

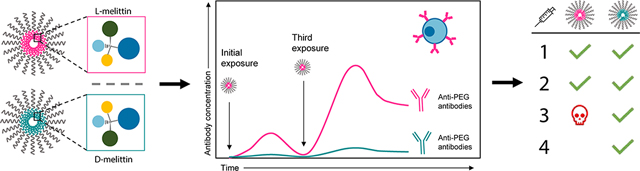
Abstract
The generation of anti-PEG antibodies in response to PEGylated proteins, peptides, and carriers significantly limits their clinical applicability. IgM antibodies mediate the clearance of these therapeutics upon repeat injection, resulting in toxicity and hindered therapeutic efficacy. We observed this phenomenon in our polymer platform, virus-inspired polymer for endosomal release (VIPER), which employs pH-sensitive triggered display of a lytic peptide, melittin, to facilitate endosomal escape. While the polymer-peptide conjugate was well tolerated after a single injection, we observed unexpected mortality upon repeat injection. Thus, the goal of this work was to enhance the safety and tolerability of VIPER for frequent dosing. Based on previous reports on anti-PEG antibodies and the adjuvant activity of melittin, we characterized the antibody response to polymer, peptide, and polymer-peptide conjugates after repeat-dosing and measured high IgM titers that bound PEG. By substituting the L-amino acid peptide for its D-amino acid enantiomer, we significantly attenuated the anti-PEG antibody generation and toxicity, permitting repeat-injections. We attempted to rescue mice from L-melittin induced toxicity by prophylactic injection of platelet activating factor (PAF) antagonist CV-6209, but observed minimal effect, suggesting that PAF is not the primary mediator of the observed hypersensitivity response. Overall, we demonstrated that the D-amino acid polymer-peptide conjugates, unlike L-amino acid polymer-peptide conjugates, exhibit good tolerability in vivo, even upon repeat administration, and do not elicit the generation of anti-PEG antibodies.
Keywords: Melittin, drug delivery, hypersensitivity, anti-PEG antibodies, polymer-peptide conjugates
Graphical Abstract
2. Introduction
PEGylated proteins and peptides are clinically-successful delivery formulations and among the highest revenue therapeutics on the market.1 However, anti-PEG antibodies, which can result in increased drug clearance, hypersensitivity responses, and reduced efficacy, remain a significant clinical hurdle.2,3 Recent studies have reported toxicity following repeat-administration of PEG-containing therapeutics, which has been linked with accelerated blood clearance (ABC) mediated by an anti-PEG antibody response, resulting in rapid clearance of PEGylated carriers, complement activation, and anaphylactic reaction.4–8 For example, a significant fraction of patients receiving PEGylated urate oxidase (38%) developed anti-PEG antibodies after injection, significantly hindering therapeutic efficacy.9 Furthermore, the presence of anti-PEG antibodies has been closely associated with rapid clearance of PEG-asparaginase (ASP), rendering the therapy ineffective.6 In fact, pre-existing anti-PEG antibodies have been identified as a risk factor to predict patient reactions to PEG-ASP, emphasizing the clinical importance of anti-PEG antibodies.10 Seminal work by Richter and Akerblom first revealed that anti-PEG antibodies are generated after injection of animals with PEG-conjugated proteins, but not with free PEG.11 These studies and others reveal that the conjugated biologics act as adjuvants in inducing anti-PEG antibodies; indeed, the extent and presence of anti-PEG antibodies generally correlates with the immunogenicity of conjugated protein.12–14 These findings from the history of PEGylated proteins reveal important immunogenicity considerations for the growing suite of polymer-protein and polymer-peptide conjugates that are in preclinical development for biologics delivery.15,16 While other anti-polymer antibodies have been identified (e.g., against silicone breast implants), anti-PEG antibodies are the best studied and this work may be generalized for other polymer-conjugates.17
We incorporated PEG into our polymer platform, virus-inspired polymer for endosomal release (VIPER), which facilitates pH-triggered, intracellular delivery of therapeutic cargos.18,19 Briefly, VIPER comprises a hydrophilic and hydrophobic block that self-assemble into micelles. The lytic peptide melittin is conjugated to the hydrophobic block, which undergoes a sharp phase transition at acidic pH for triggered display and endosomal rupture. Thus, melittin is shielded at physiological pH 7.4 but is exposed at endosomal pH 5.7, rupturing the endosome for cargo delivery to the cell cytosol. In contrast to previous iterations of VIPER, this work utilized a PEG hydrophilic block instead of pOEGMA. We observed that this formulation was well tolerated following a single intravenous (i.v.) injection, but triggered unexpected mortality upon repeat-dosing of melittin-containing micelles. The goal of this work was to improve the safety and tolerability of VIPER for frequent i.v. dosing. While this work specifically focused on VIPER, a peptide-conjugate wielding the immunogenic peptide melittin, these findings can be broadly applied to other polymer-peptide conjugates with biologically-active peptides.
Because mortality was only observed upon repeat-dosing, we posited that toxicity was associated with an adaptive immune response rather than the inherent lytic activity of melittin. Specifically, we hypothesized that melittin acted as an adjuvant to induce an antibody response against the polymer carrier, resulting in anti-PEG antibody generation and toxicity A growing body of literature utilizes the adjuvant activity of melittin in vaccines to markedly enhance antibody titers.20,21 Yet, this immunogenic activity of melittin can be reduced by replacing L-amino acids with D-amino acids, resulting in lower antibody generation.22–24 Broadly, D-amino acid substitutions can reduce peptide:MHC affinity and subsequent presentation efficiency to T and B cells, reducing immunogenicity in vivo and attenuating IgG and IgM antibody response.24,25 Applying these findings to our polymer-peptide platform, we hypothesized that utilizing D-melittin instead of L-melittin would diminish anti-PEG antibody generation, reducing immunogenicity and permitting repeat-dosing.
In this work, we compared the in vitro activity and in vivo safety of L- and D-melittin VIPER micelles. First, we validated comparable peptide and micelle activity in vitro by cytotoxicity and hemolysis assays, and confirmed endosomal rupture. Next, we compared the maximum tolerated dose (MTD) of L-melittin VIPER-micelles (LMM) and D-melittin VIPER-micelles (DMM). Using an immunodeficient nonobese diabetic-severe combined immunodeficiency (nod-scid) mouse model, we confirmed that LMM-toxicity was associated with an immune response. We then investigated antibody generation against peptide, micelles, and polymer upon repeat-injection of LMM or DMM via enzyme linked immunosorbent assay (ELISA). Finally, we evaluated the efficacy of a platelet-activating factor (PAF) antagonist to rescue mice from toxicity. Ultimately, we demonstrated that while LMM and DMM behaved similarly in vitro, DMM promoted an enhanced safety profile in vivo. Mice treated with DMM exhibited a higher MTD and tolerated four injections, whereas mice receiving LMM only tolerated two injections. This can be attributed to the generation of anti-PEG antibodies: LMM induced a robust IgG and IgM antibody response against PEG, whereas DMM did not. Overall, we showed that polymer-peptide conjugates with D-amino acid peptides mitigate the production of anti-carrier antibodies and are safe for frequent dosing. This work has major implications for protein- and peptide-PEG conjugates, as we demonstrate that reducing the immunogenicity of the biologic can reduce generation of anti-polymer antibodies.
3. Results
3.1. Synthesis of melittin micelles
The polymer was synthesized as by reversible addition-fragmentation chain-transfer (RAFT) polymerization of 2-diisopropylaminoethyl methacrylate (DIPAMA) and pyridyl disulfide ethyl methacrylate (PDSEMA) using PEGylated macro chain transfer agents (CTAs) (Figure 1A). In contrast to previous iterations of VIPER, we used a PEG block instead of pOEGMA, as PEG confers increased solubility and is commercially available. The pH-sensitive block comprised DIPAMA, which transitions sharply from hydrophobic to hydrophilic at acidic pH, copolymerized with PDSEMA, which enables conjugation with thiolated peptides. The block copolymers self-assemble into micelles at physiological pH 7.4, but disassemble into polymer chains below endosomal pH 6.3. Thus, peptides conjugated to DIPAMA are shielded at pH 7.4, but are exposed upon cellular internalization into endosomes.
