

Abstract
Polymorphisms in TMEM106B, a gene on chromosome 7p21.3 involved in lysosomal trafficking, correlates to worse neuropathological, and clinical outcomes in frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) with TDP‐43 inclusions. In a small cohort of C9orf72 expansion carriers, we previously found an atypical, neuroglial tauopathy in cases harboring a TMEM106B rs1990622 A/A genotype. To test whether TMEM106B genotype affects the risk of developing atypical tauopathy under a recessive genotype model (presence versus absence of two major alleles: A/A vs. A/G and G/G). We characterized the atypical tauopathy neuropathologically and determined its frequency by TMEM106B rs1990622 genotypes in 90 postmortem cases with a primary diagnosis of FTLD/ALS‐TDP [mean age at death 65.5 years (±8.1), 40% female]. We investigated the effect of this new atypical tauopathy on demographics and clinical and neuropsychological metrics. We also genotyped TMEM106B in an independent series with phenotypically similar cases. Sixteen cases (16/90, 17.7 %) showed the temporal‐predominant neuro‐astroglial tauopathy, and 93.7% of them carried an A/A genotype (vs. ~35% in a population cohort). The odds ratio of FTLD/ALS‐TDP individuals with the A/A genotype showing neuro‐astroglial tauopathy was 13.9. Individuals with this tauopathy were older at onset (p = 0.01). The validation cohort had a similarly high proportion of rs1990622 A/A genotype. TDP‐43 and tau changes co‐occur in a subset of neurons. Our data add to the growing body of evidence that TMEM106B polymorphisms may modulate neurodegeneration. A distinctive medial temporal predominant, 4‐repeat, neuro‐astroglial tauopathy strongly correlates to TMEM106B A/A genotype in FTLD/ALS‐TDP cases.
Keywords: frontotemporal dementia, frontotemporal lobar degeneration, neuropathologic diagnosis, TDP‐43 proteinopathy, tauopathy, TMEM106B, polymorphism, Rs1990622, genetic association, risk factor
noFTLD‐TDP cases with a TMEM106b A/A genotype have a much‐increased odds to show a distinct temporal‐predominant neuro‐astroglial tauopahty.
1. INTRODUCTION
TAR DNA binding protein 43 (TDP‐43) is the major pathological protein in frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) (1, 2, 3). FTLD/ALS‐TDP can be sporadic or hereditary, the latter most commonly due to C9orf72 expansion and TARDBP or progranulin (GRN) mutations.
As a group, FTLD‐TDP encompasses at least four distinct neuropathological subtypes (A‐D) featuring distinct patterns of cell vulnerability, topographical distribution, and inclusion types. For instance, type C is frequently associated with prominent degeneration in anterior temporal regions and often manifests as a semantic variant primary progressive aphasia. In contrast, type B encompasses most cases developing motor neuron disease. Notably, age at clinical onset and clinical phenotypes vary within the same FTLD‐TDP subtype. The clinical heterogeneity is striking even in kindreds carrying the same autosomal dominant mutations. Affected individuals within the same C9orf72 kindred may manifest behavioral variant frontotemporal dementia (bvFTD), amyotrophic lateral sclerosis (ALS), primary progressive aphasia (PPA), and even parkinsonism or a psychiatric syndrome (4).
Genome‐wide association studies (GWAS) and mechanistic studies have suggested that the single‐nucleotide polymorphisms (SNPs) rs1020004, rs6966915, and rs1990622 in transmembrane protein 106B (TMEM106B), a gene located on chromosome 7p21.3, may modify the clinical phenotype and pathological burden of FTLD/ALS‐TDP cases (5, 6, 7, 8, 9, 10). For instance, GRN mutation carriers and, to a lesser extent, C9orf72 expansion carriers harboring TMEM106B rs1990622 risk genotype (A/A) are more likely to develop worse clinical symptoms and manifest the disease at an earlier age (11, 12, 13). Also, rs1990622 A/A genotype is associated with a higher risk of developing limbic‐predominant age‐related TDP‐43 encephalopathy (LATE) (14) and associated with hippocampal sclerosis of aging (15) in individuals lacking FTLD pathology (13, 16). Finally, FTLD patients with a CHMP2B mutation and TMEM106B rs1990622 A/A genotype tend to present a more severe clinical presentation (17, 18). Although the mechanisms underlying the interaction between TMEM106B polymorphisms and TDP‐43 proteinopathy remain elusive, some studies suggest that overexpression of TMEM106B mRNA lead to a failure of the lysosomal system (17, 19) and, in turn, impacts lysosomal‐dependent mechanisms (20, 21) that are essential to degrade and clear abnormal proteins involved in neurodegeneration.
In a previous clinicopathological study of 17 individuals with C9orf72 expansions (4), we incidentally found an atypical pattern of phosphorylated tau (p‐tau Ser 202) deposits featuring granular neuronal cytoplasmic, diffuse granular immunopositivity in astrocytic processes, grains and patchy accumulation of thin threads, more prominent in limbic and paralimbic areas in two of the cases. Although this tauopathy partially resembled argyrophilic grain disease (AGD) (22), or aging‐related tau astrogliopathy (ARTAG) (23), because of its granular aspect or presence of fuzzy astrocytes, respectively, the distribution and overall morphological features were atypical for AGD and ARTAG. A literature search detected that the atypical tauopathy we observed most closely resembled the atypical tauopathy described by Kovacs and colleagues in 2011, a series of seven cases (24). A TMEM106B rs1990622 A/A genotype emerged as the only difference we could identify between the two cases with the atypical tauopathy and the other 15 cases in our C9orf72 series.
Here, to test the hypothesis that this distinctive tauopathy is associated with TMEM106B rs1990622 A/A genotype, we analyzed an extended cohort of 90 cases with a primary diagnosis of FTLD‐TDP or ALS‐TDP, regardless of the C9orf72 status from the University of California, San Francisco (UCSF) Memory and Aging Center (MAC) Neurodegenerative Disease Brain Bank (NDBB). Next, we aimed to validate the results in the independent cohort described by Kovacs and colleagues (24). Finally, we sought to identify the targeted atypical tauopathy correlates by performing an in‐depth clinical, neuropsychological, and genetic analysis of the NDBB/UCSF cohort.
2. SUBJECTS AND METHODS
Consent for brain donation was obtained from all subjects or their surrogates following the Declaration of Helsinki, and the UCSF Committee on Human Research approved the donations. This study had an exploratory and a replication stage. For the exploratory stage, postmortem human brain tissue was obtained from the NDBB. For the replication stage, postmortem human brain tissue was sourced by Dr. Gabor Kovacs (24). All participants or their surrogates provided written consent to participate in research.
2.1. UCSF participants (exploratory stage)
Cases were selected based on neuropathological diagnoses and the availability of genetic analysis (Figure 1). The NDBB collected 502 cases between 2007 and 2018, of which 461 had TMEM106B rs1990622 genotyping available. Inclusion criteria were: at least one research visit at the UCSF MAC, complete neuropathological assessment, availability of archival postmortem brain tissue for further analysis, genetic screening (see below), and TMEM106B rs1990622 genotyping.
FIGURE 1.
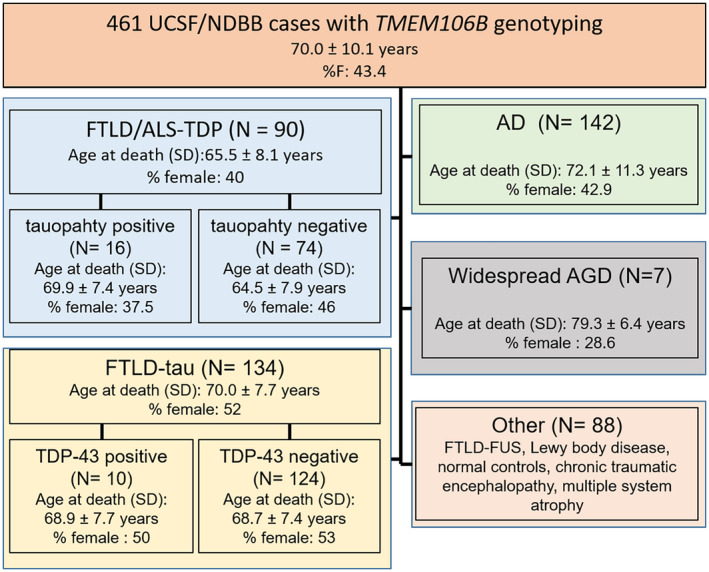
Case ascertainment by the neuropathological group. All cases were sourced by the UCSF/ Neurodegenerative Disease Brain Bank. AD, Alzheimer's disease; AGD, argyrophilic grain disease; ALS, amyotrophic lateral sclerosis; FTLD, frontotemporal lobar degeneration; MND, motor neuron disease; FUS, Fused in Sarcoma protein; TAT, temporal‐predominant neuro‐astroglial tauopathy; TDP, TAR DNA binding protein
2.2. Clinical assessment at the UCSF MAC
Clinical assessments of all patients at the UCSF MAC included a complete clinical history, physical examination, neuropsychological evaluation, and structural MRI brain imaging. Data collected during visits were analyzed for clinical diagnosis and include demographic features, the age of symptom onset, first disease symptom, cognitive, behavioral, and motor symptoms developed throughout the disease, and neurological examination findings. Clinical narratives were supplemented by standardized, prospectively applied research instruments (25). The clinical diagnosis for each patient was rendered at a multidisciplinary consensus meeting individually based on the reviewing clinical history, physical examination, neuropsychological evaluation, and structural MRI brain imaging (26, 27). As the working diagnosis for a participant sometimes changed in the face of new information over subsequent visits, we use the last diagnosis rendered before death in the present study. Upon death, all participants underwent an extensive dementia‐oriented postmortem assessment. Clinical diagnoses of bvFTD were made according to the prevailing international consensus criteria at the time of clinical evaluation (2, 3). Clinical diagnosis of Amyotrophic Lateral Sclerosis (ALS) was made according to El Escorial criteria (28). The presence of motor neuron disease (MND) in patients with bvFTD was assessed by a neurologist based on the neurological examination, usually supported by electrodiagnostic testing.
