

Abstract
In sepsis and trauma, pathogens and injured tissue provoke a systemic inflammatory reaction which can lead to overwhelming inflammation. Concurrent with the innate hyperinflammatory response is adaptive immune suppression that can become chronic. A current key issue today is that patients who undergo intensive medical care after sepsis or trauma have a high mortality rate after being discharged. This high mortality is thought to be associated with persistent immunosuppression. Knowledge about the pathophysiology leading to this state remains fragmented. Immunosuppressive cytokines play an essential role in mediating and upholding immunosuppression in these patients. Specifically, the cytokines Interleukin-10 (IL-10), Transforming Growth Factor-β (TGF-β) and Thymic stromal lymphopoietin (TSLP) are reported to have potent immunosuppressive capacities. Here, we review their ability to suppress inflammation, their dynamics in sepsis and trauma and what drives the pathologic release of these cytokines. They do exert paradoxical effects under certain conditions, which makes it necessary to evaluate their functions in the context of dynamic changes post-sepsis and trauma. Several drugs modulating their functions are currently in clinical trials in the treatment of other pathologies. We provide an overview of the current literature on the effects of IL-10, TGF-β and TSLP in sepsis and trauma and suggest therapeutic approaches for their modulation.
Keywords: IL-10, transforming growth factor β, thymic stromal lymphopoietin, immunosuppression, chronic critical illness
Immune Perturbation in Sepsis and Trauma
Infection and injury can lead to an increase of alarm signals that signal the presence of invading pathogens or injured tissue. The so called `danger hypothesis` was developed by Matzinger et al. stating that alarmins are either Pathogen-Associated Molecular Patterns (PAMPs) or Damage-Associated Molecular Patterns (DAMPs) (1). Either may trigger inflammation by recognition through Pattern Recognition Receptors (PPRs) on innate and adaptive immune cells (2). Within these cells, the nuclear translocation of the transcription factor Nuclear Factor kappa beta (NF-κB) promotes pro-inflammatory cytokine production to include IL-1 and TNF-α (3). These cytokines act upon a variety of target cells (4) that can begin a cascade of increasing pro-inflammatory cytokines, such as IL-6, IL-8, and MIF (5). Increased production of these cytokines can result in the induction of fever (6), the upregulation of adhesion receptors like ICAM-1 on endothelial cells (7), increased L-selectin (CD62L) on lymphocyte cells (8) and neutrophil mobilization. Excessive inflammation can lead to excessive damage tissue and organs, e.g. by the release of radicals by neutrophils (9) to include the lungs, heart, kidney, liver and brain (10). In addition, disseminated intravascular coagulation (DIC) and thrombocytopenia (11) can occur, which are known to contribute to multiple organ dysfunction syndrome (MODS) (12). This cytokine driven initial inflammation can lead to early death if not compensated (13).
Concurrent with the pro-inflammatory response, there is evidence of a number of immune suppressive events occurring. These include the loss of conventional T lymphocytes (14) simultaneous with the lack of regulatory T cell apoptosis (15), the increased number of myeloid suppressor cells (16) coupled with the down regulation of monocyte HLA-Dr (17), and a loss of dendritic cells (18). Additionally, there are increases in immune suppressive cytokines such as IL-10, TGF-ß, and TSLP. Significant immune suppression can impair the proper clearance of pathogens leading to higher bacterial loads both systemically and in tissues. According to a recent clinical observational cohort study, septic patients can develop chronic critical illness, with a 6-month survival of 63%, and continue to demonstrate cytokine profiles of chronic inflammation, as well as biomarker profiles characteristic for persistent immunosuppression (19). Additionally, these patients very often require treatment in long term acute care facilities, and by one year after hospital discharge half of them have died and another quarter remains bedridden (20). Elder patients are more likely to develop these immune disbalances (21). It is therefore key to better understand these pathophysiologies as they become an increasing problem for aging societies (22). It is beyond the scope of this review to detail the entire host response to sepsis and this topic is covered in a number of other reviews (23–25). Here, this review focuses upon release, impact, and potential therapy of a limited number of potent anti-inflammatory cytokines.
