

Abstract
Myeloid-derived suppressor cells (MDSCs) are immune suppressive cells that massively accumulate under pathological conditions to suppress T cell immune response. Dysregulated cell death contributes to MDSC accumulation, but the molecular mechanism underlying this cell death dysregulation is not fully understood. We report here that neutral ceramidase (N-acylsphingosine amidohydrolase, ASAH2) is highly expressed in tumor-infiltrating MDSCs in colon carcinoma and acts as a MDSC survival factor. To target ASAH2, we performed molecular docking based on human ASAH2 protein structure. Enzymatic inhibition analysis of identified hits determined NC06 as an ASAH2 inhibitor. Chemical and NMR analysis determined NC06 as 7-chloro-2-(3-chloroanilino)pyrano[3,4-e][1,3]oxazine-4,5-dione. NC06 inhibits ceramidase activity with IC50 of 10.16–25.91 μM for human ASAH2 and 18.6–30.2 μM for mouse Asah2 proteins. NC06 induces MDSC death in a dose-dependent manner and inhibition of ferroptosis decreased NC06-induced MDSC death. NC06 increases glutathione synthesis and decrease lipid ROS to suppress ferroptosis in MDSCs. Gene expression profiling identified p53 pathway as the Asah2 target in MDSCs. Inhibition of Asah2 increased p53 protein stability to up-regulate Hmox1 expression to suppress lipid ROS production to suppress ferroptosis in MDSCs. NC06 therapy increases MDSC death and reduces MDSC accumulation in tumor-bearing mice, resulting in increased activation of tumor-infiltrating CTLs and suppression of tumor growth in vivo. Our data indicate that ASAH2 protects MDSCs from ferroptosis through destabilizing p53 protein to suppress the p53 pathway in MDSCs in the tumor microenvironment. Targeting ASAH2 with NC06 to induce MDSC ferroptosis is potentially an effective therapy to suppress MDSC accumulation in cancer immunotherapy.
Keywords: Asah2, MDSCs, Ferroptosis, p53, Hmox1, Ceramide, Glutathione, Lipid ROS
Introduction
Tumor-promoting inflammation and avoiding immune destruction are enabling characteristic and emerging hallmark of cancer, respectively (1). A key component of the inflammation process is generation and activation of myeloid cells, such as neutrophils, monocytes, and macrophages (2, 3), which are the essential functional mediators of the inflammation process. Persistence of such inflammation drives tumorigenesis, particularly colon tumorigenesis in humans, for which a causal relationship to chronic inflammation has been well-established (4). The chronic inflammatory tumor microenvironment in turn deregulates myeloid cell lineage differentiation and maturation, resulting in an accumulation of immature immunosuppressive myeloid cells, primarily the myeloid-derived suppressive cells (MDSCs) which not only directly enhance tumor growth and progression through secreting growth-promoting mediators (5, 6) but also suppress T and NK cell functions, thereby assisting tumors in avoiding destruction by the immune system (7, 8). Due to their potent immune suppressive activities and abundance in the tumor microenvironment, MDSCs are a major barrier to immunotherapies and are key targets in human cancer immunotherapy (9).
Myeloid cells are also a major source of programmed death ligand 1 (PD-L1), and therefore are major targets of PD-1 blockade immunotherapy (10, 11). However, MDSCs are heterogeneous in PD-L1 expression. Subsets of tumor-infiltrating MDSCs express PD-L1 whereas other subsets of tumor-induced MDSCs do not express PD-L1 (12). Therefore, current immune checkpoint blockade immunotherapies, such as anti-PD-1 immunotherapy, may only partially block MDSC-mediated immune suppression. This is consistent with the clinical observation that MDSC accumulation is correlated with resistance to nivolumab treatment in human cancer patients (13, 14).
To target MDSCs, various therapeutic approaches are actively being tested in preclinical and clinical studies. These include depletion or inhibition of their suppressive activity, differentiation, and accumulation (9). However, none of these strategies are specific to MDSCs and some of them, such as chemotherapy, have side effects. The fact that MDSCs massively accumulate in tumor-bearing host suggests that MDSCs could have dysregulated death pathways (15, 16). Several cellular death pathways have been identified to play important roles in MDSC survival and accumulation (17–22). More importantly, targeting the death receptor TNF-related apoptosis-inducing ligand receptor 2 (TRAIL-R2) selectively with an agonistic antibody eliminated MDSCs in tumor-bearing mice and human cancer patients (23), demonstrating the significant clinical promise of targeting MDSC-specific death pathways for MDSC suppression. Here, we identified ferroptosis, an iron-dependent accumulation of lipid reactive oxygen species (ROS) and resultant regulated cell death, as a MDSC death pathway, and determined that MDSCs upregulate neutral ceramidase (N-acylsphingosine amidohydrolase, Asah2) which catalyzes sphingolipid metabolism, to resist ferroptosis. Importantly, we developed an Asah2-selective small molecule inhibitor NC06 that activates the p53-Hmox1 pathway to induce MDSC ferroptosis to suppress MDSC accumulation to activate T cell tumor infiltration to suppress tumor growth in vivo.
Materials and Methods
Mice, human tumor specimens and cells.
BALB/c and C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Six to twenty-week old male and female mice were used. All mice were housed and maintained in accordance with an approved protocol by Augusta University Institutional Animal Use and Care Committees (#2008–0162). J774M cells were generated and characterized as previously described (12). Mouse colon carcinoma CT26 cell line were obtained from American Type Culture Collection (ATCC, Manassas, VA) in 2013 and stored in liquid nitrogen as aliquots. Mouse mesothelioma AB1 cell line were obtained from Sigma-Aldrich (St Luis, MO) in 2019 and stored in liquid nitrogen as aliquots. Cells were used within 30 passages. ATCC characterized these cells by morphology, immunology, DNA fingerprint, and cytogenetics. Murine colon carcinoma MC38 cells were provided by Dr. Jeffrey Schlom (National Cancer Institute, Bethesda, MD) and characterized as previously described (24). Cells were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA) plus 10% fetal bovine serum (FBS)(Cat# SH30396.03, GE Healthcare, Chicago, IL). Cells are tested for mycoplasma every two months, and all cells used in this study were mycoplasma-negative. Human colorectal carcinoma progression tissue microarray specimens were provided by the Cooperated Human Tissue Network (University of Virginia, Charlottesville, VA). Healthy donor blood specimens were collected from consented donors in Augusta Shepeard Community Blood Center. Blood specimens of colon cancer patients were collected from de-identified colon cancer patients in Georgia Cancer Center. Blood samples were collected from patients without chemotherapy for at least 7 days. All studies involved human specimens were approved by Augusta University Institute Review Board (Approval ID: 1461188-1).
Mouse tumor models.
To generate spontaneous colon carcinoma tumor, C57BL/6 mice were treated with three cycles of Azoxymethane (AOM)/Dextran sodium sulfate (DSS) as previously described (6). To establish subcutaneous tumor models, CT26 cells (2×105 cells/mouse) were injected into the right flanks of BALB/c mice. For experimental lung metastasis models, colon carcinoma CT26, mesothelioma AB1 (2×105 cells/mouse) and mammary carcinoma 4T1 (2×104 cells/mouse) were injected into BALB/c mice. Tumor-bearing mice were treated with vehicle PEG300 (Sigma-Aldrich) and NC06 (dissolved in PEG300), respectively, every two days for 3–5 times by Intraperitoneal injection. Tumor size was measured in two dimensions with a digital micrometer caliper at the indicated time points. Tumor size was calculated by the formula: (tumor length × tumor width2)/2. To quantify lung tumor nodules, lungs were inflated with 15% India ink solution and fixed in Fekete’s Solution (58% Ethanol, 8.7% formaldehyde, and 4.3% glacial acid) as previously described (25).
Human colorectal carcinoma genomic datasets.
ASAH2 expression Datasets in human primary tumor (n=380) and normal colon tissues (n=51) were extracted from TCGA Colon and Rectal Cancer (COADREAD) ploy A+ IlluminaHiSeq pancan normalized RNA seq datasets using UCSC Xena Cancer Genomics Browser (University of California, Santa Cruz, CA). The datasets were analyzed by GraphPad Program (GraphPad Software, San Diego, CA).
Molecular docking.
Structures in the NCI Diversity Set V were supplied in 2D form. Three-dimensional representations were generated with computer program obprep from the Openbabel suite (26). Docking of these structures was done with computer program Autodock Vina 2. 0 (27)(The Scripps Research Institute, San Diego, CA) to predict binding modes of a drug candidate against the protein target using a box centered on the ligand binding site. The figure-of-merit in the scoring output of Autodock Vina is expressed as a binding affinity in kcal/mol, with a standard error of 2.85 kcal/mol. Human Asah2 protein structure was used for molecular docking (28).
Chemical characterization of NC06.
High Resolution Mass Spectrometry was performed by TSQ MS system (Thermo fisher Scientific). For fourier-transform infrared spectrum, compound was directly measured using the attenuated total reflection method on a Nicolet™ iS™ 5 FTIR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). The FTIR spectrum were measured between 4000–400 cm−1 at room temperature. For nuclear magnetic resonance, the 1H and 13C experiments were recorded using Bruker-500 NMR spectrometer (Bruker Co., Leipzig, Germany) equipped with a 5 mm BBO probe. NMR sample was prepared by dissolving 5 mg of nC06 in 500 µl DMSO-d6. The 1H and 13C chemical shift values were reported on the δ scale in ppm relative to DMSO-d6 (2.50 ppm for 1H NMR and 39.52 ppm for 13C NMR respectively).