2.3. Genetics
DNA was extracted from blood using standard protocols. Screening for (likely) pathogenic variants in the most common causative genes for Mendelian forms of FTD and Alzheimer's disease (MAPT, C9orf72, GRN, TARDBP, FUS, PSEN1, PSEN2, and APP) was performed as previously described (29). Genotypes for TMEM106B rs1990622 were obtained using a commercially available Taqman SNP assay (C__11171598_20).
2.4. General neuropathological assessment at the UCSF/MAC
Twenty‐six standard tissue blocks covering dementia‐related regions of interest were dissected from the fixed slabs, and basic and immunohistochemical stains were applied following standard diagnostic procedures developed for patients with dementia as previously described. Immunohistochemistry was performed using antibodies against TDP‐43 (rabbit, 1:2000, Proteintech Group), hyperphosphorylated tau (CP‐13, S202, mouse, 1:250, courtesy of P. Davies), ß‐amyloid (1‐16, mouse, clone DE2, 1:500, Millipore), alpha‐synuclein (LB509, mouse, 1:5000, courtesy of J. Trojanowski and V. Lee), FUS (HPA008784, rabbit, 1:1500, Millipore). Moreover, subjects with a primary tauopathy also underwent immunohistochemistry against 3‐repeat and 4‐repeat tau species (RD3 and RD4, mouse, 1:250, Millipore, Billerica, MA, USA) to determine AD‐related Braak and Braak staging (30) or phenotyping non‐conforming/atypical tauopathies. All immunohistochemical runs included positive control sections to exclude technical factors as a cause of absent immunoreactivity. Neuropathological diagnosis followed currently accepted guidelines (31, 32).
The primary neuropathological diagnosis classified participants into FTLD major molecular classes (FTLD‐tau, FTLD‐TDP‐43, or FTLD‐FUS) and subtypes (33, 34), ALS (28), Alzheimer's disease (32), Lewy body disease (35), chronic traumatic encephalopathy (36), argyrophilic grain disease (22), or healthy controls (Figure 1).
FTLD‐TDP cases were subtyped into five categories. FTLD‐TDP type A features neuronal cytoplasmic inclusions (NCIs) and threads concentrated in the external cortical layers (Figure S1A). In contrast, type B shows more granular NCIs distributed throughout all cortical layers (Figure S1B). Type C displays long dystrophic neurites in the most affected areas with NCIs restricted to the ventral striatum (Figure S1C). Finally, type D features neuronal nuclear inclusions and manifest in patients with valosin‐containing protein (VCP) mutation (23, 33). When the FTLD‐TDP findings did not fit into a defined (A–D) subtype, we adopted the term "unclassifiable," usually in cases associated with a C9orf72 expansion or in cases with too few inclusions (FTLD‐TDP‐U). Furthermore, we used the term “unspecified FTLD‐tau” in cases of non‐familial FTLD‐tau failing to fit a defined subtype (31).
Co‐pathologies were noted, including Alzheimer's disease neuropathological changes (ADNC) (32), cerebrovascular disease, Lewy body disease (35), cerebral amyloid angiopathy, hippocampal sclerosis, argyrophilic grain disease (AGD) (22), Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE‐NC) (14).
2.5. Project‐specific neuropathological analysis
Initially, we anticipated including all 461 NBDD cases with TMEM106B genotyping in the full neuropathological study. However, it proved challenging to identify our targeted atypical tauopathy in the context of late stages of AD or classical primary tauopathies as there is no specific antibody differentiating tau inclusions from different tauopathies. Also, we excluded FTLD‐FUS and Lewy body disease from the full analysis because of small sample sizes. Therefore, we screened the following groups for the atypical tauopathy: FTLD‐TDP or ALS‐TDP (n = 90) and widespread AGD (n = 7). We included an AGD group to test how precisely we could distinguish our targeted atypical tauopathy from AGD based on immunohistochemistry for p‐tau. Although showing a different topographical distribution (the atypical tauopathy is more prominent in the inferior temporal gyrus, relatively sparing the hippocampus, especially CA2), both display a prominent p‐tau‐positive granular background.
We adopted the description by Kovacs and colleagues to identify our targeted atypical tauopathy, which includes neuronal and astroglial tau cytoplasmic inclusions and a tau‐positive granular background (24). Most neuronal cytoplasmic inclusions should be diffuse with a perinuclear accentuation, and some resemble neurofibrillary tangles. The astrocytic processes should show a diffuse granular immunopositivity and patchy accumulation of thin threads not overlapping with amyloid‐β deposition, reminiscent but less extensive than those seen in corticobasal degeneration, and variable perinuclear accentuation reminiscent also of granular/fuzzy astrocytes in gray matter ARTAG. Oligodendroglial coiled bodies should be scarce (Figure 2). Two experienced observers (LTG and EJK) blinded to the clinical and neuropathological diagnosis determined the presence of the atypical tauopathy in consensus. For cases screened as positive for the atypical tauopathy (n = 16, Table S1), additional slides of the same three blocks were immunostained for RD3 (specific for 3‐repeat tau, mouse, 1:250, Millipore), RD4 (specific for 4‐repeat tau, mouse, 1:250, Millipore), hyperphosphorylated tau (AT100, Thr212, Ser214, MN1060, mouse, 1:800 Thermo Fisher), hyperphosphorylated tau (T231, Thr231, rabbit, ab151559, 1:800, Abcam), T18 (specific for tau oligomers, rabbit, 1:1000, courtesy of R. Kayed), Tau C3 (specific for truncated tau at D421, mouse, 1:500, Invitrogen), and acetylated tau (K274, acetyl‐Lys274‐tau, 1:500, rabbit, gift from L. Gan (37)). AGD lacks acetyl‐Lys274‐tau inclusions (37). We also reviewed additional histological sections (anterior cingulate cortex, middle frontal gyrus, angular gyrus, precentral gyrus, and striate cortex) immunostained for CP‐13 to determine the topographical extent of the atypical tauopathy. Before staining, all sections were coded for blinded rating. Positive (AD cases) and negative controls (replacing primary antibody for PBS) were run for all immunoreactions.
FIGURE 2.