Mediators of Immunosuppression
The initial upregulation of inflammation after onset of infection or injury results in the activation of the innate immune system, mostly of neutrophils and macrophages via Toll like receptors (TLRs) (26, 27). Orchestrated by T cells, they maintain inflammation by the release of pro-inflammatory cytokines such as TNF-α, IL-6, IL-8, MCP-1, MIP-1α (28, 29), among which IL-6 plays a prominent role as it is associated with injury severity (30), MODS and death (31). At the same time, immunosuppression is induced (13), which is strongly driven by anti-inflammatory cytokine release (32). Immunosuppression is characterized by the induction of immunosuppressive functions and partial impairment of pro-inflammatory functions of innate and adaptive immune cells: lymphopenia occurs as the number of circulating lymphocytes decreases due to apoptosis of CD4 and CD8 T cells, B cells, and NK cells (33), Regulatory T cells numbers increase after sepsis or trauma (34, 35), T cells become exhausted and anergic (36, 37), monocytes, macrophages and dendritic cells become impaired in their ability to mount a proinflammatory response (22, 38, 39), neutrophil dysfunction ensues (40) and increased release and activation of immunosuppressive Myeloid Derived Suppressor Cells (MDSCs) is triggered (41). These cells, along with other tissue cells, such as endothelial cells, potently mediate immunosuppression by the release of anti-inflammatory cytokines. The cytokines IL-10, TGF-β and TSLP all exert potent anti-inflammatory functions (42–44). In trauma and sepsis, IL-10 is considered a key immunosuppressive cytokine and correlates with injury severity and outcomes (45) and is widely examined in sepsis and trauma studies. However, it has been demonstrated that all three cytokines also exert pro-inflammatory functions under certain conditions (46–48). This seems contradictory as they are considered key regulators of immunity. In the following chapters, we will review their immunosuppressive mechanisms and dynamics in sepsis and trauma. Finally, the pathological conditions in sepsis and trauma that may drive a detrimental release of these cytokines will be examined.
Immunosuppressive Cytokines: Il-10, TGF-β, and TSLP
IL-10
How Does IL-10 Affect Immune Cells?
With its potent anti-inflammatory functions IL-10 is considered an important regulatory cytokine protecting the host from exaggerated inflammation, autoimmunity and allergy (49, 50). It is a V-shaped homodimer and has a molecular weight of 37 kDa (51) and it is a key member of the IL-10 superfamily which consists of IL-19, IL-20, IL-22, IL-24, IL-26 and the type III IFN-γ subfamily (51).
In sepsis and trauma, IL-10 mainly affects immune cells rather than tissue cells, which do not express the IL-10-receptor (IL-10R) to the same extent. Tissue cells are more affected by other member of the IL-10 super family, particularly IL-22 (50). Many variants of the IL-10 gene exist, and an increasing body of literature suggests that IL-10 gene polymorphisms play an important role in different pathological conditions (52). Tumor cells can also produce IL-10 and it is thought that this allows the tumor to mitigate an effective immune response (51).
The IL-10 receptor is a composition of the IL-10R2 and IL-10R1 subunits, with the latter being expressed mainly on immune cells. The expression of the receptor differs from cell type to cell type. For example, some monocytes and macrophages express very high amounts of the receptor, and the expression in memory T cells is higher than in naïve CD4 T cells (53). The IL-10 cytokine binds to IL-10R1, which subsequently forms a complex with IL-10R2. It is of note that IL-10 can bind to IL-10R1 alone but not to IL-10R2. However, the affinity for the complex is much higher than for IL-10R1 alone (54). The IL-10 receptor complex then phosphorylates STAT3 via Jak1 and Tyk2 activation but also STAT1 and STAT5. Among these, STAT3 seems to be most important for modulating the downstream transcription of target genes (42, 51, 54). It is of importance that STAT3 also acts downstream from IL-6, so the resulting modulation depends on A) the cell type, B) the competing stimuli the cell receives and C) the duration of the stimuli (42, 51, 54). Inflammation is potently suppressed by IL-10 through inhibition of NF-κB DNA binding activity (53). IL-10 also hinders TLR signaling through induction of inhibitory miRNAs targeting MyD88-dependent TLR4 signaling and through promoting the ubiquitin-mediated degradation of IRAK1/4 and TRAF6. MyD88-independent attenuation was also reported (55). On a translational level, IL-10 leads to an increase of tristetrapoline, a protein which downregulates mRNA of pro-inflammatory cytokines and can be down-regulated itself in TLR-dependent pathways (55). However, IL-10R binding also limits its own effect by inducing suppressor of cytokine signaling (SOCS)3, which forms a negative feedback loop by inhibiting, amongst others, STAT3 phosphorylation (56). IL-10 also suppresses the production of pro-inflammatory cytokines TNF-α, IL-1, IL-5, IL-8, IL-12, GM-CSF, MIP-1α and MIP-2α in monocytes, macrophages, neutrophils and NK cells (57).
A wide range of immune cells can produce IL-10, such as dendritic cells (DCs), macrophages, mast cells, natural killer cells (NK), eosinophils, neutrophils, B cells, CD8 T cells, type 1 T-helper cells (Th1), Th2, and Th17 CD4 T cells, regulatory T cells, TCRαβ+ double negative (CD4- CD8-) T cells (DN T cells), and myeloid derived suppressor cells (MDSCs) (49, 50, 58).
