has a propensity to form tetramers and higher molecular weight oligomers in vitro (23). A serine/arginine-rich motif (residues 184–196) was shown to be important for N protein oligomerization (31). Since constructs containing residues 211–422 or 285–422 of SARS N do not form oligomers larger than dimers (23), it is possible that the serine/arginine-rich motif located outside the dimerization domain is necessary to mediate further association of N protein dimers. Recently, the C-terminal 45 residues of the mouse hepatitis virus N protein were shown to be the major determinant for interaction with the M protein (32). Association of the N protein with the M protein may also play a role in the assembly of the nucleocapsid core into a progeny virion.
RNA viruses have high rates of sequence divergence and genome recombination, and thus it is often a challenge to study the evolutionary relationships among viruses. Structural studies have become an important method for revealing distant relationships among viruses. Among all lipid-enveloped RNA viruses, high resolution structures of the proteins that package the viral genome into virions, often termed the nucleocapsid or capsid proteins, are currently available from five virus families (Fig. 4 ) (Retroviridae are not included in this discussion because their replication pathway is through a DNA intermediate (33)). Although functionally equivalent, the size and structure of nucleocapsid proteins are remarkably diverse among these five virus families. The nucleoprotein of borna disease virus (BDV), a single-stranded RNA virus in the family Bornaviridae, contains 370 residues and folds into an S-shaped molecule consisting of 16 helices and two short β-strands (34). In the crystal lattice, four subunits of BDV N interact extensively to form a homotetramer, suggesting that the functional unit of BDV N is a tetramer (Fig. 4A). Both Semliki Forest virus (genus Alphavirus, family Togaviridae) and dengue virus (genus Flavivirus, family Flaviviridae) are small single-stranded RNA viruses with icosahedral symmetry. It has been suggested that they have a common ancestor because the structures of one of their surface glycoproteins are remarkably similar (35, 36, 37). Inside the lipid bilayer, however, the nucleocapsid cores of these two viruses are quite different. An alphavirus core consists of 240 copies of the capsid protein arranged in a T = 4 icosahedral lattice (38). Each capsid protein has ∼270 residues, of which the N-terminal ∼120 residues are flexible. The remaining 150 residues adopt a chymotrypsin-like fold and form homodimers in the crystal (39, 40) (Fig. 4B). In contrast, cryoelectron microscopy reconstructions of flaviviruses did not show a defined structure of the capsid core (41, 42), suggesting that the core either is disordered or has a different symmetry from the surface icosahedral lattice. In addition, the exact copy number of the capsid protein in the core is unclear. The C-terminal 100 residues of flavivirus capsid proteins fold into a four-helical structure with no similarity to that of an alphavirus capsid protein (43, 44) (Fig. 4C).
FIGURE 4.

Structural diversity of enveloped RNA virus nucleocapsid proteins. Representative known structural domains of nucleocapsid proteins of Bornaviridae (BDV), Togaviridae (SFV), Flaviviridae (dengue), Arteriviridae (PRRSV), and Coronaviridae (SARS-CoV) are shown. One subunit in the oligomer is colored based on its secondary structure elements: blue (α-helices), yellow (β-strands), and green (coils); the other subunits are colored in gray. A, the crystallographic tetramer of BDV nucleocapsid protein (1N93) (34). B, Semliki Forest virus nucleocapsid protein (1VCP). Only one of the two crystallographic dimers is shown (40). C, NMR structure of dimeric dengue capsid protein (1R6R) (43). D, PRRSV nucleocapsid protein (1P65) crystallographic dimer (30). E, structure of the C-terminal dimerization domain, residues 270–370, of SARS-CoV nucleocapsid protein (2GIB). F, NMR structure of the N-terminal domain of SARS-CoV nucleocapsid protein (1SSK), residues 49–178 (16). The figures were prepared with PyMOL.
It is striking that, within the group of enveloped RNA viruses, no two nucleocapsid proteins are known to be structurally similar except those of Arteriviridae and Coronaviridae (Fig. 4). The structural similarity between the N proteins of SARS-CoV and PRRSV provides valuable information for understanding the evolutionary links between corona- and arteriviruses. Although these viruses were grouped in the same order, Nidovirales, their structural proteins were previously thought to be unrelated due to the marked difference in their sizes and lack of sequence similarity (15). In this study, a common fold of the dimerization domains of Nidovirales N proteins is observed, suggesting a possible common origin of these two proteins. The amino acid sequence and gene size diversity may have resulted from extensive mutation and RNA recombination during evolution. It is also important to note that the assembled cores of corona- and arteriviruses have completely different structures. PRRSV N protein interacts with the RNA genome to form a spherical, possibly icosahedral core (45), whereas coronaviruses are known to have helical nucleocapsids (46). It was previously suggested that the ancestral virus had an icosahedral nucleocapsid core, and the larger N protein of coronavirus, freed from its icosahedral package constraints, allowed the coronavirus genome to become larger during evolution (47).