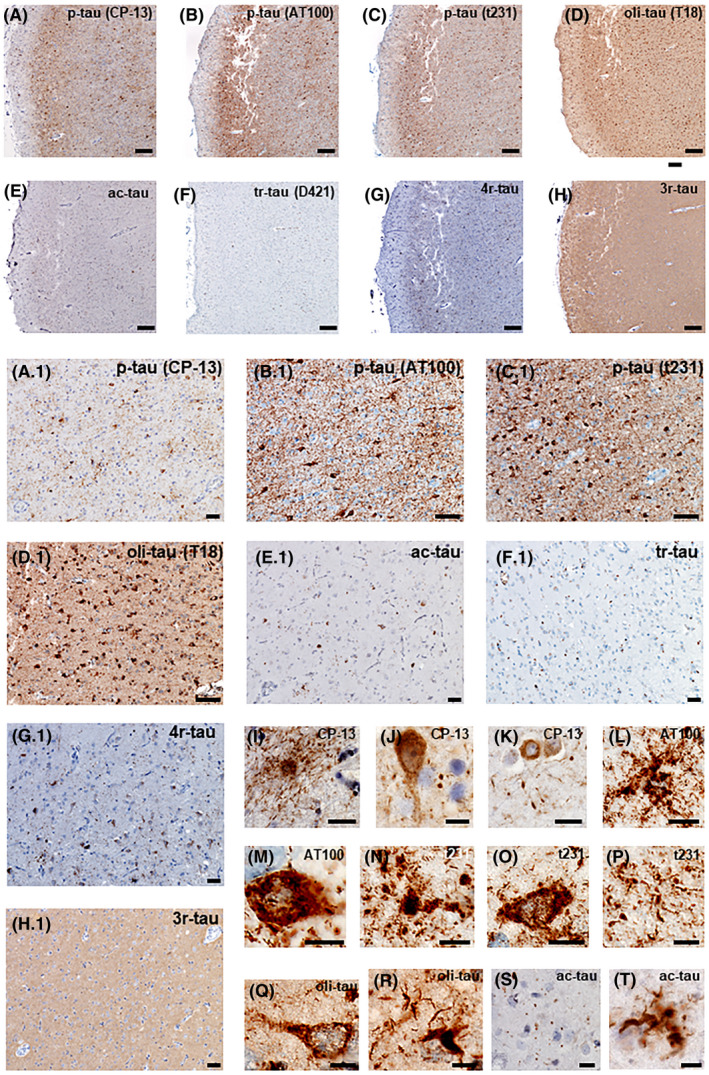
Neuropathological features of TMEM106B‐associated neuro‐astroglial tauopathy. Case #8, female, 69 years old. Serial histological sections from the inferior temporal gyrus immunostained for several antibodies against pathological tau. (A and A.1) Immunostaining against phospho‐tau, Ser 202 (p‐tau CP‐13). (B and B.1) Immunostaining against phospho‐tau, Thr212, Ser214 (p‐tau AT100). (C and C.1) Immunostaining against phospho‐tau, Thr231 (p‐tau t231). (D and D.1) Immunostaining against oligomeric tau (oli‐tau T18). Note neuronal and astroglia inclusions in a granular background. (E and E.1) Immunostaining against acetylated‐tau (acetyl‐Lys274‐tau) shows neuronal and glial inclusions. Although less abundant, it shows both neuronal and glial inclusions. (F and F.1) Immunostaining against truncated tau (Tau C3, D421). No inclusion was detected, only unspecific staining. (G and G.1) Immunostaining against 4‐repeat tau (RD4). (H and H.1) Immunostaining against 3‐repeat tau (RD3). No inclusion was detected. D and D1) High‐magnification examples of astroglial inclusions (I, L, N, P, R, T), neuronal inclusions (J, K, M, O, Q), and granular background (J, S) detected by several tau antibodies. Scale bars: A, B, C, D, E, F, G, H: 500 µm; A.1, B.1, C.1, D.1, E.1, F.1, G.1, H.1: 20 µm; J–T: 5 µm
To determine whether TPD‐43 (rabbit, 1:2000, Proteintech Group, Chicago, IL, USA) and hyperphosphorylated tau (CP‐13, S202, mouse, 1:250, courtesy of P. Davies) inclusions co‐occur in the same neurons, we immunostained cases representative of FTLD‐TDP type B, ALS‐TDP, and FTLD‐TDP type C using double‐labeled immunofluorescence protocols in a Discovery Ventana Ultra autostainer. Our antibody choice labels both physiological and pathological TDP‐43, thus enabling visualizing if neurons with p‐tau changes show TDP‐43 nuclear exclusion despite lacking TDP‐43 cytoplasmic inclusions.
Finally, we immunostained inferior temporal gyrus sections against phospho‐TDP (TAR‐5P, rat, 1:500, courtesy of M. Neumann) to interrogate if phosphorylation pattern varied between cases positive or negative for comorbid neuro‐astroglial tauopathy.
2.6. Statistical analysis
Although only 97 (AGD = 7, FTLD/ALS‐TDP = 90) out of 461 cases (21%) underwent screening for the targeted atypical tauopathy, all cases were used for correlation analyses.
First, using the whole sample (n = 461), we assessed if an association between the targeted atypical tauopathy and TMEM106B rs1990622 genotypes could be false due to a possible skewed distribution of samples genotyped for TMEM106B in the UCSF‐NBDD cohort or subgroups. We compared the distribution of the rs1990622 genotypes between a population‐based sample (38) and the NDBB cohort with available genetic information, and then between this same UCSF‐NDBB cohort and neuropathological subgroups (Table 1).
TABLE 1.
Comparison of TMEM106B rs1990622 genotypes between UCSF/NDBB cohort and a population‐based cohort and UCSF/NDBB total cohort and subgroups and with literature data
| A/A n(%) | A/G n(%) | G/G n(%) | p vs. REF | |
|---|---|---|---|---|
| Population(N = 5020) a | 1637 (32.6) | 2485 (49.5) | 898 (17.9) | REF |
| NDBB cohort (N = 461) | 167 (36.2) | 222 (48.2) | 72 (15.6) | 0.22 |
| NDBB cohort b (N = 461) | 167 (36.2) | 222 (48.2) | 72 (15.6) | REF |
| FTLD/ALS‐TDP plus tauopathy (n = 16) | 15 (93.7) | 1 (6.3) | – | 0.001 |
| FTLD/ALS‐TDP w/o tauopathy (n = 74) | 23 (31.1) | 38 (51.3) | 13 (17.6) | 0.68 |
| FTLD‐tau (n = 134) | 55 (41) | 59 (44) | 20 (15) | 0.59 |
| FTLD‐tau plus FTLD‐TDP (n = 10) | 5 (50.0) | 4 (40.0) | 1 (10.0) | 0.65 |
| AD (n = 142) | 39 (27.5) | 77 (54.2) | 26 (18.3) | 0.15 |
| AGD (n = 7) | – | 5 (71.4) | 2 (28.6) | 0.36 |
Abbreviations: AD, Alzheimer's disease; AGD, argyrophilic grain disease; ALS, amyotrophic lateral sclerosis; FTLD, frontotemporal lobar degeneration.
Genotype frequency within a UK control population of 5020 people from the Wellcome Trust Case Control Consortium (38).
FTLD/ALS‐TDP‐TAT and other path groups (FTLD/ALS‐TDP, FTLD‐TAU, AGD, AD) vs. NDBB cohort with TMEM106B genotyping as reference (REF) group. The results are not significant at p > 0.05.
For the subsequent analyses, we only included the 90 FTLD/ALS‐TDP cases. Our working hypothesis was that rs1990622 genotypes would affect the risk of developing the target atypical tauopathy under a recessive genotype model (presence versus absence of two major alleles: A/A vs. A/G and G/G) in FTLD/ALS‐TDP cases. We performed univariate analyses on all variables, means, and standard deviations to describe continuous variables and frequencies and percentages for categorical variables. Data analysis included demographic features: age of symptom onset, clinical diagnosis, and age at death. The characteristics of the sample were compared by TMEM106B genotype using one‐way Analysis of Variance (ANOVA) for continuous variables and Chi‐squared or Fisher's exact test for categorical data. Non‐parametric analyses (Mann‐Whitney U and Kruskal Wallis) were used when the sample size did not satisfy the requirements for parametric analyses. We then investigated the odds ratio of showing the targeted atypical tauopathy by TMEM106B genotype, using binary logistic regression models, adjusted for age of disease onset, sex, and disease duration. The model predicted the targeted atypical tauopathy by TMEM106B genotype alone and after adding important covariates.
Next, to probe a possible effect of the targeted atypical tauopathy on clinical and neuropsychological outcomes, we compared demographics, clinical and neuropathological characteristics, and neuropsychological performance (as described below) between cases positive and negative for the targeted atypical tauopathy.
2.7. Neuropsychological testing
Normative data were calculated using an age‐matched sample (mean age: 67.48, SD = 8.25, range: 39‐89) of 295 cognitively normal controls (58.64% female) recruited at the University of California, San Francisco's (UCSF) Memory and Aging Center as part of the Hillblom Healthy Aging Study, a deeply phenotyped longitudinal cohort. Visits included neuropsychological and neurologic examination in addition to an MRI scan. The participants' status as cognitively intact was confirmed via a consensus meeting that included a board‐certified neuropsychologist and neurologist. To further ensure that our normative sample comprised clinically intact controls, we included only those with a stable Clinical Dementia Rating Scale (CDR) of 0 (i.e., clinically normal) who also had a Mini‐Mental State Examination (MMSE) that was above 26 and that had not declined by more than 1 point across follow‐up visits.
2.8. Cognitive measures
Executive function was assessed using a backward digit span task, modified trail making test(24) Stroop Color‐Word Interference (39, 40), the Delis‐Kaplan Executive Function System(D‐KEFS) Design Fluency (Filled Dots Condition) (39), and a lexical fluency task (number of words per minute that begin with a specific letter). Visuospatial function was assessed using the Benson figure copy (25). Episodic memory was measured using the 10‐minute free recall of the Benson figure (25), and the short delay and long delay recall scores from a nine‐item list‐learning task, the short‐form of the California Verbal Learning Test (CVLT‐II) (25). Language tests included a 15‐item version of the Boston Naming Test and animal fluency (41).
Composite scores were calculated for the domains of episodic memory, executive function, and language. Consistent with published methodology (42), scores for each test were first transformed to z‐scores based on the means and standard deviations of the normative sample. Within each cognitive domain, z‐scores were averaged across the subtests. Participants were required to have all episodic memory and language tests to create a composite score. Given the larger number of executive function tests, we required 3 of 5 measures to be present and took the average z‐score of those completed tests. A visuospatial composite was not created because only one measure of interest was included.