Figure 1:
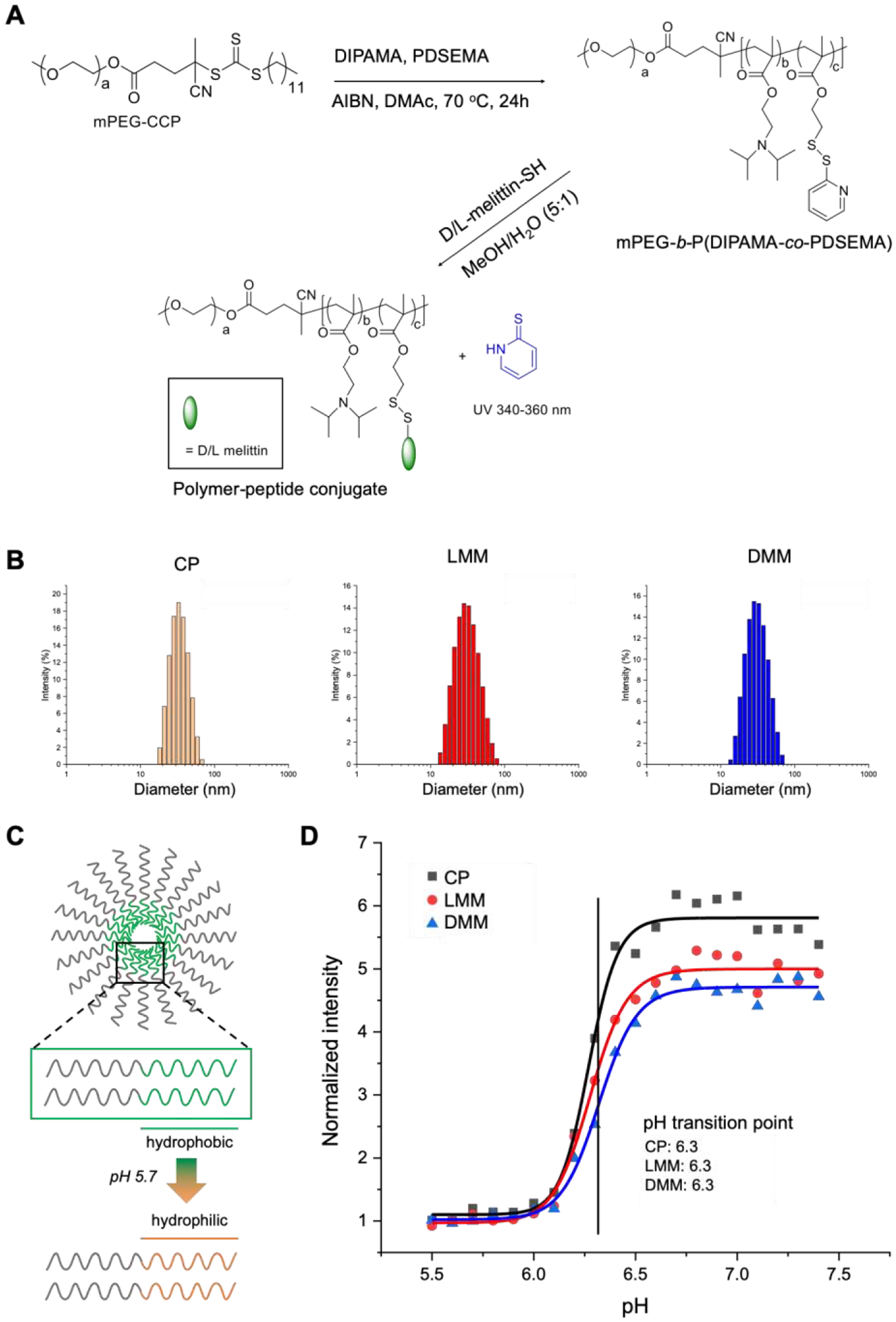
Polymer synthesis of micelles. A) Micelles were synthesized by RAFT polymerization of PEG, DIPAMA, and PDSEMA. L- or D-melittin was conjugated onto the polymer via disulfide exchange, yielding L-melittin micelles (LMM) or D-melittin micelles (DMM). Micelles without peptide are denoted as control polymer (CP). B) The hydrodynamic diameter of micelles were assessed by dynamic light scattering (DLS) and was determined to be 34.6 ± 9.9, 32.9 ± 12.5, and 32.2 ± 11.0 nm for CP, LMM, and DMM, respectively. C) A schematic demonstrating the phase transition of DIPAMA, which switches from hydrophobic to hydrophilic at acidic pH. This enables pH-triggered display of melittin for endosomal escape. D) The transition point of micelles was determined to be pH 6.3 for CP, LMM, and DMM.
The molar ratio of ethylene glycol, DIPAMA, and PDSEMA was found to be 113:40:2 by 1H NMR (Supplemental Figure 1). Therefore, the polymer was determined to be PEG113-b-p(DIPAMA40-co-PDSEMA2). This polymer structure without conjugated peptide is referred to as control polymer (CP). L or D-melittin was incorporated by disulfide exchange at a polymer:peptide feeding ratio of 1:1.5, yielding L-melittin or D-melittin micelles (LMM or DMM, respectively) with a final peptide loading content of 16.2 wt%. UV absorbance at 353 nm was used to monitor conjugation kinetics, which showed that the conjugation reaction occurred rapidly in the mixture solvent of methanol and water (V:V 5:1) (Supplemental Figure 2). We confirmed that polymer and polymer-peptide conjugates self-assembled into micelles at pH 7.4 with hydrodynamic diameters of 34.6 ± 9.9, 32.9 ± 12.5, and 32.2 ± 11.0 nm for CP, LMM, and DMM, respectively (Figure 1B) (Table 1). Critical micellar concentration (CMC) of the micelles was assessed using the Nile red method (ex/em 557/625 nm), and was determined to be 0.017, 0.027, and 0.030 mg/mL for CP, LMM, and DMM, respectively (Table 1) (Supplemental Figure 3). Micelles were tested for pH-sensitivity in buffers with various pH, and the pH transition point was determined to be pH 6.3, which is consistent with that of p(DIPAMA) (Figure 1C, D). Lastly, micelles demonstrated long term stability for 48 hours in the presence of serum (10% FBS) at 37 °C (Supplemental Figure 3). Overall, these data show that the synthesized micelles have comparable physical properties independent of peptide conjugation, similar to findings in our previous work.19
Table 1:
Characterization of micelles. The hydrodynamic diameter of micelles was determined by DLS. The critical micelle concentration was determined by Nile Red.
| Diameter (nm) | CMC (mg/mL) | |
|---|---|---|
| CP | 34.6 ± 9.9 | 0.017 |
| LMM | 32.9 ± 12.5 | 0.027 |
| DMM | 32.2 ± 11.0 | 0.030 |
3.2. In vitro activity of L- and D-melittin peptides and micelles is comparable
Because melittin is a lytic peptide, we validated in vitro activity of melittin and micelles by measuring cytotoxicity and blood hemolytic activity. To determine cytotoxicity, we incubated RAW 264.7 cells with peptide and micelles for 24h and measured viability by an MTS/PMS assay. Both L- and D-melittin peptides and micelles demonstrated comparable toxicity, as indicated by similar half maximal inhibitory concentrations (IC50) (Figure 2A–B) (Table 2). To assess hemolytic activity, we incubated human red blood cells (RBCs) with peptides and micelles at pH 6.4 and 7.4 and evaluated lysis (Table 2).26 We expected peptides to have similar hemolytic activity regardless of pH, whereas we expected micelles to only be lytic at acidic pH (Figure 2C–F). While peptides demonstrated slightly higher hemolytic activity at neutral pH 7.4 compared to acidic pH 6.4, this slight difference could be attributed to peptide aggregation, which is influenced by salt concentration and pH of the buffer.27 We observed ~100% lysis by LMM and DMM at pH 6.4, and no lysis at pH 7.4, confirming that VIPER successfully shields melittin at physiological pH and only triggers display of melittin at acidic pH. Both LMM and DMM exhibited comparable hemolytic concentrations for 50% RBC lysis (HC50) (Figure 2E–F). Lastly, we assessed the ability of LMM and DMM to escape the endosome in a Gal8-GFP-RAW 264.7 reporter cell line (Figure 2G).28 Gal8-GFP is constitutively expressed throughout the cell cytoplasm. Upon endosomal disruption, Gal8-GFP redistributes and binds to the inner face of endosomal membranes; disrupted endosomes are expressed as green punctate in cells. Micelles (12.5 μM peptide) were incubated with cells for 16–18 h at 37 °C, fixed, and stained for nuclei. Cells were imaged on a confocal microscope. Both LMM and DMM induced GFP+ punctate, confirming that both formulations disrupt the endosome. Micelles without melittin (CP) had no lytic or cytotoxic activity (Supplemental Figure 4).
Figure 2:
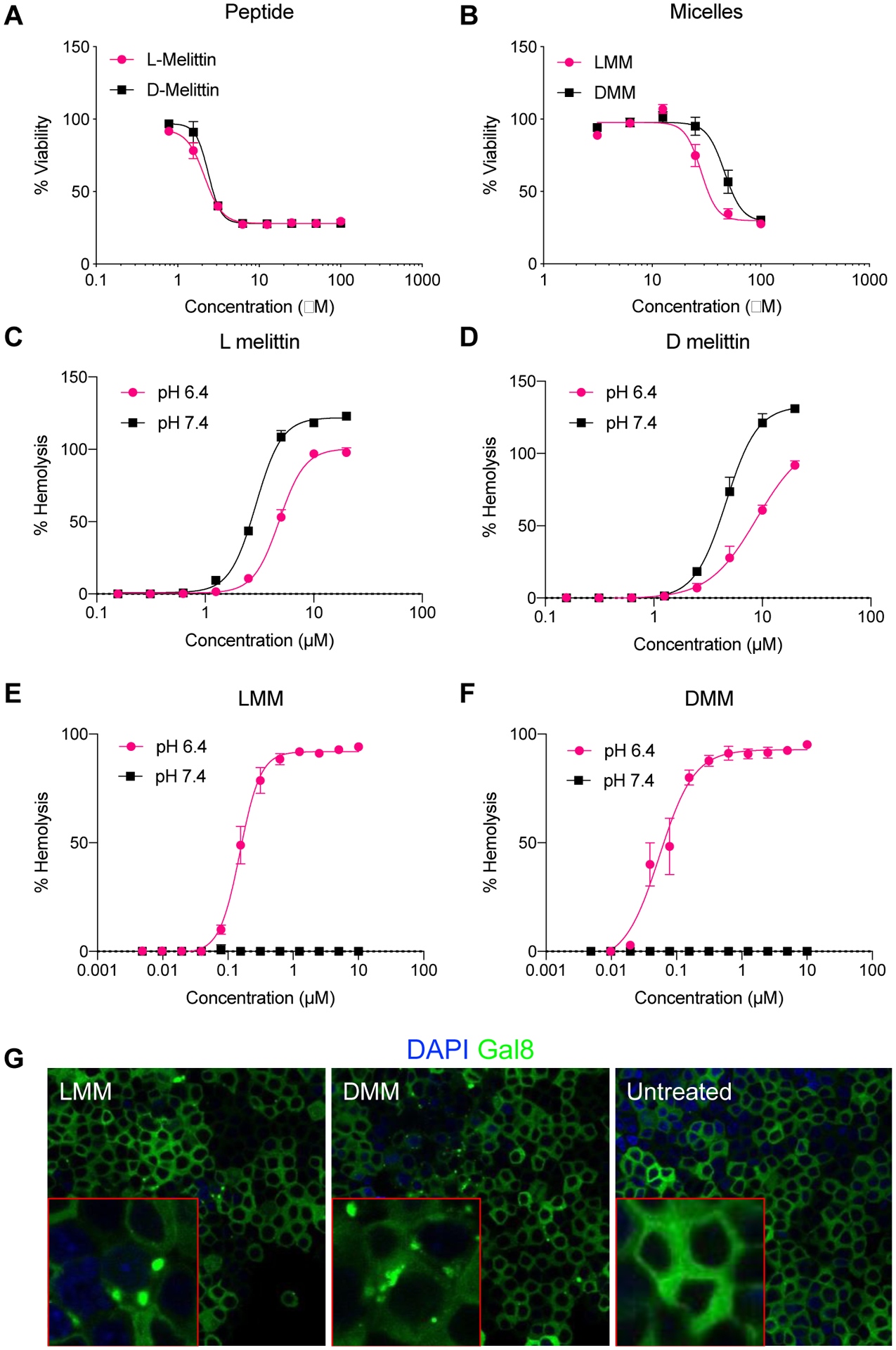
In vitro activity of L- and D-melittin peptides and micelles. A-B) L- and D-melittin peptide (A) and micelles (B) were incubated with RAW 264.7 cells for 24 h and viability was assessed. C-F) Hemolytic activity of peptide (C-D) and micelles (E-F) was evaluated against RBCs at pH 6.4 and 7.4. G) Endosomal disruption by LMM and DMM was evaluated in Gal8-GFP-RAW 264.7 cells. Endosomal disruption is expressed as GFP+ (green) punctates as Gal8-GFP binds to the inner face of endosomal membranes. Image insets (outlined in red) are single-cell magnification images.
