

Abstract
Objectives
We and others have previously shown that epigallocatechin gallate (EGCg) inhibits the activity of an important virulence factor, leukotoxin (LtxA), produced by the oral bacterium Aggregatibacter actinomycetemcomitans, suggesting the potential use of this molecule as an anti-virulence strategy to treat periodontal infections. Here, we sought to better understand the effects of EGCg on toxin secretion and A. actinomycetemcomitans pathogenicity in a co-culture model.
Methods
We used a quantitative immunoblot assay to determine the concentrations of LtxA in the bacterial supernatant and on the bacterial cell surface. Using a co-culture model, consisting of A. actinomycetemcomitans and THP-1 cells, we studied the impact of EGCg-mediated changes in LtxA secretion on the toxicity of A. actinomycetemcomitans.
Key findings
EGCg increased production of LtxA and changed the localization of secreted LtxA from the supernatant to the surface of the bacterial cells. In the co-culture model, a single low dose of EGCg did not protect host THP-1 cells from A. actinomycetemcomitans-mediated cytotoxicity, but a multiple dosing strategy had improved effects.
Conclusions
Together, these results demonstrate that EGCg has important, but complicated, effects on toxin secretion and activity; new dosing strategies and comprehensive model systems may be required to properly develop these anti-virulence activities.
Keywords: catechins, epigallocatechin gallate, leukotoxin, Aggregatibacter actinomycetemcomitans, virulence
Graphical Abstract
Graphical Abstract.
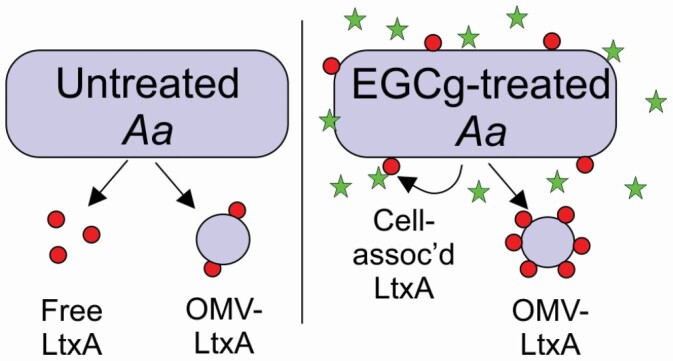
Epigallocatechin gallate increases the amount of leukotoxin produced, but inhibits its release while increasing its association with the bacterial cell.
Introduction
To combat the problem of antibiotic resistance, new strategies for treating bacterial infections, such as targeting the activity of virulence factors, have been proposed.[1, 2] An anti-virulence approach focuses on disabling the activity of the specific molecules produced by the pathogenic bacteria that give them an advantage over other bacteria and/or the host. Rather than kill the bacteria, the goal of this approach is to eliminate this advantage, thereby weakening the pathogen and enabling clearance by the host immune system or competition by the healthy microbiome.[3, 4]
We and others have found that some catechins, polyphenolic molecules isolated from tea leaves, particularly green tea leaves,[5] exhibit anti-virulence behaviour at low concentrations[6–11] in addition to their antibiotic activity. [9, 12–15] The United States Food and Drug Administration (FDA) classifies these molecules are generally recognized as safe (GRAS). The most abundant catechins in green tea extract include (−)-catechin (C), (−)-epicatechin (EC), (−)-epigallocatechin (EGC), (−)-epicatechin gallate (ECg), (−)-epigallocatechin gallate (EGCg), and (−)-gallocatechin gallate (GCg).[5]
In particular, we have demonstrated that galloylated catechins have strong inhibitory activity against the leukotoxin (LtxA) produced by Aggregatibacter actinomycetemcomitans.[10, 11]A. actinomycetemcomitans is a Gram-negative organism associated with aggressive forms of periodontitis.[16–20] As part of its colonization strategy, A. actinomycetemcomitans secretes LtxA, a protein toxin that kills immune cells, thus reducing the host immune response to the infection.[21–24] Because strains of A. actinomycetemcomitans that are more closely associated with the disease, such as the JP2 clone, have been reported to release more LtxA,[16, 19, 25] it has been proposed that the toxin’s immune-modulating activity plays an important role in disease progression.[22, 24, 26] Thus, inhibition of toxin activity represents a viable strategy to reduce the pathogenicity of A. actinomycetemcomitans without the use of antibiotics.
Fresh, oral isolates of A. actinomycetemcomitans are highly fimbriated and form colonies with a rough surface, while passaged strains, such as JP2, are nonfimbriated and form smooth colonies.[27] In both rough and smooth strains, LtxA is secreted across both the inner and outer membranes via a Type 1 secretion system (T1SS).[28, 29] However, the localization of the toxin differs significantly in the two types of strains. In rough strains, the secreted toxin associates primarily with the bacterial surface, and in smooth strains, such as JP2, the toxin is predominately released into the culture supernatant,[28] where it is able to interact with host immune cells.
As outer membrane vesicles (OMVs) are released by the bacteria, LtxA on the surface of the bacterial cells becomes localized to the surface of the vesicles.[30] This OMV-associated form of the toxin is also delivered to and active against host cells.[30–32] In its “free” form, LtxA binds to both cholesterol[33, 34] and an integrin receptor, lymphocyte function-associated antigen-1 (LFA-1)[35–38] on the host cell membrane to trigger internalization and subsequent cell death.[39] In the OMV-associated form, LtxA is delivered in cholesterol- and LFA-1-independent manner.[30]
Saito et al. demonstrated that catechins, including epigallocatechin gallate (EGCg), inhibit the activity of OMV-associated LtxA.[8] Similarly, we found that several galloylated catechins, including EGCg, inhibit the toxicity of “free” LtxA. Our work demonstrated that EGCg dramatically changes the secondary structure of LtxA, resulting in decreased affinity for the host cell membrane, particularly cholesterol,[10] and a corresponding increased affinity for the bacterial cell membrane.[11] We demonstrated that in the presence of EGCg, LtxA is still secreted through the T1SS by JP2, but the catechin alters its release by promoting association with the bacterial cell surface rather than release into the extracellular space.[11]
All of these studies suggest the strong potential of EGCg as an effective anti-virulence strategy for treating A. actinomycetemcomitans infections. While each study on its own is important, no study has yet investigated the complicated, combined effects of EGCg on A. actinomycetemcomitans proliferation, and toxin production/secretion, and the resulting impact on host cells. For this reason, we undertook the current study to investigate how EGCg-mediated alterations in LtxA secretion affect the release profiles of LtxA and the resulting pathogenicity of A. actinomycetemcomitans.