Ceramidase activity assays:
Ceramidase activity assay was based on the previously described method (29). Ceramide template RBM14C16 (0.1 mM, Avantilipids, cat#860856) were incubated in Asah2 buffer (50 mM Hepes, 150 mM NaCl, 1% sodium cholate, pH 7.4) or acid ceramidase buffer (25 mM sodium acetate buffer, pH 4.5) with recombinant mouse Asah2 protein, human ASAH2 protein, and human ASAH1 protein (Abnova, Cat # H00000427-P01) as described above at 37oC for 3h. Reactions were stopped by adding 25 μl/well of methanol and then 100 μl/well NaIO4 solution (2.5 mg/ml in 100 mM Glycine-NaOH, PH 10.6) and incubated at 37°C for 1h in the dark. Fluorescence was measured at excitation and emission wavelength of 355 and 460, respectively.
Gene expression analysis.
siRNA was obtained from Santa Cruz Biotech. Total RNA was isolated from cells using the GeneJET RNA isolation kit according to the manufacturer’s instructions (Fisher Scientific). RNA concentrations were measured using a NanoDrop spectrophotometer (Thermo Scientific, Mansfield, TX). RNA purity was determined based on OD260/OD280 ratio and only samples with ratio >1.90 were used. cDNA was synthesized from total RNA using the ThermoScript RT-PCR kit (Cat#11146–016, Invitrogen) with the substitution of ThermoScript reverse transcriptase by M-MLV reverse transcriptase (Cat #M170B, Promega, Madison, WI). PCR was performed in triplicates in the StepOne Plus Real-Time PCR System (Applied Biosystems). Gene expression was determined as ΔΔCT with β-actin as internal normalization control.
DNA microarray.
Cells were treated with NC06 at 10 μM for 24 hour for total RNA preparation. The mouse gene 2.0 ST array (Affymetrix, Santa Clara, CA) was used for the gene expression profiling. Total RNA samples were processed using the Ambion WT expression kit (Life Technologies, Calsbad, CA) and a GeneChip WT terminal labeling kit (Affymetrix). The synthesized sense strand cDNAs were fragmented and biotin-labeled using a GeneChip WT terminal labeling kit. The labeled cDNAs were hybridized onto the arrays using Affymetrix GeneChip fluidics station 450 systems. The expression data were imported into Partek GS version 6.6 using a standard import tool with GeneChip robust multiarray analysis normalization. The differential expressions were calculated using ANOVA of Partek package. The entire dataset is deposited to NCBI GEO database (Accession #: GSE163399, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE163399)
BM-MDSC differentiation.
Bone marrow cells were isolated from BALB/c mice and cultured in RMPI medium with 10% FBS and recombinant mouse GM-CSF (40 ng/ml, Biolegend) for 3–6 days.
Ceramide analysis.
Endogenous ceramide was measured by Virginia Commonwealth University Lipidomics/Metabolomics Core using high-performance liquid chromatography-mass spectrometry approach (LC-MS/MS). Ceramide level was normalized to the total phospholipid contents.
T cell proliferation assay.
Single cells were prepared from spleen of tumor-free BALB/c mice and used to purify CD3+ T cells with the MojoSort mouse CD3 T cell isolation kit (Biolegend) according to the manufacturer’s instructions. For T cell proliferation assay, 96-well culture plate was coated with anti-mouse CD3 (8 μg/ml) and anti-mouse CD28 MAbs (10 μg/ml) overnight. The purified T cells were labeled with CFSE (Life Technologies) and then seeded in the plate at a density. J774M cells were added to the T cell culture at various rations and cultured for 72h. Cells were analyzed by flow cytometry.
Flow cytometry analysis:
J774M and BM-MDSCs were treated with NC06, Z-DEVD, Necropstatin-1 (Nec-1), Ferrostatin-1 (Fer-1) for various time. The cells cells were then stained with propidium iodide (PI) and Annexin V in Annexin V-binding buffer [10 mM Hepes (pH 7.4), 140 mM NaCl, and 2.5 mM CaCl2) and analyzed in a FACS Caliber (BD Biosciences, San Diego, CA). To analyze cells from tumors, and spleens of mice, single cells were prepared from spleens and tumors as previously described (12). Cells were stained with DAPI (Biolegend) and monoclonal antibodies that are specific for CD45.2, CD8, PD-1, CD11b, and Gr1 (Biolegend). Mouse bone marrow and spleen cells were stained with CD11b- and Gr1-specific mAb. To analyze human MDSCs, peripheral blood mononuclear cells were prepared from peripheral blood samples of healthy donors and colon cancer patients, centrifuged at 3,000 RPM for 10 min to collect buffy coat. Red cells were lysed. The cells were stained with DAPI, CD11b-, CD33-, HLA-DR5-specific mAbs (Biolegend), followed by intracellular staining using Alexa Fluor 647-conjugated anti-human neutral ceramidase antibody (Santa Cruz) and the Cytofix/Cytoperm Plus intracellular staining kit (BD Biosciences). The stained cells were analyzed in a LSR Fortessa cytometer (BD Biosciences). Data were analyzed with FlowJo V10 program (Flow Jo, Ashland, OR).
Immunohistochemistry.
Human colon carcinoma specimens were stained with anti-Asah2 (Santa Cruz). The AOM-DSS-induced mouse colon carcinoma tissues were stained with anti-Asah2. The tissues were then probed with mouse specific IHC polymer detection kit (Cat# ab209101, Abcam) according to the manufacturer’s instructions. The stained tissues were counterstained with hematoxylin (Richard-Allan Scientific, Kalamazoo, MI).
Statistical analysis.
All statistical analysis was performed using SAS 9.4 (SAS Institute Inc., Cary, NC) and statistical significance was assessed using an alpha level of 0.05. A repeated-measures mixed model with fixed effects of treatment groups and day, as well as all two- and three-factor interactions between these effects, was used to examine changes in tumor size over time. A one- or two-factor ANOVA with fixed effects of F5446 and anti-PD-1 groups and the two-factor
Results
Asah2 expression and function in tumor-infiltrating MDSCs.
ASAH2 expression datasets were extracted from the TCGA database. Comparison of Datasets from human colorectal carcinoma to datasets from normal colon tissues revealed that ASAH2 mRNA levels are significantly higher in tumor tissues than the normal colon (Fig. 1A). Immunohistochemical analysis of human and the AOM-DSS-induced mouse colon carcinoma tissues validated that Asah2 protein level is highly expressed in tumor cells (Fig. 1B). However, ASAH2 protein is also detected in tumor-infiltrating immune cells in both human and mouse colon carcinoma (Fig. 1B). MDSCs are a major subset of immune cells in the tumor microenvironment. To determine ASAH2 expression level in MDSCs, peripheral blood mononuclear cells were prepared from healthy donors and colon cancer patients. CD11b+CD33+HLA-DR5− MDSCs were analyzed for ASAH2 protein level by intracellular staining (Fig. 1C). Quantification of ASAH2 protein level in these MDSCs indicates that ASAH2 protein level is significantly higher in MDSCs from colon cancer patients that cells with the same phenotype from healthy donors (Fig. 1D).
Figure 1. Asah2 expression is upregulated in tumor-infiltrating MDSCs.
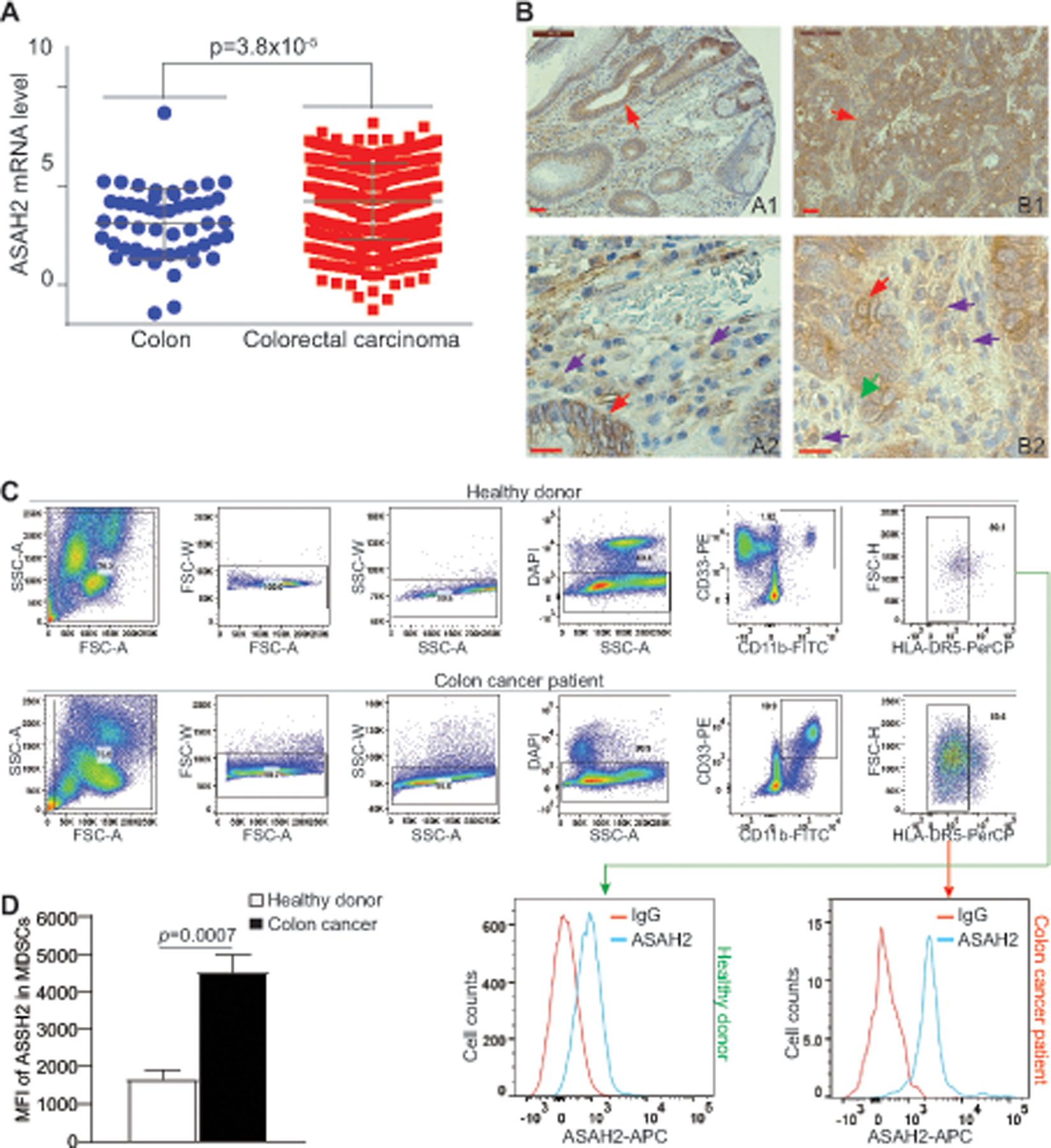
A. Data sets of ASAH2 relative mRNA level in human colorectal carcinoma specimens (n=380) and non-neoplastic colon (n=51) were extracted from TCGA database and plotted. The ASAH2 mRNA levels were compared between the colon and tumor specimens using Graphpad Prism. Each dot represents ASAH2 relative mRNA level of one patient. B. Human colon carcinoma (A1) and the AOM-DSS-induced mouse colon tumors (B2) were stained with ASAH2-specific antibody. A2 shows magnified image of A1 and B2 shows magnified image of B1. (Scale bar: brown line at the top left=50 μM). C. Peripheral blood mononuclear cells from healthy donors (n=5) and colon cancer patients (n=7) were stained with CD11b-, CD33-, HLA-DR, and ASAH2-specific mAbs, followed by DAPI staining. The live cells were gated for ASAH2 level in MDSCs (CD11b+CD33+HLA-DR5− cells). Shown are gating strategies. D. Quantification of ASAH2 MFI.