Table 2:
Cytotoxic and hemolytic activity of melittin. The half maximal inhibitory concentration (IC50) of L- and D-melittin peptide and micelles was determined against RAW 264.7 cells after 24 h incubation. The hemolytic concentration that lysed 50% of red blood cells (RBCs) (HC50) was determined against human RBCs after 1h incubation.
| Viability IC50 (μM) | Hemolysis HC50 (μM) | ||
|---|---|---|---|
| pH 6.4 | pH 7.4 | ||
| Peptide | |||
| L-melittin | 2.13 ± 0.1 | 4.80 ± 0.1 | 2.92 ± 0.5 |
| D-melittin | 2.38 ± 0.1 | 8.89 ± 0.7 | 4.63 ± 0.1 |
| Micelles | |||
| LMM | 28.23 ± 2.7 | 0.15 ± 0.004 | N/A |
| DMM | 46.42 ± 2.0 | 0.05 ± 0.004 | N/A |
3.3. Incorporation of D-melittin increases maximum tolerated dose (MTD)
Due to similar in vitro behavior, we expected LMM and DMM to exhibit similar in vivo activity. We did not measure safety of peptide alone, as this has been reported previously.24,29 We determined the MTD by injecting micelles i.v. at 10, 20, 30, or 40 mg/kg (with respect to (w.r.t.) melittin) into normal mice and recording survival and weight for 14 days. The MTD of DMM was twice that of LMM; mice tolerated DMM at a dose up to 20 mg/kg, whereas LMM was tolerated only up to 10 mg/kg. Several mice (2/4) survived DMM injection at 30 mg/kg, whereas no mice (0/4) tolerated LMM at the same dose. Most mice (3/4) survived LMM injection at 20 mg/kg, but all mice (4/4) survived DMM at the same dose (Figure 3A–B). Cohorts receiving 20 mg/kg of LMM or 30 mg/kg of DMM exhibited weight loss in the days immediately following injection, but weight rapidly recovered within 4 days (Figure 3C–D). No mice survived injection of 40 mg/kg of either LMM or DMM. Together, these results indicate there is some acute toxicity for both peptide-micelle analogues at high concentrations, likely due to the lytic activity of melittin, but DMM are better tolerated overall. Based on survival and weight loss, we determined the MTD of LMM and DMM as approximately 10 and 20 mg/kg, respectively (Table 3).
Figure 3:
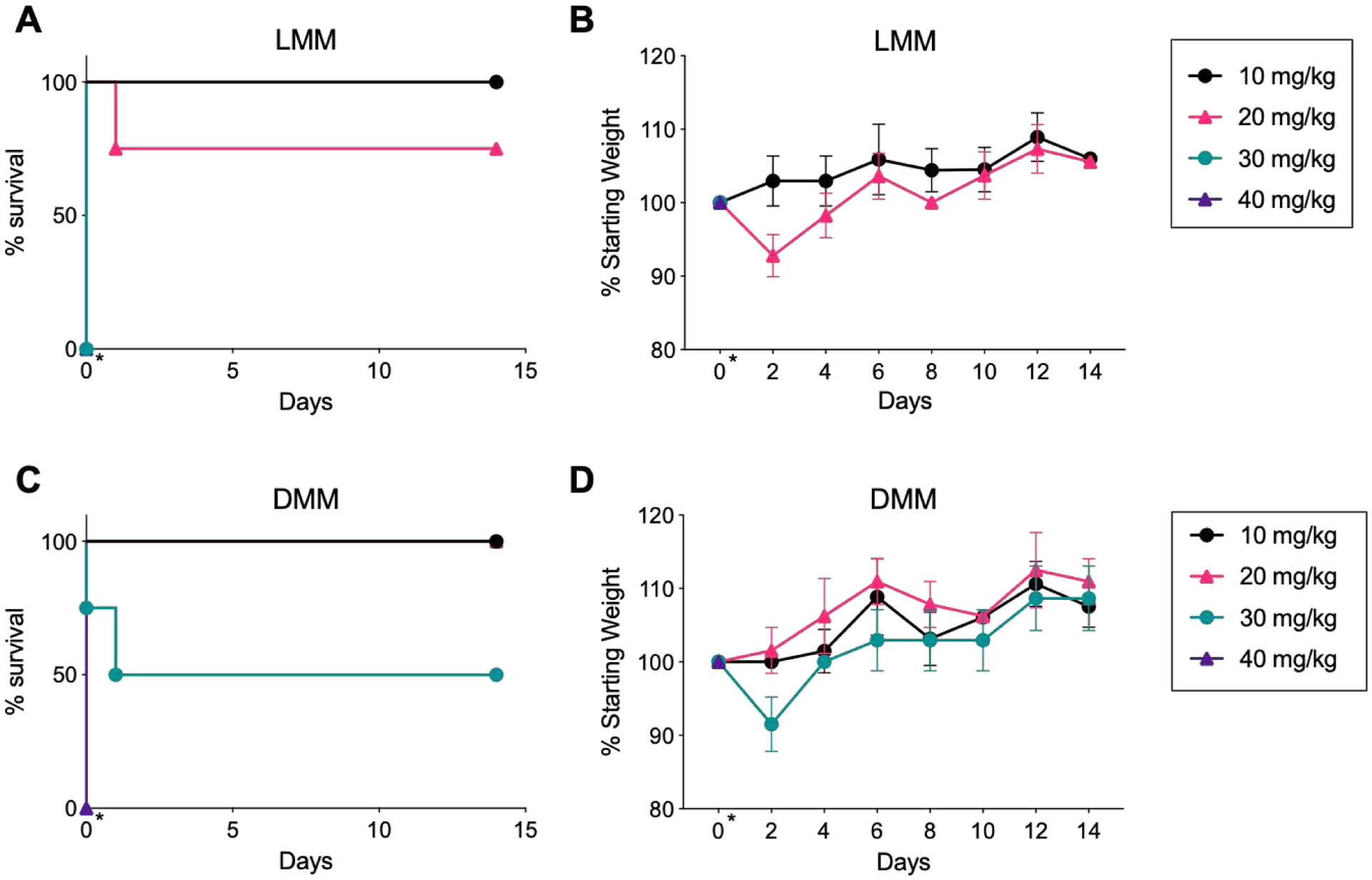
MTD determination of LMM and DMM. A-B) Survival and weight of normal mice injected with LMM at 10, 20, 30, or 40 mg/kg. The day of injection (day 0) is indicated with an asterisk (*). Weight was measured for 14 days following injection. C-D) Survival and weight of mice injected with DMM at 10, 20, 30, or 40 mg/kg. (n = 4 mice/group)
Table 3:
Death of mice after LMM or DMM injection. Based on survival and weight loss, the MTD of LMM and DMM was determined as approximately 10 and 20 mg/kg, respectively. (n = 4 mice/group)
| Death of mice | ||
|---|---|---|
| Dose | LMM | DMM |
| 10 mg/kg | 0/4 | 0/4 |
| 20 mg/kg | 1/4 | 0/4 |
| 30 mg/kg | 4/4 | 2/4 |
| 40 mg/kg | 4/4 | 4/4 |
3.4. Incorporation of D-melittin enables safe, repeated dosing of micelles
Next, we investigated the safety of repeat injections of melittin micelles in normal mice. Mice were injected i.v. with LMM or DMM at 5 mg/kg (w.r.t. to melittin) every 4th day for a total of 4 injections. This dose was chosen as it is below the MTD of both peptide-micelles, enabling us to evaluate the safety of micelles without attributing death to acute toxicity. While the cumulative dosage is the MTD of LMM, we hypothesized that the micelles would demonstrate less toxicity than a single bolus when accounting for clearance and recovery time between injections. While all mice tolerated the first two injections well, as indicated by maintenance of weight, none of the mice (0/6) in the LMM cohort survived the 3rd injection (Figure 4A–B). Some mice (2/6) died within the first 2 hours following injection; the remaining mice (4/6) died within the next 24 hours. Mice exhibited signs of anaphylaxis, such as lethargy, loss of activity, and depressed breathing. All mice (6/6) in the DMM cohort survived and tolerated the remaining 3rd and 4th injections well. Mice injected with DMM exhibited no adverse effects, even at 21 days past the 4th injection. We hypothesized that the 3rd injection of LMM triggered the observed toxicity and mortality, as no weight loss or abnormal behavior was observed in mice following the 2nd injection.