Addendum—While this work was in progress, the structure of the nucleocapsid protein of another coronavirus, the avian infectious bronchitis virus (IBV), was determined by Drs. H. Jayaram and B. V. V. Prasad at Baylor College of Medicine (unpublished data). The coordinates of the IBV N protein were kindly provided to us by them for phasing via molecular replacement. However, they were unnecessary since the experimental multiwavelength anomalous dispersion phasing was sufficient to build the initial model.
Acknowledgments
We thank Dr. M. G. Rossmann for many helpful discussions and Drs. C. Gustafson, R. J. Kuhn, M. G. Rossmann, E. G. Strauss, and J. H. Strauss for critical reading of the manuscript. We also thank the beamline staff at SBC 19-ID at the Advanced Photon Source for assistance with data collection and the Purdue Cancer Center for X-ray and DNA sequencing facilities.
Footnotes
The abbreviations used are: SARS, severe acute respiratory syndrome; SARS-CoV, SARS-associated coronavirus; PRRSV, porcine reproductive and respiratory syndrome virus; SeMet, selenomethionine; BDV, borna disease virus; Bis-Tris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1.Stadler K., Masignani V., Eickmann M., Becker S., Abrignani S., Klenk H.D., Rappuoli R. Nat. Rev. Microbiol. 2003;1:209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai M.M., Cavanagh D. Adv. Virus. Res. 1997;48:1–100. doi: 10.1016/S0065-3527(08)60286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lai M.M.C., Holmes K.V. Fields Virology. 2001;1:1163–1203. Lippincott-Raven Publishers, Philadelphia and New York. [Google Scholar]
- 4.Sturman L.S., Holmes K.V., Behnke J. J. Virol. 1980;33:449–462. doi: 10.1128/jvi.33.1.449-462.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babcock G.J., Esshaki D.J., Thomas W.D., Jr., Ambrosino D.M. J. Virol. 2004;78:4552–4560. doi: 10.1128/JVI.78.9.4552-4560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kubo H., Yamada Y.K., Taguchi F. J. Virol. 1994;68:5403–5410. doi: 10.1128/jvi.68.9.5403-5410.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bos E.C., Heijnen L., Spaan W.J. Adv. Exp. Med. Biol. 1995;380:283–286. doi: 10.1007/978-1-4615-1899-0_45. [DOI] [PubMed] [Google Scholar]
- 8.Krueger D.K., Kelly S.M., Lewicki D.N., Ruffolo R., Gallagher T.M. J. Virol. 2001;75:2792–2802. doi: 10.1128/JVI.75.6.2792-2802.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosch B.J., van der Zee R., de Haan C.A., Rottier P.J. J. Virol. 2003;77:8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vennema H., Godeke G.J., Rossen J.W., Voorhout W.F., Horzinek M.C., Opstelten D.J., Rottier P.J. EMBO J. 1996;15:2020–2028. doi: 10.1002/j.1460-2075.1996.tb00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He R., Leeson A., Andonov A., Li Y., Bastien N., Cao J., Osiowy C., Dobie F., Cutts T., Ballantine M., Li X. Biochem. Biophys. Res. Commun. 2003;311:870–876. doi: 10.1016/j.bbrc.2003.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tahara S.M., Dietlin T.A., Nelson G.W., Stohlman S.A., Manno D.J. Adv. Exp. Med. Biol. 1998;440:313–318. doi: 10.1007/978-1-4615-5331-1_41. [DOI] [PubMed] [Google Scholar]
- 13.Snijder E.J., Meulenberg J.J.M. Fields Virology. 2001;1:1205–1220. Lippincott-Raven Publishers, Philadelphia and New York. [Google Scholar]
- 14.Snijder E.J., Meulenberg J.J. J. Gen. Virol. 1998;79:961–979. doi: 10.1099/0022-1317-79-5-961. [DOI] [PubMed] [Google Scholar]
- 15.Cavanagh D. Arch. Virol. 1997;142:629–633. [PubMed] [Google Scholar]
- 16.Huang Q., Yu L., Petros A.M., Gunasekera A., Liu Z., Xu N., Hajduk P., Mack J., Fesik S.W., Olejniczak E.T. Biochemistry. 2004;43:6059–6063. doi: 10.1021/bi036155b. [DOI] [PubMed] [Google Scholar]
- 17.Fan H., Ooi A., Tan Y.W., Wang S., Fang S., Liu D.X., Lescar J. Structure (Camb.) 2005;13:1859–1868. doi: 10.1016/j.str.2005.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masters P.S. Arch. Virol. 1992;125:141–160. doi: 10.1007/BF01309634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelson G.W., Stohlman S.A. J. Gen. Virol. 1993;74:1975–1979. doi: 10.1099/0022-1317-74-9-1975. [DOI] [PubMed] [Google Scholar]
- 20.Peng D., Koetzner C.A., McMahon T., Zhu Y., Masters P.S. J. Virol. 1995;69:5475–5484. doi: 10.1128/jvi.69.9.5475-5484.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson G.W., Stohlman S.A., Tahara S.M. J. Gen. Virol. 2000;81:181–188. doi: 10.1099/0022-1317-81-1-181. [DOI] [PubMed] [Google Scholar]