Development of an Asah2-selective small molecule inhibitor: NC06.
One of the functions of Asah2 is to regulate survival of colon tumor cells (30). Our above finding that Asah2 is overexpressed in tumor-infiltrating MDSCs indicates that Asah2 is potentially a molecular target for MDSC suppression. To target Asah2, we capitalized on the recently determined human ASAH2 protein structure (28). We used the molecular docking program Autodock Vina (27) to screen the NCI small molecule virtual library for ASAH2 inhibitors. The top 23 hits were selected and then tested for inhibition of human ASAH2 protein enzymatic activity in vitro. The compound with the highest inhibitory activity was characterized for its chemical structure. A high resolution mass spectrum with low mass error was acquired. The high-resolution mass spectrum of the compound showed that the molecular ion constitutes the base peak ([M+H]+) at m/z=324.9778 (Fig S1A). Using FTIR, the presence of major functional groups in the compound was also confirmed (Fig S1B). A broad absorption peak at 3075 cm−1 represented C-H bond vibration. In addition, a strong and broad characteristic absorption, corresponding to the C=O stretch was exhibited at 1725 cm−1. An absorption peak was also observed at 3279 cm−1, which might be assigned to the stretching vibration of N–H. Figures S1C and S1D show the 1H and 13C spectra of this compound, respectively. Thirteen distinct 13C peaks were observed in the 13C NMR spectrum. Combining the analysis results of FTIR, 1H and 13C NMR, and high resolution mass spectrometry, the chemical structure of the synthetic compound (97-chloro-2-(3-chloroanilino) pyrano[3,4-e][1,3]oxazine-4,5-dione) is confirmed and named as NC06 (Fig. 2A).
Figure 2. Development of ASAH2-selective small molecule inhibitor NC06.
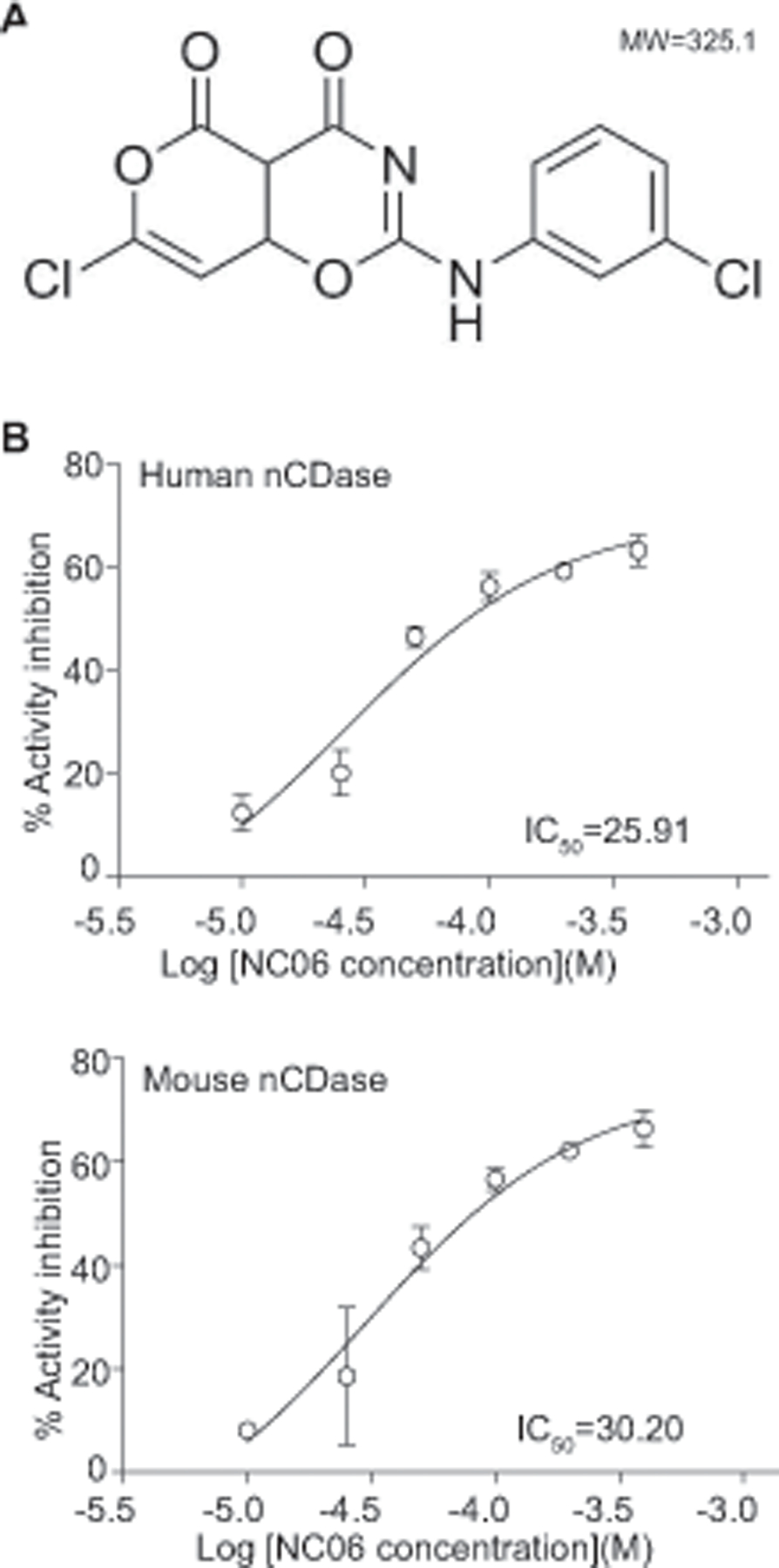
A. NC06 chemical structure. B. RBM14C16 was used as substrate and incubated with recombinant human (top panel) and mouse (bottom panel) Asah2 proteins in the presence of NC06 at the indicated concentrations. The IC50 was determined using GraphPad Prism. The enzyme activity assays were performed twice.
Molecular docking of NC06 revealed that NC06 has a high binding affinity to the catalytic site of human ASAH2 protein. In vitro enzymatic activity inhibition assays with recombinant human proteins determined that NC06 has an IC50 of 25.91 μM for human ASAH2, and 30.20 μM for mouse Asah2 depending on the ceramide substrates (Fig. 2B). Acid ceramidase (ASAH1) is another ceramide that catalyzes ceramide metabolism (31, 32) However, NC06 exhibited no significant inhibitory activity against human acid ceramidase ASAH1 protein (Fig. 3), demonstrating selectivity for ASAH2. As expected, inhibition of Asah2 with NC06 increased ceramide accumulation in MDSCs (Fig. S2).
Figure 3. NC06 does not inhibit Asah1.
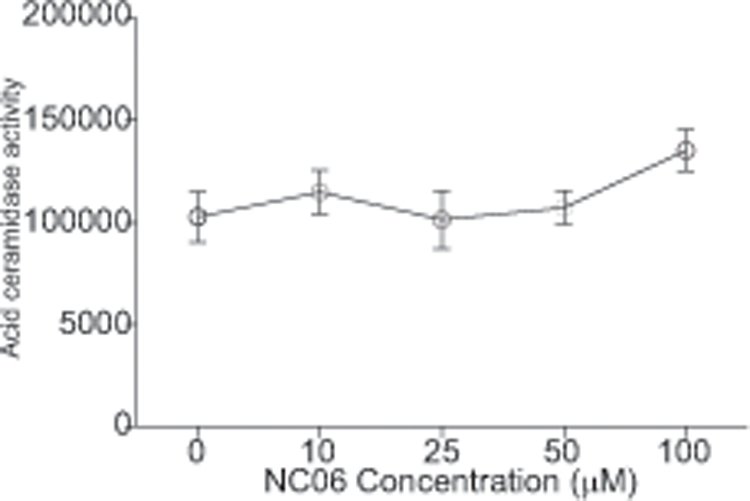
C12-ceramide was used as substrate and incubated with recombinant human Asah1 proteins in the presence of NC06 at the indicated concentrations. IC50 was determined using GraphPad program. The enzymatic activity assays were performed twice.