The effect of IL-10 on innate and adaptive immune cells differs. Macrophages and monocytes are suppressed by IL-10 through the down regulation of MHC II and costimulatory molecule expression (CD80, CD86, and CD40), both constitutively and in response to IFN-γ (49, 51, 59, 60). This prevents macrophages and monocytes from presenting their respective antigens to Th1 T cells, which in turn do not become activated to orchestrate an immune response. Additionally, the NO production of macrophages is reduced, adhesion to endothelial cells is hindered and the immunosuppressive M2-type is induced (51, 54). Interestingly, IL-10 promotes macrophage phagocytosis of cell debris (54).
Unlike macrophages, neutrophils need to be primed in order to become responsive to IL-10. Neutrophils express IL-10R2 constitutively but can only form the IL-10R complex when IL-10R1 gets upregulated after stimulation. Experimentally, this was determined to take about 4 h (61). Therefore, an immediate response to IL-10 is not possible in non-primed circulating neutrophils. Another important effect that can be seen upon local administration of IL-10 in inflammatory models is that IL-10 prevents the recruitment of neutrophils to the site of injury. Neutrophil migration was not directly hindered by IL-10, but the production of chemoattractants by macrophages or the affected organ tissue was decreased (61, 62). The inhibition of neutrophil apoptosis has been shown to be counter-regulated by IL-10 in severe sepsis (63). Further, it has been demonstrated that IL-10 can inhibit the production of TNF-α and IL-1β in neutrophils when stimulated with LPS or by the phagocytosis of bacteria (64). Further, ROS production seems to be diminished by IL-10 (54). However, further experiments are needed to systematically elucidate the impact of IL-10 on neutrophil functionality. Interestingly, under certain conditions IL-10 induces its own production in neutrophils and macrophages, which is thought to be a positive feedback loop in innate immunosuppression (65). This is of interest as neutrophils themselves are contributing significantly to IL-10 production as shown in a murine model of abdominal sepsis (66).
In dendritic cells, MHC II expression and therefore antigen presentation and activation of naïve CD4 T cells is impaired by IL-10 (54). This potently inhibits CD4 T cell functions. It also diminishes the production of T cell pro-inflammatory cytokines, such as IL-2 and IFN-γ (51). Further, IL-10 blocks proliferation of Th17 cells and inhibits Th2 cells (42, 67). It also promotes the proliferation and function of Regulatory T cell subsets. The generation of Type-1 regulatory T cells (Tr1 T cells) from naïve T cells is induced by IL-10 and it promotes survival and function of CD4+ FoxP3+ Tregs (49, 67). For example, Treg expression of FoxP3 appears to be IL-10 dependent (68). Interestingly, IL-10 suppresses thymic lymphocyte apoptosis in sepsis (69) and has stimulatory effects on CD8 T cells. When stimulated with IL-4 and/or IL-2, CD8 T cells showed increased proliferation (51). Recent murine studies showed increased IFN-γ production by CD8 T cells upon IL-10 administration (53, 70). In clinical trials it could even be shown that modified IL-10 reduces tumor burden in renal cell cancer (71). The application of modified IL-10 enhanced intratumoral CD8 T cell expression of IFN-γ and Granzyme B (42, 71) and thereby promoted anti-tumor cytolysis. However, a final conclusion cannot be drawn yet as IL-10 does also have tumor-supportive functions, i.e., enabling tumor cells to escape an effective immune response (51).
Another pro-inflammatory function of IL-10 can be seen in B cells, as it promotes their survival, proliferation and differentiation into plasma cells (51, 53). Conflicting data exists about the effect of IL-10 on NK cells. It has been shown that IL-10 suppresses TNF-α and IFN-γ production by NK cells, but promotion of IFN-γ production has also been reported in in vivo and in vitro models (51, 53, 72). Most likely, the predominant conditions, timing and dose in the respective organ or tissue lead to increased or diminished functionality.
Different effects following systemic or local administration of IL-10 could also be utilized therapeutically. Two murine trauma studies showed that IL-10 locally administered via inhalation in systemic inflammation attenuated pulmonary inflammation with little or no systemic side effects (62, 73). Altogether, additional research will be needed to elucidate the seemingly paradoxical findings regarding the effect of IL-10 on multiple types of immune cells. The effects of IL-10 depending on the mode of administration are highlighted in Figure 1 .
Figure 1.

The effect of therapeutic IL-10 modulation depends on the mode of administration and dynamics. The systemic or local application of recombinant IL-10 have experimentally shown beneficial effects. (A) Inhaled IL-10 attenuated pulmonary inflammation by preventing detrimental neutrophil recruitment into the lung (61, 62, 73). (B) Moreover, IL-10 suppresses thymic lymphocyte apoptosis in sepsis (69) and has stimulatory effects on CD8 T cells (53, 70) and B cells (51, 53). Both modes of application might represent future therapeutic approaches in humans. (C) The dynamics of the immune perturbation in trauma and sepsis was modeled in recent literature (74). In general a time dependent modulation of IL-10 levels might restore immune hemostasis (75–77). Animal models suggest a beneficial effect of early IL-10 application (76) and the correlation of elevated IL-10 levels and injury severity suggests beneficial effects by IL-10-blockage in later phases (31, 45, 78). The figure contains adapted graphics from Les Laboratoirs Servier - Medical Art under the terms of the Creative Commons Attribution License (CC BY) for non-commercial use. The use, distribution or reproduction in other forums is permitted: https://creativecommons.org/licenses/by/3.0/legalcode, last accessed July 30th, 2020.