Figure 4:
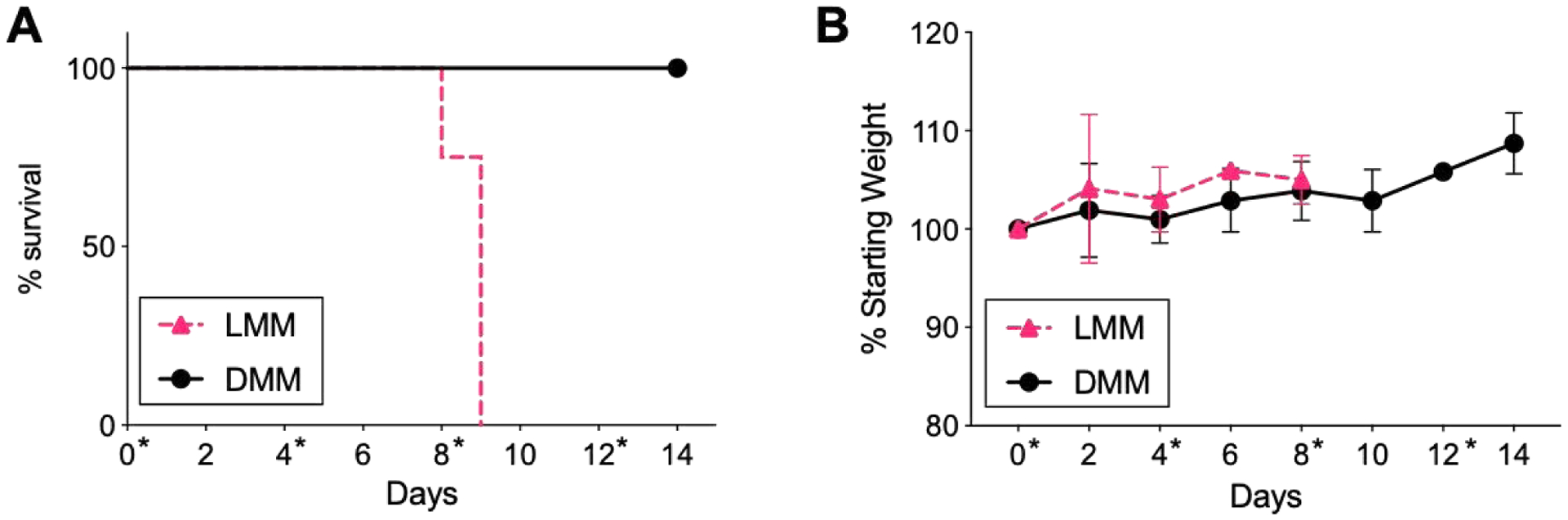
Survival and weight of mice receiving repeat injections of LMM and DMM. A) Mice were injected with LMM or DMM at 5 mg/kg every 4 days, for a total of 4 injections. All mice(6/6) receiving LMM died after the 3rd injection; all mice (6/6) receiving DMM tolerated all 4 injections well. Days of injection (0, 4, 8, 12) are indicated with an asterisk (*). B) Mice weight was recorded following injection with LMM or DMM. (n = 6 mice/group)
3.5. The adaptive immune response is attenuated in DMM-treated mice
Because mice receiving LMM died following the 3rd injection, but exhibited no adverse effects between injections, we hypothesized that an adaptive immune response was triggered upon receiving the 3rd treatment. This immune response was activated specifically by LMM, since we did not observe any deaths with repeat injections of DMM. To confirm that the immune system played a role in the death of mice upon repeat-injection, we injected normal and immune-deficient nod-scid gamma (NSG) mice with LMM at 2, 4, 6, and 8 mg/kg every 4th day. Both mice are on a Balb/c background, accounting for differences that could be attributed to mouse strain.23 We investigated a wider range of concentrations to better understand the influence of individual injection dose on toxicity upon repeat injection. Normal mice injected at doses above 5 mg/kg exhibited weight loss after the second injection, while no weight loss was observed in mice receiving doses below 5 mg/kg (Figure 5A). Due to this extreme weight loss, mice at 6 and 8 mg/kg did not receive the 3rd and 4th injections. NSG mice did not exhibit weight loss, regardless of dose, and tolerated repeat injections at 6 and 8 mg/kg (Figure 5B).
Figure 5:
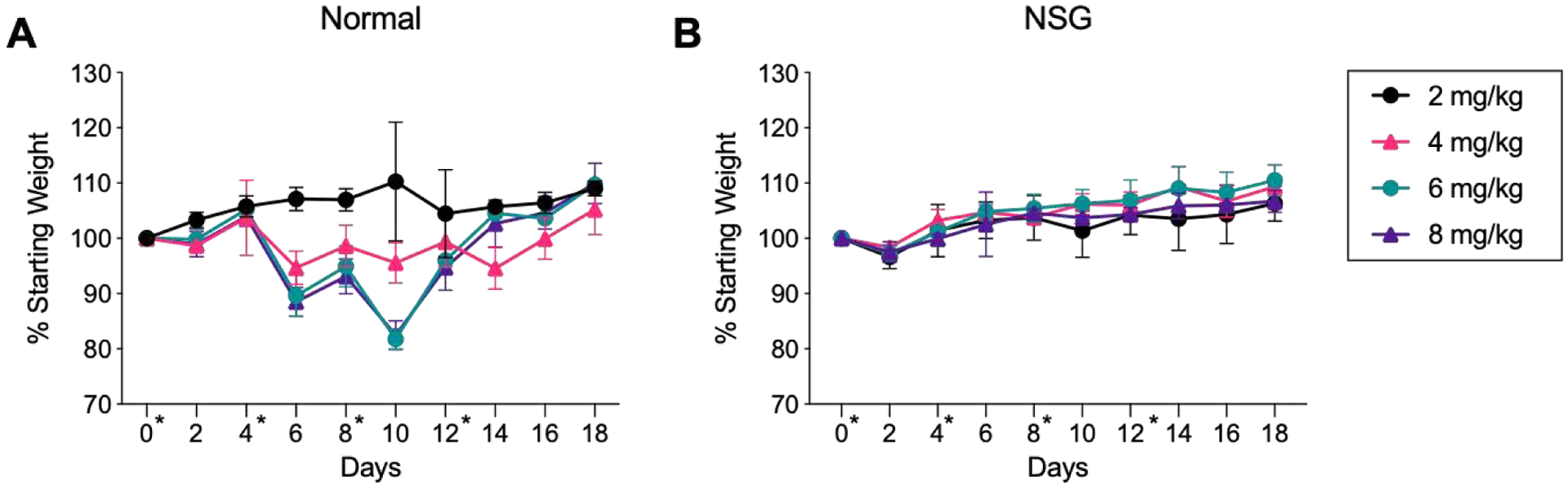
LMM injections in normal and NSG mice. Normal (A) or NSG (B) mice were injected i.v. with LMM at 2, 4, 6, and 8 mg/kg. Normal mice were not administered the 3rd or 4th injection at 6 or 8 mg/kg due to weight loss. Days of injection (0, 4, 8, 12) are indicated with an asterisk (*). (n = 4 mice/group)
Because normal mice experienced weight loss but NSG mice were unaffected, this study supported our hypothesis that an inflammatory adaptive immune response was responsible for adverse effects of LMM dosing. Therefore, we next investigated antibody generation following repeat injection of micelles. Because prior results indicated that mice tolerated the first two injections well, we investigated antibody titers immediately after the 3rd injection. Mice were injected i.v. with LMM or DMM micelles at 5 mg/kg every 4th day. Immediately following the 3rd injection, mice were sacrificed and serum was assessed for IgG and IgM antibodies against micelles, polymer, and peptide via ELISA. IgG and IgM antibodies were evaluated because of their role in binding to PEGylated conjugates and mediating type II hypersensitivity reactions. LMM-treated mice generated significant IgG and IgM antibody titers against LMM, CP, and 10k PEG (Figure 6A–F). In contrast, antibodies from DMM-treated mice were often below the limit of detection. Antibodies in LMM or DMM-treated mice primarily bound to polymer, with very little binding to peptide, confirming that LMM induced anti-PEG antibodies (Figure 6G–H). Control mice that did not receive peptide-micelles generated negligible levels of IgG or IgM antibodies (Supplemental Figure 5).
Figure 6:
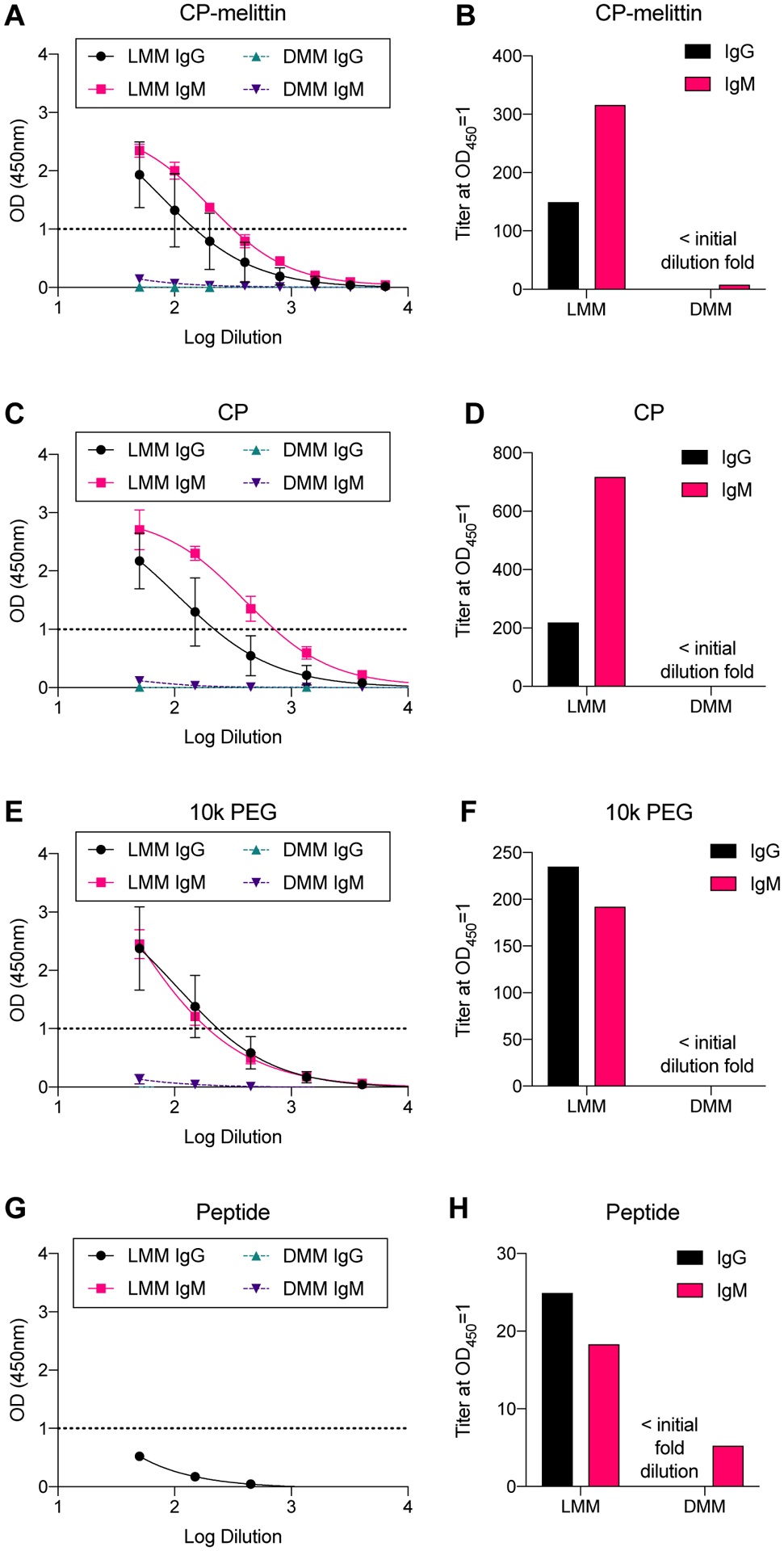
IgG and IgM antibodies against micelles, polymer, and peptide. Mice were injected i.v. with LMM or DMM (5 mg/kg). Immediately after the 3rd injection, mice were sacrificed and serum was analyzed for IgG and IgM antibodies against micelles (A,B), CP (C,D), 10k PEG (E,F), or peptide (G,H).