Inhibition of Asah2 induces MDSC cell death
To determine the function of Asah2 in MDSC survival, we made use of the mouse MDSC-like cell line J774M (12). J774M cells has a CD11b+Gr1+ phenotype (Fig. 4A) and exhibits potent suppressive activity against T cell activation and proliferation (Fig. 4B & C). NC06 treatment induced J774M cell death in a dose-dependent manner (Fig. 4D). A complimentary approach was then used to validate Asah2 function in MDSC survival. MDSCs were induced from BM cells using GM-CSF. The BM-MDSCs were then treated with NC06. Similar to the results observed in J774M cells, NC06 induced BM-MDSC cell death in a dose-dependent manner (Fig. 4E).
Figure 4. Asah2 regulates MDSC cell death.
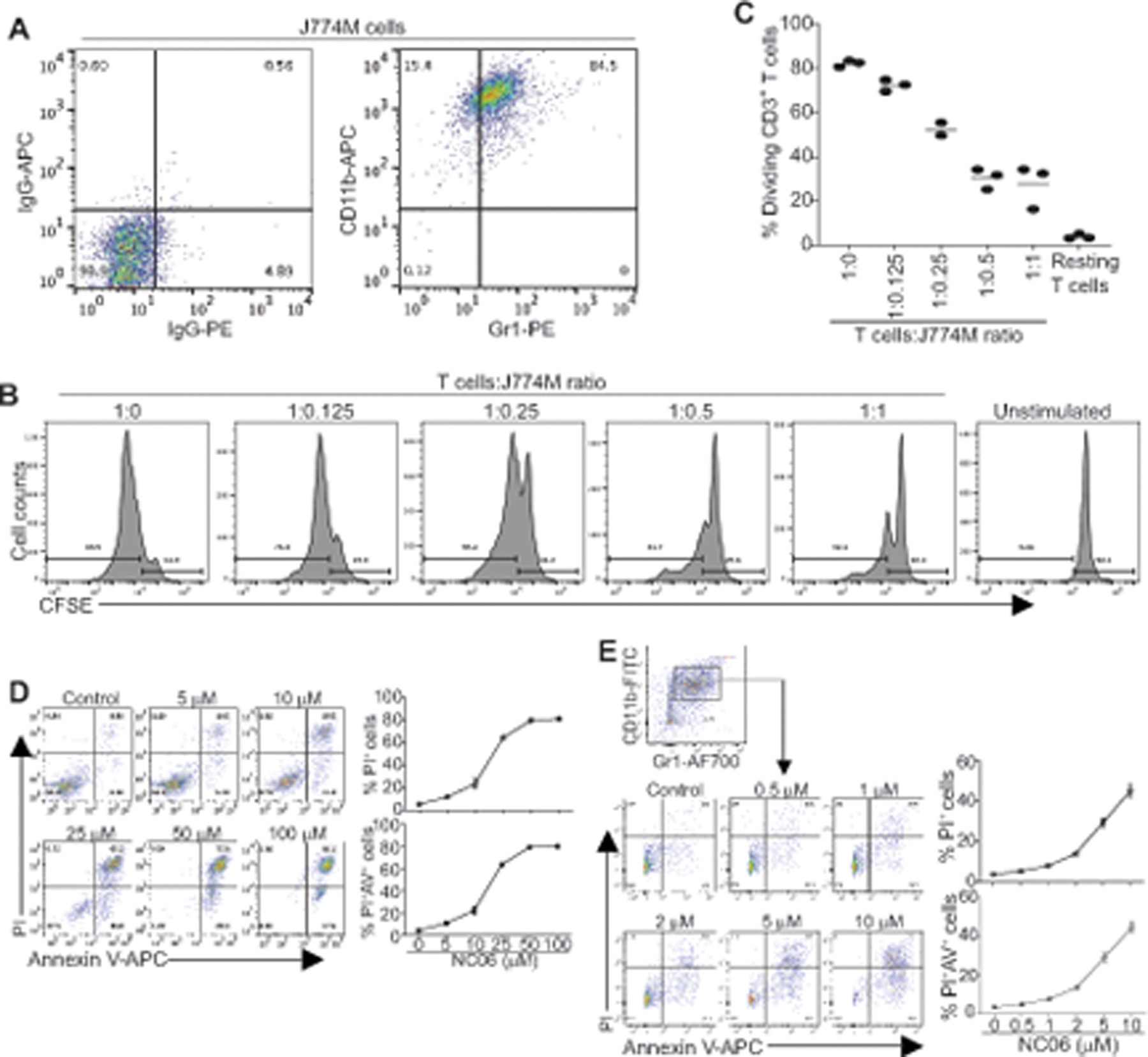
A. Phenotype of MDSC-like mouse cell line J774M. B. Purified CD3+ T cells were labeled with CFSE and seeded in anti-CD3 and anti-CD28-coated 96-well plate (1.5×105 cells/well). J774M cells were added to the cultured T cells at the indicated ratio and cultured for 72h. Cells were collected and stained with CD3-specific mAb. CD3+ cells were gated and analyzed for CFSE intensity. C. Quantification of CD3+ T cell CFSE intensity as shown in B. Vertical line: mean. D. J774M cells were cultured in the presence of NC06 at the indicated concentrations for 24h. Cells were then stained with PI and Annexin V and analyzed by flow cytometry. Both % PI+ and PI+ Annexin V+ cells were calculated. Shown are representative data of one of three experiments. E. BM cells were cultured in the presence of GM-CSF for 5 days to induce MDSCs (BM-MDSCs). BM-MDSCs were then cultured in the presence of NC06 at the indicated concentrations for 24h. Cells were stained with CD11b-, Gr1-specific antibodies and Annexin V, followed by PI staining. The stained cells were analyzed by flow cytometry. CD11+Gr1+ cells were gated and analyzed for PI+ and PI+ Annexin V+ cells.
Asah2 regulates ferroptosis in MDSCs
MDSC turnover is regulated by multiple cellular death pathways. Thus, we next sough to determine which cell death pathway(s) are regulated by Asah2. J774M cells were treated with NC06 in combination with inhibitors for apoptosis, necroptosis, and ferroptosis. Only inhibition of ferroptosis significantly decreased NC06-induced J774M cell death (Fig. 5A). A similar phenomenon was observed in BM-MDSCs (Fig. 5B). Ferroptosis is a recently identified metabolic stress-induced iron- and lipid peroxidation dependent regulated cell death that is caused by overproduction of the toxic lipid-reactive oxygen species (ROS) (33, 34). Glutathione (GSH) is a key substrate for glutathione peroxidase 4-mediated detoxification of lipid hydroperoxide and production of ROS to protect cells from ferroptosis (33, 35). Consistently, treatment of J774M and BM-MDSCs with NC06 significantly decreased glutathione level (Fig. 5C), and increased lipid ROS levels as measured by BODIPY C11 staining (Fig. 5D) in these MDSCs. Strikingly, although glutathione levels were significantly decreased in NC06-treated cells, uptake of cystine, the precursor for glutathione, was also decreased by NC06 treatment (Fig. S3), suggesting that NC06 targets glutathione synthesis downstream of cysteine. Taken together, our data indicate that Asah2 regulates ferroptosis in MDSCs.
Figure 5. Asah2 regulates MDSC ferroptosis.