Method
Chemicals
Bovine albumin serum (BSA) and epigallocatechin gallate (EGCg) were purchased from Sigma‐Aldrich (St. Louis. MO). Nunc MaxiSorp flat-bottom plates, N‐(3‐triethylammoniumpropyl)‐4‐(6‐4‐(diethylamino) phenyl) hexatrienyl) pyridinium dibromide (FM 4‐64), Goat anti‐mouse IgG (H + L), Tween-20, and 1-Step Ultra TMB-ELISA substrate solution were purchased from ThermoFisher Scientific (Waltham, MA). Goat‐anti‐mouse IgG, Human ads‐HRP (GAM‐HRP) was purchased from Southern Biotech (Birmingham, AL).
Mammalian cell culture
RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 0.05 mM 2-mercaptoethanol, and 1% Pen-Strep (100 U/mL penicillin and 100 μg/mL streptomycin) was prepared to culture THP-1 cells (ATCC).
Bacterial cell culture
The JP2 strain of A. actinomycetemcomitans, a smooth strain that expresses LtxA, was grown in A. actinomycetemcomitans growth media (AAGM) anaerobically in a candle jar[40] at 37°C under 5% CO2.
LtxA purification
The purification of LtxA from JP2 strain supernatant was adapted from a published protocol.[41] Briefly, the supernatant from the late exponential phase of A. actinomycetemcomitans culture was collected by removing A. actinomycetemcomitans by two consecutive centrifugations at 10 000 × g for 10 min, followed by filtering the supernatant through a 0.22 μm filter. The bacteria-free supernatant was run through a cation exchange column, HiTrap SP HP (GE Healthcare), and then the attached LtxA was eluted from the column with a linear gradient of NaCl. One-millilitre fractions were collected up to 80 tubes, and the tubes with the most protein, determined by the absorbance at 280 nm (A280), were analyzed for their purity, identity and activity using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE), western blot, and cytotoxicity experiments, respectively.[11]
OMV purification
The OMVs were purified using ultracentrifugation. The A. actinomycetemcomitans culture was grown to the late exponential phase with or without 5 μg/mL of EGCg supplementation and then centrifuged at 10 000 × g for 10 min twice to pellet the bacteria. Any remaining bacteria were removed by filtering the supernatant through a 0.45 μm filter. The bacteria‐free supernatant was ultracentrifuged at 105 000 × g for 30 min, and the pellet containing the OMVs was resuspended in deionized water and ultracentrifuged again under the same conditions. The final OMV pellet was resuspended in 2 mL of phosphate‐buffered saline (PBS), and any remaining bacteria were removed by centrifuging the sample at 5000 × g for 5 min, twice. The protein concentration of the OMVs determined by measuring the A280 of the samples on a NanoDrop spectrophotometer.
LtxA immunoblot
An immunoblot assay was developed to quantify the amount of LtxA in the bacterial supernatant or associated with the bacterial cells during different growth phases of A. actinomycetemcomitans. In parallel, two A. actinomycetemcomitans cultures were grown to the late exponential phase, either with or without 5 µg/mL of EGCg in the media. 1 mL of each culture was centrifuged at 16 000 × g for 2 min. Both the pellet and supernatant were collected. The pellet, which was used to quantify the amount of cell-associated LtxA, was dissolved in 1 mL of PBS. To 500 µL of the supernatant, 1 mL of cold ethanol was added, and the solution was incubated at −80°C for 5 min before centrifugation at 16 000 × g for 15 min at 4°C.[28] The resulting pellet was collected and dissolved in 500 µL of PBS.
Because EGCg treatment decreases the binding of our monoclonal anti‐LtxA antibody,[11] we used two separate nitrocellulose blots, one containing an untreated LtxA standard and the samples from untreated A. actinomycetemcomitans and a second containing an EGCg‐treated LtxA standard and samples from EGCg‐treated A. actinomycetemcomitans. On the first nitrocellulose membrane, 2 µL of purified LtxA at a concentration of 50 µg/mL, serially diluted by a factor of two was spotted. Below this row of standards, 2 µL of the pellet and supernatant samples collected at different time points (3, 6, 12, 24 h) from the untreated A. actinomycetemcomitans culture were dotted on the second and fourth row. To prevent the overexposure of the dots, dilutions (10×) of those samples were dotted on the third and fifth rows. On the second nitrocellulose membrane, everything was kept the same except the standard consisted of 2 µL of LtxA at a concentration of 50 µg/mL, pretreated with 2 µg/mL of EGCg, and the following rows contained samples from EGCg-treated A. actinomycetemcomitans. Both membranes were incubated in separate chambers with an anti‐LtxA antibody (mAb 107)[42] followed by a horseradish peroxidase-labelled secondary antibody (GAM‐HRP). The intensities of each experimental spot were compared to that of the corresponding standard (LtxA with or without EGCg) using ImageJ.
Dynamic light scattering (DLS)
The size distribution of OMVs purified from A. actinomycetemcomitans grown with or without EGCg was compared using DLS. The system used was ALV/ CGS-3 compact goniometer. The OMVs were dissolved in filtered PBS (0.2 μm), then placed in the chamber and measured for 2 min at a wavelength of 638.2 nm with a scattering angle of 90°. The size distributions of each OMV population was calculated using the ALV software assuming a spherical particle[43] with a membrane thickness (r*) of 5 nm.[44] We defined those OMVs with a diameter of less than 200 nm as “small” OMVs and those greater than or equal to 200 nm as “large” OMVs, as previously reported.[30]
Quantification of lipid concentration of OMVs
The lipid concentrations of the OMVs purified from untreated A. actinomycetemcomitans and 5 µg/mL of EGCg-treated A. actinomycetemcomitans were determined using the lipophilic dye, FM 4‐64. The OMVs were incubated with the FM 4‐64 dye for 15 s. The fluorescence intensity at a wavelength of 640 nm was measured, using an excitation wavelength of 515 nm. The emission intensities of three independent OMV samples from each group were averaged.