The statistical software R (www.r‐project.org) was used to conduct statistical analyses. An alpha level < 0.05 was considered statistically significant in two‐tailed tests.
2.9. Validation cohort
To investigate if our results would replicate in an independent cohort, we genotyped TMEM106B rs1990622 in a group of seven cases showing our targeted atypical tauopathy published by Kovacs and colleagues (24). Five out of the seven cases feature some degree of TDP‐43 proteinopathy. We received DNA samples that underwent the same processing as the UCSF cases.
3. RESULTS
3.1. Neuropathological features of our targeted temporal‐predominant neuro‐astroglial tauopathy
All 16 cases (Table S1) identified as our targeted atypical tauopathy featured neuronal and astroglial tau cytoplasmic inclusions and a tau‐positive granular background (Figure 2). Three antibodies against p‐tau (CP‐13, AT100, t231) labeled neuronal inclusions that were finely granular and diffusely distributed in the cytoplasm and proximal axonal segment. Astroglial inclusions mostly resembled granular or fuzzy astrocytes (GFA). However, immunostaining for p‐tau also labeled a minority of other glial forms, either resembling astrocytic plaques or ramified astrocytes. Immunostaining for oligomeric tau also showed strong positivity in glia and neurons and highlighted threads and grains (Figure 2D,D.1,Q,R). Immunostaining for acetylated tau (ac‐tau) showed less conspicuous but definitively positive inclusions in neurons, glia, and grains (Figure 2E,E.1,S,T). Finally, Tau C3 antibody against truncated tau D421 showed no pathology (Figure 2F). Figure 2F.1 illustrates unspecific staining around, especially around vessels (the immunoassay was overdeveloped to guarantee that pathological changes would be unveiled, if present). Across cases, the tauopathy burden was variable, but overall, it was higher in the inferior temporal gyrus, followed by the entorhinal cortex. In 2/3 of the cases, a low tauopathy burden was recognized in the hippocampal formation and amygdala. The targeted atypical tauopathy showed positivity for 4‐repeat tau specific, but not to 3‐repeat tau specific antibodies (Figure 2G,G.1,H,H.1).
3.2. AGD vs. our targeted atypical tauopathy
In most cases, it was straightforward to distinguish AGD from our targeted atypical tauopathy since the former is more prominent in the hippocampal formation, and the latter usually featured a higher burden of lesions in the inferior temporal gyrus than in the hippocampal formation. The distinction between atypical tauopathy and AGD was more difficult in widespread AGD cases. Thus, we included in the screening seven cases with a primary diagnosis of widespread AGD and a low burden of AD neuropathologic changes. Blinded neuropathological analysis based on CP‐13 (p‐tau Ser 202) immunostaining was enough to differentiate all AGD cases from the FTLD/ALS‐TDP plus atypical tauopathy cases. In a second step, we detected a strong positivity for acetyl‐Lys274‐tau in all atypical tauopathy cases (Figure 2). In contrast, AGD inclusions are negative for acetyl‐Lys274‐tau (37), reinforcing that despite some morphological and topographical overlapping, our targeted atypical tauopathy and widespread AGD can be differentiated.
Analysis of cases immunostained for TDP‐43 and p‐tau unveiled that a proportion of neurons harbors p‐tau show TDP‐43 nuclear exclusion and cytoplasmic inclusions (Figure 3A,B, and Figure S2). However, both neurons with TDP‐43 inclusions but lacking p‐tau and neurons with p‐tau inclusions lacking TDP‐43 changes were also found in the FTLD‐TDP type B case. We failed to find neurons with TDP‐43 nuclear exclusion in the ALS‐TDP and FTLD‐TDP‐type C cases (Figure 3C,D, and Figure S2). We also failed to find obvious differences in the pattern of phospho‐TDP staining between FTLD‐TDP cases (types A, B, and C) positive and negative for the neuro‐astroglial tauopathy (Figure S1).
FIGURE 3.
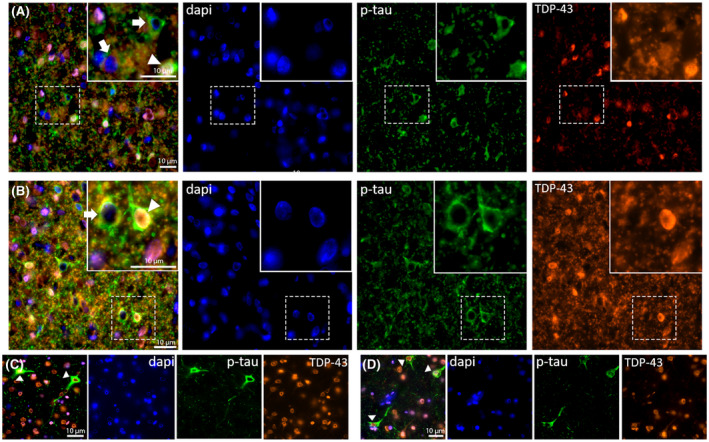
Double‐labeled immunostaining against phospho‐tau (p‐tau‐CP13) and TDP‐43. Inferior temporal gyrus. (A and B) two different fields of a case with a primary diagnosis of FTLD‐TDP type B (Case #8, female, 69 years old). Note the presence of neurons harboring either phospho‐tau (p‐tau) only, mislocalized TDP‐43 only, or both. (C and D) two different fields of cases with a primary diagnosis of ALS‐TDP type B (Case #5, male, 50 years old). Despite the presence of neurons containing p‐tau inclusions, we failed to observe any neurons with TDP‐43 nuclear exclusion
3.3. TMEM106B rs19906220 A/A genotype is enriched in FTLD/ALS‐TDP cases with comorbid atypical tauopathy
The distribution of TMEM106B rs1990622 genotypes in the NBDD total cohort (n = 461), is similar to a population‐based cohort from the UK study (n = 5020) (38) (Table 1).
Next, we used the UCSF‐NBDD cohort with TMEM106B genotyping as a reference to examine if different neuropathological subgroups showed a different distribution of rs1990622 genotypes. We used the following subgroups: #1‐FTLD/ALS‐TDP plus the targeted atypical tauopathy (n = 16); #2‐FTLD/ALS‐TDP lacking tauopathy (n = 74), #3‐FTLD‐tau lacking comorbid TDP‐43 proteinopathy (n = 134); #4‐FTLD‐tau plus contributing/primary FTLD‐TDP (43) (n = 10); #5‐AD (n = 142) #6‐widespread AGD (22, 37), lacking any other co‐primary or contributing neuropathological diagnosis (n = 7). We grouped ALS and FTLD cases because the number of ALS cases were too low for statistical analysis (Table S2). Compared to the rest of the NBDD cohort, cases with FLTD/ALS‐TDP with the targeted atypical tauopathy were highly enriched for rs1990622 A/A genotype (37, 38). No significant differences were found for any other group (Table 1).
3.4. Within the FLTD/ALS‐TDP group, the presence of the targeted atypical tauopathy was the only significant difference among TMEM106B rs1990622 genotypes
Among the 90 FTLD/ALS‐TDP cases, 61.1% of cases (n = 55) were sporadic, whereas 25 patients had a C9orf72 repeat expansion, eight a GRN mutation, and two a pathogenic TARDBP variant (44). We examined possible demographic, clinical, and neuropathological differences among TMEM106B rs1990622 genotypes (A/A, A/G, G/G) in FTLD/ALS‐TDP cases. Groups of cases with different genotypes (A/A, A/G, and G/G) had a similar age of onset, duration, and gender distribution. We also did not detect differences in the frequency of familial vs. sporadic cases, brain weight at death, clinical phenotype, or neuropathological subtype (Table 2). The only significant statistically significant difference was the presence of our targeted atypical tauopathy (p < 0.001). We noticed a trend for a higher frequency of hippocampal sclerosis in the A/A genotype group (Table 2).
TABLE 2.