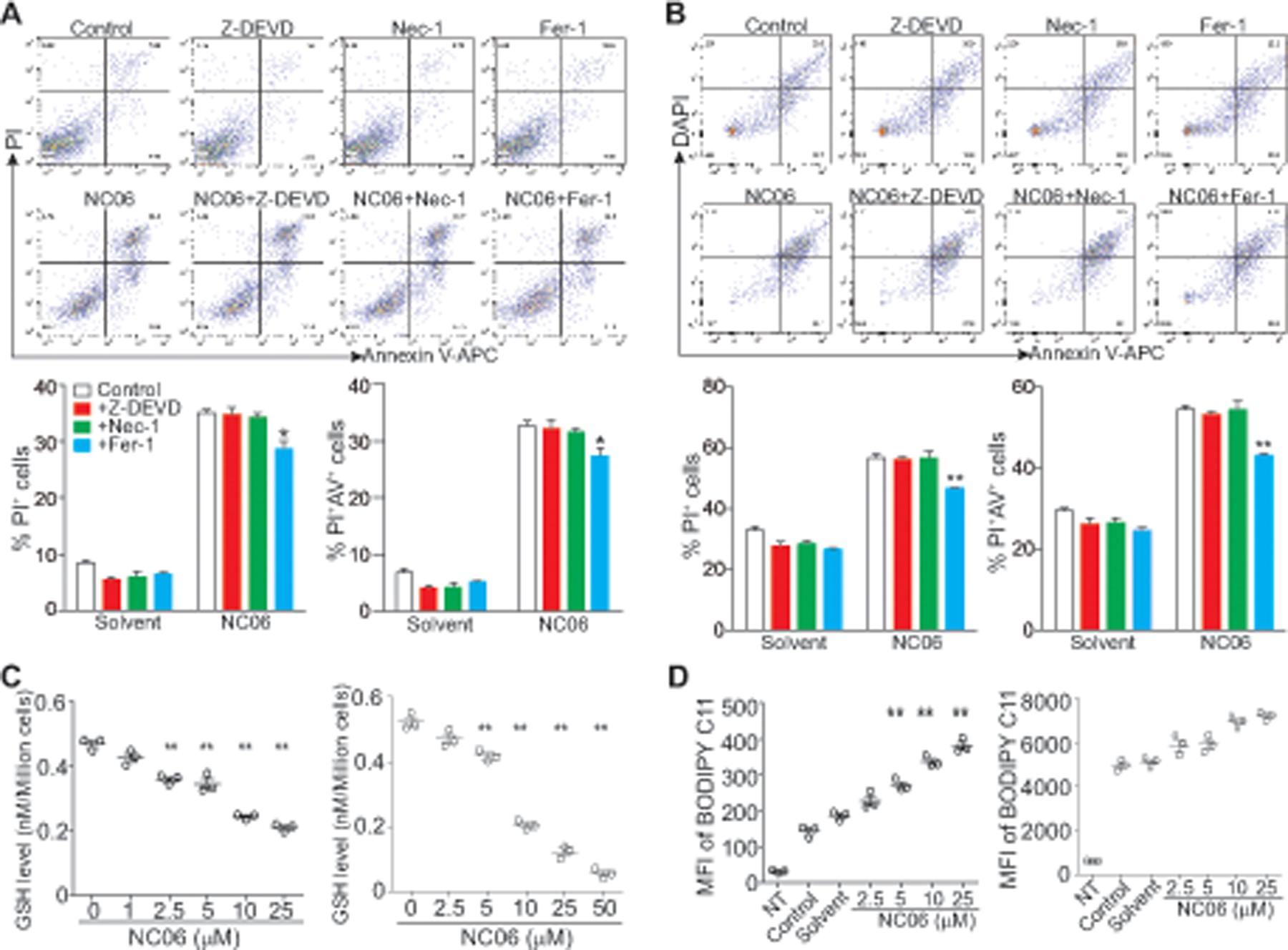
A. J774M cells were cultured in the presence of NC06 (25 μM) plus Z-DEVD (10 μM), Nec-1 (10 μM) and Fer-1 (10 μM) for 24h. Solvent, Z-DEVD, Nec-1, and Fer-1 alone were used as controls. Cells were stained with PI and Annexin V and analyzed by flow cytometry. Cell death was quantified and presented at the bottom panel. Column: mean, bar: SD. * p<0.05. Shown are representative results of one of three experiments. B. BM-MDSCs were treated as in A. Cells were stained with CD11b-, Gr1-specific antibodies and Annexin V, followed by staining with PI and analysis by flow cytometry. CD11b+Gr1+ cells were gated and analyzed for PI+ and Annexin V+ cells. Cell death was quantified and presented at the bottom panel. Column: mean, bar: SD. * p<0.05. C. J774M (left panel) and BM-MDSCs were treated with NC06 at the indicated concentrations for 24h. GSH levels were then determined using the GSH-Glo Glutathione Assay kit. ** p<0.01. D. J774M cells (left panel) and BM-MDSCs (right panel) were treated with NC06 as indicated for 24h. Cells were then incubated with BODIPY C11 for 30 min and analyzed by flow cytometry for quantifying lipid ROS level as measured by BODIPY C11 intensity. Shown is BODIPY C11 MFI.** p<0.01.
NC06 suppresses MDSC accumulation in tumor-bearing mice.
We next determined whether the above observation that inhibiting Asah2, thereby inducing MDSC death, can be extended to MDSC suppression in vivo. Colon carcinoma cells CT26 were transplanted to mice to establish colon tumors. We first validated MDSC accumulation in CT26 tumor-bearing mice. Analysis of spleen cells (Fig. 6A) and bone marrow cells (Fig. 6B) indicates that the level of CD11b+Gr1+ MDSCs is significantly higher in the spleen and bone marrow in the tumor-bearing mice than in the tumor-free mice (Fig. 6A & B). Because there is a correlation between MDSC accumulation level and tumor sizes (36–38), we allowed mice to develop extensive tumor burden before NC06 therapy. In this way, NC06 is not expected to change tumor size significantly but it will enable us to unmask the direct effect of NC06 on MDSCs. NC06 therapy did not significantly decrease the size of the established tumors (Fig. 6C). However, NC06 therapy significantly increased MDSC cell death in the tumor-bearing mice in vivo (Fig. 6D). Consistent with increased MDSC turnover, NC06 therapy significantly decreased MDSC accumulation in tumor-bearing mice (Fig. 6E & F). Decreased MDSC accumulation is correlated with increased CD8+ cytotoxic T lymphocytes (CTL) tumor infiltration (Fig. 6G). The majority of these tumor-infiltrating CTLs are also PD-1+ (Fig. 6H). We therefore conclude that targeting Asah2 is an effective strategy to suppress MDSC accumulation to increase CTL tumor infiltration and activation in tumor-bearing mice.
Figure 6. Inhibition of Asah2 increases MDSC cell turnover and suppresses MDSC accumulation in tumor-bearing mice.
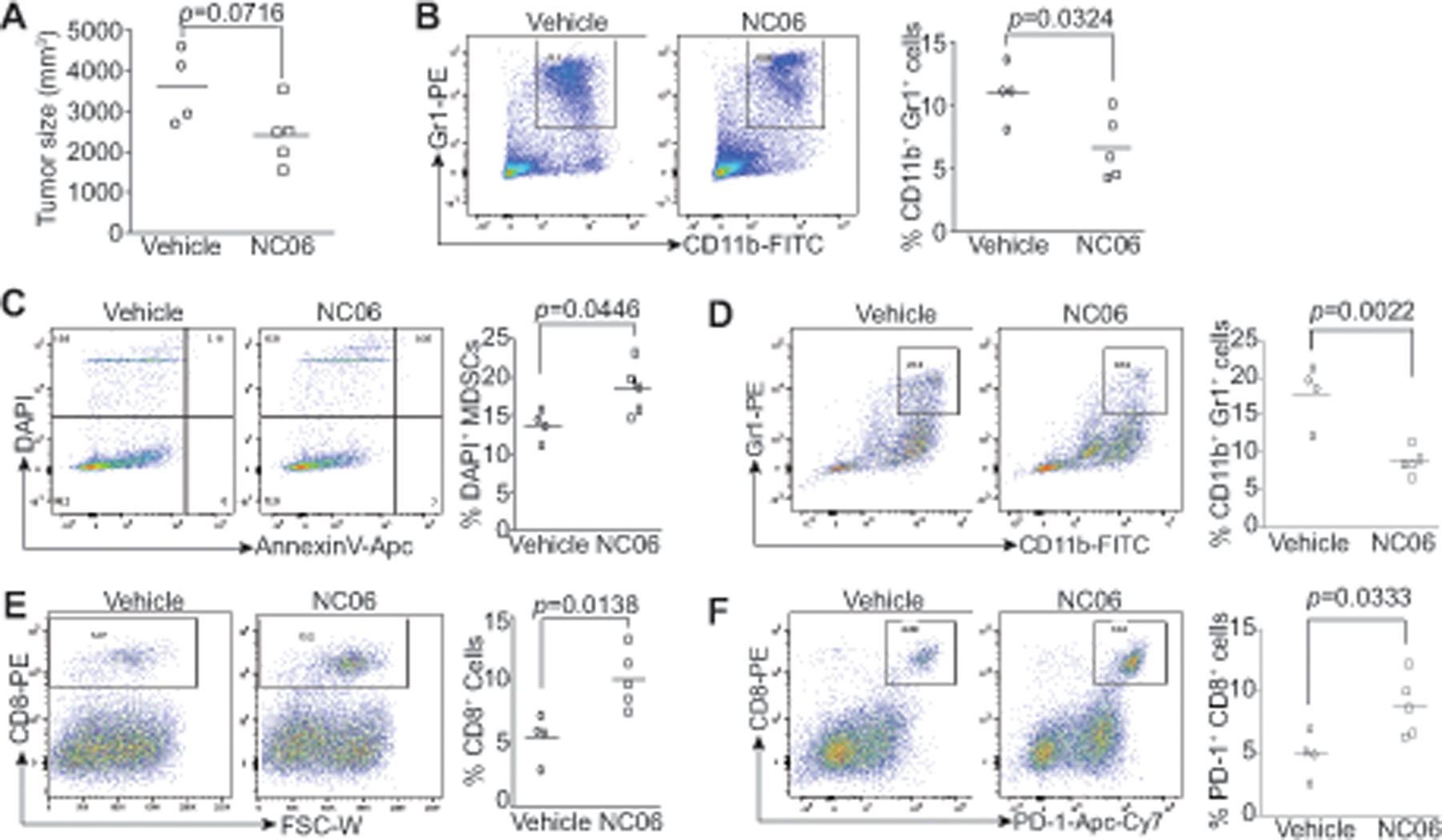
A & B. Sinle cells were prepared from spleen (A) and bone marrow of tumor-free (TF, n=5) and CT26 tumor-bearing mice (TB, n=5). Cells were stained with CD11b- and Gr1-specific mAbs and analyzed by flow cytometry. Shown are representative dot plots (left panels) and quantification of MDSCs (right panel). C. CT26 cells were injected to BALB/c mice s.c. Tumor-bearing mice, after 21 days, were treated with vehicle (n=4) or NC06 (n=5, 20 mg/kg body weight) by i.p. injection for 3 times every 2 days. Tumor size quantification is presented. D. Representative dot plots showing MDSCs in spleens of tumor-bearing mice (left panel) and MDSC quantification (right panel). E. Representative dot plots showing MDSC cell death in spleens and quantification. F. Representative dot plot showing tumor-infiltrating MDSCs (left panel) and quantification (right panel). G & H. Representative dot plots showing levels of tumor-infiltrating CD8+ cells (E) and CD8+PD-1+ cells (F).