Quantification of LtxA concentration of OMVs
The amount of LtxA associated with OMVs purified from untreated A. actinomycetemcomitans or EGCg-treated A. actinomycetemcomitans was quantified using an ELISA. The OMVs from both A. actinomycetemcomitans cultures were serially diluted and added to the bottom of a Nunc MaxiSorp flat-bottom plate. Purified LtxA, with or without EGCg treatment, was added in separate rows, as a standard. The standard and OMVs were incubated in the plate for 3 h, and then the wells were washed five times with ELISA wash buffer (EWB), which was composed of Tris-buffered saline (25 mM Tris and 150 mM NaCl at pH 7.2), 0.1% BSA, and 0.05% Tween-20. After the wash, the wells were blocked for 1 h with a blocking buffer consisting of EWB containing 1% BSA. The wells were incubated with 100 µL of mAb107[45] overnight at 4°C, then washed with EWB five times. Next, the primary antibody was labelled with GAM-HRP for 1 h at room temperature, then washed with EWB five times. After the wash, 1-Step Ultra TMB-ELISA substrate solution was added and incubated for 15 to 30 min. The reaction was stopped with 50 µL of 2 m sulfuric acid. The absorbance was measured at 450 nm, and the amount of LtxA associated with each OMV population was quantified from the standard to relate LtxA concentration to the measured absorbance.
Cytotoxicity measurements
A. actinomycetemcomitans was cultured with or without EGCg at 5 μg/mL, and the cells were harvested at either the early (6 h) or late exponential phase (24 h). The pellet and supernatants were collected as described previously. 10 µL of each sample or the same volume of PBS was added to 0.1 mL of THP-1 cells at a concentration of 2 × 106 cells/mL. The cells were incubated for 24 h at 37°C under 5% CO2, before the viability was measured using a Trypan blue exclusion assay. The viability was normalized to the viability of the untreated (PBS only) control sample.
THP-1 cell and A. actinomycetemcomitans co-culture
An A. actinomycetemcomitans culture was grown to the late exponential phase before one mL of the sample was centrifuged. 10 µL of the supernatant was added to 0.1 mL of THP-1 cells at a density of 2 × 106 cells/mL. The coculture was grown for 24 h with three different methods of EGCg addition. (1) 5 µg/mL of EGCg was added at the start of the co-culture. (2) 50 µg/mL of EGCg was added at the start of the co-culture. (3) 5 µg/mL of EGCg was added at the 0, 6 and 13 h time points, where the growth of A. actinomycetemcomitans is doubled or tripled relative to the initial time point. The EGCg concentrations and timepoints were determined from our previous study of A. actinomycetemcomitans growth with or without EGCg treatment.[11] All co-culture systems were incubated for 24 days at 37 °C under 5% CO2. The cell viability was determined using a Trypan blue assay, and the viability was normalized to the negative control in which THP-1 cells were incubated with AAGM only.
Statistical analysis
All data are presented as the mean ± standard deviation. Statistical analysis was performed using the Kruskal–Wallis test, followed by the Dunn’s posthoc test, where p values less than 0.01 were considered to be statistically significant.
Results
EGCg inhibits release of LtxA during the exponential phase
We previously reported that EGCg, at a concentration of 50 μg/mL or greater inhibits the growth of A. actinomycetemcomitans strain JP2. At a concentration of 5 μg/mL, EGCg has no effect on A. actinomycetemcomitans JP2 growth but inhibits the release of LtxA by the bacterial cells during early growth phases by promoting its reassociation with the bacterial membrane immediately after secretion by the T1SS.[11] To obtain a better understanding of the effect of EGCg on toxin secretion throughout growth, we expanded those studies here. As before, a subinhibitory concentration (5 μg/mL) of EGCg was added at the beginning of the A. actinomycetemcomitans culture. One-millilitre samples were collected from the culture at the 3, 6, 12, and 24 h time points, and the pellet and supernatant were separated using cold ethanol precipitation.[28]
The LtxA concentrations in the untreated and EGCg-treated A. actinomycetemcomitans cultures were quantified using a dot blot. The dot intensities (Supplementary Figure S1) were quantified, and the concentration of LtxA was calculated based on the respective standards. As shown in Figure 1, in both the untreated and EGCg-treated A. actinomycetemcomitans cultures, the amount of LtxA in both the supernatant and pellet increased over time. The total LtxA production in the EGCg-treated culture was almost twice that of the untreated culture at all time points (Figure 1A). However, the amount of LtxA released into the supernatant in the EGCg-treated culture was reduced relative to the untreated culture (Figure 1B). In the EGCg-treated culture, much of the LtxA remained associated with the bacterial cells (Figure 1C). Thus, as shown in Figure 1D, EGCg treatment altered the ratio of cell-associated versus secreted toxin, with more LtxA being released in untreated cultures and more being cell-associated in EGCg-treated cultures.
Figure 1.

Quantification of LtxA concentrations in supernatant and cell pellet. An overnight culture of A. actinomycetemcomitans was grown with or without 5 µg/mL of EGCg. 1 mL samples from untreated A. actinomycetemcomitans and EGCg-treated A. actinomycetemcomitans cultures were collected at 3, 6, 12, and 24 h time points. The pellet was collected by centrifuging the sample at 3000 × g for 5 min, and the supernatant was collected by cold ethanol precipitation. Both the pellet and supernatant were dissolved in 1 mL of PBS, and an immunoblot assay was employed to quantify the amount of LtxA in each sample at different time points. (A) The total concentration of LtxA in the untreated A. actinomycetemcomitans (grey) and EGCg-treated A. actinomycetemcomitans (black) cultures. (B) The concentration of LtxA that is associated (parallel lines) with untreated A. actinomycetemcomitans or EGCg-treated A. actinomycetemcomitans. (C) The concentration of LtxA in the supernatant (diagonal lines) of untreated A. actinomycetemcomitans or EGCg-treated A. actinomycetemcomitans. (D) The ratio of LtxA in the pellet (parallel lines) to the supernatant (diagonal lines) of untreated A. actinomycetemcomitans (grey) or EGCg-treated A. actinomycetemcomitans (black). The data are from a representative (N = 3) blot.
EGCg affects A. actinomycetemcomitans OMV composition but not production
Because we observed increased amounts of LtxA associated with the bacterial cells, rather than released into the supernatant in EGCg-supplemented cultures, we hypothesized that the OMVs produced in these cultures might contain more LtxA than those produced in untreated cultures. To study this, OMVs were purified from either untreated or EGCg-treated (5 μg/mL) A. actinomycetemcomitans cultures at the late exponential phase. The purified OMVs were characterized using dynamic light scattering (DLS), FM 4‐64 fluorescence and ELISA to analyze the OMV size distributions, lipid concentrations, and LtxA concentrations, respectively.
Figure 2A and B shows the size distributions of the OMVs purified from untreated and EGCg-treated cultures, respectively. We have previously reported that A. actinomycetemcomitans JP2 strain produces OMVs of two distinct sizes, “small” (less than 250 nm in diameter) and “large” (greater than 250 nm in diameter).[30] The size distribution of the OMVs was not affected by EGCg treatment; the populations consisted of approximately 87% small OMVs and 13% large OMVs in both cultures.
