

Abstract
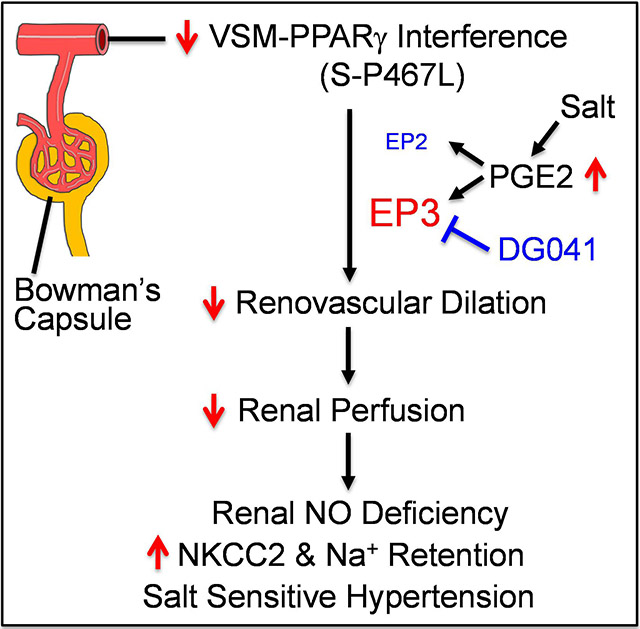
We previously showed that impaired vasodilation in systemic and renal vessels contributes to salt-sensitive hypertension in a mouse model of impaired PPARγ function. We determined the mechanisms mediating impaired salt-induced vasodilation and whether improved vasodilation attenuates augmented hypertension in response to salt. Mice selectively expressing a PPARγ dominant negative mutation in vascular smooth muscle (S-P467L) exhibited salt-sensitive hypertension and severely impaired vasodilation in systemic and renal vessels. High salt diet (HSD) fed S-P467L and control mice displayed comparable levels of renal oxidative stress markers. Pre-incubation with Tempol, a superoxide dismutase mimetic, or Calphostin C, a protein kinase C (PKC) inhibitor failed to improve salt-induced impairment of vasodilation in S-P467L mice, arguing against a role of oxidative stress or PKC activity. Inhibition of Rho kinase partially rescued impaired vasodilation in HSD-fed S-P467L mice suggesting a contribution of the RhoA/Rho Kinase pathway. HSD selectively increased synthesis of prostaglandin E2 (PGE2) in S-P467L aorta. Expression of E-prostanoid 3 (EP3) receptor mRNA was increased in aorta from chow- and high salt-fed S-P467L mice. Pharmacological inhibition of cyclooxygenase 2 or blockade of EP3 completely normalized the impaired vasodilation and EP3 antagonism induced larger decreases in systolic blood pressure in HSD-fed S-P467L mice. In conclusion, interference with PPARγ in vascular smooth muscle causes activation of the PGE2/EP3 signaling pathway in systemic and renal vasculature resulting in salt-induced impairment of vasodilation and salt-sensitive hypertension. PGE2/EP3 axis maybe a druggable target to prevent salt-sensitive hypertension in chronic conditions associated with decreased PPARγ activity.
Keywords: PPARγ, vasodilation, blood pressure, salt-sensitivity, prostaglandin E2
Graphical Abstract
Introduction
Increased renal vascular resistance (RVR) and blunted renal blood flow (RBF) during salt loading are hallmarks of salt-sensitive hypertension.1,2 This phenomenon, initially observed in Dahl salt-sensitive (SS) rats half a century ago, has been confirmed in humans and other experimental models.3-7 We recently showed that mice selectively expressing a dominant negative P467L mutation in peroxisome proliferator activated receptor γ (PPARγ) in vascular smooth muscle (termed S-P467L) failed to vasodilate in both systemic and renal vessels during salt loading and exhibited salt-sensitive hypertension.8 Our data suggested that impaired vasodilation in systemic vessels likely contributed to the hypertension through increased peripheral vascular resistance. In the renal circulation, impaired vasodilation restricted RBF which was associated with decreased natriuresis/diuresis capacity and enhanced sodium retention in S-P467L mice fed high salt diet (HSD). The enhanced sodium retention in HSD-fed S-P467L mice was mediated by a lack of salt-induced suppression of Na+-K+−2Cl− co-transporter (NKCC2) as furosemide corrected the impaired natriuresis/diuresis and abolished salt-induced hypertension. These observations suggest that impaired vasodilation portends renal dysfunction by restricting renal perfusion in the pathogenesis of SS hypertension.8 However, the mechanisms underlying the salt-induced vascular dysfunction in S-P467L mice are not fully understood.
RVR and RBF are regulated by vasoactive molecules such as prostaglandin E2 (PGE2). In salt resistant (SR) normotensive humans and wild type animals, dietary salt stimulates renal production of PGE2,9,10 which decreases RVR and increases RBF by acting on the dominant vasodilatory E-Prostanoid 2 (EP2) and E-Prostanoid 4 (EP4) receptors.11 Blunted PGE2 production in response to salt loading increases RVR and accelerates the development of hypertension in spontaneous hypertensive rats (SHR).6 Salt-induced increases in RVR is associated with a decrease in urinary PGE2 in SHR, while stimulation of prostaglandin synthesis by cicletanine prevents the increases in RVR and attenuates salt-induced hypertension. The protective effects of cicletanine are abolished by indomethacin, a non-specific inhibitor of cyclooxygenase, suggesting that the COX-PGE2-EP2/4 axis decreases salt sensitivity by restraining renovascular tone and promoting renal perfusion. Thus, decreased renal PGE2 bioavailability promotes salt-induced hypertension through a renal hemodynamic mechanism.
Alterations in PGE2 receptor signaling can also raise RVR and promote SS hypertension. In wildtype mice, PGE2 induces vasodilation in systemic and pre-glomerular resistance vessels through EP2/4.11,12 However, in EP2-deficient mice PGE2 induces potent vasoconstrictive and pressor effects by acting on E-prostanoid 3 (EP3) receptor.11,12 Of note, preservation of the depressor effects of EP4 in EP2-deficient mice is not sufficient to counteract the pressor effects of EP3 activation. As a result, EP2-deficient mice are SS and develop profound hypertension when fed HSD.13 These observations suggest that during EP2 deficiency, EP1/3 become dominant and evoke pressor effects in part through impaired vasodilation and augmented vasocontraction. Thus, the net effect of PGE2 on arterial pressure and vascular function is determined by the relative expression of PGE2 receptors. EP3 receptor expression is increased by NF-κB which is antagonized by PPARγ-mediated suppression of NF-κB.14 Moreover, interference with PPARγ in vascular smooth muscle promotes NF-κB activity by preventing nuclear export of p65.15 Therefore, we hypothesized that interference with PPARγ promotes systemic and renovascular dysfunction and salt sensitivity via upregulation of EP3 receptor. Here we provide evidence supporting the concept that altered PGE2 signaling impairs systemic and renal vascular dilation in response to salt loading. In keeping with this, EP3 antagonism in vivo decreased blood pressure in HSD-fed S-P467L mice.