Characteristics of the FTLD/ALS‐TDP a sample by TMEM106B genotype (n = 90)
| A/A | A/G | G/G | p | |
|---|---|---|---|---|
| n = 38 | n = 39 | n = 13 | ||
| Demographics | ||||
| Age at onset, mean (SD) in years b | 58.9 (7.5) | 54.8 (11.6) | 54.7 (13.3) | 0.92 |
| Disease duration, mean (SD) in years b | 6.9 (3.9) | 10.0 (6.4) | 7.8 (5.2) | 0.11 |
| Female sex, n (%) c | 19 (52.8) | 12 (33.3) | 5 (13.9) | 0.22 |
| Brain weight (g), mean (SD) b | 1160 (198) | 1161 (190) | 1116 (122) | 0.27 |
| Genetics | ||||
| C9orf72, n (%) c | 7 (28.0) | 13 (52.0) | 5 (20.0) | 0.22 |
| GRN, n (%) b | 5 (62.5) | 3 (37.5) | – | 0.42 |
| TARDBP, n (%) | 1 (50.0) | 1 (50.0) | – | – |
| % Sporadic, n (%) c | 25 (45.5) | 22 (40.0) | 8 (14.5) | 0.69 |
| Clinical groups c No. (%) | 0.22 | |||
| AD type dementia | 0 | 0 | 1 (100) | – |
| bvFTD | 12 (52.2) | 9 (39.1) | 2 (8.7) | 0.12 |
| bvFTD‐MND or ALS | 14 (41.2) | 16 (47.1) | 4 (11.7) | 0.31 |
| CBS/PSPS | 3 (100) | 0 | 0 | – |
| nfPPA | 2 (50.0) | 1 (25.0) | 1 (25.0) | 0.25 |
| svPPA | 7 (28.0) | 13 (52.0) | 5 (20.0) | 0.32 |
| Neuropathological diagnosis c No. (%) | 0.09 | |||
| FTLD‐TDP; +/‐ MND (C9orf72) | 6 (26.1) | 12 (52.2) | 5 (21.7) | 0.18 |
| FTLD‐TDP, type A, (C9orf72) | 1 (50.0) | 1 (50.0) | – | – |
| FTLD‐TDP; +/‐ MND (TARDBP) | 1 (50.0) | 1 (50.0) | – | – |
| FTLD‐TDP, type A (GRN) | 5 (37.5) | 3 (62.5) | – | 0.42 |
| FTLD‐TDP, type A, sporadic | 7 (87.5) | – | 1 (12.5) | – |
| FTLD‐TDP; +/‐ MND, type B or ALS‐TDP, sporadic | 12 (48.0) | 11 (44.0) | 2 (8.0) | 0.46 |
| FTLD‐TDP, type C, sporadic | 6 (27.2) | 11 (50.0) | 5 (22.7) | 0.18 |
| Hippocampal sclerosis as co‐pathology d | 11 (64.7) | 4 (23.5) | 2 (11.8) | 0.06 |
Abbreviations: ALS, amyotrophic lateral sclerosis; bvFTD, behavioral variant frontotemporal dementia; CBS/PSPS, corticobasal syndrome/progressive supranuclear palsy syndrome; C9orf72, chromosome 9 open reading frame 72; GRN, Progranulin; FTLD, frontotemporal lobar degeneration; MND, motor neuron disease (motor neuron–related signs referred to the presence of 1 or more of reduced muscle power, muscle atrophy, and fasciculations); nfPPA, non‐fluent Primary Progressive Aphasia; svPPA, semantic variant Primary Progressive Aphasia.
Includes FTLD/ALS‐TPD cases with and without TAT; TARDBP, 43‐kDa transactive response (TAR)‐DNA‐binding protein.
One‐way ANOVA.
Fisher's exact test.
Presence of hippocampal sclerosis as co‐pathology. Therefore numbers are not mutually exclusive with previous primary pathological diagnostic categories.
Next, we investigated whether the rs1990622 A/A genotype is associated with a higher risk of developing our targeted atypical tauopathy in the FTLD/ALS‐TDP sample (Table 3). Using binary logistic regression models predicting our targeted atypical tauopathy by A/A genotype, we found that, in our series, individuals with FTLD/ALS‐TDP and an A/A genotype had an odds ratio (OR) of 13.9 (3.7‐51.9 95% confidence interval (CI)) to develop the targeted atypical tauopathy. This association was even stronger after controlling for age of disease onset, sex, or specific mutations (C9orf72 or GRN), suggesting that this association is unlikely to reflect a demographic or another genetic factor commonly associated with FTLD/ALS‐TDP.
TABLE 3.
Association between the TMEM106B rs1990622 A/A genotype and neuro‐astroglial tauopahty in FTLD/ALS‐TDP (n = 90)
| OR | 95% CI | p | |
|---|---|---|---|
| Crude | 13.9 | 3.7–51.9 | <0.0001 |
| Multivariate a | 31.3 | 3.9–256.1 | <0.0001 |
Reference group: G/G + A/G.
Adjusted for age at disease onset, gender, and disease duration
3.5. Cases positive for temporal‐predominant neuro‐astroglial tauopathy were older at symptoms onset
Neuro‐astroglial tauopathy‐positive and neuro‐astroglial tauopathy‐negative cases (all with FTLD/ALS‐TDP) showed similar demographics, clinical phenotype, global clinical measures, and neuropsychological performance adjusted by CDR, except that those with comorbid neuro‐astroglial tauopathy had a moderately older (p = 0.01) age at onset compared with tauopathy‐negative FTLD/ALS‐TDP cases (Table 4). There was a trend of a better global executive function performance in neuro‐astroglial tauopathy‐positive cases, but the analysis lacks the power to draw definitive conclusions (p = 0.06).
TABLE 4.
Demographics and Clinical Characteristics of pure FTLD/ALS‐TDP and FTLD/ALS‐TDP plus neuro‐astroglial tauopathy
| FLTD/ALS‐TDP only (n = 74) | FLTD/ALS‐TDP plus tauopathy (n = 16) | p | |
|---|---|---|---|
| Demographics | |||
| Age at onset, mean (SD) in years | 55.4 (8.3) | 61.9 (7.9) | 0.01 |
| Female sex, N (%) | 30 (40.5) | 6 (37.5) | 0.82 |
| Disease duration, mean (SD), in years | 8.7 (6.0) | 8.1 (5.1) | 0.68 |
| Global cognitive measures | |||
| MMSE a | 22.6 (5.5) | 24.3 (3.3) | 0.19 |
| CDR b | 1.4 (0.8) | 1.1 (0.8) | 0.32 |
| GDS c | 8.8 (5.9) | 5.6 (4.6) | 0.11 |
| Clinical diagnosis, No. (%) | 0.30 | ||
| AD type dementia | 1 (100) | 0 | – |
| bvFTD | 18 (78.3) | 5 (21.7) | 0.45 |
| bvFTD‐MND or ALS | 28 (82.4) | 6 (17.6) | 0.71 |
| CBS/PSPS | 1 (33.3) | 2 (66.7) | – |
| nfPPA | 4 (100) | 0 | – |
| svPPA | 22 (88.0) | 3 (22.0) | 0.63 |
| Neuropsychological performance (SD) d | |||
| Executive funtion (EF) | |||
| Modified Trail Making Test | 41.6 (24.8) | 34.6 (18.7) | 0.31 |
| Stroop color naming | 55.9 (22.3) | 62.3 (19.9) | 0.42 |
| Stroop inhibition | 31.3 (19.2) | 36.2 (12.3) | 0.11 |
| Digit span task forward | 5.3 (1.8) | 6.2 (2.1) | 0.26 |
| Digit span task backward | 3.7 (1.6) | 4.4 (2.1) | 0.27 |
| Design fluency task | 5.2 (3.4) | 7.0 (3.9) | 0.18 |
| D‐words, lexical fluency | 6.0 (4.9) | 8.3 (6.4) | 0.29 |
| Verbal fluency animal category | 9.3 (6.9) | 11.4 (9.6) | 0.47 |
| Total EF composite Z‐score | −2.0 (1.3) | −1.2 (1.3) | 0.01 |
| Visuospatial | |||
| Benson copy | 13.9 (2.8) | 14.8 (2.2) | 0.21 |
| Memory | |||
| Benson delayed recall | 8.5 (4.2) | 9.2 (3.5) | 0.60 |
| CVLT short delay recall | 4.2 (2.9) | 5.0 (3.1) | 0.40 |
| CVLT delayed recall | 3.3 (3.1) | 3.4 (3.4) | 0.87 |
| Total Memory composite Z‐score | −3.0 (2.2) | −2.4 (1.9) | 0.42 |
| Language | |||
| Repetition | 4.4 (0.9) | 4.6 (0.7) | 0.34 |
| Syntax comprehension | 3.9 (1.3) | 3.7 (1.4) | 0.62 |
| Boston Naming Test | 9.9 (4.9) | 10.2 (5.1) | 0.98 |
| Total language composite Z‐score | −2.8 (2.3) | −2.9 (2.8) | 0.94 |
| Calculations | 3.7 (1.5) | 4.0 (1.1) | 0.50 |
| Affect matching | 9.7 (3.5) | 10.5 (4.1) | 0.51 |
| Neuropathological diagnosis, No. (%) | 0.14 | ||
| FTLD‐TDP; +/‐ MND (C9orf72) | 21 (28.4) | 2 (12.5) | 0.34 |
| FTLD‐TDP, type A, (C9orf72) | 1 (1.4) | 1 (6.3) | 0.14 |
| FTLD‐TDP; +/‐ MND (TARDBP) | 1 (1.4) | 1 (6.3) | 0.14 |
| FTLD‐TDP, type A (GRN) | 6 (8.1) | 2 (12.5) | 0.27 |
| FTLD‐TDP, type A, sporadic | 5 (6.7) | 3 (18.7) | 0.06 |
| FTLD‐TDP; +/‐ MND type B or ALS‐TDP, sporadic | 20 (27.0) | 5 (31.2) | 0.33 |
| FTLD‐TDP, type C, sporadic | 20 (27.0) | 2 (12.5) | 0.28 |
| Hippocampal sclerosis as co‐pathology e | 11 (64.7) | 6 (35.3) | 0.07 |
All values indicate the means and standard deviation, derived from a general linear model comparing the 2 subgroups. No pairwise comparisons were statistically significant between groups. Neuropsychological test performances were controlled for CDR in this model.