Figure 2.

Characterization of OMVs purified from untreated or EGCg-treated A. actinomycetemcomitans. OMVs were purified from A. actinomycetemcomitans strain JP2 with or without EGCg supplementation. The size distribution of OMVs from untreated A. actinomycetemcomitans (A) and EGCg-treated A. actinomycetemcomitans (B) was studied using DLS. The percentage of OMVs with specific sizes is shown. Data from one representative run (N = 3) are presented. (C) The lipid content of each OMV fraction was quantified using the FM 4‐64 dye, where the emission intensity is proportional to the total lipid content in each OMV fraction. Data are presented as the mean (N = 5) ± standard deviation. (D) The amount of LtxA associated with the OMVs was quantified using ELISA. The intensities of twelve measurements were averaged and the data was normalized to the amount of lipid content in each OMV fraction. Then the values were then normalized to the amount of LtxA per lipid of OMVs purified from untreated A. actinomycetemcomitans. Data are presented as the mean (N = 12) ± standard deviation. The level of significance, relative to untreated control was determined using a Kruskal Wallis test, followed by a Dunn’s posthoc test. NS, not significant; ****, P < 0.0005.
Because the size distributions were similar, we could compare the lipid content of each OMV sample, using FM 4–64 fluorescence intensity, to determine differences in the total number of OMVs released. This dye exhibits increased fluorescence upon intercalation into the lipid bilayer; thus, fluorescence intensity can be used as a measure of lipid concentration.[46] As shown in Figure 2C, the fluorescence intensity of OMVs purified from the EGCg-treated A. actinomycetemcomitans culture was slightly lower than the intensity of OMVs purified from the untreated A. actinomycetemcomitans culture. However, the difference was statistically insignificant, indicating that EGCg treatment has no effect on the amount of OMVs produced by A. actinomycetemcomitans.
Finally, the amount of LtxA per OMV was calculated using an ELISA with an anti-LtxA antibody.[45] Untreated and EGCg-treated standards were prepared separately to enable quantification of the concentration of LtxA in each OMV sample. The ELISA intensity was normalized based the respective standard and then divided by the amount of lipid concentration (FM 4–64 dye intensity) to enable comparison of the amount of LtxA per OMV in each culture. As shown in Figure 2D, approximately six-fold more LtxA was associated with the OMVs purified from the EGCg-treated A. actinomycetemcomitans culture than in OMVs purified from the untreated culture.
EGCg decreases the toxicity of secreted LtxA but not cell-associated LtxA
To assess the toxicity of cell-associated LtxA and secreted LtxA at the early and late exponential phases, samples of untreated and EGCg-treated A. actinomycetemcomitans were collected at the six- and 24 h time points of growth, and the pellet and the supernatant were separated. These samples were then incubated with THP-1 cells for 24 h, and the number of viable cells was measured using a Trypan blue assay. As shown in Figure 3, all of the cell pellet samples, containing A. actinomycetemcomitans-associated LtxA, were strongly active against THP-1 cells, regardless of the collection time or supplementation with EGCg. At the 6 h time point, the supernatant from the untreated A. actinomycetemcomitans culture caused a slight decrease in THP-1 cell viability while the supernatant from the EGCg-treated A. actinomycetemcomitans culture caused no decrease in THP-1 cell viability. At the 24 h time point, this effect was more pronounced, with the supernatant from the untreated A. actinomycetemcomitans culture killing approximately 60% of the THP-1 cells and the supernatant from the EGCg-treated culture causing no cell death.
Figure 3.

Cytotoxicity of A. actinomycetemcomitans pellet and supernatant. A. actinomycetemcomitans was grown with or without EGCg, and a 1-mL of the sample from each batch was collected at the six- and 24 h time points. The supernatant (S) and pellet (P) were collected. The cytotoxicity of each was tested using a Trypan blue assay after incubating each sample with 2 × 106 cells/mL of THP-1 cells for 24 h. Data are presented as the mean (N = 5) ± standard deviation. The levels of significance were determined using a Kruskal–Wallis test, followed by a Dunn’s posthoc test. All marked statistical levels are relative to untreated control unless otherwise noted. NS, not significant; *, P < 0.01; ***, P < 0.001; ****, P < 0.0005.
Effective dosing strategy reduces the pathogenicity of A. actinomycetemcomitans
Previously, we had demonstrated that EGCg inhibits the activity of purified LtxA[10]; however, the results presented here demonstrated that EGCg has a more complicated effect on LtxA, increasing the total amount of toxin that is produced and changing its location. The results in Figure 3 indicate that the LtxA that is associated with the bacterial cell membrane remains active. We, therefore, sought to more fully study the effect of EGCg treatment on A. actinomycetemcomitans pathogenicity by conducting a co-culture experiment, consisting of A. actinomycetemcomitans and THP-1 cells, supplemented with different concentrations of EGCg in either a single or multiple doses.
In this co-culture model, three separate cultures were established, all containing A. actinomycetemcomitans and THP-1 cells. In the first group, EGCg was added to the co-culture at a concentration of 5 μg/mL, the concentration we have previously found to inhibit LtxA activity, at the beginning of the incubation.[10] In the second group, EGCg was added to the co-culture at a concentration of 50 μg/mL, a concentration that inhibits A. actinomycetemcomitans growth.[10] For the third group, EGCg was added to the co-culture at a concentration of 5 μg/mL at the 0, 6, and 13 h time points (for a total addition of 15 μg/mL). These time points correspond to the times at which the population of A. actinomycetemcomitans has doubled or tripled relative to the starting concentration.[11] In each group, the positive control consisted of THP-1 and A. actinomycetemcomitans cells plus PBS (instead of EGCg), and the negative control consisted of THP-1 cells plus bacterial growth medium (instead of A. actinomycetemcomitans) and PBS (instead of EGCg). The data were normalized so that the negative control corresponded to 100% viability and the positive control to 0% viability.
As shown in Figure 4, the addition of 5 μg/mL of EGCg to the co-culture at the initial time point had no effect on THP-1 cell viability. The addition of 50 μg/mL of EGCg inhibited A. actinomycetemcomitans-mediated cytotoxicity, resulting in a THP-1 cell viability of approximately 90%. Interestingly, in the multiple dosing strategy, where only 15 μg/mL was used total, the cytotoxicity of A. actinomycetemcomitans was also reduced, resulting in approximately 60% THP-1 cell viability.
Figure 4.