Asah2 represses the p53 pathway to suppress ferroptosis
The above finding that Asah2 regulates ferroptosis is not entirely surprising since ceramide is known to regulate various cell death pathways (39–45). In addition, ferroptosis is caused by lipid peroxidation (46) and Asah2 regulates sphingolipid ceramide metabolism (47). To identify the molecular link between Asah2 and ferroptosis, we then performed genome-wide gene expression profiling in MDSCs. J774 M cells were treated with NC06 and the cells were analyzed by DNA microarray. Gene Set Enrichment Analysis (GSEA) of the differentially expressed genes between control and NC06-treated cells revealed that the most dominant enrichment is genes of the p53 pathway based on Gene Ontology (GO) terms (Fig. 7A & B; the entire dataset is deposited to GEO database). Strikingly, the p53 mRNA level in J774M cells was not significantly altered by NC06 treatment (Fig. 7A & C). However, NC06 treatment increased p53 protein level in a dose-dependent manner (Fig. 7D). qPCR analysis validated that NC06 regulates the expression of p53 pathway genes such as Slc7a11, Notch, and Hmox1 (Fig. 7E). Notably, two of the p53 pathway genes, Slc7a11 and Hmox1, are known to regulate ferroptosis (48, 49) and are regulated by Asah2 in ferroptosis in MDSCs (Fig. 7E). Consistent with the finding that inhibition of Asah2 with NC06 decreased GSH level (Fig. 5C), silencing Asah2 decreased GSH level in J774M cells (Fig. S4 & 7F). Furthermore, silencing p53 significantly increased GSH levels in J774M cells (Fig. 7F). Consistent with the GSH level changes, silencing Asah2 increased lipid ROS and silencing p53 decreased lipid ROS levels in MDSCs (Fig. 7G).
Figure 7. Asah2 represses the p53 pathway in MDSCs.
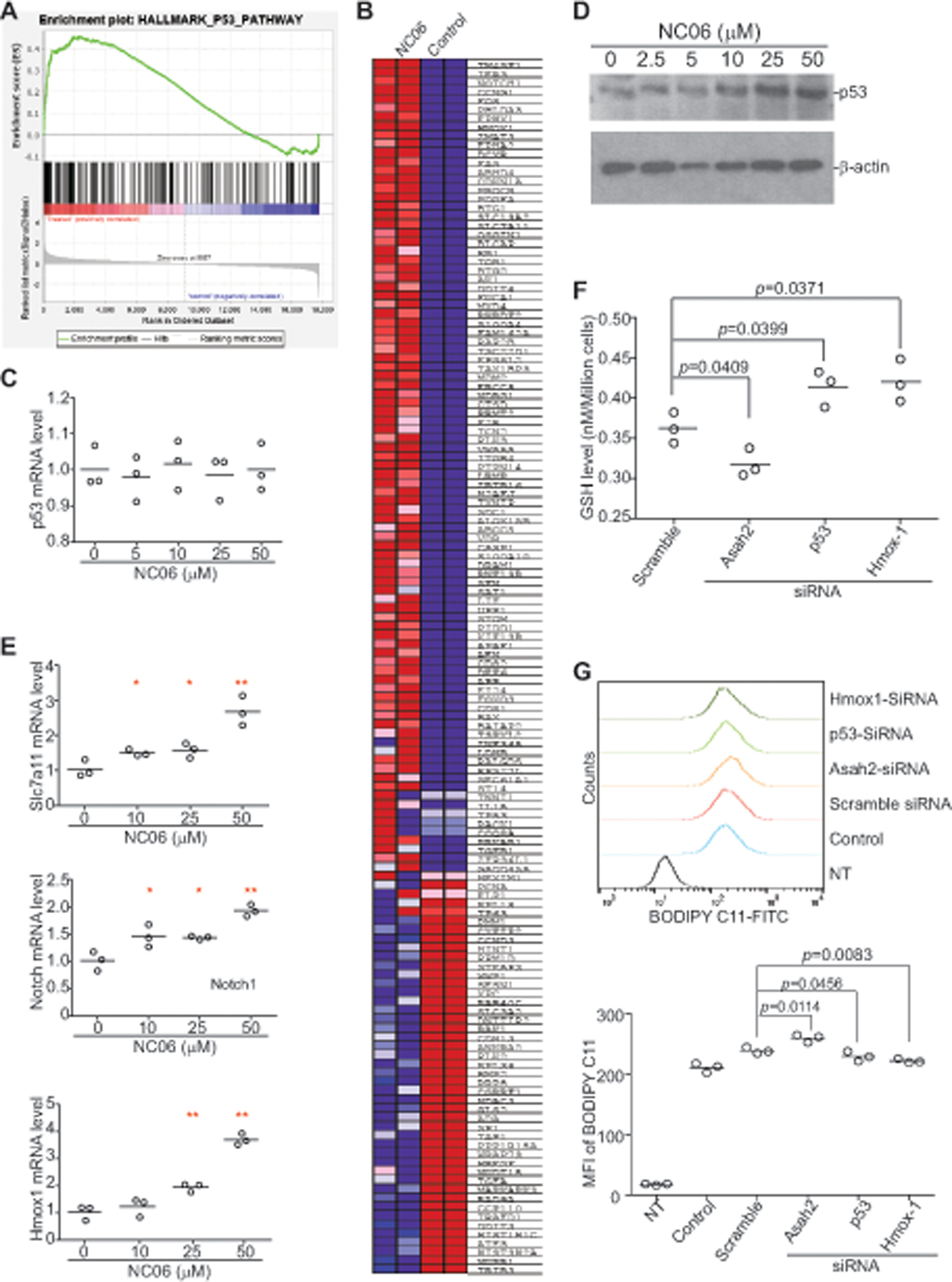
A. J774M cells were treated with NC06 (10 μM) for 24h and analyzed for genome-wide gene expression profiles by DNA microarray. Shown is the Gene Set Enrichment Analysis (GSEA) showing the enrichment plot of the p53 pathway. All GSEA false discovery rate q-values <0.0001. B. square graph showing the differentially expressed p53 pathway genes based on Gene Ontology (GO) terms. C. qPCR analysis of p53 expression in J774M cells after NC06 treatment for 24h at the indicated concentrations. D. Western blotting analysis of p53 protein in J774M cells treated with NC06 as indicated for 24h. The experiments were performed twice. E. J774M cells were treated with NC06 at the indicated concentrations for 24h and analyzed by qPCR using the indicated gene-specific primers. F. J774M cells were transfected with scramble and siRNAs that are specific for Asah2, p53 and Hmox1, respectively, for 24h. The cells were then measured for GSH level by flow cytometry. G. J774M cells were transfected with scramble and siRNAs that are specific for Asah2, p53 and Hmox1, respectively, for 24h. Cells were then analyzed for lipid ROS by flow cytometry. NT: No staining negative control cells for flow cytometry; control: no transfection.
Hmox1 degrades heme and releases free iron to generate oxidized lipids in the mitochondria membrane to induce ferroptosis (50, 51). The expression of Hmox-1 is regulated by p53 (49, 52). Consistently, NC06 treatment increased Hmox1 expression in a dose-dependent manner in J774M cells (Fig. 7E). Silencing Hmox1 increased glutathione level (Fig. 7F) and decreased lipid ROS level in MDSCs (Fig. 7G). Our data thus indicate the Asah2 represses Hmox1 expression to reduce lipid ROS to suppress ferroptosis.
Targeting Asah2 to suppress tumor development in vivo.
Our above findings determined that Asah2 suppresses ferroptosis to promote MDSC survival and accumulation. MDSCs are not only a potent immune suppressor but also a promoter for tumor growth and progression (6, 53). NC06 therapy therefore may be expected to suppress tumor development. To test this hypothesis, 4T1 mouse mammary carcinoma cells were injected i.v. into syngeneic BALB/c mice, and the tumor-bearing mice were treated with NC06 in the early stage of tumor development. NC06 significantly decreased 4T1 tumor growth in the mouse lungs (Fig. 8A). To determine whether this efficacy can be extended to other types of tumors, mouse colon tumor CT26 and mesothelioma AB1 cells were injected into syngeneic BALB/c mice. The tumor-bearing mice were then treated with NC06. As observed in the 4T1 tumor model, NC06 also significantly decreased colon and mesothelioma tumor growth in the lungs in the tumor-bearing mice (Fig. 8B & C). In summary, NC06 is an effective therapeutic agent in suppression of tumor growth in preclinical mouse models.
Figure 8. NC06 suppresses tumor growth in vivo.