Methods
Top Guidelines:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Experimental Animals:
All protocols were approved by the Animal Care and Use Committees at the Medical College of Wisconsin and University of Iowa. Care of the mice used in this study met the standards set forth by the National Institutes of Health (NIH) guidelines. Transgenic mice selectively expressing a human hypertension-causing mutation in PPARγ (P467L) in vascular smooth muscle (S-P467L) were previously described.16 S-P467L mice were maintained by successive generations of backcross breeding to C57BL/6 mice. Age- and sex-matched non-transgenic (NT) littermates were used as negative controls. Only males were studied as we previously showed that the phenotype of salt-induced impaired vasodilation is markedly attenuated in female S-P467L mice.8
Some experiments were completed at the University of Iowa and all mice were maintained on a standard laboratory rodent chow diet containing 0.8% sodium chloride or 0.3% sodium (the NIH-31 Modified Open Formula Mouse/Rat Sterilizable Diet, Teklad Catalog # 7013) at baseline. During the 4-week study, some animals received a HSD containing 4% sodium chloride equivalent to 1.57% sodium (Teklad Catalog # TD. 03095), while others remained on standard chow diet (Supplemental Figure S1A). We previously showed that this regimen induces severe impairment of vasodilation and progressive rise in arterial pressure in response to salt in the S-P467L mice.8 Carotid artery and renal segmental arteries were studied because these vessels exhibit severe impairment of vasodilation capacity in response to HSD, and these phenotypes were previously confirmed in the basilar artery, a cerebral resistance vessel.8
Additional experiments were performed at the Medical College of Wisconsin. All mice were weaned to a low salt diet containing 0.3% sodium chloride equivalent to 0.1% sodium (Teklad Catalog # 2920X). Both NT controls and S-P467L mice consumed the standard low salt chow diet at baseline until they were provided with the 4% HSD (Teklad Catalog # TD. 03095) for 5 weeks.
Measurement of blood pressure, oxidative stress, vascular function, mass spectrometry, and quantitative real-time RT-PCR are described in the online supplement 8,17-20.
Statistics:
All results were expressed as mean ± SEM. Statistical analysis of the data was performed using GraphPad Prism 8.4. Daily and hourly time course data of blood pressure, heart rate and activity as well as vascular function data were analyzed by Two-way ANOVA with repeated measurements. Mean data of telemetry recordings, mass spectrometry, gene expression, and U46619-induced contraction were analyzed by Two-way ANOVA. Tukey’s or Sidak’s multiple comparison procedures were performed for pairwise comparisons. Grubb’s test was used to assess the presence of outliers. One outlier was detected in the blood pressure experiment and is indicated in the figure legend. P < 0.05 was considered significant.
Results
Role of superoxide, protein kinase C, and RhoA/Rho kinase activity.
Salt stimulates the generation of reactive oxygen species (ROS) through NADPH oxidases (Noxs) which regulate the function of multiple organ/systems including the kidney and blood vessels in salt-sensitive hypertension.21,22 Therefore, we first determined whether markers of oxidative stress were increased in HSD-fed NT and S-P467L mice using the protocol illustrated in Supplemental Figure S1. 24-hour urine samples were collected in metabolic cages during week 4 and were assayed for 8-isoprostane, a marker of oxidative stress.21 Urinary 8-isoprostane was not different between genotypes and no effect of salt was observed (Supplemental Figure S1). Consistent with this, renal cortical and medullary superoxide levels as determined by the dihydroethidium (DHE)-HPLC method were not different between NT and S-P467L mice and neither was altered by chronic high salt consumption (Supplemental Figure S1C-D).17 In support of the biochemical measures, pre-incubation with Tempol (1 mM), a superoxide dismutase mimetic, failed to improve the impaired vasodilation responses to acetylcholine (ACh) in the carotid arteries of S-P467L mice with chow or HSD (Figure 1A). Nonlinear regression of ACh dose response curves revealed that Tempol did not change EC50 and failed to improve the decreased Emax in S-P467L mice fed chow or HSD (Supplemental Table S1A). Together these data suggest that superoxide-mediated oxidative stress does not play a role in salt-induced vasodilatory dysfunction in S-P467L mice.
Figure 1. Vasodilation: Role of Superoxide, Protein Kinase C, and Rho kinase.

Dose-dependent vasodilation (following precontraction with thromboxane A2 receptor agonist U46619) in response to acetylcholine (ACh). Vessel segments were pre-incubated with pharmacological inhibitors or vehicle for 30 min prior to U46619. Effects of A) superoxide dismutase mimetic Tempol (1 mmol/L, n=4-8), B) a protein kinase C inhibitor Calphostin C (50 nmol/L, n=4-6), and C) a Rho kinase inhibitor Y27632 (1 μmol/L, n=4-8) were shown in curves with diamonds. Curves with filled circles (chow) or squares (HSD) represent vehicle-treated vessel segments. Data are presented as mean ± SEM. Two-way ANOVA Repeated Measurements (RM) was performed to determine whether two curves were different (main-group effect, denoted by statistical symbols on the right of the curve). †p<0.05, S-P467L Chow Diet vs NT Chow Diet; *p<0.05, S-P467L HSD vs NT HSD; #p<0.05, Y-27632-treated VS vehicle-treated. Dose response curves were analyzed by nonlinear regression to generate EC50 and Emax values listed in Supplemental Table S1.
We have previously demonstrated that smooth muscle specific expression of the P467L mutation in PPARγ resulted in increased protein kinase C (PKC) activity in the mesenteric artery and that pharmacological inhibition of PKC diminished the enhanced myogenic tone in these vessels.23 Moreover, salt has been recently shown to activate PKC via sodium calcium exchanger-mediated calcium influx.24 Therefore, we sought to determine whether PKC activity contributed to the salt-induced vasodilatory dysfunction in S-P467L mice. Pre-incubation with a highly specific PKC inhibitor Calphostin C (50 nmol/L) did not improve the impaired vasodilation responses in the carotid artery of S-P467L mice on chow diet or HSD (Figure 1B). Calphostin C did not restore the EC50 shift nor did it correct the dampened Emax in S-P467L vessels (Supplemental Table S1B). Thus, the salt-induced impairment in vasodilation in S-P467L mice was not due to enhanced PKC activity.
RhoA/Rho Kinase signaling is enhanced in blood vessels of S-P467L mice on normal salt diet due to impaired ubiquitination and degradation of RhoA via the Cullin3-Ring-Ligase (CRL3) ubiquitin ligase complex.18,25 Pharmacological inhibition of Rho kinase activity eliminates enhanced contraction, restores impaired vasodilation and reduces arterial pressure in S-P467L mice fed standard chow diet.18,25 To determine whether salt worsened the dysfunction in vasodilation in S-P467L mice by further stimulating the RhoA/Rho kinase pathway, we tested the effect of a Rho kinase inhibitor Y27632 (1 μmol/L) on vasodilation properties at the end of 4 weeks. Pre-incubation with Y27632 had minimal effects in the carotid artery of NT mice fed chow or HSD (Figure 1C and Supplemental Table S1C). Y27632 modestly improved Emax of ACh-induced dilatation (Figure 1C and Supplemental Table S1C) and normalized Emax of SNP-induced dilatation in chow fed S-P467L mice (Supplemental Figure S2). In carotid arteries isolated from HSD-fed S-P467L mice, Y27632 partially restored ACh-induced (Figure 1C, Table S1C) and SNP-induced (Figure S2) dilatation as evidenced by improved Emax values and Supplemental. These data suggest that RhoA/Rho kinase-dependent mechanisms may contribute to the impaired vasodilation in HSD-fed S-P467L mice, but additional mechanisms are likely to be involved.
Role of the Cyclooxygenase and PGE2/EP3 receptor axis.
We next examined the cyclooxygenase (COX)/prostaglandin E2 (PGE2)/E-prostanoid receptor pathway because increased PGE2 production in response to a salt load has been reported to be a compensatory mechanism in salt-resistant normotensive humans and wild type animals (Figure 2A).9,10 Because the oxidation of arachidonic acid by COX is the first step in the biogenesis of prostaglandins and HSD has been reported to induce Cox-2 expression,9 we sought to determine whether salt-induced impairment in vasodilation involved changes in the expression of Cox-1 and/or Cox-2. There was no change in the transcription of the constitutively expressed Cox-1 gene among the four groups (Table 1). However, Cox-2 was significantly downregulated in S-P467L aortas while salt had no effect (Pgenotype = 0.037 in two-way ANOVA). This confirmed reports that PPARγ activation regulates Cox-2 expression in vascular smooth muscle cells and other cell types.26-28 We hypothesized that decreased Cox-2 expression in S-P467L aortas is the result of decreased PPARγ activation caused by the expression of dominant negative P467L mutation in PPARγ. Pre-incubation with a non-selective COX inhibitor indomethacin (10 μM) did not alter U46619-induced pre-contraction (Figure 2B), but markedly improved ACh-induced vasodilation (Figure 2C), and normalized SNP-induced vasodilation (Figure 2D) in carotid arteries of chow- or HSD-fed S-P467L mice.