A. actinomycetemcomitans toxicity against THP-1 cells. Ten-µL of an overnight culture of A. actinomycetemcomitans was cultured with 100 µL of 2 × 106 THP-1 cells/mL for 24 h with or without of EGCg at 37°C under 5% CO2. For the first and second group, 5 µg/mL and 50 µg/mL of EGCg were added to the cultures, respectively. For the third group, 5 µg/mL of EGCg was added at the 0 , 6 and 13 h time points. After 24 h of incubation, the THP-1 cell viability was measured using a Trypan blue assay. Data are presented as the mean (N = 3) ± standard deviation. The levels of significance were determined using a Kruskal Wallis test, followed by a Dunn’s posthoc test. All marked statistical levels are relative to untreated control unless otherwise noted. NS, not significant; ****, P < .0005.
Discussion
Because of the association of LtxA with clinical outcomes, we have focused on developing strategies to inhibit the activity of LtxA, as an alternative or adjuvant strategy for treatment.[10, 11, 47–50] In particular, we propose that EGCg and related catechins hold great promise for this purpose as they are readily available and easily accessible. We have demonstrated that EGCg has strong anti-LtxA activity at concentrations well below its minimum inhibitory concentration (MIC); at such sub-inhibitory concentrations, the risk of development of resistance is much lower. Thus, EGCg at these low concentrations could represent a possible long-term preventive strategy in at-risk patients in addition to its possible therapeutic use.
In this work, we have focused on the JP2 clone of A. actinomycetemcomitans, which differs from other types of A. actinomycetemcomitans in that it contains a 530-bp deletion in the promoter region of the LtxA gene operon[51]; this mutation causes significantly increased LtxA production compared to non-JP2 types of A. actinomycetemcomitans. This clone of A. actinomycetemcomitans has been associated with a significantly higher progression of clinical attachment loss in adolescents than other, non-JP2 types of A. actinomycetemcomitans,[25, 52] suggesting an association between leukotoxin production and disease progression.[19] This clone, which arose in the Mediterranean region of Africa, [53] seems to be passed through families and is now often detected in adolescent patients of African descent.[54–56]
Here, we studied how the presence of EGCg affects LtxA production, secretion, and activity throughout the growth of the A. actinomycetemcomitans JP2 clone and the resulting effect on A. actinomycetemcomitans toxicity against host cells. The results demonstrate that EGCg increases the amount of LtxA that is produced and alters its localization after secretion; in the absence of EGCg, most LtxA is released into the supernatant, while in the presence of EGCg, most LtxA reassociates with the bacterial cell surface. As a result of this change in localization, EGCg has significant effects on the release of OMV-associated LtxA. In EGCg-supplemented A. actinomycetemcomitans, the same number of OMVs are released, but the concentration of LtxA on the OMVs is significantly increased. We then demonstrated, in a co-culture experiment, that the localization of LtxA has significant implications in the activity of EGCg in inhibiting A. actinomycetemcomitans-mediated toxicity against THP-1 cells.
Our finding that EGCg promotes binding of LtxA to the surface of the bacterial cells is consistent with previous reports that culture conditions affect secretion or cell surface association of the toxin. When LtxA was first identified, several reports suggested that the toxin was not released into the supernatant, but was instead associated with the cell surface.[57–61] However, later work identified two genes that encoded for proteins that were homologous to proteins involved in the secretion of a related toxin, Escherichia coli α-hemolysin (HlyA).[62, 63] Ultimately, it became clear that LtxA is sensitive to conditions of the culture, including pH and composition[28, 64]; the differing results were likely due to variations in experimental protocols. Consensus now is that LtxA is a secreted protein, released through the T1SS, but after release, culture conditions determine the amount of LtxA that is released into the supernatant versus reassociated with the bacterial cell membrane.[22, 24, 26] Additionally, localization varies between smooth and rough strains.[28] This work further demonstrates that the presence of even small amounts of EGCg can have large effects on LtxA localization throughout bacterial growth, with significant consequences on host cell viability.
While a number of studies have demonstrated that catechins can affect the activity of bacterial toxins,[8, 10, 65, 66] few have investigated the effect of these molecules on toxin secretion. In one study, epicatechin gallate (ECg) was reported to inhibit the secretion of an unrelated toxin, α-toxin, from Staphylococcus aureus.[67] Importantly, this work measured the decrease in the toxin concentration of the supernatant using an immunoblot, but the authors did not investigate if any toxin had localized to the surface of the bacteria.[67] Thus, it is quite possible that ECg might actually increase the total amount of toxin produced, but change its release profile, as we have observed in this work with EGCg and LtxA. It should be noted that we have also found that EGCg alters LtxA conformation and, as a result, epitope accessibility. We have accounted for this problem by using LtxA or LtxA+EGCg standards in all immunoblots to enable quantitative comparisons. We strongly caution against the use of immunoblots without such quantitative standards. An earlier report demonstrated that EGCg and gallocatechin gallate (GCg) decreased the secretion of verotoxins (Shiga toxins) 1 and 2 (VT1 and VT2/Stx1 and Stx2) by enterohemorrhagic E. coli (EHEC). A corresponding increase in the amount of cell-associated Stx1, but not Stx2, was observed.[6] The authors hypothesized but did not demonstrate, that the catechins inhibited the secretion of Stx1 and Stx2 from the bacterial cytosol to the extracellular environment. As we have observed in this work, it is possible that the catechins did not affect the secretion of the toxins, but rather altered their affinity for the bacterial cell surface. While these toxins are unrelated to LtxA, we believe that our work demonstrates the need to fully investigate toxin concentrations and localization when determining the activity of catechins and other anti-virulence molecules. The results presented in this paper demonstrate that the EGCg-mediated changes in release profiles have significant implications in the toxicity of the bacterial cells.