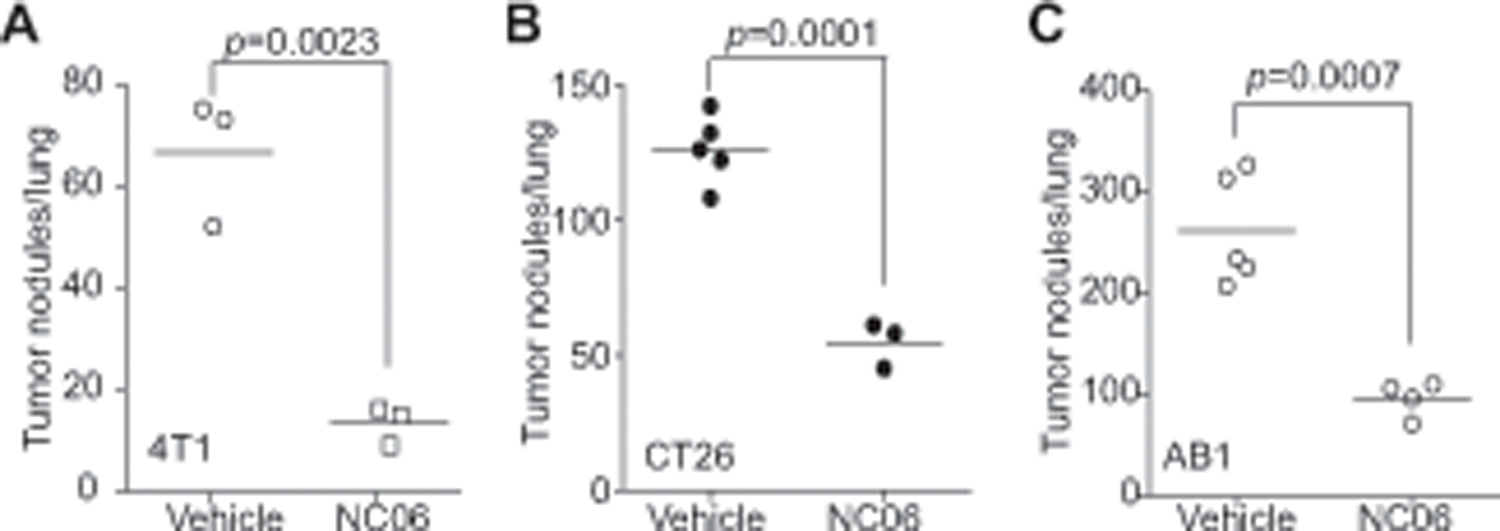
A. 4T1 cells (1×104/mouse) were injected i.v. to BALB/c mice. The mice were then treated with vehicle (n=3) and NC06 (20 mg/kg body weight) 3 days after tumor cell injection every 2 days for 7 times. Mice were sacrificed 17 days after tumor injection for every 2 days for 7 times. Mice were sacrificed and lungs were inflated with ink. The tumor nodules were counted. B & C. CT26 (B) and AB1 (C) cells (2×105 cells/mouse) were injected i.v. to mice. The tumor-bearing mice were treated and analyzed for lung tumors as in A.
Discussion
MDSCs are a major population of immune cells in the tumor microenvironment. In this study, we observe that colon tumor-infiltrating MDSCs overexpress Asah2. Asah2 is a key ceramidase that mediates sphingolipid metabolism (47) and regulates colon tumor cell apoptosis and autophagy (30). However, we observed that Asah2 protects MDSCs from ferroptosis. Our findings thus indicate that Asah2 function may be cell type-dependent and reveal a previously uncharacterized function of Asah2 in MDSC survival in the tumor microenvironment. Triggering ferroptosis is considered one of the best strategies to circumvent drug-resistant cancer (54) and to enhance the efficacy of T cell-based immunotherapy (55). In this study, we extend the ferroptosis-based anti-cancer target from tumor cells to MDSCs. MDSCs use dysregulation of ferroptosis as one mechanism of survival that allow them to accumulate in the tumor microenvironment. Accordingly, we show that induction of MDSC ferroptosis with a pharmacological agent effectively suppresses MDSC accumulation to increase tumor-infiltration CTL activation, resulting in tumor suppression.
The tumor suppressor p53 is a master regulator of ferroptosis and p53 can function as either a ferroptosis suppressor or activator depending on level of oxidative stress in tumor cells (34, 51). Under cellular stress, p53 represses the expression of SLC7A11 to suppress cystine uptake and decrease extracellular cysteine, the rate-limiting precursor for glutathione that is essential for GPX4 function in suppression of lipid peroxidation and lipid ROS generation (48, 56). On the other hand, p53 can activate p21 transcription to delay the onset of ferroptosis in response to subsequent cystine deprivation in tumor cells (34). P53 also can directly regulate Hmox1 expression (49), and Hmox1 can induce Fe2+ overproduction and resultant lipid ROS (50, 51, 54). In this study, we observe that inhibiting Asah2 induces ferroptosis in MDSCs. However, inhibiting Asah2 did not alter p53 expression level in MDSCs. Instead, genome-wide gene expression profiling reveals that inhibiting Asah2 activates the p53 pathway, suggesting that Asah2 may regulate p53 stabilization to suppress the p53 pathway in MDSCs (34), which requires further study. Although inhibiting Asah2 significantly decreases glutathione level and increases lipid ROS levels in MDSCs, cystine uptake actually increases after inhibition of Asah2, suggesting that Asah2 acts on a target between extracellular cysteine synthesis and glutathione synthesis. It is known that Hmox1 regulates glutathione synthesis (57). It is therefore likely that the Asah2-p53-Hmox1 axis regulates glutathione synthesis downstream of cysteine and that Hmox1 represses glutathione synthesis. The precise mechanism underlying Asah2 repression of glutathione synthesis while enhancing cysteine uptake requires further study.
We propose that the canonical cystine-Slc5A11-GSH-lipid ROS pathway (48) protects MDSCs from ferroptosis to accumulate under pathological conditions such as cancer. MDSCs overexpress Asah2 in order to degrade sphingolipid ceramide (46) to decrease membrane lipid peroxidation potential. Targeting Asah2 stabilizes p53 to activate the p53 pathway, including up-regulation of Hmox1, a gene that causes overproduction of Fe2+ (50, 57). Overproduction of Fe2+ may not only repress glutathione synthesis to decrease glutathione (58) to increase lipid ROS (59, 60), but may also enhance membrane ceramide lipid peroxidation and resultant lipid ROS(61). These Homx1-mediated two lipid ROS generation pathways leads to ferroptosis of MDSCs.
A limitation of this study is the use of the J774M cell line as a MDSC model. Although we determined that J774M cells phenotypically and functionally resemble MDSCs in in vitro assays. It cannot completely recapitulate the biological aspects of tumor-induced MDSCs in vivo. More studies are needed to determine the function of Asah2 in ferroptosis and MDSC accumulation in the tumor microenvironment under physiopathological conditions. Nevertheless, our findings that MDSCs from human colon cancer patients overexpress Asah2 and that Asah2 protects MDSCs from ferroptosis indicate that Asah2 in a molecular target for suppression MDSCs. Accordingly, the Asah2-selective small molecule inhibitor NC06 effectively suppresses MDSC accumulation thereby facilitating an increase in CTL tumor infiltration in colon tumor-bearing mice. Furthermore, NC06 therapy suppresses colon carcinoma, mammary carcinoma, and mesothelioma in preclinical mouse models. NC06 is a novel MDSC-targeting agent and the encouraging results reported herein further support its development and evaluation in cancer immunotherapy.
Supplementary Material
Key points.
ASAH2 is overexpressed in MDSCs in human colon cancer patients
The Slc5A11-GSH-GPX4-lipid ROS-ferroptosis pathway is regulated by Asah2 in MDSCs
Inhibition of Asah2 by NC06 suppresses MDSC accumulation in tumor-bearing mice
Acknowledgement
We thank Dr. Kimya Jones at Georgia Esoteric Molecular Labs (Augusta, GA) for assistance in immunohistochemical staining, and Dr. Thomas Albers for assistance in molecular docking. We thank Dr. Jeremy Allgood at Virginia Commonwealth University Lippidomics/Metabilics Core for assistance in ceramide analysis.
Grant supports from the National Cancer Institute CA227433 and CA133085 (to K.L.), and from the Veterans Affairs CX001364 (to K.L.).