Figure 2. Vasodilation: Role of Cyclooxygenase.

A) Schematic representation of the cyclooxygenase (COX)/prostaglandin E2 (PGE2)/E-prostanoid receptors pathway and pharmacological agents targeting different components of the pathway. PTGES1-3, prostaglandin E synthase 1-3; HPGD, 15-Hydroxyprostaglandin Dehydrogenase. B) Vasoconstriction was measured in carotid artery in response to U46619 (60 nmol/L) preincubated in the presence or absence of the non-specific COX inhibitor indomethacin (Indo, 30 min pre-incubation at 10 μmol/L)(n=8-11 as indicated). C-D) Effects of indomethacin on ACh-induced and SNP-induced vasodilation in the carotid artery (C, n=6-9; D, n=7-8). Curves with diamonds denote indomethacin-treated vessel segments. Curves with filled circles (chow) and squares (HSD) represent vehicle-treated vessel segments. Data are plotted as mean ± SEM. Two-way ANOVA RM was performed to determine whether two curves there different (main-group effect, denoted by statistical symbols on the right of the curve). †p<0.05, S-P467L chow diet vs NT chow diet; *p<0.05, S-P467L HSD vs NT HSD; #p<0.05, Indomethacin-treated vs vehicle-treated. Dose response curves were analyzed by nonlinear regression to generate EC50 and Emax values listed in the insets.
Table 1. Expression of Selected Genes in the COX/PGE2/E-Prostanoid Receptor Pathway in the Aorta.
Relative mRNA expression of selected genes in PGE2 biogenesis including cyclooxygenase-1 (Cox-1), cyclooxygenase-2 (Cox-2); and prostaglandin E synthase (Ptges) 1-3 and PGE2 degradation (15-hydroxyprostaglandin dehydrogenase, Hpgd) as well as E-prostanoid receptors (EP1-4, also known as prostaglandin E2 receptor, Ptger1-4) were determined by quantitative real-time RT-PCR from aorta of the indicated mouse strains at the end of the 4-week study. Two-way ANOVA was performed with ΔCt values to test the effects of genotype (pgenotype) and diet (pdiet).
| Target Gene |
Two-way ANOVA | Ctrl. | NT Chow | NT HSD | S-P467L Chow | S-P467L HSD | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pgenotype | Pdiet | Pinteraction | ΔCt | Mean (Lower-Upper) |
n | Mean (Lower-Upper) |
n | Mean (Lower-Upper) |
n | Mean (Lower-Upper) |
n | |
| Cox-1 | 0.09 | 0.73 | 0.80 | 0.7 | 1.00 (0.89-1.12) | 5 | 1.01 (0.93-1.10) | 6 | 0.79 (0.71-0.89) | 8 | 0.86 (0.76-0.96) | 8 |
| Cox-2 | 0.037 | 0.53 | 0.95 | 3.7 | 1.00 (0.81-1.23) | 5 | 1.10 (0.94-1.30) | 6 | 0.71 (0.62-0.82) | 8 | 0.81 (0.74-0.89) | 8 |
| Ptges-1 | 0.09 | 0.001 | 0.27 | 2.6 | 1.00 (0.98-1.02) | 4 | 0.59* (0.54-0.64) | 5 | 1.08 (0.98-1.19) | 8 | 0.81 (0.72-0.91) | 8 |
| Ptges-2 | 0.49 | 0.36 | 0.54 | 1.8 | 1.00 (0.95-1.05) | 5 | 0.88 (0.82-0.94) | 6 | 0.99 (0.92-1.07) | 8 | 0.97 (0.88-1.07) | 8 |
| Ptges-3 | 0.22 | 0.19 | 0.85 | 0.4 | 1.00 (0.90-1.11) | 5 | 0.84 (0.76-0.94) | 6 | 0.85 (0.79-0.92) | 8 | 0.75 (0.66-0.86) | 8 |
| Hpgd | 0.15 | 0.38 | 0.78 | 3.7 | 1.00 (0.78-1.29) | 5 | 1.20 (1.05-1.37) | 6 | 0.83 (0.75-0.92) | 8 | 0.93 (0.80-1.07) | 8 |
| EP1 | <0.001 | 0.96 | 0.94 | 10.8 | 1.00 (0.94-1.06) | 5 | 0.99 (0.83-1.19) | 4 | 1.63 (1.41-1.88) | 7 | 1.62 (1.39-1.87) | 7 |
| EP2 | 0.007 | 0.12 | 0.65 | 12.9 | 1.00 (0.93-1.08) | 5 | 0.68 (0.61-0.77) | 4 | 1.59 (1.38-1.83) | 7 | 1.30 (1.04-1.63) | 7 |
| EP3 | 0.03 | 0.51 | 0.75 | 7.1 | 1.00 (0.81-1.24) | 5 | 0.77 (0.68-0.87) | 4 | 1.62 (1.27-2.06) | 7 | 1.49 (1.11-1.98) | 7 |
| EP4 | 0.16 | 0.57 | 0.64 | 6.8 | 1.00 (0.89-1.12) | 5 | 1.01 (0.83-1.24) | 4 | 1.43 (1.22-1.67) | 7 | 1.41 (0.97-1.41) | 7 |
p<0.05, HSD compared to Chow Diet, Tukey’s multiple comparisons test, two-way ANOVA. ΔCt indicates the average of ΔCt values in the NT Chow Diet group. Group fold change relative to NT chow were calculated as 2^(−ΔΔCt) using the Livak method. Data are presented as mean values with upper and lower SEM in the parentheses. One outlier in NT Chow and one in NT HSD group were excluded from Ptges-1 expression based on Grubb’s test.
To rule out the potential off-target effects of indomethacin, we tested highly specific inhibitors targeting COX-1 (SC560, 100 nM) and COX-2 (Celecoxib, 1 μM) in the carotid artery and renal segmental artery in additional studies. Pre-incubation with SC560 had no effect in vessels from HSD-fed NT mice, but partly improved ACh-induced vasodilation in carotid and renal vessels of HSD-fed S-P467L mice as evidenced by improved Emax values (Figure 3A). In comparison, the selective COX-2 inhibitor Celecoxib completely normalized impaired vasodilation and restored ACh Emax in both the carotid and renal segmental arteries (Figure 3B). Together, these data suggest that salt-induced impairment of vasodilation in S-P467L mice likely involves the enzymatic activity of COX-2.
Figure 3. Vasodilation: Roles of the COX-2 and E-Prostanoid-3 Receptor.