- 22.Chang C.K., Sue S.C., Yu T.H., Hsieh C.M., Tsai C.K., Chiang Y.C., Lee S.J., Hsiao H.H., Wu W.J., Chang C.F., Huang T.H. FEBS Lett. 2005;579:5663–5668. doi: 10.1016/j.febslet.2005.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu I.M., Gustafson C.L., Diao J., Burgner J.W., II Li, Zhang Z., Chen J.J. J. Biol. Chem. 2005;280:23280–23286. doi: 10.1074/jbc.M501015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otwinowski Z., Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.Terwilliger T.C. Methods Enzymol. 2003;374:22–37. doi: 10.1016/S0076-6879(03)74002-6. [DOI] [PubMed] [Google Scholar]
- 26.Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S., Read R.J., Rice L.M., Simonson T., Warren G.L. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 27.Collaborative Computational Project N. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994:760–763. [Google Scholar]
- 28.Jones T.A., Zou J.Y., S. W. CowanKjeldgaard. Acta Crystallogr. Sect. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 29.Laskowski R.A., Macarthur M.W., Moss D.S., Thornton J.M. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 30.Doan D.N., Dokland T. Structure (Camb.) 2003;11:1445–1451. doi: 10.1016/j.str.2003.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He R., Dobie F., Ballantine M., Leeson A., Li Y., Bastien N., Cutts T., Andonov A., Cao J., Booth T.F., Plummer F.A., Tyler S., Baker L., Li X. Biochem. Biophys. Res. Commun. 2004;316:476–483. doi: 10.1016/j.bbrc.2004.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurst K.R., Kuo L., Koetzner C.A., Ye R., Hsue B., Masters P.S. J. Virol. 2005;79:13285–13297. doi: 10.1128/JVI.79.21.13285-13297.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goff S.P. Retroviridae: the Viruses and Their Replication. 2001;2:1871–1940. Lippincott Williams and Wilkins, Philadelphia and New York. [Google Scholar]
- 34.Rudolph M.G., Kraus I., Dickmanns A., Eickmann M., Garten W., Ficner R. Structure (Camb.) 2003;11:1219–1226. doi: 10.1016/j.str.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 35.Pletnev S.V., Zhang W., Mukhopadhyay S., Fisher B.R., Hernandez R., Brown D.T., Baker T.S., Rossmann M.G., Kuhn R.J. Cell. 2001;105:127–136. doi: 10.1016/s0092-8674(01)00302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rey F.A., Heinz F.X., Mandl C., Kunz C., Harrison S.C. Nature. 1995;375:291–298. doi: 10.1038/375291a0. [DOI] [PubMed] [Google Scholar]
- 37.Lescar J., Roussel A., Wien M.W., Navaza J., Fuller S.D., Wengler G., Rey F.A. Cell. 2001;105:137–148. doi: 10.1016/s0092-8674(01)00303-8. [DOI] [PubMed] [Google Scholar]
- 38.Zhang W., Mukhopadhyay S., Pletnev S.V., Baker T.S., Kuhn R.J., Rossmann M.G. J. Virol. 2002;76:11645–11658. doi: 10.1128/JVI.76.22.11645-11658.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi H.K., Tong L., Minor W., Dumas P., Boege U., Rossmann M.G., Wengler G. Nature. 1991;354:37–43. doi: 10.1038/354037a0. [DOI] [PubMed] [Google Scholar]
- 40.Choi H.K., Lu G., Lee S., Wengler G., Rossmann M.G. Proteins. 1997;27:345–359. doi: 10.1002/(sici)1097-0134(199703)27:3<345::aid-prot3>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 41.Kuhn R.J., Zhang W., Rossmann M.G., Pletnev S.V., Corver J., Lenches E., Jones C.T., Mukhopadhyay S., Chipman P.R., Strauss E.G., Baker T.S., Strauss J.H. Cell. 2002;108:717–725. doi: 10.1016/s0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y., Corver J., Chipman P.R., Zhang W., Pletnev S.V., Sedlak D., Baker T.S., Strauss J.H., Kuhn R.J., Rossmann M.G. EMBO J. 2003;22:2604–2613. doi: 10.1093/emboj/cdg270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma L., Jones C.T., Groesch T.D., Kuhn R.J., Post C.B. Proc. Natl. Acad. Sci. U. S. A. 2004;101:3414–3419. doi: 10.1073/pnas.0305892101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dokland T., Walsh M., Mackenzie J.M., Khromykh A.A., Ee K.H., Wang S. Structure (Camb.) 2004;12:1157–1163. doi: 10.1016/j.str.2004.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brinton-Darnell M., Plagemann P.G. J. Virol. 1975;16:420–433. doi: 10.1128/jvi.16.2.420-433.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Macneughton M.R., Davies H.A. J. Gen. Virol. 1978;39:545–549. doi: 10.1099/0022-1317-39-3-545. [DOI] [PubMed] [Google Scholar]
- 47.Godeny E.K., Chen L., Kumar S.N., Methven S.L., Koonin E.V., Brinton M.A. Virology. 1993;194:585–596. doi: 10.1006/viro.1993.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gouet P., Robert X., Courcelle E. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barton G.J. Protein Eng. 1993;6:37–40. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]