We also investigated liver toxicity following repeat injection of micelles via hematoxylin and eosin (H&E) staining and liver enzyme (alanine transaminase (ALT), aspartate transaminase (AST)) activity in serum. Mice were injected with either PBS, LMM, or DMM at 5 mg/kg on days 0 and 4. Mice were sacrificed 6 hours after receiving the 2nd injection to avoid the mortality observed after the 3rd injection with LMM. Liver H&E was completed and no abnormalities in the liver were observed in any of the mice (Figure 7A–C). This suggests that hepatotoxicity is not a factor in the observed deaths. ALT and AST levels were within the normal range for all groups as well, further supporting this conclusion30,31 (Figure 7D–E).
Figure 7:
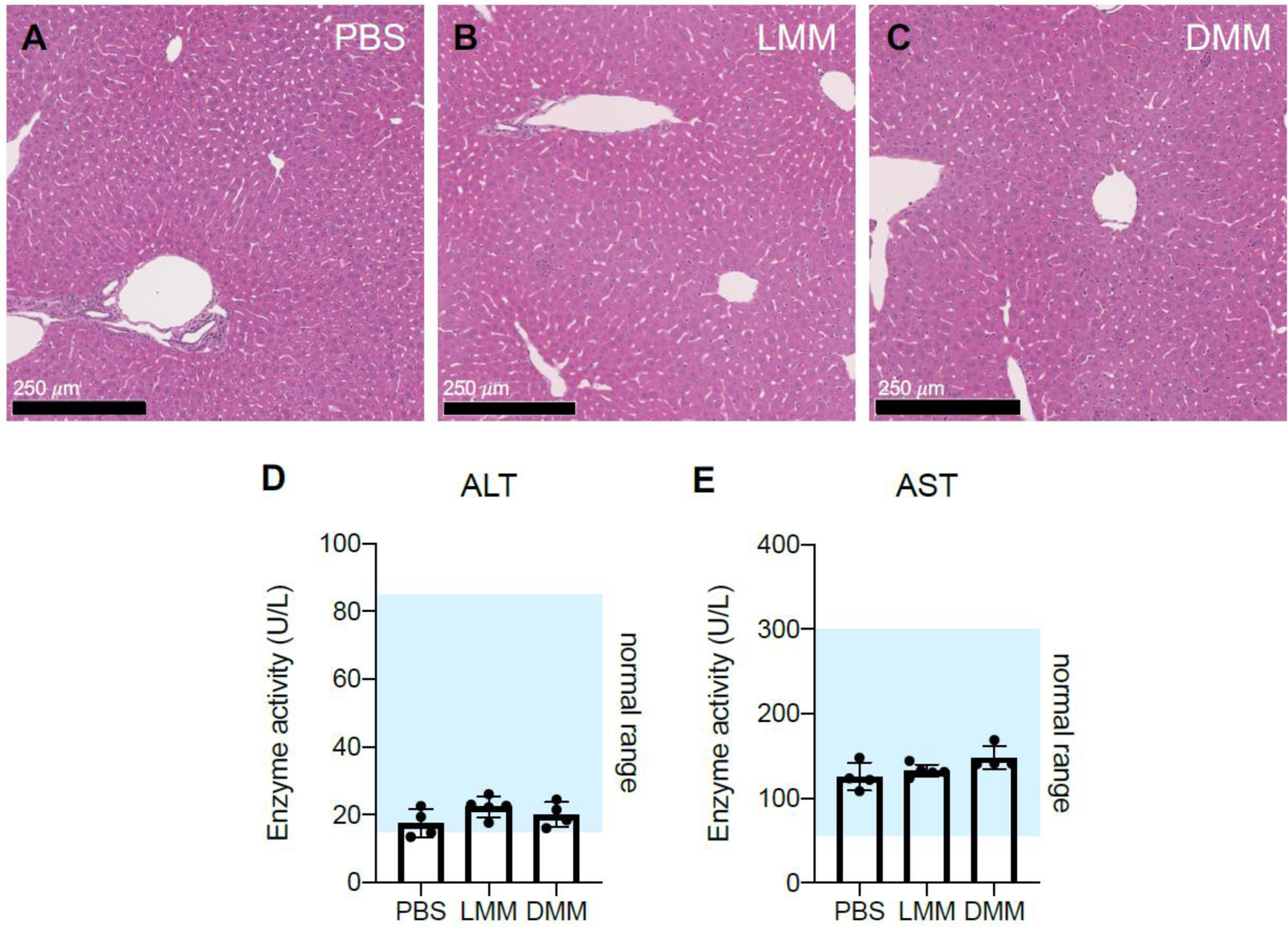
Liver toxicity after micelle injection was evaluated by H&E and ALT/AST activity. Mice were injected i.v. with PBS, LMM, or DMM (5 mg/kg) on days 0 and 4. Six hours after the 2nd injection, mice were sacrificed and livers and serum were collected for H&E staining (A-C) or liver enzyme (ALT, AST) analysis (D, E). H&E images are at 10X magnification (n = 3 mice/group). (For ALT/AST analysis, n = 4–5 mice/group)
3.6. PAF receptor antagonist extends survival but does not rescue mice
Based on micelle toxicity and immune response, we hypothesized that the platelet activating factor (PAF) receptor could be associated with both acute and adaptive toxicity. PAF is a phospholipid signaling molecule that plays a central role in normal and pathological responses, particularly inflammation, allergy, and shock.32,33 Its receptor, PAFR, is expressed on the surface of many cells, including platelets, macrophages, and neutrophils. PAF has been implicated in the immune response against lipid nanoparticles, in which mice exhibited signs of acute toxicity and shock-like symptoms (e.g., edema, hypovolemia). Prophylactic blockade of the PAFR can rescue mice and completely prevent immune-mediated toxicity against lipid nanoparticles.34–36
We observed shock-like symptoms in mice following injection of a high single dose or repeat-dosing of micelles that we linked to an immune response. Thus, we next attempted to rescue mice with an intraperitoneal (i.p.) injection of PAFR antagonist CV-6209 prior to micelle injection, as reported.34,35 We sought to rescue normal Balb/c mice from LMM-associated toxicity prior to either a single injection at 30 mg/kg or a 3rd injection at 5 mg/kg. The dose of 30 mg/kg was selected because it is above the MTD of both LMM and DMM. Furthermore, some mice (3/4) survived treatment with LMM at 20 mg/kg, but none (0/4) survived treatment at 30 mg/kg. Thus, this selected dose would ensure that rescue could be attributed to pre-treatment with the PAF antagonist. We also investigated the effect of prophylactic treatment prior to the 3rd injection because of the toxicity at this dosing regimen; neither the first or second injections were toxic at the selected dose. CV-6209 was unable to rescue mice from acute toxicity at 30 mg/kg; all mice (4/4) died within 1 hour of LMM injection. However, CV-6209 did exhibit some prophylactic effect on mice receiving repeat-injections. Mice who did not receive CV-6209 died within 30–60 minutes of injection. Mice receiving CV-6209 exhibited some signs of toxicity but remained alert, active, and responsive. Yet, all treated mice died within 24 hours. Overall, CV-6209 was unable to rescue mice from acute toxicity, but did extend survival by several hours in mice receiving repeat-injections.
4. Discussion and Conclusion
Here, we report the replacement of L-amino acid peptides with D-amino acid peptides in polymer-peptide conjugates to attenuate anti-polymer antibody generation. Specifically, we replaced L-melittin for D-melittin in our VIPER platform, which enhanced safety in vivo by increasing the MTD and reducing anti-PEG antibody generation, ultimately enabling frequent repeat-dosing. While these findings are specific to VIPER, they can be applied to other peptide-polymer systems.