A-B) Effects of a selective COX-1 inhibitor SC560 (1 μmol/L) and a selective COX-2 inhibitor Celecoxib (1 μmol/L) on ACh-induced vasodilation in carotid artery and renal segmental arteries (A, n=5-6; B, n=5-6). C) PGE2 content in whole aortas was detected by mass spectrometry at the end of 4 weeks (re-plotted in ng/mg protein from Table 2, Two-way ANOVA Pgenotype: 0.15; Pdiet: 0.09; Pinteraction: 0.10, n=6). D) E-Prostanoid-3 receptor (EP3) gene expression in the aorta at the end of 4 weeks (Two-way ANOVA Pgenotype: 0.04; Pdiet: 0.66; Pinteraction: 0.78, n=4-7 as indicated). The same dataset was re-plotted from Table 1 (group fold change) to show individual fold changes. PGE2 content and EP3 expression were analyzed with two-way ANOVA. The effect of PPARγP467L mutation on EP3 is indicated by Pstrain. E) Dose-dependent vasodilation of carotid artery and renal segmental artery (following precontraction with thromboxane A2 receptor agonist U46619) in response to acetylcholine (n=6). Effects of an EP3 antagonist DG041 (30 min preincubation at 100 nmol/L) were tested. Data are plotted as mean ± SEM. Curves with diamonds denotes drug-treated vessel segments. Curves with squares represent vehicle-treated vessel segments. In vasodilation responses, two-way ANOVA RM was first performed to determine whether two curves there different (main-group effect, denoted by statistical symbols on the right of the curve). When the main-group effect was not significantly different, Tukey’s multiple comparison procedures were performed for comparisons at different concentrations of ACh (statistical symbols above the curves). *p<0.05, S-P467L HSD vs NT HSD; #p<0.05 Drug-treated vs vehicle-treated. Dose response curves were analyzed by nonlinear regression to generate EC50 and Emax values listed in the insets.
To determine whether PGE2, a prostaglandin downstream of COX, was implicated in the pathogenesis of SS hypertension in S-P467L mice, we first examined the gene expression of enzymes involved in the biogenesis and metabolism of PGE2 in the aorta of NT and S-P467L animals in week 4 (Table 1). Salt significantly suppressed the expression of prostaglandin E synthase 1 (Ptges-1) in NT controls but not in S-P467L mice (Pdiet = 0.001 in two-way ANOVA), resulting in higher Ptges-1 mRNA levels in HSD-fed S-P467L mice than in HSD-fed NT controls. There was no change in the expression of prostaglandin E synthase 2 (Ptges-2), prostaglandin E synthase 3 (Ptges-3), or 15-hydroxyprostaglandin dehydrogenase (Hpgd), an enzyme that degrades PGE2 to its inactive form. We next measured levels of prostaglandins in the aorta using liquid chromatography tandem mass spectrometry (LC-MS/MS). HSD selectively increased aortic PGE2 in S-P467L mice, but not in NT controls (Table 2). There was also a trend for increases in PGD2 and PGF2α levels in aorta from HSD-fed S-P467L mice, while 8-iso PGF2α was largely undetectable (Table 2). As a negative control, the expression of leukotriene B4 (LTB4), another arachidonic acid metabolite generated via the distinct lipoxygenase pathway, was not altered. Of 6 samples studied, an increase in PGE2 was detected in 4 samples (Figure 3C). Although the increase was not statistically significant, it was consistent with changes in Ptges-1 expression and thus lead us to mechanistically examine the role of PGE2 in mediating vasodilation in response to HSD.
Table 2. Mass Spectrometry Analysis of Selected Arachidonic Acid Metabolites.
Aorta samples were harvested at the end of 4 weeks and were cleaned free of peri-adventitial fat. Selected vasoactive metabolites of arachidonic acid were detected by LC-MS/MS analysis (pg/mg protein, n=6). Data are presented as mean ± SEM. The effects of genotype, diet and interaction in two-way ANOVA are indicated by Pgenotype, Pdiet and Pinteraction. n.s. signifies not significant; N.D. signifies not detectable and zero value was entered for data analysis.
| Metabolite | Two-way ANOVA | NT Chow | NT HSD | S-P467L Chow | S-P467L HSD | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pgenotype | Pdiet | Pinteraction | Mean | S.E. | Mean | S.E. | Mean | S.E. | Mean | S.E. | |
| PGE2 | 0.15 | 0.09 | 0.10 | 776.3 | 157.3 | 776.9 | 203.6 | 690.7 | 161.9 | 1891.3 | 613.4 |
| PGD2 | 0.17 | 0.13 | 0.11 | 760.6 | 168.5 | 731.5 | 230.6 | 684.3 | 164.7 | 1840.9 | 637.4 |
| PGF2α | 0.25 | 0.12 | 0.15 | 271.2 | 63.8 | 299.5 | 87.4 | 221.6 | 47.4 | 759.4 | 322.8 |
| 8-Iso PGF2α | 0.52 | 0.52 | 0.11 | 35.3 | 26.4 | N.D. | N.D. | 15.2 | 15.2 | ||
| LTB4 | 0.15 | 0.69 | 0.96 | 202.8 | 10.9 | 197.0 | 12.9 | 183.7 | 17.4 | 179.2 | 3.6 |
PGE2 normally dilates blood vessels and promotes natriuresis via the dominant E-prostanoid 2 receptor (EP2).9,11 However, it induces vasopressor effects and raises arterial pressure by acting on E-prostanoid 3 receptor (EP3) when the dominant EP2 receptor is deleted.13 Because the expression of EP3 receptor is suppressed in response to PPARγ activation,14 we determined whether the dominant negative PPARγ P467L mutation altered the expression profile of E-prostanoid receptors after week 4. We found that EP1, EP2 and EP3 were upregulated in the aortas of S-P467L mice compared to NT controls regardless of diet (Pgenotype <0.05 in two-way ANOVA, Table 1, Figure 3D), while EP4 expression was not altered by genotype or diet. Although the vasodilatory EP2 was significantly upregulated in S-P467L aortas, the vasoconstrictive EP1 and EP3 were much more abundant than EP2 at the messenger RNA level as indicated by the mean ΔCt values (vs glyceraldehyde 3-phosphate dehydrogenase, GAPDH) (Table 1, 12.9 for EP2; 10.8 for EP1; and 7.1 for EP3).
To investigate whether PGE2/EP3 receptor axis was functionally involved in salt-induced impaired vasodilation, we tested a selective EP3 receptor antagonist DG041 (100 nM) in the carotid artery and renal segmental arteries isolated from NT HSD and S-P467L HSD mice. Pre-incubation with DG041 had no effect in NT vessels but completely rescued the impaired vasodilation and corrected ACh EC50/Emax values in carotid and renal arteries from HSD-fed S-P467L mice, suggesting that salt-induced vascular dysfunction was mediated by activation of EP3 receptor (Figure 3E).
Salt-Sensitive Hypertension and BP-Lowering Effects of EP3 Antagonism
NT and S-P467L mice were implanted with blood pressure radiotelemeters at 12-16 weeks of age and were allowed 10 days for recovery (Figure 4A). Baseline blood pressure was measured for 5 consecutive days during which time all mice consumed a low salt chow diet (0.3% sodium chloride and 0.1% sodium). After this, both NT controls and S-P467L mice received 4% HSD for 5 weeks. Daily averages of blood pressure, heart rate, and activity during baseline and the first 4 weeks of HSD were plotted in Figure 4B and Supplemental Figure S3. NT mice in response to HSD exhibited mild changes in systolic blood pressure (SBP, Baseline 129.1 ± 0.9 mmHg vs HSD 133.0 ± 1.1 mmHg, Δ = 3.9 mmHg, Figure 4C) and mean blood pressure (MBP, Baseline 113.8 ± 1.1 mmHg vs HSD 117.5 ± 0.9 mmHg, Δ = 3.7 mmHg, p<0.05, Supplemental Figure S3A). In contrast, S-P467L mice exhibited progressive increases in SBP and MBP in response to chronic high salt, resulting in a 6.3 mmHg increase in SBP (Baseline 133.7 ± 0.8 mmHg vs HSD 140.0 ± 1.7 mmHg, p<0.05, Figure 4C) and a 5.1 mmHg increase in MBP (Baseline 116.3 ± 0.6 mmHg vs HSD 121.4 ± 1.4 mmHg, p<0.05, Supplemental Figure S3A). There was an overall effect of salt on diastolic blood pressure (DBP) in all mice receiving HSD (Pdiet<0.05, Two-way ANOVA), but Sidak’s multiple comparison tests did not detect significant HSD-induced changes in DBP in NT or S-P467L mice (Supplemental Figures S3B). S-P467L mice exhibited a trend towards increased pulse pressure (Supplemental Figures S3C) and significantly increased heart rate (Supplemental Figures S3D) compared to NT controls at baseline, and both parameters remain markedly elevated at the end of week 4.8,29 Of note, HSD significantly suppressed heart rate in S-P467L mice but not in NT controls, consistent with salt-induced changes in blood pressure. Activity was not different between groups and was not changed by HSD (Supplemental Figures S3E). Together, these data indicate that S-P467L mice in response to 4-week HSD developed SS hypertension defined as a change in blood pressure of at least 5% or 5 mmHg.30
Figure 4. Salt-Sensitive Hypertension and BP-Lowering Effects of EP3 Antagonism.