Abbreviations: AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; bvFTD, behavioral variant frontotemporal dementia; CBS/PSPS, corticobasal syndrome/progressive supranuclear palsy syndrome; CDR, Clinical Dementia Rating; CVLT, California Verbal Learning Test–short form; C9orf72, chromosome 9 open reading frame 72; FTLD, frontotemporal lobar degeneration; GDS, Geriatric Depression Scale; GRN, Progranulin; MMSE, Mini‐Mental State Examination; MND, motor neuron disease (motor neuron–related signs referred to the presence of 1 or more of reduced muscle power, muscle atrophy, and fasciculations); nfPPA, non‐fluent Primary Progressive Aphasia, svPPA, semantic variant Primary Progressive Aphasia; TARDBP, 43‐kDa transactive response (TAR)‐DNA‐binding protein.
Scores range from 0 to 30, with higher scores indicating better cognition.
Scores range from 0 to 3, with higher scores indicating more advanced dementia.
Scores range from 0 to 30, with higher scores indicating more significant depression.
For all neuropsychological performance tests, higher scores indicate better performance.
Presence of Hippocampal Sclerosis as co‐pathology. Therefore numbers are not mutually exclusive with previous primary pathological diagnostic categories.
3.6. TMEM106B rs1990622 A/A genotype is also enriched in the validation cohort
We genotyped TMEM106B at rs1990622 in the seven cases described by Kovacs and colleagues that featured an atypical tauopathy, with similar features to the ones we used to identify neuro‐astroglial tauopathy in our cohort (24). The mean age at death was 83.8 years (Male = 3, Female = 4). Five of the cases had some degree of TDP‐43 proteinopathy. Although these cases lacked a comprehensive clinical history, clinical presentations combined dementia with psychiatric symptoms and/or parkinsonism. Six out of the seven cases had DNA available for genotyping, and, of those, five carried the A/A genotype (Table 5). Of the two cases lacking TDP‐43 proteinopathy, one had the rs1990622 A/G genotype, and the other was a carrier of a non‐pathogenic MAPT N255N variant.
TABLE 5.
Replication Cohort. Sociodemographic, neuropathological, and genetic findings in seven cases diagnosed as a complex tauopathy (7) a
| Case number | |||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| Age at death | 82 | 83 | 83 | 84 | 84 | 94 | 77 |
| Sex | M | M | F | F | F | F | M |
| TDP‐43 proteinopathy | − | + | − | + | + | + | + |
| MAPT variant | na | − | N255N b | − | − | − | na |
| APOE genotype | na | ε3/3 | ε4/3 | ε3/3 | ε3/2 | ε3/3 | na |
| TMEM106B rs1990622 genotype | A/G | A/A | A/A | A/A | A/A | A/A | na |
Abbreviation: na, data not available.
Link to original cohort description: (https://link.springer.com/article/10.1007%2Fs00401‐011‐0819‐x).
MAPT N255N is not considered a pathogenic variant (https://www.alzforum.org/mutations/mapt‐n255n).
4. DISCUSSION
We combined clinical, pathological, and genetic evidence to explore the relationship between TMEM106B polymorphisms (rs1990622) and the presence of a temporal‐predominant atypical neuro‐astroglial tauopathy in subjects with FTLD/ALS‐TDP as the primary diagnosis. We found that homozygosity for rs1990622 major allele A is associated with a high odds for developing this neuro‐astroglial tauopathy in individuals with FTLD/ALS‐TDP, regardless of clinical phenotype or heritable versus sporadic etiology. Our findings were replicated in an independent sample (24). In the replication series, five out of six cases featuring an atypical tauopathy, which features modeled the criteria we adopted to identify neuro‐astroglial tauopathy in our cohort, carried the TMEM106B rs1990622 A/A genotype.
In our FTLD/ALS series, over 90% of neuro‐astroglial tauopathy‐positive cases carried the TMEM106B rs1990622 A/A genotype. FTLD/ALS‐TDP cases with the A/A genotype were 13 times more likely to show neuro‐astroglial tauopathy than those with an A/G or G/G genotype. In contrast, the TMEM106B rs1990622 genotype distribution for all the other neuropathological groups (AD, FTLD‐tau, or AGD) tested was similar to a normal population distribution (38).
TMEM106B polymorphisms represent a risk factor for FLTD (45, 46), and its association with worse pathological and clinical outcomes seems to be more robust in GRN mutation carriers. Some studies suggest that TMEM106B polymorphisms increase the odds of TDP‐43 pathology in older individuals without FTLD (13). TMEM106B has been implicated in modulating degenerative changes regardless of the disease pathology (47), and TMEM106B and APOE interact to increase the risk of developing late‐onset AD and hippocampal sclerosis (48, 49, 50). Also, the TMEM106B rs1990622 minor allele G, deemed to be a protective allele, is associated with a lower burden of p‐tau in individuals with CTE (51). However, to the best of our knowledge, no previous studies have reported an association between TMEM106B polymorphism and increased odds for a specific tauopathy, especially with an odds ratio of 31.3 after multivariate analysis. Actually, except for AD and AGD, a primary FTLD‐TDP rarely co‐occurs with a primary tauopathy (52, 53), and, to date, such overlap has not been linked to a specific polymorphism.
Several factors may have contributed to our findings. First, TMEM 106B is a type‐II glycoprotein localized to late endosomes/lysosomes, and it plays a role in the maintenance of lysosomal storage, morphology, and function. Its overexpression causes enlarged lysosomes, which delays the degradation of endocytic cargo (45), suggesting a pathway to modify FLTD‐TDP pathophysiology (54, 55). Some studies have shown that TMEM106B may control the dendritic trafficking of lysosomes and functionally interact with microtubule‐associated protein 6 (MAP6) (54). Furthermore, TMEM106B has been associated with axonal transport of LAMP1‐positive organelles in motoneurons and axonal sorting at the initial segment, providing further insight into how TMEM106B affects lysosomal proteolysis and degradative capacity in neurons (56, 57).
Also, TMEM106B polymorphism may function as a risk factor that might enhance neuronal toxicity associated with neurodegeneration with endosomal defects, such as ESCRT (endosomal sorting complexes required for transport) dysfunction‐associated neurodegeneration (17). TMEM106B polymorphism may also be implicated in neuronal number variations in normal brains (58). It is still under debate whether TMEM106B polymorphisms produce changes in the level of TMEM106B or its function. However, recent studies(17, 18) have shown that SNPs in the coding region of TMEM106B might affect the cellular signaling and protein to protein interactions involved in the endolysosomal pathway. Therefore, Jun and colleges (17) suggest that TMEM106B might have general effects on neurodegenerative diseases associated with defects in the endolysosomal pathway, as in Alzheimer's disease and Parkinson's disease. Furthermore, recent studies by Nho and colleges (12) found a strong correlation between the TMEM106B SNP (rs1990622) and Hippocampal Sclerosis of Aging, providing further evidence of the general role of TMEM106 polymorphisms in neurodegeneration. Here, we found a tread for comorbid hippocampal sclerosis in the TMEM106B A/A genotype group.
In any case, further studies are needed to elucidate why this neuro‐astroglial tauopathy is predominantly medial temporal, even in cases for which TDP‐43 pathology is absent in this region, such as the ALS‐TDP cases. Maybe the correlation between this neuro‐astroglial tauopathy and rs1990622 is entirely independent of the presence of FTLD/ALS‐TDP pathology. In fact, the age‐related nature of this neuro‐astroglial tauopathy and its occurrence in FTLD/ALS‐TDP with broad‐spectrum clinical, genetic, and pathological features favors this hypothesis. Although we identified partial overlapping between TDP‐43 and p‐tau changes in FTLD‐TDP type B, the pattern was unclear, and a significant number of neurons either have TDP‐43 or p‐tau changes alone. It is possible that TDP‐43 and p‐tau co‐occurrence was incidental, mainly because we failed to find TDP‐43 changes in inferior temporal gyrus with neuro‐astroglial tauopathy from ALS‐TDP and FTLD‐TDP‐type C cases. Noteworthy, previous studies (59) have described an increased propensity for tau in both FTLD‐C9ORF72 and sporadic FTLD compared with FTLD‐GRN. These differences were most marked in limbic regions. The extent to which such findings are related to a direct effect of C9ORF72 mutations in protein degradation that favors the accumulation of multiple different proteins or confounded by the presence of TMEM106B A/A is yet to be determined.