Dynamics in Trauma and Sepsis
The immune perturbation in trauma and sepsis is characterized by the dichotomy of a potent pro-inflammatory upregulation of immunity and immunosuppression (13). During immunosuppression, anti-inflammatory cytokines including IL-10 are released. In search for a marker predicting the severity and outcome of the systemic inflammation, IL-10 has been studied in many animal and clinical studies. There are three broad categories that have been investigated: 1) traumatic injury (mostly burn) 2) sepsis and 3) major surgery, which all lead to a systemic inflammation.
Although studies examining trauma and sepsis do not examine the same clinical conditions leading to systemic inflammation, they share many similarities in the expression of IL-10. Some studies did not show significant differences in IL-10 serum expression between healthy controls, septic patients and patients with septic shock (75, 79). However, the major body of clinical studies examining sepsis does demonstrate this phenomenon.
Huang et al. quantified IL-10 levels in 106 burn patients and showed increased IL-10 levels in septic vs non-septic patients (45). In another study Stensballe et al. examined 265 trauma patients. They found a significant increase of IL-10 levels in patients not surviving 30 days and a positive correlation of IL-10 levels and the injury severity score (ISS) (78). A further multicenter study including 54 polytrauma patients found that elevated IL-10 levels and an elevated IL-6/IL-10 ratio were directly proportional with MODS and mortality (31). In a clinical trial, Chen et al. studied 37 thermally injured patients with a total burn surface area >20% and found increased IL-10 levels in patients who developed sepsis versus those that did not (80).
The team around Gogos et al. included 65 patients with severe sepsis in their trial and found a positive association between serum IL-10 levels, and a high IL-10 to TNF-α ratio and death (81). In another large clinical study of 153 patients with severe sepsis and septic shock, the levels of IL-10 were significantly higher in patients that died within 48h but not for patients that died between 48h and 28 days or after 28 days (82). A separate clinical study by Frencken et al. included 708 patients with severe sepsis or septic shock in the ICU and demonstrated that higher IL-10 levels were positively associated with mortality (83). Another sizable study from Van Vught et al. in 2016 found overexpression of IL-10 signaling genes in septic patients admitted to the ICU. They did not find differences in the gene expression of IL-10 signaling between patients who later developed ICU-acquired infection compared to those who did not (84). Serum IL-10 levels were not measured, and translational and post-translational regulation of IL-10 expression was observed (85). From these results we extrapolate that sepsis appears to increase IL-10 production and signaling. Whether this indicates adverse outcomes in terms of secondary infection in septic patients has to be examined by assessment of their serum levels.
In cases of sterile trauma associated with major surgery, 28 out of 40 studies found increased systemic IL-10 levels in patients (28). An additional review by Easton and Balogh found a similar conclusion (86).
Together, these studies overwhelmingly suggest that detrimental outcome and elevated IL-10 production are associated. Further, some studies provide interesting additional findings. First, IL-10 levels correlate with the severity of the injury: elevated IL-10 serum levels correlated with the ISS of trauma patients (78), were directly proportional with MODS and mortality in a trauma population (31), showed linear correlation with the increasing percentage of burned total body surface area (45) and the IL-10/lymphocyte ratio was even correlated with the APACHE II score in severe septic patients (87). Secondly, the increase in IL-10 can be seen almost immediately after the onset of trauma, sepsis or surgery. Blood draws of the aforementioned studies were conducted either at admission to the ER or ICU (78, 83) or within 24 h after onset of the trauma, disease or surgery (45, 82, 87). This is of clinical relevance if IL-10 is used to assess the immunological disturbance in a patient at admittance in the ER or the ICU.
Effects and Drivers of Pathological Release in Trauma and Sepsis
The regulatory anti-inflammatory functions are considered a protective counter-regulation to prevent excessive inflammation in the balance of immune hemostasis. However, IL-10 does make the host more susceptible to overwhelming infection if it impairs a proper immune response, and thus has been associated with worse outcomes in infection and cancer (54, 88). It is currently unclear when and how supportive stimulating functions and protective anti-inflammatory regulation deteriorate under pathologic conditions. Therefore, three major questions should be addressed in future studies. A) Which stimuli drive IL-10 expression and how are these signals transmitted in the cells? B) What are the dynamics of IL-10 expression and when does IL-10 become detrimental to an orchestrated clearance of bacteria and cell debris? C) Which cell type drives IL-10 expression in which diseases?