We first validated that both L- and D- peptide and micelles possess similar physical properties (e.g., diameter, pH transition, and CMC) and biologic behavior in vitro, as demonstrated by comparable cytotoxicity, hemolysis, and endosomal disruption. These findings are consistent with those by Boeckle, et. al., who showed that PEI conjugates with D-melittin exhibited similar lytic activity to conjugates with L-melittin, but offered the advantage of being non-immunogenic.37 Differences between the peptide analogues were primarily observed in vivo, as the MTD of DMM (20 mg/kg) was twice that of LMM (10 mg/kg). Significantly, DMM enabled frequent, repeat-dosing, whereas LMM resulted in premature mortality. We first confirmed that toxicity was immune-related by administering LMM to both normal and immunodeficient NSG mice and evaluating weight loss. NSG mice lack mature T and B cells, whereas normal mice have complete immune functionality. While normal mice rapidly lost weight following two repeat-injections, NSG mice maintained weight and tolerated all four injections. Thus, we posited that LMM induced anti-PEG antibodies in normal mice, which mediated toxicity and death upon repeat-injection. While anti-PEG antibodies can be generated in both a T cell dependent (TD) and independent (TI) mechanism, it is likely that the immune response raised by VIPER is TD. A report by Mima, et. al. demonstrated that the immunogenic conjugate PEG-OVA induced anti-PEG antibodies, whereas free PEG did not. This immune response was determined to be TD, as antibodies were generated in normal mice but not in T cell deficient mice.12 These results are parallel to our findings, as our peptide-polymer platform raised anti-PEG antibodies, but polymer micelles without peptide did not. In contrast, antibodies generated in a TI manner trigger antibody formation regardless of cargo, as Ichihara, et. al. showed that empty PEGylated liposomes elicited anti-PEG IgM in normal and T cell deficient mice.38
We next evaluated the antibody response to micelles upon repeat injection. In PEGylated platforms, IgM is the primary antibody generated and is responsible for the rapid clearance of PEGylated liposomes, protein-, and peptide-conjugates.12,39,40 ELISA analysis of generated antibodies exhibited the specificity of anti-PEG IgM antibodies for the terminal methoxy or the backbone of the polymer.41,42 In this work, we observed that LMM-treated mice generated robust IgG and IgM antibody response against micelles and polymer, while DMM-treated mice generated a negligible antibody response. Antibodies from LMM-treated mice bound both LMM and CP (no melittin) micelles, suggesting that the polymer, rather than the peptide, is the antigen target. We confirmed this by assessing antibody binding against 10k PEG, which validated that antibodies bound the polymer. These anti-PEG antibodies demonstrated higher binding to polymer micelles than to 10k PEG, which has also been observed with other clones of anti-PEG antibodies (data not shown). This phenomenon could perhaps be explained by micelle structure, which could increase avidity and facilitate a higher observed binding affinity. Lastly, antibodies showed negligible binding to peptide without polymer. These results support our hypothesis that induced toxicity is due to a hypersensitivity response against LMM, in which L-melittin acts as an adjuvant to elicit antibodies against the PEG in the polymer backbone. The first two injections prime the immune system to generate antibodies against the carriers, and the third injection triggers the generation of IgG and IgM antibodies against the PEG backbone.43 In contrast, DMM did not generate an antibody-based immune response and was well tolerated in four repeat-injections. These results are supported by the literature, as the adjuvant activity of L-melittin and the reduced immunogenicity of D-melittin have been reported.5,20,24 Neither formulation elicited liver toxicity, as evaluated by H&E liver staining and ALT/AST enzyme activity in serum.
Lastly, we investigated the role of PAF in toxicity following acute and repeat-dosing of LMM, as PAFR has been implicated in lipid nanoparticle-associated toxicity.34–36 Thus, we investigated the effect of prophylactic treatment with PAF antagonist CV-6209 prior to LMM injection of either a single injection (30 mg/kg) or repeat-injection (3rd treatment). While CV-6209 was unable to entirely rescue mice, it extended survival in mice receiving a repeat-injection, suggesting that the PAFR is associated with LMM-toxicity. The inability to completely prevent mortality could be attributed to insufficient dose, requirement of a longer time period between CV-6209 and micelle injection, or involvement of other immune cascades that we have not yet identified.
Overall, we demonstrated that exchanging a natural peptide for its D-amino acid enantiomer mitigates the generation of anti-polymer antibodies, enabling safe, repeat-dosing of polymer-peptide conjugates. This phenomenon can be explained by the enhanced resistance to proteolytic degradation of D-peptides, preventing processing by antigen presenting cells and subsequent recognition and antibody generation by T and B cells.24 Another report suggests that the lack of immunogenicity of D-amino acid analogues is due to enzymatic resistance, prolonging retention and circulation in vivo.44 This delayed degradation could result in toxicity if the melittin is unable to be cleared from the body. However, we did not observe adverse effects at 7 or 14 days after repeat- or MTD injections, respectively.
Our work has additional implications for anti-cancer therapeutics. While VIPER employs melittin to facilitate endosomal escape, this lytic peptide has been used extensively in traditional medicines and cancer applications.45–48 Melittin disrupts cell membranes to induce immunogenic cell death by releasing intracellular contents (e.g., tumor associated antigens) and stimulating T cell and natural killer cell immunity.49–51 However, free peptide induces extensive non-specific hemolysis and severe off-target toxicity, and suffers from poor pharmacokinetics, requiring an appropriate drug delivery vehicle to realize its therapeutic potential.52,53 Our findings can be applied to these carrier formulations, as we have demonstrated a strategy to enhance the safety of melittin, significantly attenuating the generation of anti-PEG antibodies and enabling safe, repeated dosing.
In conclusion, our findings add to the repertoire of strategies to enhance the safety of PEG-containing therapeutics. There is an increasing need to address this issue as the occurrence of anti-PEG in the healthy population has rapidly grown from ~0.2% to ~72% in the past four decades.6,54,55 While Yang, et. al. found that the majority of people have low levels of anti-PEG antibodies, this study underscored the importance of pre-screening patients prior to administration of PEG-therapeutics. Another strategy to enhance the safety of PEG-conjugates is to suppress anti-PEG antibody generation, as we have shown here with D-amino acid peptide substitution. Approaching the problem from another side, Sherman, et. al. characterized the immunogenicity of different PEG polymers and demonstrated that methoxy PEG was more immunogenic than hydroxy PEG, as methoxy PEG elicited higher antibody titers.56 This strategy was effective in protein-polymer conjugates with interferon, uricase, and albumin. For cases in which pre-existing anti-PEG antibodies are already present, McSweeney, et. al. demonstrated that saturating anti-PEG antibody binding with infusion of 40 kDa restored PEGylated liposome circulation time.57 Yet, despite these advances in characterizing the immunogenicity of PEGylated conjugates, there remains a need to better understand how the composition of PEGylated conjugates affects the generation and specificity of anti-PEG antibodies. Further discernment of the relationship between PEG architecture and immunogenicity is critical in order to create the next generation of biocompatible PEG-conjugates.
5. Experimental Procedures
Materials.
PEG macro CTA was ordered from Sigma. PDSEMA was synthesized as described previously.58 L- and D-melittin (GIGAVLKVLTTGLPALISWIKRKRQQC) peptides were synthesized through solid phase peptide synthesis on a microwave peptide synthesizer (Liberty Blue CEM) using L- or D-amino acids, respectively. Peptides were cleaved from resin in a trifluoroacetic acid (TFA) cocktail with 5% dimethoxybenzene 2.5% triisopropylsilane and 2.5% ethanedithiol, and 2.5% deionized water. Crude peptide was precipitated twice in cold diethyl ether and purified by reverse-phase HPLC using 0.1% TFA water and acetonitrile. Peptide molecular mass was determined by MALDI-TOF (University of Washington Department of Medicinal Chemistry Mass Spectrometry Center) in a CHCA:DHB 2:1 matrix. All other chemicals were purchased from Sigma and used as received.
Polymer synthesis.
Block copolymer PEG113-b-p(DIPAMA40-co-PDSEMA2) was prepared with RAFT polymerization. In brief, PEG macro CTA (1000 mg, 0.182 mmol), DIPAMA (1850 mg, 7.58 mmol), PDSEMA (190 mg, 0.89 mmol) and azobisisobutyronitrile (AIBN) (3 mg, 0.018 mmol) were dissolved in 15 mL dimethylacetamide (DMAc), purged in argon, and immersed in an oil bath at 70°C.18 After 24h, the polymerization was quenched with liquid nitrogen and the polymer was purified by dialysis against methanol for 2 days (yield: 80%). Synthesized peptides were conjugated to the PDSEMA monomer via disulfide exchange reaction in a mixture of methanol and water (5:1) at a polymer:peptide molar ratio of 1:1.5. After 8 h, the polymer-peptide conjugates were purified by dialysis against DI water for 2 days. To prepare the micelles, the polymers or polymer-peptide conjugates were first dissolved in acidic phosphate buffer (pH 4.0), and the pH was adjusted to pH 7–8. All micelles were sterile filtered using a 0.22 mm pore filter.
Polymer characterization.
Polymer was characterized by 1H NMR on a Bruker AV 300 in deuterated chloroform (CDCl3). Peptide conjugation with PDSEMA was monitored by UV at 353 nm for the release of 2-thiopyridine. Micelle size was assessed by dynamic light scattering (DLS) at 0.5 mg/mL in PBS 7.4. Critical micellar concentration (CMC) of the micelles was determined using a Nile red method (ex/em 557/625 nm) with 0.5 μg/mL of dye. The CMC was determined as the inflection point on the emission curve.
Cell culture.
RAW 264.7 macrophages were obtained from ATCC and cultured in RPMI 1640 (Gibco) supplemented with 10% FBS (Gibco). For toxicity assays, macrophages were seeded at 15k cells/well in a 96-well plate. Cells were cultured with peptide or micelles for 24 hours and viability was determined by MTS/PMS (Promega) by plate reader. The Gal8-RAW 264.7 cell line was generated through similar means as described.28 Briefly, RAW cells were co-transfected with plasmids containing a transposable Gal8-GFP construct and PiggyBac transposon (gift of Prof. Jordan Green) using Lipofectamine 2000 (3:1 molar ratio transposon:transposase plasmid). Cells were sorted for the top 5% brightest GFP+ singlet cell events using a FACS Aria sorter (BD), expanded, and sorted three more times to yield a population of Gal8-GFPhigh cells.
Hemolysis assays.