Like many bacterial toxins, LtxA is released in a “free” form as well as in an OMV-associated form.[30–32] Our previous results have demonstrated that A. actinomycetemcomitans, strain JP2, releases two types of OMVs. One population is small (~100 nm) and lacks LtxA while the second population is much larger (~350 nm) and is enriched in LtxA.[30] Because EGCg promoted LtxA association with the bacterial cell surface, we studied the resulting effects on the size distribution and LtxA composition of the OMVs. Interestingly, we found that EGCg had no effect on the ratio of large to small OMVs. OMV production is known to be sensitive to environmental stresses,[68] and we, therefore, studied the total amount of OMVs produced in the absence and presence of 5 μg/mL of EGCg. Our results demonstrated that the amount of OMVs produced was similar. However, the increased amount of LtxA on the surface of the bacteria in the presence of EGCg resulted in approximately six times more LtxA being associated with the OMVs purified from EGCg-treated A. actinomycetemcomitans relative to untreated A. actinomycetemcomitans. Thus, EGCg treatment did not affect the size or number of OMVs released but did promote the release of OMVs containing much more LtxA. We hypothesized that this change would affect the overall activity of A. actinomycetemcomitans against host cells, even though our previous results demonstrated that EGCg inhibits free LtxA activity,[10] and Saito et al. showed that EGCg inhibits the activity of OMV-associated LtxA.[8]
Therefore, we designed a co-culture experiment in which to more accurately test the effect of EGCg on LtxA-mediated cytotoxicity. This set of co-culture experiments demonstrated that a high concentration of EGCg (50 μg/mL) inhibited host cell death, likely by killing the bacterial cells. However, the subinhibitory concentration of EGCg (5 μg/mL), which we had previously demonstrated to effectively inhibit LtxA activity, did not protect host cells. We hypothesized that one possible explanation of this lack of inhibition is the continued production of LtxA by A. actinomycetemcomitans throughout the co-culture experiment. Whereas in our previous in vitro experiments, we added a fixed amount of LtxA and EGCg to a host cell culture, in the co-culture experiment, the amount of LtxA is constantly increasing as the A. actinomycetemcomitans cells grow and divide, while the amount of EGCg is fixed. Thus, with time, the EGCg:LtxA ratio decreases to a point where EGCg is no longer able to inhibit LtxA activity. To address this temporal variation, we, therefore, devised a dosing strategy in which we found that multiple doses of EGCg were much more effective in promoting an anti-virulence effect. Further refinement would enable the identification of an optimal dosing strategy.
While we set out to study the effect of EGCg on A. actinomycetemcomitans and LtxA, we believe this work highlights two important caveats in the anti-virulence field. (1) To study the anti-virulence effects of a particular molecule, more realistic/less reductionist approaches are required to account for all changes enacted by the molecule. For example, as we have shown here, EGCg greatly decreased the amount of LtxA in the bacterial supernatant and inhibited the activity of that free LtxA; however, it increased the total amount of LtxA produced and increased the activity of the bacterial cells themselves. (2) To demonstrate the effectiveness of an anti-virulence strategy, dosing strategies should be considered to account for temporal changes in the system. A major complication in the design of anti-virulence strategies is that virulence factor expression is temporal, a factor that must be considered in the development of these types of strategies. While these caveats complicate the study of these molecules, we expect that such strategies are essential for the successful development of new therapeutic options to treat bacterial diseases.
Conclusion
In conclusion, this work demonstrated that EGCg has significant effects on LtxA secretion and localization in A. actinomycetemcomitans JP2, which are important to consider when studying this molecule as an anti-virulence agent. While we and others have reported that EGCg inhibits LtxA activity, our demonstration that the molecule increases the amount of LtxA produced raises important concerns about its possible therapeutic use. Our results demonstrate the importance of studying the effect of these molecules in more realistic systems and considering dosing strategies when designing anti-virulence agents. We anticipate that EGCg and its derivatives could have useful applications as anti-virulence drugs, but further work must be done to completely elucidate the relevant effects of these molecules.
Author Contributions
EHC and ACB conceived and designed the study. EHC performed the experiments and analysed data. EHC and ACB wrote the manuscript. The authors agreed to be accountable for all aspects of the work, ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors approved the final version of the manuscript.
Funding
This work was supported by the National Institutes of Health (DE025275) and the National Science Foundation (1554417).
Conflict of Interest
None declared.
Data Availability
Some or all data, models, or code generated or used during the study are available from the corresponding author by request.
Supplementary Material
Contributor Information
En Hyung Chang, Department of Chemical and Biomolecular Engineering, Lehigh University, Bethlehem, PA, USA.
Angela C Brown, Department of Chemical and Biomolecular Engineering, Lehigh University, Bethlehem, PA, USA.
References
- 1. Rasko DA, Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nature Reviews Drug Discovery 2010; 9: 117–128. [DOI] [PubMed] [Google Scholar]
- 2. Krueger E, Brown AC. Inhibition of bacterial toxin recognition of membrane components as an anti-virulence strategy. Journal of Biological Engineering 2019; 1: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clatworthy AE, Pierson E, Hung DT. Targeting virulence: A new paradigm for antimicrobial therapy. Nature Chemical Biology 2007; 9: 541–548. [DOI] [PubMed] [Google Scholar]
- 4. Cegelski L, Marshall GR, Eldridge GRet al. The biology and future prospects of antivirulence therapies. Nature Reviews Microbiology 2008; 1: 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choi O, Yahiro K, Morinaga Net al. Inhibitory effects of various plant polyphenols on the toxicity of staphylococcal α -toxin. Microbial Pathogenesis 2007; 5GÇô6: 215–224. [DOI] [PubMed] [Google Scholar]
- 6. Sugita-Konishi Y, Hara-Kudo Y, Amano Fet al. Epigallocatechin gallate and gallocatechin gallate in green tea catechins inhibit extracellular release of Vero toxin from enterohemorrhagic Escherichia coli O157:H7. Biochimica et Biophysica Acta (BBA) - General Subjects 1999; 1472: 42–50. [DOI] [PubMed] [Google Scholar]
- 7. Hirasawa M, Takada K, Otake S. Inhibition of acid production in dental plaque bacteria by green tea catechins. Caries Research 2004; 265: 270. [DOI] [PubMed] [Google Scholar]