It is known that DAMPs and PAMPs induce IL-10 production by a variety of receptors, that different bacterial pathogens provoke different expression patterns and that IL-10 regulating transcription factors differ in different T cells subsets, macrophages and neutrophils (54). In addition, IL-10 expression is also regulated post-transcriptionally (51). In myeloid cells, TLR-induction, including 2, 4, 5, 7 and 9, drives IL-10 production (54, 89). It is important to differentiate between macrophages and neutrophils in the downstream signaling as they differ. A study conducted by Tamassia et al. found that the MyD88-independent pathway is not mobilized after TLR4 stimulation in human neutrophils (90).
Further, the understanding the dynamics of IL-10 expression is essential. Several studies showed time-dependent effects of IL-10 administration or inhibition in sepsis models (75–77). Given the fact that IL-10 expression is upregulated quickly after trauma and sepsis, as described in the previous section, it seems likely that in the beginning, lower levels of IL-10 may stimulate bacterial clearance but impair functions when the serum levels exceed a certain threshold. A murine study showed increased survival in septic mice when treated with IL-10 up to 6h after the onset of sepsis, which extended the therapeutic window to conduct surgical infectious site control until 30 h after the commencement of sepsis (76).
CD4+FoxP3+Tregs are considered an important source of IL-10 in certain sepsis models (45). It is likely that the dynamic of the apoptosis of T cells in general, and the corresponding proportional changes in the T cell population affect proper IL-10 production and distribution of IL-10 (91). It is important to know which cell type contributes the most to IL-10 production in different diseases. As CD4+FoxP3+Tregs reveal an increase in frequency after trauma it is likely that they significantly contribute to IL-10 production (45). However, T cell apoptosis and murine sepsis models question this assumption. In murine abdominal sepsis, neutrophils are significant producers of IL-10 (66, 92). Of note, in murine burn models, a significant number of IL-10 producing neutrophils were found in the spleen post-injury (54). It has been shown that not only murine- but also human neutrophils can produce IL-10 (65).
Besides CD4+FoxP3+Tregs and neutrophils, also macrophages can produce IL-10 (93) and TGF-β (94) after phagocyting apoptotic antigen presenting cells (APC’s), a process coined efferocytosis (95). In this process APC’s, such as neutrophils clear bacteria via phagocytosis, become apoptotic and then are ingested by macrophages (96). This serves to resolve the inflammation, tissue repair and restores immune homeostasis (94, 97). Efferocytosis has been shown be impaired in sepsis, and if promoted seems to improve outcome in sepsis (98, 99) and trauma (100). In some (98, 101) but not all (99) sepsis and infection models an increase of IL-10 was shown. What remains to elucidate is whether macrophage efferocytosis contributes to chronic immunosuppression by the release of IL-10 and TGF-β. To our knowledge no studies have yet been conducted examining their role in long term (>7 days) immunosuppression after trauma or sepsis.
To assess which cell type contributes the most to IL-10 production is technically difficult. However, this should be addressed when assessing IL-10 mediated effects in the respective pathological condition, as it is necessary for evaluating any potential therapeutic approaches. Modulation of IL-10 has shown promising results in several clinical trials on autoimmune diseases (42). However, the chronic nature of these diseases and the steadier production of IL-10 varies from the highly dynamic and heterogenous pathology in systemic inflammation due to sepsis or trauma. This may provide some understanding on why past attempts to modulate IL-10 in murine sepsis models did provide conflicting results regarding their outcome (69, 102, 103). Human interventional experimental endotoxemia trials (104, 105) and in vitro functional assessment of the innate and adaptive immune system of sepsis patients (46), both modulating IL-10 levels, could not yet provide a clear rationale for IL-10 modulation in a specific inflammatory disease that would allow clinical trials modulating IL-10 production in sepsis and trauma. A better understanding of the dynamics and the producing cell types would significantly improve diagnostics and therapeutic advances.