Nevertheless, the lack of reports describing pathological changes resembling this neuro‐astroglial tauopathy in individuals with minimal neurodegeneration levels, even in cohorts with an older age of death than our cohort, challenges the hypothesis that this neuro‐astroglial tauopathy is independent of FTLD/ALS‐TDP pathology. Also, TMEM106B rs3173615 minor allele G was less frequent in CBD with comorbid TDP‐43 than in CBD alone (61). We attempted to interrogate this question by examining non‐FTLD‐TDP samples, but, without a specific marker, attempts to distinguish between this neuro‐astroglial tauopathy and cases with a primary diagnosis of AD or other primary tauopathies, except for AGD, lack rigor.
This neuro‐astroglial tauopathy has some resemblance to ARTAG and AGD and could represent a combination of both, rather than an independent neuropathological entity. Several factors support that this tauopathy is different from ARTAG and/or AGD. First of all, there is a neuronal component in addition to the glial component. Second, both the neuronal inclusions and granular background are positive for acetylated tau, which would be atypical for AGD (49). Third, the distribution is different than in AGD, with sparing of CA2 and highest pathology burden in inferior temporal gyrus.
Furthermore, other findings support the interpretation that this neuro‐astroglial tauopathy is different from ARTAG and/or AGD. First, Kovacs et al. series show a similar tauopathy, also in cases with TDP‐43 proteinopathy (53). Second, none of the widespread AGD cases had a TMEM A/A genotype (albeit, this could be by chance because the numbers are small). Third, we failed to find an association between TMEM106B genotype and the presence of ARTAG in a series of published 83 AD cases (60).
This study has strengths as well as limitations. Although our FTLD/ALS‐TDP cohort is well‐characterized and relatively large, the sample represents a wide variety of clinical, genetic, and pathological subgroups, decreasing the power analysis, as reflected by the large confidence intervals resulted from the analysis between rs1990622 A/A genotype and the neuro‐astroglial tauopathy. Also, the outcome variable (FTLD/ALS‐TDP plus tauopathy) is somewhat infrequent in the entire NDBB series, which can yield imprecise estimations of odds ratio, especially after corrections. Lack of power may also explain why we could not identify any clinical and neuropsychological correlates of the neuro‐astroglial tauopathy. However, even with this small sample size, we could detect a very high odds ratio that the neuro‐astroglial tauopathy associates with TMEM106B rs1990622 A/A genotype, an association replicated in a small, albeit well‐characterized and independent, series.
Despite its limitations, this study investigates and confirms, for the first time, an independent association between a TMEM106B variant and a distinct neurodegenerative change, in this case, a clearly defined tauopathy, highlighting the importance of rs1990622 as a genetic determinant of neurodegeneration. Although the molecular consequences of TMEM106 polymorphisms and potential mode of actions into tau metabolism remain unclear, our findings advance the field by proving strong evidence of a link among TDP‐43, tau pathology, and aging processes possibly mediated by a TMEM106B polymorphism that can be addressed in future mechanistic studies.
5. CONCLUSIONS
We have provided evidence of a strong association between a distinctive medial temporal predominant, 4‐repeat, neuro‐astroglial tauopathy with a TMEM106B rs1990622 A/A genotype in cases with TDP‐43 proteinopathies in two independent cohorts. Our data add to the growing body of evidence that TMEM106B polymorphisms may modulate neurodegeneration and open avenues for mechanistic studies on the role of TMEM106B modulation of tau pathology. Methods advancement permitting, further studies should determine if this association is seen in cases that feature other neurodegenerative conditions than FLTD/ALS‐TDP.
CONFLICT OF INTEREST
Jorge J. Llibre‐Guerra, Suzee E. Lee, Claudia K Suemoto, Gabor G. Kovacs, Anna Karydas, Adam Staffaroni, Elisa De Paula Franca Resende, Eun‐Joo Kim, Eliana Marisa Ramos, Kevin J. Wojta, Shirley Pang, Lorenzo Pasquini, Salvatore Spina, Joel Kramer, Ji‐Hye Hwang, Isabel E. Allen, William W. Seeley, Bruce L. Miller, and Lea T. Grinberg: reports no conflict of interest related to this manuscript.
AUTHOR CONTRIBUTIONS
Jorge J. Llibre‐Guerra and Lea Tenenholz Grinberg had full access to all the data in the study and take responsibility for the integrity of the data and the data analysis accuracy. Study concept and design: Lea T. Grinberg, William W. Seely, Jorge J. Libbre Guerra. Acquisition, analysis, or interpretation of data: Jorge J. Llibre‐Guerra, Suzee E. Lee, Alexander J. Ehrenberg, Anna Karydas, Eliana Marisa Ramos, Adam Staffaroni, Ji‐Hye Hwang, Elisa De Paula Franca Resende, Shirley Pang, Eun J. Kim, Kevin Wojta, Salvatore Spina, Joel Kramer, Isabel E. Allen, Gabor Kovacs, Lea T. Grinberg. Drafting of the manuscript: Jorge J. Llibre‐Guerra, Isabel E. Allen, Lea T. Grinberg Critical revision of the manuscript for important intellectual content: All authors. Statistical analysis: Jorge J. Llibre‐Guerra, Claudia K Suemoto, Isabel E. Allen, Alex J. Ehrenberg, Giovanni Coppola, Lorenzo Pasquini. Obtained funding: Lea T. Grinberg, William W. Seeley, Bruce L. Miller. Project administration: Jorge J. Llibre‐Guerra, Lea T. Grinberg. Study supervision: Lea T. Grinberg.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Participants or their surrogates provided written consent of participating in research at the UCSF/MAC and donated their brains to the UCSF/NDBB between 2007 and 2016.
Supporting information
ACKNOWLEDGMENTS
We thank the families of the brain donors, the physicians, and the Autopsy service staff for their unconditional support. We thank Giovanni Coppola from UCLA for technical support on the genetic analysis. We thank Peter Davies, Rafez Kayed, and Manuella Neumann for providing primary antibodies.
Funding information
Grants that supported this study: NIH‐NIA K24AG053435 (Neuropathological changes underlying clinical heterogeneity in Alzheimer disease), Global Brain Health Institute (GBHI), NIH‐NIA (P01AG019724, Frontotemporal Dementia: Genes, Images, and Emotions, Bruce L.Miller), NIH‐NIA (P50AG023501, New Approaches to Dementia Heterogeneity, Bruce L Miller). Suzee E. Lee is supported by NIH‐NIA‐K23AG039414 (Detecting presymptomatic disease in inherited frontotemporal dementia, Suzee Eurie Lee) NIH R01 AG058233. Salvatore Spina is supported by NIH K08AG052648 (Neuropathological basis of brain network dysfunction, Salvatore Spina). Adam Staffaroni is supported by NIA‐NIH_K23AG061253 and Larry L. Hillblom Foundation: 2018‐A‐025‐FEL. Gabor G. Kovacs is supported by the Rossy Foundation and the Edmond J. Safra Foundation, and the Bishop Karl Golser Award. The funding sources had no role in the study's design and conduct; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
DATA AVAILABILITY STATEMENT
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
References
- 1. Ferrari R, Hernandez DG, Nalls MA, Rohrer JD, Ramasamy A, Kwok JB, et al. Frontotemporal dementia and its subtypes: a genome‐wide association study. Lancet Neurol. 2014;13(7):686–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–54. [DOI] [PubMed] [Google Scholar]
- 3. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shinagawa S, Naasan G, Karydas AM, Coppola G, Pribadi M, Seeley WW, et al. Clinicopathological study of patients with C9ORF72‐associated frontotemporal dementia presenting with delusions. J Geriatr Psychiatry Neurol. 2015;28(2):99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cruchaga C, Graff C, Chiang HH, Wang J, Hinrichs AL, Spiegel N, et al. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol. 2011;68(5):581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC, et al. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 2014;127(3):407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pottier C, Ravenscroft TA, Sanchez‐Contreras M, Rademakers R. Genetics of FTLD: overview and what else we can expect from genetic studies. J Neurochem. 2016;138(Suppl 1):32–53. [DOI] [PubMed] [Google Scholar]
- 8. Van Deerlin VM, Sleiman PM, Martinez‐Lage M, Chen‐Plotkin A, Wang LS, Graff‐Radford NR, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP‐43 inclusions. Nat Genet. 2010;42(3):234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van der Zee J, Van Broeckhoven C. TMEM106B a novel risk factor for frontotemporal lobar degeneration. J Mol Neurosci. 2011;45(3):516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Zee J, Van Langenhove T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R, et al. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain. 2011;134(Pt 3):808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus‐Hernandez M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76(5):467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hernandez I, Rosende‐Roca M, Alegret M, Mauleon A, Espinosa A, Vargas L, et al. Association of TMEM106B rs1990622 marker and frontotemporal dementia: evidence for a recessive effect and meta‐analysis. J Alzheimers Dis. 2015;43(1):325–34. [DOI] [PubMed] [Google Scholar]