- 8. Saito M, Tsuzukibashi O, Takada K. Anticytotoxic effect of green tea catechin on Aggregatibacter actinomycetemcomitans vesicles. International Journal of Oral-Medical Sciences 2012; 2: 101–105. [Google Scholar]
- 9. Okubo S, Sasaki T, Hara Yet al. Bactericidal and anti-toxin activities of catechin on enterohemorrhagic Escherichia coli. The Journal of the Japanese Association for Infectious Diseases 2015; 3: 211–217. [DOI] [PubMed] [Google Scholar]
- 10. Chang EH, Huang J, Lin Zet al. Catechin-mediated restructuring of a bacterial toxin inhibits activity. Biochimica et Biophysica Acta (BBA) - General Subjects 2019; 1: 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang EH, Giaquinto P, Huang Jet al. Epigallocatechin gallate inhibits leukotoxin release by Aggregatibacter actinomycetemcomitans by promoting association with the bacterial membrane. Molecular Oral Microbiology 2020; 1: 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Friedman M, Henika PR, Levin CEet al. Antimicrobial activities of tea catechins and theaflavins and tea extracts against Bacillus cereus. Journal of Food Protection 2006; 2: 354–361. [DOI] [PubMed] [Google Scholar]
- 13. Kohda C, Yanagawa Y, Shimamura T. Epigallocatechin gallate inhibits intracellular survival of Listeria monocytogenes in macrophages. Biochemical and Biophysical Research Communications 2008; 2: 310–315. [DOI] [PubMed] [Google Scholar]
- 14. Osterburg A, Gardner J, Hyon SHet al. Highly antibiotic-resistant Acinetobacter baumannii clinical isolates are killed by the green tea polyphenol (-)-epigallocatechin-3-gallate (EGCg). Clinical Microbiology and Infection 2009; 4: 341–346. [DOI] [PubMed] [Google Scholar]
- 15. Bai L, Takagi S, Ando Tet al. Antimicrobial activity of tea catechin against canine oral bacteria and the functional mechanisms. Journal of Veterinary Medical Science 2016; 9: 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zambon JJ, Slots J, Genco RJ. Serology of oral Actinobacillus actinomycetemcomitans and serotype distribution in human periodontal disease. Infection and Immunity 1983; 1: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zambon JJ. Actinobacillus actinomycetemcomitans in human periodontal disease. Journal of Clinical Periodontology 1985; 12: 1–20. [DOI] [PubMed] [Google Scholar]
- 18. Zambon JJ, Haraszthy VI, Hariharan Get al. The microbiology of early-onset periodontitis: Association of highly toxic Actinobacillus actinomycetemcomitans strains with localized juvenile periodontitis. Journal of Periodontology 1996; 3: 282–290. [DOI] [PubMed] [Google Scholar]
- 19. Haraszathy VI, Hariharan G, Tinoco EMBet al. Evidence for the role of highly leukotoxic Actinobacillus actinomycetemcomitans in the pathogenesis of localized juvenile and other forms of early-onset periodontitis. Journal of Periodontology 2000; 71: 912–922. [DOI] [PubMed] [Google Scholar]
- 20. Fine DH, Markowitz K, Furgang Det al. Aggregatibacter actinomycetemcomitans and its relationship to initiation of localized aggressive periodontitis: Longitudinal cohort study of initially healthy adolescents. Journal of Clinical Microbiology 2007; 12: 3859–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Diaz R, Ghofaily LA, Patel Jet al. Characterization of leukotoxin from a clinical strain of Actinobacillus actinomycetemcomitans. Microbial Pathogenesis 2006; 2: 48–55. [DOI] [PubMed] [Google Scholar]
- 22. Johansson A. Aggregatibacter actinomycetemcomitans leukotoxin: A powerful tool with capacity to cause imbalance in the host inflammatory response. Toxins 2011; 3: 242–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Höglund Åberg C, Haubek D, Kwamin Fet al. Leukotoxic activity of Aggregatibacter actinomycetemcomitans and periodontal attachment loss. PLOS ONE 2014; 8: e104095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krueger E, Brown AC. Aggregatibacter actinomycetemcomitans leukotoxin: From mechanism to targeted anti-toxin therapeutics. Molecular Oral Microbiology 2020; 3: 85–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haubek D, Ennibi O-K, Poulsen Ket al. Risk of aggressive periodontitis in adolescent carriers of the JP2 clone of Aggregatibacter (actinobacillus) actinomycetemcomitans in Morocco: A prospective longitudinal cohort study. The Lancet 2008; 9608: 237–242. [DOI] [PubMed] [Google Scholar]
- 26. Kachlany SC. Aggregatibacter actinomycetemcomitans leukotoxin:From threat to therapy. Journal of Dental Research 2010; 6: 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Y, Liu A, Chen Cet al. Genetic basis for conversion of rough-to-smooth colony morphology in Actinobacillus actinomycetemcomitans. Infection and Immunity 2005; 6: 3749–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kachlany SC, Fine DH, Figurski DH. Secretion of RTX leukotoxin by Actinobacillus actinomycetemcomitans. Infection and immunity 2000; 11: 6094–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crosby JA, Kachlany SC. TdeA, a TolC-like protein required for toxin and drug export in Aggregatibacter (actinobacillus) actinomycetemcomitans. Gene 2007; 1–2: 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nice JB, Balashova NV, Kachlany SCet al. Aggregatibacter actinomycetemcomitans leukotoxin is delivered to host cells in an lfa-1-independent manner when associated with outer membrane vesicles. Toxins 2018; 10: 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kato S, Kowashi Y, Demuth DR. Outer membrane-like vesicles secreted by Actinobacillus actinomycetemcomitans are enriched in leukotoxin. Microbial Pathogenesis 2002; 1: 1–13. [DOI] [PubMed] [Google Scholar]
- 32. Demuth D, James D, Kowashi Yet al. Interaction of Actinobacillus actinomycetemcomitans outer membrane vesicles with HL60 cells does not require leukotoxin. Cellular Microbiology 2003; 2: 111–121. [DOI] [PubMed] [Google Scholar]
- 33. Fong KP, Pacheco CMF, Otis LLet al. Actinobacillus actinomycetemcomitans leukotoxin requires lipid microdomains for target cell cytotoxicity. Cellular Microbiology 2006; 11: 1753–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brown AC, Balashova NV, Epand RMet al. Aggregatibacter actinomycetemcomitans leukotoxin utilizes a cholesterol recognition/amino acid consensus site for membrane association. Journal of Biological Chemistry 2013; 32: 23607–23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dileepan T, Kachlany SC, Balashova NVet al. Human CD18 is the functional receptor for Aggregatibacter actinomycetemcomitans leukotoxin. Infection and Immunity 2007; 10: 4851–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kieba IR, Fong KP, Tang HYet al. Aggregatibacter actinomycetemcomitans leukotoxin requires β -sheets 1 and 2 of the human CD11a β-propeller for cytotoxicity. Cellular Microbiology 2007; 11: 2689–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nygren P, Balashova NV, Brown ACet al. Aggregatibacter actinomycetemcomitans leukotoxin causes activation of lymphocyte function-associated antigen 1. Cellular Microbiology 2019: e12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ristow LC, Tran V, Schwartz KJet al. The extracellular domain of the β2 integrin β subunit (CD18) is sufficient for Escherichia coli hemolysin and Aggregatibacter actinomycetemcomitans leukotoxin cytotoxic activity. mBio 2019; 4: e01459-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Korostoff J, Wang JF, Kieba IRet al. Actinobacillus actinomycetemcomitans leukotoxin induces apoptosis in HL-60 cells. Infection and Immunity 1998; 9: 4474–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kachlany SC, Fine DF, Figurski DH. Purification of secreted leukotoxin (LtxA) from Actinobacillus actinomycetemcomitans. Protein Expression and Purification 2002; 3: 465–471. [DOI] [PubMed] [Google Scholar]