Potential Therapeutic Modulation of IL-10
Regarding the dynamics and functionality of IL-10, we conclude that two therapeutic modulations of IL-10 in trauma and sepsis seem promising: First, inhalation of IL-10 to treat Acute Respiratory Distress Syndrome (ARDS). This pathology occurs from pulmonary infection or systemic insults such as systemic inflammation in trauma and sepsis (106). Neutrophil infiltration potently contributes to pulmonary inflammation in ARDS and leads to significant pulmonary damage (106). In two different murine trauma models inhaled IL-10 attenuated pulmonary inflammation without systemic immunosuppression by preventing the recruitment of neutrophils to the site of injury through reducing the production of chemoattractants by macrophages or the affected organ tissue (61, 62, 73)
Secondly, systemic administration of recombinant IL-10 for the treatment of sepsis can be considered. However, this approach must be carefully executed as the systemic effects are not yet fully understood. Animal studies show that the effects of IL-10 modulation depend on the injury dynamic in sepsis (75–77, 102). Murine abdominal sepsis studies suggest that the administration of IL-10 within hours after septic onset may have beneficial effects (76, 102). An endotoxemia model in primates seem to support these findings, as early intravenous injection of recombinant human IL-10 significantly decreased TNF-α, IL-6, IL-8 and IL-12 release (107). If this results in a better outcome still has to be examined in further studies, as another murine abdominal sepsis model did not find a beneficial effect of IL-10 administration on mortality (108). In the acute phase IL-10 is suggested to promote protective immunosuppressive effects, whereas in later phases of sepsis increased IL-10 is depicted as a marker for detrimental outcome in many human studies (45, 78, 81, 83). Blockage of IL-10 production or function might constitute a therapeutic approach at this point, as some animal models suggest (102). Pharmaceutic drugs for IL-10 modulation are already in clinical trials. An overview of current IL-10 modulating drugs is provided in Table 1 . These might be considered to use to augment IL-10 function in very acute sepsis phases (Pegilodecakin: PEGylated recombinant human IL-10 (LY3500518 or AM0010); Phase 3 NCT02923921: pancreatic cancer) or to impair its function in later phases (AS101: tellurium based small compound with immune-modulating characteristics attributed to direct inhibition of IL-10; Phase 2 NCT00926354: treatment of thrombo-cytopenia in solid tumor patients). How a potential dynamic modulation of IL-10 might look like is outlined in Figure 1 .
Table 1.
Clinical Trials targeting IL-10.
| Cytokine | Drug | Sponsor | Description | Indication | Clinical trial |
|---|---|---|---|---|---|
| IL-10 | Prevascar | Renovo | Recombinant human IL-10 | Wound healing | Phase 2 NCT00984646 |
| AS101 | BioMAS Ltd | Tellurium based small compound with immune-modulating characteristics attributed to direct inhibition of IL-10 | Treatment of thrombo-cytopenia in solid tumor patients | Phase 2 NCT00926354 |
|
| T-allo10 | Stanford University | Donor-derived CD4+ T cells which contain a high proportion of host alloantigen specific Tr1 cells | Therapy to prevent GvHD and induce graft tolerance in hematopoietic stem cell transplant | Phase 1 NCT03198234 |
|
| Pegilodecakin (LY3500518 or AM0010) | Eli Lilly and Company | PEGylated recombinant human IL-10 | Pancreatic cancer | Phase 3 NCT02923921 |
Before application of any kind of IL-10 modulation, a very detailed and dynamic assessment of the individual’s immune status must be conducted.
TGF-β
How Does TGF-β Affect Immune Cells?
The TGF-β cytokine exits in three isoforms (β1, β2 and β3) and steers multiple immune processes such as immunosuppression and peripheral homeostasis in autoimmunity and infection (109, 110). Leukocytes mainly express the TGF-β1 isoform (48). It regulates a number of different immune cells and importantly T cells. Their function is mediated by the TGF-β receptor complexes type I and II with TGF-β binding first to type II, then I. Signals of these receptors are mediated by the SMAD transcription factors (43).
Many genetic knockout studies in the past have shown that TGF-β has an important influence on T cells. It can impair T cell proliferation and differentiation, except for Tregs whose proliferation is paradoxically stimulated by TGF-β. It inhibits the proliferation and differentiation of Th1, Th2 and cytotoxic T cells by suppressing, amongst others, the transcription factors T-bet, Gata-3 and c-myc. Further, IL-2 expression as a crucial stimulus for T cell proliferation is suppressed by TGF-β as well (48). TGF-β also promotes the development of Th17, Th9 and T follicular helper cells in conjunction with other Interleukins, but blocks the development of Th1 and Th2 cells (109). The ability of TGF-β to suppress T cell proliferation depends on the activation status of the T cell, which was demonstrated in a study by Cottrez and Groux (111). The proliferation and cytokine secretion in resting, but not in activated T cells is inhibited by TGF-β1. The latter down regulate TGF-β receptor type II, which interestingly could be upregulated again by IL-10 restoring their responsiveness to TGF-β1 (111). TGF-β regulation of T cell survival appears to be dependent on CD28 co-stimulation. In the presence of CD28 agonist TGF-β inhibits T cell apoptosis and enhances their expansion. In its absence, TGF-β inhibits the TCR-stimulated proliferation of naïve T cells (48). Moreover, TGF-β1 supports survival of T cells with memory and effector functions (48), although this seems to contradict its role in the above-mentioned regulation of T cell differentiation.
The role of TGF-β in the regulation of Tregs cells appears to be less contradictory. TGF-β is necessary for the induction of thymic (109) and peripheral Tregs, which develop by antigen-specific stimulation in the presence of TGF-β (112). TGF-β induces the expression of FoxP3 and the production of IL-10 in Tregs (113, 114). Tregs themselves secrete TGF-β forming a positive feedback-loop (50, 115).