A) A separate cohort of mice were first maintained on a low salt chow diet (LSD, 0.3% salt) since weaning and at 12 weeks of age were shifted to a 4% HSD for 5 weeks. Blood pressure was continuously recorded with radiotelemetry at baseline and during these 5 weeks. DG-041 was injected (20 mg/kg/day, subcutaneous) for 4 consecutive days. B) Establishment of salt-sensitive hypertension in S-P467L mice during the first 4 weeks of HSD. Per institutional animal facility regulations, cages were changed once a week (on days 1, 8, 15, and 22). N=10-11. *p<0.05, S-P467L HSD vs NT HSD by two-way ANOVA repeated measurements. C) Average SBP at baseline and at the end of 4 weeks (HSD days 26-28) were plotted. Two-way ANOVA Pgenotype: <0.0001; Pdiet: 0.001; Pinteraction: 0.32. Sidak’s multiple comparisons, *p<0.05, S-P467L vs NT; #p<0.05, S-P467L HSD vs S-P467L LSD. See Figure S3 for DBP, MBP, pulse pressure, HR and activity). D-E) Hourly SBP for first two days in week 5 (HSD days 29-30) was consolidated and plotted in 24-hour format (solid blue and solid red). During HSD days 31-34, mice received a subcutaneous injection of DG041 (20 mg/kg/day) at 10AM each day for 4 consecutive days (see protocol in panel A). Averages of SBP during the last two days of DG041 administration (open squares, see supplemental Figure 4 for daily effects) were plotted against the pre-treatment SBP. DG041 (plasma half-life = 4-6 hours, efficacy window = 2-6 hours post s.c. administration) caused transient dipping of BP in S-P467L mice but not in NT controls. F) SBP in the efficacious window between 12PM and 4PM was plotted to show the effect of DG-041. N=10-11. Two-way ANOVA Pgenotype: 0.002; PDG041: <0.0001; Pinteraction: 0.022. *p<0.05 S-P467L vs NT before DG041; Sidak’s multiple comparisons, #p<0.05 S-P467L after vs before DG041. One outlier was eliminated from the NT group as determined by a Grubb’s test.
To evaluate the BP-lowering effects of EP3 antagonism, we administered DG041 (20 mg/kg body weight, s.c. at 10AM) once a day for 4 consecutive days during week 5 (HSD days 31-34), after the establishment of salt-sensitive hypertension (Figure 4A). Hourly SBP prior to DG041 administration (HSD days 29-30, Supplemental Figure S4, solid squares) was compared to 24-hour tracings from days 31, 32, 33 and 34 (Supplemental Figure S4, open squares). Following the first two DG041 injections (HSD days 31 and 32) there were transient increases of blood pressure in both groups likely due to handling stress (Supplemental Figure S4A-B). As mice adapted to the handling stress, there was no change in blood pressure after the third and fourth injections (HSD days 33-34) in NT controls (Supplemental Figure S4C-D). In S-P467L mice, the third and fourth doses of DG041 induced a significant decrease of blood pressure 3-4 hours after administration (Supplemental Figure S4C-D), consistent with the 4-6 hours plasma half-life of DG041 when administered via the subcutaneous route.20 The night-time blood pressure before and after DG041 was superimposable confirming the short efficacy window of DG041. Hourly blood pressure during the final two days of DG041 administration (HSD days 33-34) was averaged and plotted against 24-hour tracings from HSD days 29-30 (Figure 4D-E). To quantitate the transient BP-lowering effects of DG041, blood pressure between 12PM-4PM in Figure 4D-E was averaged for each mouse and then plotted in Figure 4F. DG041 significantly decreased SBP in S-P467L mice (136.5 ± 2.1 mmHg before vs 125.4 ± 2.3 mmHg after) but not in NT controls (125.4 ± 1.3 mmHg before vs 121.0 ± 0.7 mmHg after). Thus, this data provides evidence for a role of PGE2/EP3 pathway in mediating increased blood pressure in S-P467L mice in response to high salt.
Discussion
The data in this paper provide evidence supporting the concept that interference with PPARγ activity in vascular smooth muscle results in activation of PGE2/EP3 signaling in the vasculature, which in turn causes salt-induced impairment of vasodilation in S-P467L mice. Compensatory increases in PGE2 in response to high salt intake have been documented in both human studies and animal models.9,10 In the present study, aortic PGE2 levels were increased in 4 out of 6 S-P467L mice fed HSD. Although this did not reach statistical significance, it led us to investigate the vasoactive role of PGE2 using pharmacological methods. The depressor effect of intravenously infused PGE2 is mediated predominantly by the activation of the vasodilatory EP2 receptor in normal blood vessels.11 Global deletion of EP2 receptor completely reverses the vascular actions of PGE2 by abolishing the dominant vasodilatory effect of EP2 which unleashes the vasoconstrictive effects of EP1/3 receptors.11 For this reason, systemic infusion of PGE2 in EP2 receptor deficient mice increases blood pressure by activating EP1/3 receptors despite the preservation of a lesser vasodilatory EP4 receptor.11 Interestingly, like S-P467L mice, EP2-deficient mice also exhibit mild elevation of systolic blood pressure at baseline but develop profound systolic hypertension when fed HSD.13 Consistent with a role of PGE2/EP3 axis in our model, the salt-induced impairment of vasodilation responses in S-P467L mice was completely rescued by COX-2 inhibition and EP3 antagonism. The activation of PGE2/EP3 axis in vessels of HSD-fed S-P467L mice might require a two-hit mechanism involving a) increased PGE2 synthesis, and b) enhanced sensitivity to PGE2 due to increased expression of EP3.
Our data suggest that there was a modest decrease in Cox-2 expression, but this did not impair the formation of PGE2 and other prostaglandins in S-P467L vessels. Interestingly, COX-2 is not the rate-limiting enzyme in PGE2 biosynthesis.31 Further loss of COX enzymatic activity caused by pharmacological inhibition may have normalized vasodilation through blockade of arachidonic acid oxidation and subsequent PGE2 production. A trend towards increased prostaglandin levels was only observed in S-P467L vessels under high salt conditions, suggesting this was a combined effect of the PPARγ dominant negative mutation and salt. In fact, HSD suppressed prostaglandin E synthase-1 mRNA expression in NT littermates, but not in S-P467L mice. Consistent with this, PPARγ activation by rosiglitazone blocks interleukin-1β-induced upregulation of prostaglandin E synthase-1 and PGE2 formation, supporting a role of PPARγ in transcriptional repression of the PGE2 biogenesis.32 The expression of 15-hydroxyprostaglandin dehydrogenase was not changed by salt or PPARγ mutation suggesting that PGE2 degradation mechanism was not affected.
The EP3 receptor is a G protein-coupled receptor (GPCR) which interacts with G12/13, and ligand-induced activation of the EP3 receptor stimulates the RhoA/ROCK signaling via G12/13.33,34 The ROCK inhibitor, Y27632, has been shown to completely abolish EP3-induced arterial contraction.34 We have previously shown that RhoA levels are elevated in the vascular smooth muscle of S-P467L mice due to impairment of CUL3-mediated ubiquitylation and turnover of RhoA.25,35 In the present study, ROCK inhibition by Y27632 partially restored impaired vasodilation in HSD-fed S-P467L mice, suggesting that elevated RhoA levels and ROCK activity may amplify the deleterious effects of EP3 receptor activation in causing the salt-induced vascular dysfunction in this model.