Hemolysis assays were conducted as described.26 Human blood was obtained in accordance with guidelines established by the University of Washington Institutional Review Board. Briefly, human blood was washed in 150 mM NaCl and washed twice. Blood was transferred to PBS pH 7.4, washed, and resuspended in PBS at each pH to be tested (pH 6.4, 7.4). Blood was diluted 1:50 and 190 μl of diluted blood was plated in a V-bottom 96 well plate. Samples were incubated with peptide in appropriate pH at 37°C for 1h. Supernatant was transferred to a clear, flat bottom 96-well plate and absorbance (541 nm) was detected on a plate reader. Triton X-100 20% (w/v) was used as a positive control, and PBS at appropriate pH was used as a negative control.
Imaging.
N01 coverslips were coated with bovine collagen I (Thermo) (45 μg/mL) for 1h at room temperature. Gal8-RAW 264.7 cells were plated (300–400k) onto coverslips overnight, and were incubated with peptide or micelles for 16–18h in complete media. Cells were fixed in 4% PFA and stained with DAPI (1:1000). Slides were imaged on a confocal microscope with a 63X oil immersion objective (Leica SP8X). For H&E imaging, livers were submitted to the UW Histology & Imaging Core for tissue processing and staining.
Animal studies.
All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Washington and were conducted in accordance use and regulations. Female Balb/c mice (6–8 weeks) were ordered from Charles River Laboratories. Particles were injected i.v. (intravenous) via tail vein injections at indicated doses. Mice were injected every 4th day. Mice were humanely euthanized when euthanasia criteria were met (e.g., hunched, squinting, low to no activity, depressed respiration). Terminal blood draws were collected from the vena cava after drug overdose or CO2 euthanization. Serum was collected in serum separator tubes (BD) by allowing blood to coagulate for 30 minutes at room temperature, followed by centrifugation at 1000 xg for 10 minutes at 4 °C. Serum was stored at −80°C. For PAF rescue studies, mice were injected with PAF antagonist CV-6209 (Cayman) by intraperitoneal (i.p.) injection (50 μg per mouse) 5 minutes prior to micelle injection, as described.34
Enzyme linked immunosorbent assays (ELISA).
Assays were performed as previously reported.55,59 Briefly, micelles (1 μg/mL) were coated onto flat-bottom MaxiSorp 96-well plates overnight at 4 °C. Wells were washed with PBS and blocked with 5% skim milk (Difco) in PBS for 1h at room temperature. Wells were washed with PBS and incubated with serum (diluted in 2% skim milk) for 1h at room temperature on a shaker. Wells were washed twice with PBS and incubated with anti-mouse IgG-HRP or IgM-HRP secondary antibody (Jackson Laboratories) for 45 minutes at room temperature on a shaker. Wells were washed three times with PBS for 1 minute on a shaker, and incubated with TMB (Thermo) for 3–5 minutes. 2N H2SO4 was added to stop the reaction and absorbance (450 nm) was read on a plate reader. For peptide ELISAs, thiol-containing peptide (50 μg/mL) was immobilized onto maleimide coated plates (ThermoFisher) for 2h. Unreacted maleimides were blocked with free cysteine (10 mg/mL) for 1h, prior to continuing with the above ELISA protocol. Prism 8.0 was used to fit data and interpolate titer at OD450=1 using a sigmoidal, 4PL, X is log(concentration) model. Serum ALT/AST levels were determined by a kit (Sigma), following manufacturer instructions.
Supplementary Material
Highlights:
Substituting L-melittin for its enantiomer, D-melittin, enables safe, repeat-dosing of melittin peptide-polymer conjugates
Toxicity against L-melittin micelles in vivo is mediated by anti-PEG IgG and IgM antibodies
D-melittin micelles do not generate a strong IgG or IgM antibody response in vivo
5. Acknowledgements
This work was supported by NIH (1R01CA17727, R01NS064404, and U54CA199090). We thank Nathanial Peters and W. M. Keck Microscopy Center (S10 OD016240) for confocal microscopy support. We thank Kim Woodrow (UW Bioengineering) for use of her plate reader. We thank Prof. Andre Lieber for his help interpreting the liver H&E images.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
S.H.P. has submitted a patent with the U.S. Patent and Trademark Office (WO2018027164A1) related to VIPER. The other authors have no competing interests.
7. References
- 1.Kang JS, Deluca PP & Lee KC Emerging PEGylated drugs. Expert Opin Emerg Drugs 14, 363–380, doi: 10.1517/14728210902907847 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Zhang P, Sun F, Liu S & Jiang S Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation. J Control Release 244, 184–193, doi: 10.1016/j.jconrel.2016.06.040 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Q & Lai SK Anti-PEG immunity: emergence, characteristics, and unaddressed questions. Wiley Interdiscip Rev Nanomed Nanobiotechnol 7, 655–677, doi: 10.1002/wnan.1339 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsieh YC, Wang HE, Lin WW, Roffler SR, Cheng TC, Su YC, Li JJ, Chen CC, Huang CH, Chen BM, Wang JY, Cheng TL & Chen FM Pre-existing anti-polyethylene glycol antibody reduces the therapeutic efficacy and pharmacokinetics of PEGylated liposomes. Theranostics 8, 3164–3175, doi: 10.7150/thno.22164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishida T & Kiwada H Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int J Pharm 354, 56–62, doi: 10.1016/j.ijpharm.2007.11.005 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Armstrong JK, Hempel G, Koling S, Chan LS, Fisher T, Meiselman HJ & Garratty G Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer 110, 103–111, doi: 10.1002/cncr.22739 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Abu Lila AS, Kiwada H & Ishida T The accelerated blood clearance (ABC) phenomenon: clinical challenge and approaches to manage. J Control Release 172, 38–47, doi: 10.1016/j.jconrel.2013.07.026 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Elsadek NE, Hondo E, Shimizu T, Takata H, Abu Lila AS, Emam SE, Ando H, Ishima Y & Ishida T Impact of Pre-Existing or Induced Anti-PEG IgM on the Pharmacokinetics of Peginterferon Alfa-2a (Pegasys) in Mice. Mol Pharm 17, 2964–2970, doi: 10.1021/acs.molpharmaceut.0c00366 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Ganson NJ, Kelly SJ, Scarlett E, Sundy JS & Hershfield MS Control of hyperuricemia in subjects with refractory gout, and induction of antibody against poly(ethylene glycol) (PEG), in a phase I trial of subcutaneous PEGylated urate oxidase. Arthritis Res Ther 8, R12, doi: 10.1186/ar1861 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Smith CA, Panetta JC, Yang W, Thompson LE, Counts JP, Molinelli AR, Pei D, Kornegay NM, Crews KR, Swanson H, Cheng C, Karol SE, Evans WE, Inaba H, Pui CH, Jeha S & Relling MV Antibodies Predict Pegaspargase Allergic Reactions and Failure of Rechallenge. J Clin Oncol 37, 2051–2061, doi: 10.1200/JCO.18.02439 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richter AW & Akerblom E Antibodies against polyethylene glycol produced in animals by immunization with monomethoxy polyethylene glycol modified proteins. Int Arch Allergy Appl Immunol 70, 124–131, doi: 10.1159/000233309 (1983). [DOI] [PubMed] [Google Scholar]
- 12.Mima Y, Hashimoto Y, Shimizu T, Kiwada H & Ishida T Anti-PEG IgM Is a Major Contributor to the Accelerated Blood Clearance of Polyethylene Glycol-Conjugated Protein. Mol Pharm 12, 2429–2435, doi: 10.1021/acs.molpharmaceut.5b00144 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Shiraishi K & Yokoyama M Toxicity and immunogenicity concerns related to PEGylated-micelle carrier systems: a review. Sci Technol Adv Mater 20, 324–336, doi: 10.1080/14686996.2019.1590126 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webster R, Elliott V, Park BK, Walker D, Hankin M & Taupin P in PEGylated Protein Drugs: Basic Science and Clinical Applications Ch. Milestones in Drug Therapy, 127–146 (Birkhäuser Basel, 2009). [Google Scholar]
- 15.Pelegri-O’Day EM, Lin EW & Maynard HD Therapeutic protein-polymer conjugates: advancing beyond PEGylation. J Am Chem Soc 136, 14323–14332, doi: 10.1021/ja504390x (2014). [DOI] [PubMed] [Google Scholar]
- 16.Ekladious I, Colson YL & Grinstaff MW Polymer-drug conjugate therapeutics: advances, insights and prospects. Nat Rev Drug Discov 18, 273–294, doi: 10.1038/s41573-018-0005-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tenenbaum SA, Rice JC, Espinoza LR, Cuéllar ML, Plymale DR, Sander DM, Williamson LL, Haislip AM, Cluck OS, Tesser JRP, Nogy L, Stribrny KM, Bevan JÁ & Garry RF Use of antipolymer antibody assay in recipients of silicone breast implants. The Lancet 349, 449–454, doi: 10.1016/s0140-6736(96)07131-0 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Cheng Y, Yumul RC & Pun SH Virus-Inspired Polymer for Efficient In Vitro and In Vivo Gene Delivery. Angew Chem Int Ed Engl 55, 12013–12017, doi: 10.1002/anie.201605958 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peeler DJ, Thai SN, Cheng Y, Horner PJ, Sellers DL & Pun SH pH-sensitive polymer micelles provide selective and potentiated lytic capacity to venom peptides for effective intracellular delivery. Biomaterials 192, 235–244, doi: 10.1016/j.biomaterials.2018.11.004 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kind LS, Ramaika C & Allaway E Antigenic, adjuvant and permeability enhancing properties of melittin in mice. Allergy 36, 155–160, doi: 10.1111/j.1398-9995.1981.tb01830.x (1981). [DOI] [PubMed] [Google Scholar]