- 41. Reinholdt J, Poulsen K, Brinkmann CRet al. Monodisperse and LPS-free Aggregatibacter actinomycetemcomitans leukotoxin: Interactions with human β2 integrins and erythrocytes. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2013; 2: 546–558. [DOI] [PubMed] [Google Scholar]
- 42. Lally ET, Golub EE, Kieba IR. Identification and immunological characterization of the domain of Actinobacillus actinomycetemcomitans leukotoxin that determines its specificity for human target cells. Journal of Biological Chemistry 1994; 49: 31289–95. [PubMed] [Google Scholar]
- 43. Hallett FR, Watton J, Krygsman P. Vesicle sizing: Number distributions by dynamic light scattering. Biophysical Journal 1991; 2: 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alberts B, Bray D, Hopkin Ket al. Essential cell biology, New York: W.W. Norton & Company, 2014. [Google Scholar]
- 45. Lally ET, Golub EE, Kieba IR. Identification and immunological characterization of the domain of Actinobacillus actinomycetemcomitans leukotoxin that determines its specificity for human target cells. Journal of Biological Chemistry 1994; 49: 31289–31295. [PubMed] [Google Scholar]
- 46. Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. Journal of Cell Biology 1995; 5: 779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brown AC, Koufos E, Balashova NV.et al. Inhibition of LtxA toxicity by blocking cholesterol binding with peptides. Molecular Oral Microbiology 2016; 1: 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koufos E, Chang EH, Rasti ESet al. Use of a cholesterol recognition amino acid consensus (CRAC) peptide to inhibit binding to cholesterol by a bacterial toxin. Biochemistry 2016; 34: 4787–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Webb JN, Koufos E, Brown AC. Inhibition of bacterial toxin activity by the nuclear stain, Draq5™. The Journal of Membrane Biology 2016; 4: 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Krueger E, Hayes S, Chang EHet al. Receptor-based peptides for inhibition of leukotoxin activity. ACS Infectious Diseases 2018; 7: 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brogan JM, Lally ET, Poulsen Ket al. Regulation of Actinobacillus actinomycetemcomitans leukotoxin expression: Analysis of the promoter regions of leukotoxic and minimally leukotoxic strains. Infection and Immunity 1994; 2: 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haubek D, Ennibi OK, Poulsen Ket al. The highly leukotoxic JP2 clone of Actinobacillus actinomycetemcomitans and progression of periodontal attachment loss. Journal of Dental Research 2004; 10: 767–770. [DOI] [PubMed] [Google Scholar]
- 53. Haubek D, Poulsen K, Kilian Met al. Microevolution and patterns of dissemination of the JP2 clone of Aggregatibacter (Actinobacillus) actinomycetemcomitans. Infection and Immunity 2007; 6: 3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dirienzo JM, Mckay TL. Identification and characterization of genetic cluster groups of Actinobacillus actinomycetemcomitans isolated from the human oral cavity. Journal of Clinical Microbiology 1994; 1: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Haubek D, Poulsen K, Asikainen Set al. Evidence for absence in northern Europe of especially virulent clonal types of Actinobacillus actinomycetemcomitans. Journal of Clinical Microbiology 1995; 2: 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Haubek D, Poulsen K, Westergaard Jet al. Highly toxic clone of Actinobacillus actinomycetemcomitans in geographically widespread cases of juvenile periodontitis in adolescents of African origin. Journal of Clinical Microbiology 1996; 6: 1576–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tsai CC, Shenker BJ, Dirienzo JMet al. Extraction and isolation of a leukotoxin from Actinobacillus actinomycetemcomitans with polymyxin B. Infection and Immunity 1984; 2: 700–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lally ET, Golub EE, Kieba IRet al. Structure and function of the B and D genes of the Actinobacillus actinomycetemcomitans leukotoxin complex. Microbial Pathogenesis 1991; 2: 111–121. [DOI] [PubMed] [Google Scholar]
- 59. Ohta H, Kato K, Kokeguchi Set al. Nuclease-sensitive binding of an Actinobacillus actinomycetemcomitans leukotoxin to the bacterial cell surface. Infection and Immunity 1991; 12: 4599–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Berthold P, Forti D, Kieba IRet al. Electron immunocytochemical localization of Actinobacillus actinomycetemcomitans leukotoxin. Oral Microbiology and Immunology 1992; 1: 24–27. [DOI] [PubMed] [Google Scholar]
- 61. Ohta H, Hara H, Fukui Ket al. Association of Actinobacillus actinomycetemcomitans leukotoxin with nucleic acids on the bacterial cell surface. Infection and Immunity 1993; 11: 4878–4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Guthmiller JM, Kolodrubetz D, Cagle MPet al. Sequence of the lktB gene from Actinobacillus actinomycetemcomitans. Nucleic Acids Res 1990; 17: 5291–5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guthmiller JM, Kolodrubetz D, Kraig E. Mutational analysis of the putative leukotoxin transport genes in Actinobacillus actinomycetemcomitans. Microbial Pathogenesis 1995; 5: 307–321. [DOI] [PubMed] [Google Scholar]
- 64. Balashova NV, Diaz R, Balashov SVet al. Regulation of Aggregatibacter (Actinobacillus) actinomycetemcomitans leukotoxin secretion by iron. Journal of Bacteriology 2006; 24: 8658–8661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Caturla N, Vera-Samper E, Villalain Jet al. The relationship between the antioxidant and the antibacterial properties of galloylated catechins and the structure of phospholipid model membranes. Free Radical Biology & Medicine 2003; 6: 648–662. [DOI] [PubMed] [Google Scholar]
- 66. Kawashima Y. Effects of catechin gallate on bactericidal action and leukotoxic activity of Aggregatibacter actinomycetemcomitans. International Journal of Oral-Medical Sciences 2011; 1: 20–24. [Google Scholar]
- 67. Shah S, Stapleton PD, Taylor PW. The polyphenol (−)-epicatechin gallate disrupts the secretion of virulence-related proteins by Staphylococcus aureus. Letters in Applied Microbiology 2008; 2: 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Macdonald IA, Kuehn MJ. Stress-induced outer membrane vesicle production by Pseudomonas aeruginosa. Journal of Bacteriology 2013; 13: 2971–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Some or all data, models, or code generated or used during the study are available from the corresponding author by request.