Further supporting a vasopressor effect of activated PGE2/EP3 axis in SS hypertension observed in S-P467L mice was the BP-lowering actions of EP3 antagonism in vivo. Due to the short half-life of EP3 antagonist DG041 (4-6 hours) in plasma following subcutaneous administration,20 its efficacy was transient. Nevertheless, during its efficacious window (2-6 hours following administration) EP3 antagonism induced a markedly stronger depressor effect in HSD-fed S-P467L mice compared to NT controls (ΔSBP: −11.1 mmHg S-P467L vs −4.4 mmHg NT). We recognize that the depressor effect of EP3 antagonism may not be solely accounted for by the vascular actions of DG041, as EP3 activation in the brain elicits pressor responses through sympathoexcitation.36 In fact, knockdown of EP3 receptor in the brain through intracerebroventricular injection of a lentiviral vector encoding shRNA targeting EP3 decreases sympathetic outflow, prevents peripheral inflammation and lowers blood pressure in Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME)/high salt-induced hypertension.37 Therefore, EP3 antagonists with long plasma half-life and permeability across the blood brain barrier may be an effective anti-hypertensive therapy in SS hypertension. Given the limitations of the pharmacological approach, future studies are warranted to determine whether selective deletion of EP3 gene in the vascular smooth muscle on S-P467L background achieves similar vasodilatory and depressor effects as EP3 antagonism and whether these benefits are primarily mediated by ablation of smooth muscle PGE2/EP3 signaling.
Salt induces renal and vascular oxidative stress in experimental hypertension through enhanced NAD(P)H oxidase activity,21,22,38 nitric oxide synthase uncoupling,39 and/or decreased antioxidant capacity.21,39,40 In resistance vessels, superoxide scavenges NO causing decreased NO bioavailability and impaired endothelial-dependent vasodilation.40-42 In renal tubules, superoxide promotes renal sodium reabsorption through NO-dependent and NO-independent mechanisms.43,44 These abnormalities may be corrected by pharmacological or genetic ablation of superoxide generation as well as supplementation of the antioxidant capacity.38,40-42 However, in the present study HSD did not increase renal superoxide in control or S-P467L mice and superoxide mutase mimetic tempol failed to improve salt-induced vasodilation impairment, arguing against a role of superoxide in mediating the renal and vascular phenotypes in this particular model. We have previously reported that S-P467L mice in response to chronic HSD exhibit blunted renal NO levels and decreased acetylcholine-induced dilation in renal vessels.8 Thus, the apparent renal NO deficiency is not likely to be caused by increased superoxide production (or superoxide-mediated NO inactivation), but by decreased NO biogenesis. One limitation of the present study is that we did not examine the role of hydrogen peroxide, another reactive oxygen species known to play important roles in salt-sensitive hypertension.38,45 Further studies are necessary to determine whether the improvement of vasodilation in renal vessels of HSD-fed S-P467L mice by COX-2 inhibition and EP3 antagonism involves enhancement of renovascular NO bioavailability.
Genetic background and sex dimorphism play important roles in the salt sensitivity of mouse models. C57BL/6 and BALB/c strains are the most commonly used mouse strains in experimental models of hypertension.30 C57BL/6 mice exhibit SS increases of blood pressure in some studies,46,47 but are salt resistant in other studies.48,49 In comparison, while male BALB/c mice are salt resistant,45 female BALB/c mice develop SS hypertension through aldosterone-mineralocorticoid receptor-mediated endothelium-dependent mechanisms.50 The salt resistant phenotype of male BALB/c mice may be mediated in part by two mechanisms: 1) salt-induced production of hydrogen peroxide which negatively regulates sodium reabsorption in the renal proximal tubule;45 and 2) salt-induced suppression of sodium-chloride cotransporter (NCC) and epithelial sodium channel α (ENaCα) in the distal nephron.51 In our studies, the S-P467L mice were maintained on C57BL/6 background through successive backcross breeding (since 2004).16 Thus, loss of PPARγ function in vascular tissue may have enhanced the salt sensitivity of strain which is naturally salt sensitive. Because the female S-P467L mice were protected from salt-induced vascular dysfunction, we focused on male S-P467L mice which exhibit severe impairment of vasodilation and restricted renal perfusion in response to HSD.8 Interestingly, while salt suppressed NCC and NKCC2 in non-transgenic littermates, male S-P467L mice failed to exhibit salt-induced down-regulation of these sodium transporters resulting in impaired natriuresis, enhanced sodium retention, and SS hypertension that were corrected by pharmacological inhibition of NKCC2.8 In the present study, EP3 antagonism rescued impaired vasodilation and transiently attenuated salt-induced hypertension in S-P467L mice. Future studies are warranted to determine whether selective deletion of EP3 in vascular smooth muscle preserves vasodilation capacity, enhances renal perfusion and promotes renal sodium excretion during excess salt intake.
Perspectives
PPARγ is an important transcription factor regulating adipogenesis, metabolism, vascular tone, renal sodium handling, and inflammatory responses in the broad context of cardiometabolic physiology. Dominant negative PPARγ mutations cause hypertension, insulin resistance and hyperglycemia in humans. Patients with type 2 diabetes and metabolic syndrome display decreased PPARγ activity and are often salt-sensitive. Thiazolidinedione therapy is complicated by fluid retention and are no longer a first line anti-diabetic agent. Nevertheless, compelling evidence supports the concept that the benefit of PPARγ activation in the cardiovascular system outweighs the renal adverse effects. In the systemic and renal circulation, vascular PPARγ is essential for preserving vasodilation and renal blood flow in face of risk factors such as high salt intake.8 Understanding the downstream pathways that mediate these protective effects may lead to identification of druggable targets that drive the beneficial aspects of PPARγ activation without evoking the adverse effects. One such downstream pathway is the PGE2/EP3 axis which is normally suppressed by PPARγ but may be activated in chronic conditions with coincident PPARγ impairment, which may cause vasodilatory dysfunction in systemic and renal vessels leading to exaggeration of SS hypertension. Herein, the data from this study identify vascular smooth muscle PPARγ/EP3 pathway as a relevant therapeutic target in hypertensive subjects especially those complicated with type 2 diabetes and metabolic syndrome.
Supplementary Material
Novelty and Significance.
What Is New?
Impaired vasodilation in systemic and renal vessels contributes to salt-sensitive hypertension in a mouse model of impaired PPARγ function.
Vascular PPARγ impairment resulted in an upregulation of E-prostanoid (EP3) receptor while high salt diet selectively increased synthesis of prostaglandin E2 (PGE2) in S-P467L vessels, providing the basis of PGE2/EP3 activation in HSD-fed S-P467L mice.
Pharmacological inhibition of cyclooxygenase 2 or blockade of EP3 completely normalized the impaired vasodilation and EP3 antagonism decreased systolic blood pressure in high salt diet fed S-P467L mice.
What Is Relevant?
Although PPARγ interference has been associated with increased salt sensitivity of blood pressure in type 2 diabetes and metabolic syndrome, tissue-specific mechanisms are poorly understood.
Vascular PPARγ interference favors activation of the vasoconstrictive EP3 over the vasodilatory EP2 and predisposes to salt sensitive hypertension through salt-induced impairment of vasodilation in the systemic and renal circulation.