- 21.Bramwell VW, Somavarapu S, Outschoorn I & Alpar HO Adjuvant action of melittin following intranasal immunisation with tetanus and diphtheria toxoids. J Drug Target 11, 525–530, doi: 10.1080/10611860410001670080 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Paull B, Yunginger J & Gleich G Melittin: An allergen of honeybee venom. Journal of Allergy and Clinical Immunology 59, 334–338, doi: 10.1016/0091-6749(77)90056-2 (1977). [DOI] [PubMed] [Google Scholar]
- 23.King T, Kochoumian L & Joslyn A Melittin-specific monoclonal and polyclonal IgE and IgG1 antibodies from mice. J Immunol 133, 2688–2673 (1984). [PubMed] [Google Scholar]
- 24.King TP, Wade D, Coscia MR, Mitchell S, Kochoumian L & Merrifield B Structure-immunogenicity relationship of melittin, its transposed analogues, and D-melittin. J Immunol 153, 1124–1131 (1994). [PubMed] [Google Scholar]
- 25.Nakagawa Y, Kikuchi H & Takahashi H Molecular analysis of TCR and peptide/MHC interaction using P18-I10-derived peptides with a single D-amino acid substitution. Biophys J 92, 2570–2582, doi: 10.1529/biophysj.106.095208 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans BC, Nelson CE, Yu SS, Beavers KR, Kim AJ, Li H, Nelson HM, Giorgio TD & Duvall CL Ex vivo red blood cell hemolysis assay for the evaluation of pH-responsive endosomolytic agents for cytosolic delivery of biomacromolecular drugs. J Vis Exp, e50166, doi: 10.3791/50166 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bello J, Bello HR & Granados E Conformation and aggregation of melittin: dependence on pH and concentration. Biochemistry 21, 461–465, doi: 10.1021/bi00532a007 (1982). [DOI] [PubMed] [Google Scholar]
- 28.Kilchrist KV, Dimobi SC, Jackson MA, Evans BC, Werfel TA, Dailing EA, Bedingfield SK, Kelly IB & Duvall CL Gal8 Visualization of Endosome Disruption Predicts Carrier-Mediated Biologic Drug Intracellular Bioavailability. ACS Nano 13, 1136–1152, doi: 10.1021/acsnano.8b05482 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LeBeau AM, Brennen WN, Aggarwal S & Denmeade SR Targeting the cancer stroma with a fibroblast activation protein-activated promelittin protoxin. Mol Cancer Ther 8, 1378–1386, doi: 10.1158/1535-7163.MCT-08-1170 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang W-C, Shen J-J, Liou C-J, Kuo M-L, Chang Y-P, Yang R-C & Li M-L Enhancing Th1 Cell Activities in Mice by Short-term Oral Administration of Gynostemma pentaphyllum Extracts. BioFormosa 42, 9–16 (2007). [Google Scholar]
- 31.Clinical Pathology Data for BALB/C Mouse Colonies in North America, <https://www.criver.com/products-services/find-model/balbc-mouse?region=3611>
- 32.Gill P, Jindal NL, Jagdis A & Vadas P Platelets in the immune response: Revisiting platelet-activating factor in anaphylaxis. J Allergy Clin Immunol 135, 1424–1432, doi: 10.1016/j.jaci.2015.04.019 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Upton J & Vadas P Potential Therapeutic Strategies for Severe Anaphylaxis Targeting Platelet-Activating Factor and PAF Acetylhydrolase. Current Treatment Options in Allergy 1, 232–246, doi: 10.1007/s40521-014-0020-2 (2014). [DOI] [Google Scholar]
- 34.Judge A, McClintock K, Phelps JR & Maclachlan I Hypersensitivity and loss of disease site targeting caused by antibody responses to PEGylated liposomes. Mol Ther 13, 328–337, doi: 10.1016/j.ymthe.2005.09.014 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Jackson MA, Patel SS, Yu F, Cottam MA, Glass EB, Dollinger BR, Hoogenboezem EN, Patil P, Liu DD, Kelly IB, Bedingfield SK, King AR, Miles RE, Hasty AM, Giorgio TD & Duvall CL Kupffer Cell Release of Platelet Activating Factor Drives Dose Limiting Toxicities of Nucleic Acid Nanocarriers, doi: 10.1101/2020.02.11.944504 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rabinovici R, AS R, Yue T & Feuerstein G Biological responses to liposome-encapsulated hemoglobin (LEH) are improved by a PAF antagonist. Circ Shock 31, 431–445 (1990). [PubMed] [Google Scholar]
- 37.Boeckle S, Wagner E & Ogris M C- versus N-terminally linked melittin-polyethylenimine conjugates: the site of linkage strongly influences activity of DNA polyplexes. J Gene Med 7, 1335–1347, doi: 10.1002/jgm.783 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Ichihara M, Shimizu T, Imoto A, Hashiguchi Y, Uehara Y, Ishida T & Kiwada H Anti-PEG IgM Response against PEGylated Liposomes in Mice and Rats. Pharmaceutics 3, 1–11, doi: 10.3390/pharmaceutics3010001 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Ishida T & Kiwada H Anti-PEG IgM elicited by injection of liposomes is involved in the enhanced blood clearance of a subsequent dose of PEGylated liposomes. J Control Release 119, 236–244, doi: 10.1016/j.jconrel.2007.02.010 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Shiraishi K, Kawano K, Maitani Y, Aoshi T, Ishii KJ, Sanada Y, Mochizuki S, Sakurai K & Yokoyama M Exploring the relationship between anti-PEG IgM behaviors and PEGylated nanoparticles and its significance for accelerated blood clearance. J Control Release 234, 59–67, doi: 10.1016/j.jconrel.2016.05.010 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Neun BW, Barenholz Y, Szebeni J & Dobrovolskaia MA Understanding the Role of Anti-PEG Antibodies in the Complement Activation by Doxil in Vitro. Molecules 23, doi: 10.3390/molecules23071700 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saifer MG, Williams LD, Sobczyk MA, Michaels SJ & Sherman MR Selectivity of binding of PEGs and PEG-like oligomers to anti-PEG antibodies induced by methoxyPEG-proteins. Mol Immunol 57, 236–246, doi: 10.1016/j.molimm.2013.07.014 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Zolnik BS, Gonzalez-Fernandez A, Sadrieh N & Dobrovolskaia MA Nanoparticles and the immune system. Endocrinology 151, 458–465, doi: 10.1210/en.2009-1082 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sela M & Zisman E Different roles of D-amino acids in immune phenomena. FASEB J 11, 449–456, doi: 10.1096/fasebj.11.6.9194525 (1997). [DOI] [PubMed] [Google Scholar]
- 45.Terwilliger TC & Eisenberg D The structure of melittin. The Journal of Biological Chemistry 257, 6016–6022 (1982). [PubMed] [Google Scholar]
- 46.Soman NR, Baldwin SL, Hu G, Marsh JN, Lanza GM, Heuser JE, Arbeit JM, Wickline SA & Schlesinger PH Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J Clin Invest 119, 2830–2842, doi: 10.1172/JCI38842 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu X, Dai Y, Zhao Y, Qi S, Liu L, Lu L, Luo Q & Zhang Z Melittin-lipid nanoparticles target to lymph nodes and elicit a systemic anti-tumor immune response. Nat Commun 11, 1110, doi: 10.1038/s41467-020-14906-9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orsolic N Bee venom in cancer therapy. Cancer Metastasis Rev 31, 173–194, doi: 10.1007/s10555-011-9339-3 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Orsolic N, Terzic S, Sver L & Basic I Honey-bee products in prevention and/or therapy of murine transplantable tumours. Journal of the Science of Food and Agriculture 85, 363–370, doi: 10.1002/jsfa.2041 (2005). [DOI] [Google Scholar]
- 50.Yu X, Chen L, Liu J, Dai B, Xu G, Shen G, Luo Q & Zhang Z Immune modulation of liver sinusoidal endothelial cells by melittin nanoparticles suppresses liver metastasis. Nat Commun 10, 574, doi: 10.1038/s41467-019-08538-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu M, Wang H, Liu L, Wang B & Sun G Melittin-MIL-2 fusion protein as a candidate for cancer immunotherapy. J Transl Med 14, 155, doi: 10.1186/s12967-016-0910-0 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rady I, Siddiqui IA, Rady M & Mukhtar H Melittin, a major peptide component of bee venom, and its conjugates in cancer therapy. Cancer Lett 402, 16–31, doi: 10.1016/j.canlet.2017.05.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pan H, Soman NR, Schlesinger PH, Lanza GM & Wickline SA Cytolytic peptide nanoparticles (‘NanoBees’) for cancer therapy. Wiley Interdiscip Rev Nanomed Nanobiotechnol 3, 318–327, doi: 10.1002/wnan.126 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Yang Q, Jacobs TM, McCallen JD, Moore DT, Huckaby JT, Edelstein JN & Lai SK Analysis of Pre-existing IgG and IgM Antibodies against Polyethylene Glycol (PEG) in the General Population. Anal Chem 88, 11804–11812, doi: 10.1021/acs.analchem.6b03437 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen BM, Su YC, Chang CJ, Burnouf PA, Chuang KH, Chen CH, Cheng TL, Chen YT, Wu JY & Roffler SR Measurement of Pre-Existing IgG and IgM Antibodies against Polyethylene Glycol in Healthy Individuals. Anal Chem 88, 10661–10666, doi: 10.1021/acs.analchem.6b03109 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Sherman MR, Williams LD, Sobczyk MA, Michaels SJ & Saifer MG Role of the methoxy group in immune responses to mPEG-protein conjugates. Bioconjug Chem 23, 485–499, doi: 10.1021/bc200551b (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McSweeney MD, Price LSL, Wessler T, Ciociola EC, Herity LB, Piscitelli JA, DeWalle AC, Harris TN, Chan AKP, Saw RS, Hu P, Jennette JC, Forest MG, Cao Y, Montgomery SA, Zamboni WC & Lai SK Overcoming anti-PEG antibody mediated accelerated blood clearance of PEGylated liposomes by pre-infusion with high molecular weight free PEG. J Control Release 311–312, 138–146, doi: 10.1016/j.jconrel.2019.08.017 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilson JT, Keller S, Manganiello MJ, Cheng C, Lee CC, Opara C, Convertine A & Stayton PS pH-Responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano 7, 3912–3925, doi: 10.1021/nn305466z (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Su YC, Chen BM, Chuang KH, Cheng TL & Roffler SR Sensitive quantification of PEGylated compounds by second-generation anti-poly(ethylene glycol) monoclonal antibodies. Bioconjug Chem 21, 1264–1270, doi: 10.1021/bc100067t (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.