Footnotes
Compliance with ethical standards
Conflict of interest: The authors declare that they have no conflict of interest
Reference
- 1.Hanahan D, and Weinberg RA 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura K, and Smyth MJ 2020. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol 17: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, To TKJ, Schug Z, Basu S, Wang F, Ricciotti E, DiRusso C, Murphy ME, Vonderheide RH, Lieberman PM, Mulligan C, Nam B, Hockstein N, Masters G, Guarino M, Lin C, Nefedova Y, Black P, Kagan VE, and Gabrilovich DI 2019. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 569: 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lasry A, Zinger A, and Ben-Neriah Y 2016. Inflammatory networks underlying colorectal cancer. Nat Immunol 17: 230–240. [DOI] [PubMed] [Google Scholar]
- 5.Sceneay J, Parker BS, Smyth MJ, and Moller A 2013. Hypoxia-driven immunosuppression contributes to the pre-metastatic niche. Oncoimmunology 2: e22355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ibrahim ML, Klement JD, Lu C, Redd PS, Xiao W, Yang D, Browning DD, Savage NM, Buckhaults PJ, Morse HC 3rd, and Liu K 2018. Myeloid-Derived Suppressor Cells Produce IL-10 to Elicit DNMT3b-Dependent IRF8 Silencing to Promote Colitis-Associated Colon Tumorigenesis. Cell Rep 25: 3036–3046 e3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagaraj S, and Gabrilovich DI 2008. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res 68: 2561–2563. [DOI] [PubMed] [Google Scholar]
- 8.Bunt SK, Sinha P, Clements VK, Leips J, and Ostrand-Rosenberg S 2006. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol 176: 284–290. [DOI] [PubMed] [Google Scholar]
- 9.Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, Utikal J, and Umansky V 2018. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front Immunol 9: 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, Vats P, Chinnaiyan AM, Lao CD, Lawrence TS, Wicha M, Hamanishi J, Mandai M, Kryczek I, and Zou W 2018. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest 128: 805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang H, Liang Y, Anders RA, Taube JM, Qiu X, Mulgaonkar A, Liu X, Harrington SM, Guo J, Xin Y, Xiong Y, Nham K, Silvers W, Hao G, Sun X, Chen M, Hannan R, Qiao J, Dong H, Peng H, and Fu YX 2018. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J Clin Invest 128: 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu C, Redd PS, Lee JR, Savage N, and Liu K 2016. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 5: e1247135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weber J, Gibney G, Kudchadkar R, Yu B, Cheng P, Martinez AJ, Kroeger J, Richards A, McCormick L, Moberg V, Cronin H, Zhao X, Schell M, and Chen YA 2016. Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunol Res 4: 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weber R, Fleming V, Hu X, Nagibin V, Groth C, Altevogt P, Utikal J, and Umansky V 2018. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front Immunol 9: 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ostrand-Rosenberg S, Beury DW, Parker KH, and Horn LA 2019. Survival of the fittest: how myeloid-derived suppressor cells survive in the inhospitable tumor microenvironment. Cancer Immunol Immunother. [DOI] [PMC free article] [PubMed]
- 16.Haverkamp JM, Smith AM, Weinlich R, Dillon CP, Qualls JE, Neale G, Koss B, Kim Y, Bronte V, Herold MJ, Green DR, Opferman JT, and Murray PJ 2014. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity 41: 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, El-Deiry WS, Winograd R, Vonderheide RH, English NR, Knight SC, Yagita H, McCaffrey JC, Antonia S, Hockstein N, Witt R, Masters G, Bauer T, and Gabrilovich DI 2014. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest 124: 2626–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parker KH, Horn LA, and Ostrand-Rosenberg S 2016. High-mobility group box protein 1 promotes the survival of myeloid-derived suppressor cells by inducing autophagy. J Leukoc Biol 100: 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, Wu H, Xu X, Erben U, Wu P, Syrbe U, Sieper J, and Qin Z 2012. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest 122: 4094–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roth F, De La Fuente AC, Vella JL, Zoso A, Inverardi L, and Serafini P 2012. Aptamer-mediated blockade of IL4Ralpha triggers apoptosis of MDSCs and limits tumor progression. Cancer Res 72: 1373–1383. [DOI] [PubMed] [Google Scholar]
- 21.Sinha P, Chornoguz O, Clements VK, Artemenko KA, Zubarev RA, and Ostrand-Rosenberg S 2011. Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood 117: 5381–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alissafi T, Hatzioannou A, Mintzas K, Barouni RM, Banos A, Sormendi S, Polyzos A, Xilouri M, Wielockx B, Gogas H, and Verginis P 2018. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J Clin Invest 128: 3840–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dominguez GA, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, Forero A, Bendell J, Witt R, Hockstein N, Kumar P, and Gabrilovich DI 2017. Selective Targeting of Myeloid-Derived Suppressor Cells in Cancer Patients Using DS-8273a, an Agonistic TRAIL-R2 Antibody. Clin Cancer Res 23: 2942–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hodge JW, and Schlom J 1999. Comparative studies of a retrovirus versus a poxvirus vector in whole tumor-cell vaccines. Cancer Res 59: 5106–5111. [PubMed] [Google Scholar]
- 25.Zimmerman M, Hu X, and Liu K 2010. Experimental metastasis and CTL adoptive transfer immunotherapy mouse model. J Vis Exp. [DOI] [PMC free article] [PubMed]
- 26.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, and Hutchison GR 2011. Open Babel: An open chemical toolbox. Journal of cheminformatics 3: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trott O, and Olson AJ 2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Airola MV, Allen WJ, Pulkoski-Gross MJ, Obeid LM, Rizzo RC, and Hannun YA 2015. Structural Basis for Ceramide Recognition and Hydrolysis by Human Neutral Ceramidase. Structure 23: 1482–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casasampere M, Bielsa N, Riba D, Bassas L, Xu RJ, Mao CG, Fabrias G, Abad JL, Delgado A, and Casas J 2019. New fluorogenic probes for neutral and alkaline ceramidases. Journal of Lipid Research 60: 1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Barros M, Coant N, Kawamori T, Wada M, Snider AJ, Truman JP, Wu BX, Furuya H, Clarke CJ, Bialkowska AB, Ghaleb A, Yang VW, Obeid LM, and Hannun YA 2016. Role of neutral ceramidase in colon cancer. FASEB J 30: 4159–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogretmen B 2006. Sphingolipids in cancer: regulation of pathogenesis and therapy. FEBS Lett 580: 5467–5476. [DOI] [PubMed] [Google Scholar]
- 32.Ogretmen B, and Hannun YA 2004. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer 4: 604–616. [DOI] [PubMed] [Google Scholar]
- 33.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, and Stockwell BR 2012. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149: 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, and Dixon SJ 2018. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep 22: 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti SV, Toyokuni S, Woerpel KA, and Zhang DD 2017. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171: 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee WC, Hsu PY, and Hsu HY 2020. Stem cell factor produced by tumor cells expands myeloid-derived suppressor cells in mice. Sci Rep 10: 11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bosiljcic M, Cederberg RA, Hamilton MJ, LePard NE, Harbourne BT, Collier JL, Halvorsen EC, Shi R, Franks SE, Kim AY, Banath JP, Hamer M, Rossi FM, and Bennewith KL 2019. Targeting myeloid-derived suppressor cells in combination with primary mammary tumor resection reduces metastatic growth in the lungs. Breast Cancer Res 21: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinha P, Clements VK, and Ostrand-Rosenberg S 2005. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol 174: 636–645. [DOI] [PubMed] [Google Scholar]
- 39.Dany M, and Ogretmen B 2015. Ceramide induced mitophagy and tumor suppression. Biochim Biophys Acta 1853: 2834–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang W, and Ogretmen B 2014. Autophagy paradox and ceramide. Biochim Biophys Acta 1841: 783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Senkal CE, Ponnusamy S, Bielawski J, Hannun YA, and Ogretmen B 2010. Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. Faseb J 24: 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park MA, Reinehr R, Haussinger D, Voelkel-Johnson C, Ogretmen B, Yacoub A, Grant S, and Dent P 2010. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol Cancer Ther 9: 2220–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White-Gilbertson S, Mullen T, Senkal C, Lu P, Ogretmen B, Obeid L, and Voelkel-Johnson C 2009. Ceramide synthase 6 modulates TRAIL sensitivity and nuclear translocation of active caspase-3 in colon cancer cells. Oncogene 28: 1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coe GL, Redd PS, Paschall AV, Lu C, Gu L, Cai H, Albers T, Lebedyeva IO, and Liu K 2016. Ceramide mediates FasL-induced caspase 8 activation in colon carcinoma cells to enhance FasL-induced cytotoxicity by tumor-specific cytotoxic T lymphocytes. Sci Rep 6: 30816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oleinik N, Kim J, Roth BM, Selvam SP, Gooz M, Johnson RH, Lemasters JJ, and Ogretmen B 2019. Mitochondrial protein import is regulated by p17/PERMIT to mediate lipid metabolism and cellular stress. Sci Adv 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Agmon E, and Stockwell BR 2017. Lipid homeostasis and regulated cell death. Curr Opin Chem Biol 39: 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coant N, and Hannun YA 2019. Neutral ceramidase: Advances in mechanisms, cell regulation, and roles in cancer. Adv Biol Regul 71: 141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, and Gu W 2015. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520: 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meiller A, Alvarez S, Drane P, Lallemand C, Blanchard B, Tovey M, and May E 2007. p53-dependent stimulation of redox-related genes in the lymphoid organs of gamma-irradiated--mice identification of Haeme-oxygenase 1 as a direct p53 target gene. Nucleic Acids Res 35: 6924–6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, and Chang WC 2018. Heme oxygenase-1 mediates BAY 11–7085 induced ferroptosis. Cancer Lett 416: 124–137. [DOI] [PubMed] [Google Scholar]
- 51.Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, Cheng Q, Zhang P, Dai W, Chen J, Yang F, Yang HT, Linkermann A, Gu W, Min J, and Wang F 2019. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A 116: 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, and Zhang DD 2009. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell 34: 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pawelec G, Verschoor CP, and Ostrand-Rosenberg S 2019. Myeloid-Derived Suppressor Cells: Not Only in Tumor Immunity. Front Immunol 10: 1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hassannia B, Vandenabeele P, and Vanden Berghe T 2019. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 35: 830–849. [DOI] [PubMed] [Google Scholar]
- 55.Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, Xia H, Zhou J, Li G, Li J, Li W, Wei S, Vatan L, Zhang H, Szeliga W, Gu W, Liu R, Lawrence TS, Lamb C, Tanno Y, Cieslik M, Stone E, Georgiou G, Chan TA, Chinnaiyan A, and Zou W 2019. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569: 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, Sirohi K, Li X, Wei Y, Lee H, Zhuang L, Chen G, Xiao ZD, Hung MC, Chen J, Huang P, Li W, and Gan B 2018. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol 20: 1181–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chiang SK, Chen SE, and Chang LC 2019. A Dual Role of Heme Oxygenase-1 in Cancer Cells. International Journal of Molecular Sciences 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jenkins NL, James SA, Salim A, Sumardy F, Speed TP, Conrad M, Richardson DR, Bush AI, and McColl G 2020. Ferrous-glutathione coupling mediates ferroptosis and frailty in Caenorhabditis elegans. bioRxiv: 10.1101/594408. [DOI] [PMC free article] [PubMed]
- 59.Papanikolaou G, and Pantopoulos K 2005. Iron metabolism and toxicity. Toxicol Appl Pharmacol 202: 199–211. [DOI] [PubMed] [Google Scholar]
- 60.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, and Stockwell BR 2014. Regulation of ferroptotic cancer cell death by GPX4. Cell 156: 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, and Mattson MP 2004. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci U S A 101: 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.