Additionally, dendritic cells mediate the induction of peripheral Tregs via TGF-β (116). If dendritic cells themselves are exposed to TGF-β in a pro-inflammatory setting, they show impaired production of pro-inflammatory mediators (117). Similar to dendritic cells, TGF-β strongly influences monocytes and macrophages by downregulating their pro-inflammatory functions when activated (48). However, the effect of TGF-β on monocytes and macrophages strongly depends on their activation. In general, TGF-β promotes monocyte function when resting. For example, adhesion molecules, matrix metalloproteinases and IL-6 production is promoted by TGF-β in monocytes (48). In contrast, when activated monocytes become macrophages, TGF-β can have a dampening effect on their pro-inflammatory functions in that it impairs their ability to produce LPS-induced TNF-α, MIP-1α, MIP-2 and reactive oxygen and nitrogen species (48). Another important contribution to uphold inflammation is the ability of macrophages to present antigens to T cells. TGF-β1 downregulates the IFN-γ-induced expression of MHC II mRNA and antigen presentation in macrophages (118). This might serve to prevent excessive and therefore harmful activation of T cells in inflammation.
The impact of TGF-β on neutrophils is poorly understood. In cancer, TGF-β polarizes neutrophils from an antitumoral N1 type to a pro-tumoral N2 type (119). However, TGF-β can also promote neutrophil oxidant production (120). Of note, TGF-β is a strong chemoattractant for monocytes, eosinophils, mast cells and neutrophils (48). The immunosuppressive effects of TGF-β on immune cells are outlined in Figure 2 .
Figure 2.
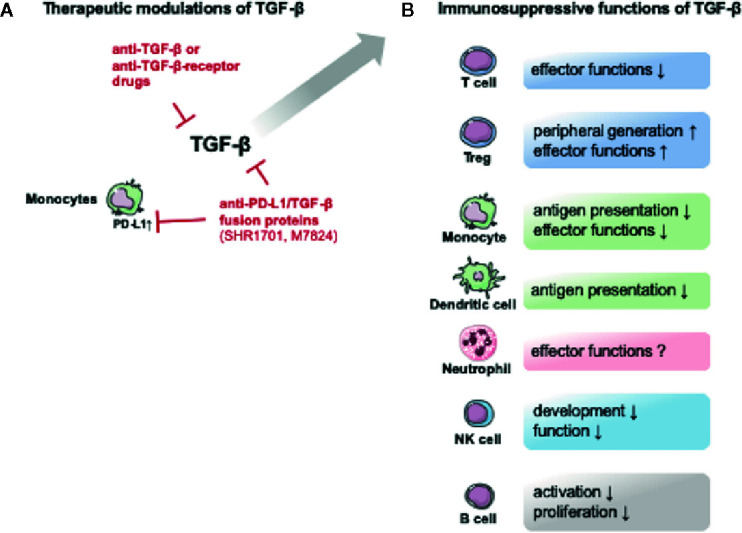
Potential therapeutic TGF-β modulating drugs and the immunosuppressive functions of TGF-β. The figure provides an overview over potential therapeutic approaches and the immunosuppressive effects of TGF-β. (A) Several drugs targeting TGF-β or its receptors are currently being tested in non-septic patients. Interestingly an anti- programmed cell death 1 ligand 1 (PD-L1)/TGF-β fusion protein is also being tested. The upregulation of PD-L1 is associated with monocyte dysfunction in post-traumatic and septic immune dysfunction (32). (B) TGF-β exerts immunosuppressive effects on both innate and adaptive immune cells. The figure contains adapted graphics from Les Laboratoirs Servier - Medical Art under the terms of the Creative Commons Attribution License (CC BY) for non-commercial use. The use, distribution or reproduction in other forums is permitted: https://creativecommons.org/licenses/by/3.0/legalcode, last accessed July 30th, 2020.
Dynamics in Trauma and Sepsis
Given the fact that TGF-β is a potent regulator of immune responses and is considered an important cytokine released by Tregs, the question arises if it is a main regulator in trauma and sepsis and if its dysfunction in these conditions contributes to a disturbed immune response.
Several studies provide contradictory findings. In 2004, Monneret et al. conducted a study examining 38 adult septic patients with septic shock and found that TGF-β levels were decreased compared to normal values and concluded that the general immunosuppressive state they found the patients in is more likely attributed to increased IL-10 levels (121). In addition, the group suggested that no prognostic information could be obtained from TGF-β levels (121). In further support of this, an additional study did not find any prognostic value of TGF-β2 to determine survival in patients with gram-positive septicemia (122). While TGF-β2 levels were increased in the non-survivor group compared to the survivor group in gram-positive septicemic patients, this difference was not statistically significant (122). Gram-negative septicemic patients showed a trend towards decreased TGF-β levels, but this was also not statistically significant (122). Due to the low numbers of patients within each group (11 or more) the study is potentially underpowered to reveal statistically significant differences (122). Of note, TGF-β1 and not TGF-β2 seems to have the largest impact on T cell function and leukocytes mainly express the TGF-β1 isoform (48). A further trial observing gastrointestinal surgery patients with and without infectious complications could not detect serum TGF-β in most of the samples (103). Burn patients examined for IL-10 and TGF-β1 serum levels showed an initial peak in serum TGF-β1 within one day post-burn and a second peak in which the serum levels of the survivors were higher than in the non-survivor group. No differences were found between patients with burned total body surface area (TBSA) of greater or less than 50% (123). However, a limitation in this study of 15 patients is that the surviving group was younger and suffered less severe burn injury with regard to the TBSA (124).