Summary
Genetic interference with PPARγ specifically in the vascular smooth muscle results in severe impairment of vasodilation in both systemic and renal vessels during high salt intake. This salt-induced impairment of vasodilation was not rescued by superoxide scavenging or PKC inhibition, but was partially attenuated by ROCK inhibition, suggesting contribution of the RhoA/ROCK pathway. Importantly, vascular PPARγ interference enhanced PGE2/EP3 signaling in high salt diet-fed S-P467L mice and pharmacological inhibition of cyclooxygenase 2 or EP3 corrected the dilatory dysfunction. Further, EP3 antagonism in vivo attenuated salt sensitive hypertension in S-P467L mice. Thus, loss of the beneficial effects of PPARγ in the vascular smooth muscle predisposes to vasodilation impairment induced by risk factors such as excess salt and increases the susceptibility to salt sensitive hypertension.
Acknowledgments
We thank Ms. Katherine H. Gotlinger at New York Medical College School of Medicine for her technical support in the LC-MS/MS studies. We thank the UI Gene Editing Core and genotyping service in the Genome Science and Precision Medicine Center (GSPMC) at MCW. This paper was supported by the U.S. National Institutes of Health (see below) and the University of Iowa. Its contents are solely the responsibility of the author(s) and do not necessarily represent the official views of the U.S. National Institutes of Health, The University of Iowa, or Medical College of Wisconsin.
Sources of Funding
This work was supported through research grants from the National Institutes of Health (NIH) to CDS (HL084207, HL144807), MLS (HL139793), SD (P01 HL129941), JLG (HL134850, 18EIA33890055), and American Heart Association (15SFRN23480000) to CDS. JW was supported by an AHA postdoctoral fellowship (17POST33660685) and currently by a NIDDK K01 (DK126792). SF was supported by an AHA predoctoral fellowship (20PRE35120137).
Footnotes
Conflict of Interest/Disclosures: CDS is a member of a Scientific Advisory Board for Ionis Pharmaceuticals. His contributions to that board are unrelated to the content of this manuscript.
References
- 1.Ganguli M, Tobian L and Dahl L. Low renal papillary plasma flow in both Dahl and Kyoto rats with spontaneous hypertension. Circ Res. 1976;39:337–341. [DOI] [PubMed] [Google Scholar]
- 2.Fink GD, Takeshita A, Mark AL and Brody MJ. Determinants of renal vascular resistance in the Dahl strain of genetically hypertensive rat. Hypertension. 1980;2:274–280. [DOI] [PubMed] [Google Scholar]
- 3.Redgrave J, Rabinowe S, Hollenberg NK and Williams GH. Correction of abnormal renal blood flow response to angiotensin II by converting enzyme inhibition in essential hypertensives. J Clin Invest. 1985;75:1285–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campese VM, Parise M, Karubian F and Bigazzi R. Abnormal renal hemodynamics in black salt-sensitive patients with hypertension. Hypertension. 1991;18:805–812. [DOI] [PubMed] [Google Scholar]
- 5.Higashi Y, Oshima T, Watanabe M, Matsuura H and Kajiyama G. Renal response to L-arginine in salt-sensitive patients with essential hypertension. Hypertension. 1996;27:643–648. [DOI] [PubMed] [Google Scholar]
- 6.Ando K, Ono A, Sato Y and Fujita T. Involvement of prostaglandins and renal haemodynamics in salt-sensitivity of young spontaneously hypertensive rats. J Hypertens. 1993;11:373–377. [DOI] [PubMed] [Google Scholar]
- 7.Sparks MA, Stegbauer J, Chen D, Gomez JA, Griffiths RC, Azad HA, Herrera M, Gurley SB and Coffman TM. Vascular Type 1A Angiotensin II Receptors Control BP by Regulating Renal Blood Flow and Urinary Sodium Excretion. J Am Soc Nephrol. 2015;26:2953–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J, Agbor LN, Fang S, Mukohda M, Nair AR, Nakagawa P, Sharma A, Morgan DA, Grobe JL, Rahmouni K, Weiss RM, McCormick JA and Sigmund CD. Failure to Vasodilate in Response to Salt Loading Blunts Renal Blood Flow and Causes Salt-Sensitive Hypertension. Cardiovasc Res. 2021;117:308–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Zhao M, He W, Milne GL, Howard JR, Morrow J, Hebert RL, Breyer RM, Chen J and Hao CM. Increased dietary NaCl induces renal medullary PGE2 production and natriuresis via the EP2 receptor. Am J Physiol Renal Physiol. 2008;295:F818–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dreisbach AW, Rice JC, Japa S, Newman JW, Sigel A, Gill RS, Hess AE, Cemo AC, Fonseca JP, Hammock BD, Lertora JJ and Hamm LL. Salt loading increases urinary excretion of linoleic acid diols and triols in healthy human subjects. Hypertension. 2008;51:755–761. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Guan Y, Schneider A, Brandon S, Breyer RM and Breyer MD. Characterization of murine vasopressor and vasodepressor prostaglandin E(2) receptors. Hypertension. 2000;35:1129–1134. [DOI] [PubMed] [Google Scholar]
- 12.Imig JD, Breyer MD and Breyer RM. Contribution of prostaglandin EP(2) receptors to renal microvascular reactivity in mice. Am J Physiol Renal Physiol. 2002;283:F415–422. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy CR, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, Magnuson MA, Oates JA, Breyer MD and Breyer RM. Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med. 1999;5:217–220. [DOI] [PubMed] [Google Scholar]
- 14.Sui X, Liu Y, Li Q, Liu G, Song X, Su Z, Chang X, Zhou Y, Liang B and Huang D. Oxidized low-density lipoprotein suppresses expression of prostaglandin E receptor subtype EP3 in human THP-1 macrophages. PLoS One. 2014;9:e110828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukohda M, Lu KT, Guo DF, Wu J, Keen HL, Liu X, Ketsawatsomkron P, Stump M, Rahmouni K, Quelle FW and Sigmund CD. Hypertension-Causing Mutation in Peroxisome Proliferator-Activated Receptor gamma Impairs Nuclear Export of Nuclear Factor-kappaB p65 in Vascular Smooth Muscle. Hypertension. 2017;70:174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halabi CM, Beyer AM, de Lange WJ, Keen HL, Baumbach GL, Faraci FM and Sigmund CD. Interference with PPAR gamma function in smooth muscle causes vascular dysfunction and hypertension. Cell Metab. 2008;7:215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dikalov S, Griendling KK and Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukohda M, Fang S, Wu J, Agbor LN, Nair AR, Ibeawuchi SC, Hu C, Liu X, Lu KT, Guo DF, Davis DR, Keen HL, Quelle FW and Sigmund CD. RhoBTB1 protects against hypertension and arterial stiffness by restraining phosphodiesterase 5 activity. J Clin Invest. 2019;130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nair AR, Agbor LN, Mukohda M, Liu X, Hu C, Wu J and Sigmund CD. Interference With Endothelial PPAR (Peroxisome Proliferator-Activated Receptor)-gamma Causes Accelerated Cerebral Vascular Dysfunction in Response to Endogenous Renin-Angiotensin System Activation. Hypertension. 2018;72:1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ceddia RP, Downey JD, Morrison RD, Kraemer MP, Davis SE, Wu J, Lindsley CW, Yin H, Daniels JS and Breyer RM. The effect of the EP3 antagonist DG-041 on male mice with diet-induced obesity. Prostaglandins Other Lipid Mediat. 2019;144:106353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitiyakara C, Chabrashvili T, Chen Y, Blau J, Karber A, Aslam S, Welch WJ and Wilcox CS. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J Am Soc Nephrol. 2003;14:2775–2782. [DOI] [PubMed] [Google Scholar]