- 13. Yu L, De Jager PL, Yang J, Trojanowski JQ, Bennett DA, Schneider JA. The TMEM106B locus and TDP‐43 pathology in older persons without FTLD. Neurology. 2015;84(9):927–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nelson PT, Smith CD, Abner EL, Wilfred BJ, Wang WX, Neltner JH, et al. Hippocampal sclerosis of aging, a prevalent and high‐morbidity brain disease. Acta Neuropathol. 2013;126(2):161–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nho K, Saykin AJ, Alzheimer's Disease Neuroimaging I, Nelson PT. Hippocampal sclerosis of aging, a common Alzheimer's disease ‘Mimic’: risk genotypes are associated with brain atrophy outside the temporal lobe. J Alzheimers Dis. 2016;52(1):373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jun MH, Han JH, Lee YK, Jang DJ, Kaang BK, Lee JA. TMEM106B, a frontotemporal lobar dementia (FTLD) modifier, associates with FTD‐3‐linked CHMP2B, a complex of ESCRT‐III. Mol Brain. 2015;8:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nicholson AM, Finch NA, Wojtas A, Baker MC, Perkerson RB 3rd, Castanedes‐Casey M, et al. TMEM106B p. T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem. 2013;126(6):781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Klein ZA, Takahashi H, Ma M, Stagi M, Zhou M, Lam TT, et al. Loss of TMEM106B ameliorates lysosomal and frontotemporal dementia‐related phenotypes in progranulin‐deficient mice. Neuron. 2017;95(2):281–96.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kao AW, McKay A, Singh PP, Brunet A, Huang EJ. Progranulin, lysosomal regulation and neurodegenerative disease. Nat Rev Neurosci. 2017;18(6):325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paushter DH, Du H, Feng T, Hu F. The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. 2018;136(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rodriguez RD, Suemoto CK, Molina M, Nascimento CF, Leite RE, de Lucena Ferretti‐Rebustini RE, et al. Argyrophilic grain disease: demographics, clinical, and neuropathological features from a large autopsy study. J Neuropathol Exp Neurol. 2016;75(7):628–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H, et al. Aging‐related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 2016;131(1):87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kovacs GG, Molnar K, Laszlo L, Strobel T, Botond G, Honigschnabl S, et al. A peculiar constellation of tau pathology defines a subset of dementia in the elderly. Acta Neuropathol. 2011;122(2):205–22. [DOI] [PubMed] [Google Scholar]
- 25. Kramer JH, Jurik J, Sha SJ, Rankin KP, Rosen HJ, Johnson JK, et al. Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol. 2003;16(4):211–8. [DOI] [PubMed] [Google Scholar]
- 26. Perry DC, Brown JA, Possin KL, Datta S, Trujillo A, Radke A, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. 2017;140(12):3329–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ranasinghe KG, Rankin KP, Pressman PS, Perry DC, Lobach IV, Seeley WW, et al. Distinct subtypes of behavioral variant frontotemporal dementia based on patterns of network degeneration. JAMA Neurol. 2016;73(9):1078–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron D . El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293–9. [DOI] [PubMed] [Google Scholar]
- 29. Ramos EM, Dokuru DR, Van Berlo V, Wojta K, Wang Q, Huang AY, et al. Genetic screen in a large series of patients with primary progressive aphasia. Alzheimers Dement. 2019;15(4):553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960–9. [DOI] [PubMed] [Google Scholar]
- 31. Cairns NJ, Bigio EH, Mackenzie IRA, Neumann M, Lee VMY, Hatanpaa KJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114(1):5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, et al. A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol. 2011;122(1):111–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies ‐ Third report of the DLB consortium. Neurology. 2005;65(12):1863–72. [DOI] [PubMed] [Google Scholar]
- 36. McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016;131(1):75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grinberg LT, Wang X, Wang C, Sohn PD, Theofilas P, Sidhu M, et al. Argyrophilic grain disease differs from other tauopathies by lacking tau acetylation. Acta Neuropathol. 2013;125(4):581–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Harding SR, Bocchetta M, Gordon E, Cash DM, Cardoso MJ, Druyeh R, et al. The TMEM106B risk allele is associated with lower cortical volumes in a clinically diagnosed frontotemporal dementia cohort. J Neurol Neurosurg Psychiatry. 2017;88(11):997–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Delis DC, Kaplan E, Kramer JH. Delis‐Kaplan Executive Function System (D‐KEFS): examiner's manual. San Antonio, TX: The Psychological Corporation; 2001. [Google Scholar]
- 40. Stroop JR. Studies of interference in serial verbal reactions. Journal of Experimental Psychology. 1935;18:643–62. [Google Scholar]
- 41. Dunn L, Dunn D. Peabody picture vocabulary test. 4th ed. Minneapolis, MN: Pearson Assessments; 2007. [Google Scholar]
- 42. Staffaroni AM, Brown JA, Casaletto KB, Elahi FM, Deng J, Neuhaus J, et al. The longitudinal trajectory of default mode network connectivity in healthy older adults varies as a function of age and is associated with changes in episodic memory and processing speed. J Neurosci. 2018;38(11):2809–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Josephs KA, Whitwell JL, Knopman DS, Hu WT, Stroh DA, Baker M, et al. Abnormal TDP‐43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology. 2008;70(19 Pt 2):1850–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moreno F, Rabinovici GD, Karydas A, Miller Z, Hsu SC, Legati A, et al. A novel mutation P112H in the TARDBP gene associated with frontotemporal lobar degeneration without motor neuron disease and abundant neuritic amyloid plaques. Acta Neuropathol Commun. 2015;3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brady OA, Zheng Y, Murphy K, Huang M, Hu F. The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet. 2013;22(4):685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Busch JI, Unger TL, Jain N, Tyler Skrinak R, Charan RA, Chen‐Plotkin AS. Increased expression of the frontotemporal dementia risk factor TMEM106B causes C9orf72‐dependent alterations in lysosomes. Hum Mol Genet. 2016;25(13):2681–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rhinn H, Abeliovich A. Differential aging analysis in human cerebral cortex identifies variants in TMEM106B and GRN that regulate aging phenotypes. Cell Syst. 2017;4(4):404–15.e5. [DOI] [PubMed] [Google Scholar]
- 48. Lu RC, Wang H, Tan MS, Yu JT, Tan L. TMEM106B and APOE polymorphisms interact to confer risk for late‐onset Alzheimer's disease in Han Chinese. J Neural Transm (Vienna). 2014;121(3):283–7. [DOI] [PubMed] [Google Scholar]
- 49. Nelson PT, Wang WX, Partch AB, Monsell SE, Valladares O, Ellingson SR, et al. Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology. J Neuropathol Exp Neurol. 2015;74(1):75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Satoh J, Kino Y, Kawana N, Yamamoto Y, Ishida T, Saito Y, et al. TMEM106B expression is reduced in Alzheimer's disease brains. Alzheimers Res Ther. 2014;6(2):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cherry JD, Mez J, Crary JF, Tripodis Y, Alvarez VE, Mahar I, et al. Variation in TMEM106B in chronic traumatic encephalopathy. Acta Neuropathol Commun. 2018;6(1):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. 2015;129(4):469–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kovacs GG, Kwong LK, Grossman M, Irwin DJ, Lee EB, Robinson JL, et al. Tauopathy with hippocampal 4‐repeat tau immunoreactive spherical inclusions: a report of three cases. Brain Pathol. 2018;28(2):274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Debaisieux S, Schiavo G. TiME for TMEM106B. EMBO J. 2014;33(5):405–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay‐Falcone D, et al. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 2011;121(3):373–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ito Y, Hartley T, Baird S, Venkateswaran S, Simons C, Wolf NI, et al. Lysosomal dysfunction in TMEM106B hypomyelinating leukodystrophy. Neurol Genet. 2018;4(6):e288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Luningschror P, Werner G, Stroobants S, Kakuta S, Dombert B, Sinske D, et al. The FTLD risk factor TMEM106B regulates the transport of lysosomes at the axon initial segment of motoneurons. Cell Rep. 2020;30(10):3506–19.e6. [DOI] [PubMed] [Google Scholar]
- 58. Li Z, Farias FHG, Dube U, Del‐Aguila JL, Mihindukulasuriya KA, Fernandez MV, et al. The TMEM106B FTLD‐protective variant, rs1990621, is also associated with increased neuronal proportion. Acta Neuropathol. 2020;139(1):45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bieniek KF, Murray ME, Rutherford NJ, Castanedes‐Casey M, DeJesus‐Hernandez M, Liesinger AM, et al. Tau pathology in frontotemporal lobar degeneration with C9ORF72 hexanucleotide repeat expansion. Acta Neuropathol. 2013;125(2):289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nolan A, Resende EDPF, Petersen C, Neylan K, Spina S, Huang E, et al. Astrocytic tau deposition is frequent in typical and atypical alzheimer disease presentations. J Neuropathol Exp Neurol. 2019;78(12):1112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Koga S, Kouri N, Walton RL, Ebbert MTW, Josephs KA, Litvan I, et al. Corticobasal degeneration with TDP‐43 pathology presenting with progressive supranuclear palsy syndrome: a distinct clinicopathologic subtype. Acta Neuropathol. 2018;136: 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.