- 22.Touyz RM, Rios FJ, Alves-Lopes R, Neves KB, Camargo LL and Montezano AC. Oxidative Stress: A Unifying Paradigm in Hypertension. Can J Cardiol. 2020;36:659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ketsawatsomkron P, Lorca RA, Keen HL, Weatherford ET, Liu X, Pelham CJ, Grobe JL, Faraci FM, England SK and Sigmund CD. PPARgamma regulates resistance vessel tone through a mechanism involving RGS5-mediated control of protein kinase C and BKCa channel activity. Circ Res. 2012;111:1446–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, Mernaugh RL, Itani HA, Loperena R, Chen W, Dikalov S, Titze JM, Knollmann BC, Harrison DG and Kirabo A. Dendritic Cell Amiloride-Sensitive Channels Mediate Sodium-Induced Inflammation and Hypertension. Cell Rep. 2017;21:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pelham CJ, Ketsawatsomkron P, Groh S, Grobe JL, de Lange WJ, Ibeawuchi SR, Keen HL, Weatherford ET, Faraci FM and Sigmund CD. Cullin-3 Regulates Vascular Smooth Muscle Function and Arterial Blood Pressure via PPARγ and RhoA/Rho-Kinase. Cell Metabolism. 2012;16:462–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bishop-Bailey D and Warner TD. PPARgamma ligands induce prostaglandin production in vascular smooth muscle cells: indomethacin acts as a peroxisome proliferator-activated receptor-gamma antagonist. FASEB J. 2003;17:1925–1927. [DOI] [PubMed] [Google Scholar]
- 27.Meade EA, McIntyre TM, Zimmerman GA and Prescott SM. Peroxisome proliferators enhance cyclooxygenase-2 expression in epithelial cells. J Biol Chem. 1999;274:8328–8334. [DOI] [PubMed] [Google Scholar]
- 28.Pontsler AV, St Hilaire A, Marathe GK, Zimmerman GA and McIntyre TM. Cyclooxygenase-2 is induced in monocytes by peroxisome proliferator activated receptor gamma and oxidized alkyl phospholipids from oxidized low density lipoprotein. J Biol Chem. 2002;277:13029–13036. [DOI] [PubMed] [Google Scholar]
- 29.Borges GR, Morgan DA, Ketsawatsomkron P, Mickle AD, Thompson AP, Cassell MD, Mohapatra DP, Rahmouni K and Sigmund CD. Interference with peroxisome proliferator-activated receptor-gamma in vascular smooth muscle causes baroreflex impairment and autonomic dysfunction. Hypertension. 2014;64:590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Felder RA, White MJ, Williams SM and Jose PA. Diagnostic tools for hypertension and salt sensitivity testing. Curr Opin Nephrol Hypertens. 2013;22:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tithof PK, Roberts MP, Guan W, Elgayyar M and Godkin JD. Distinct phospholipase A2 enzymes regulate prostaglandin E2 and F2alpha production by bovine endometrial epithelial cells. Reprod Biol Endocrinol. 2007;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kapoor M, Kojima F, Qian M, Yang L and Crofford LJ. Microsomal prostaglandin E synthase-1 deficiency is associated with elevated peroxisome proliferator-activated receptor gamma: regulation by prostaglandin E2 via the phosphatidylinositol 3-kinase and Akt pathway. J Biol Chem. 2007;282:5356–5366. [DOI] [PubMed] [Google Scholar]
- 33.Hata AN and Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103:147–166. [DOI] [PubMed] [Google Scholar]
- 34.Kraemer MP, Choi H, Reese J, Lamb FS and Breyer RM. Regulation of arterial reactivity by concurrent signaling through the E-prostanoid receptor 3 and angiotensin receptor 1. Vascul Pharmacol. 2016;84:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu J, McCormick JA and Sigmund CD. Cullin-3: Renal and Vascular Mechanisms Regulating Blood Pressure. Curr Hypertens Rep. 2020;22:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ariumi H, Takano Y, Masumi A, Takahashi S, Hirabara Y, Honda K, Saito R and Kamiya HO. Roles of the central prostaglandin EP3 receptors in cardiovascular regulation in rats. Neurosci Lett. 2002;324:61–64. [DOI] [PubMed] [Google Scholar]
- 37.Xiao L, Itani HA, do Carmo LS, Carver LS, Breyer RM and Harrison DG. Central EP3 (E Prostanoid 3) Receptors Mediate Salt-Sensitive Hypertension and Immune Activation. Hypertension. 2019;74:1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng D, Yang C, Geurts AM, Kurth T, Liang M, Lazar J, Mattson DL, O'Connor PM and Cowley AW Jr. Increased expression of NAD(P)H oxidase subunit p67(phox) in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension. Cell Metab. 2012;15:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor NE, Maier KG, Roman RJ and Cowley AW Jr. NO synthase uncoupling in the kidney of Dahl S rats: role of dihydrobiopterin. Hypertension. 2006;48:1066–1071. [DOI] [PubMed] [Google Scholar]
- 40.Park JB, Touyz RM, Chen X and Schiffrin EL. Chronic treatment with a superoxide dismutase mimetic prevents vascular remodeling and progression of hypertension in salt-loaded stroke-prone spontaneously hypertensive rats. Am J Hypertens. 2002;15:78–84. [DOI] [PubMed] [Google Scholar]
- 41.Somers MJ, Mavromatis K, Galis ZS and Harrison DG. Vascular superoxide production and vasomotor function in hypertension induced by deoxycorticosterone acetate-salt. Circulation. 2000;101:1722–1728. [DOI] [PubMed] [Google Scholar]
- 42.Zhu J, Huang T and Lombard JH. Effect of high-salt diet on vascular relaxation and oxidative stress in mesenteric resistance arteries. J Vasc Res. 2007;44:382–390. [DOI] [PubMed] [Google Scholar]
- 43.Yu L, Bao HF, Self JL, Eaton DC and Helms MN. Aldosterone-induced increases in superoxide production counters nitric oxide inhibition of epithelial Na channel activity in A6 distal nephron cells. Am J Physiol Renal Physiol. 2007;293:F1666–1677. [DOI] [PubMed] [Google Scholar]
- 44.Silva GB, Ortiz PA, Hong NJ and Garvin JL. Superoxide stimulates NaCl absorption in the thick ascending limb via activation of protein kinase C. Hypertension. 2006;48:467–472. [DOI] [PubMed] [Google Scholar]
- 45.Cuevas S, Asico LD, Jose PA and Konkalmatt P. Renal Hydrogen Peroxide Production Prevents Salt-Sensitive Hypertension. J Am Heart Assoc. 2020;9:e013818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Escano CS, Armando I, Wang X, Asico LD, Pascua A, Yang Y, Wang Z, Lau YS and Jose PA. Renal dopaminergic defect in C57Bl/6J mice. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1660–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Combe R, Mudgett J, El Fertak L, Champy MF, Ayme-Dietrich E, Petit-Demouliere B, Sorg T, Herault Y, Madwed JB and Monassier L. How Does Circadian Rhythm Impact Salt Sensitivity of Blood Pressure in Mice? A Study in Two Close C57Bl/6 Substrains. PLoS One. 2016;11:e0153472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mangrum AJ, Gomez RA and Norwood VF. Effects of AT(1A) receptor deletion on blood pressure and sodium excretion during altered dietary salt intake. Am J Physiol Renal Physiol. 2002;283:F447–453. [DOI] [PubMed] [Google Scholar]
- 49.Kopkan L, Hess A, Huskova Z, Cervenka L, Navar LG and Majid DS. High-salt intake enhances superoxide activity in eNOS knockout mice leading to the development of salt sensitivity. Am J Physiol Renal Physiol. 2010;299:F656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faulkner JL, Harwood D, Bender L, Shrestha L, Brands MW, Morwitzer MJ, Kennard S, Antonova G and Belin de Chantemele EJ. Lack of Suppression of Aldosterone Production Leads to Salt-Sensitive Hypertension in Female but Not Male Balb/C Mice. Hypertension. 2018;72:1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu G, Yu Y, Ren Z, Asico L, Jose P and Wang X. The salt-sensitivity in C57Bl/6J mice is linked to increased renal protein expressions of SLC4A4 and SLC4A5. FASEB J. 2019;33 (1 Supplement):533. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.