Other studies found elevated TGF-β levels in septic patients. One study has demonstrated elevated TGF-β1 levels in 26 patients at the time of sepsis diagnosis when compared to healthy controls (125). A more recent study from Huang et al. examined Treg function and IL-10 and TGF-β1 serum levels in 106 burned, septic and non-septic patients with a TBSA of more than 30% (45). They revealed a significant increase in TGF-β1 levels in septic patients compared to non-septic patients and in non-survivors compared to survivors (45). They also stratified the patients in three groups depending on the size of the burn. Serum TGF-β1levels were positively associated increasing TBSA (45). From all these studies, the latter examined the biggest population, therefore making it likely that there is indeed a systemic increase in TGF-β in severely injured and septic patients. This view is supported by studies examining specific organ failures in septic populations. A study from De Pablo et al. revealed that increased TGF-β1 levels were positively associated with sepsis induced ARDS with fatal outcome. The TGF-β1 levels were increased in patients 7 days after ICU admission with sepsis and ARDS compared to patients with sepsis without ARDS. In addition, patients with septic shock associated ARDS showed significantly higher TGF-β1 levels in non-survivors than in survivors (126).
In rodent sepsis models, findings concerning TGF-β serum levels are less contradictory then in human studies. Increased levels of TGF-β were found in peripheral blood of septic mice (127). Nullens et al. conducted a murine sepsis model in which lymphocyte depletion occurred in all tissues examined (spleen, mesenteric lymph node, Ileum, colon) at day seven, coinciding with increased levels of IL-10 and TGF-β (128). In a rat sepsis model, increased levels of TGF-β were observed in the circulation and in adherent splenic cells (129). Additionally, supporting the findings in the human study mentioned above from De Pablo et al., Xu et al. reported a protective role of curcumin, most likely by decreasing the expression of TGF-β1 and the SMAD3-dependent signaling pathway (130). Plasma TGF-β1 levels were significantly increased in the septic group and were downregulated by curcumin treatment. Histology showed curcumin treatment prevented some sepsis-induced lung damage (130). These results suggest that TGF-β plays a detrimental role in sepsis most likely by impairing a proper immune response. Yet TFG-β1 might exert detrimental effects on the host by other cells. A study examining the effect of TGF-β on liver cells when challenged with LPS revealed that it promotes the release of proinflammatory cytokines, e.g. IL-6, by the liver cells, which led to a higher mortality (131). Inducible TGF-β1-transgenic mice that express TGF-β1 under control of the C-reactive protein promoter were used in this study (131). Therefore, future studies will be needed to confirm this mechanism in mice that were not genetically modified.
In contrast, other studies found beneficial effects of TGF-β in sepsis. For example, treatment with TGF-β blocked endotoxin-induced hypotension and improved survival in rat models using Salmonella typhosa and Salmonella enteritidis to induce septic shock and a murine endotoxic shock model (4). Moreover, cardioprotective effects of TGF-β could be shown, as TGF-β reverses the depression of myocyte contraction in vitro (132). Another study found TGF-β1 to decrease neutrophil numbers during the onset of LPS-induced acute lung injury (133). This was due to increased apoptosis rather than reduced migration. TGF-β1 did not directly regulate neutrophil apoptosis but instead promoted IL-6 release from mast cells, which promotes neutrophil clearance (133).
Given the partially contradictory findings in the different human studies, there still lacks a definite answer to the question of whether systemic TGF-β release contributes to a systemic detrimental immune disturbance in sepsis. The unambiguous findings in animal models and the findings of Huang et al. including 106 patients, in which TGF-β serum levels were increased in septic patients (45), suggest that TGF-β does play an important role. Even more so as in the current understanding of sepsis, the action of Tregs is considered important for the outcome. Tregs are considered an important source of TGF-β. Moreover, the association of increased TGF-β levels and impaired lung function seems to be relevant in this context, considering that ARDS is a major clinical challenge in septic patients. The local effect of TGF-β in the different tissues has to be examined in the context of trauma and sepsis. This is of importance, as other diseases like rheumatoid arthritis that TGF-β acts very differently when administered locally or systemically (48). Future studies need to address the dynamic expression of this cytokine during sepsis. Several studies mentioned a peak in the beginning of sepsis development (4, 123). This might be due to an overwhelming stimulation of TGF-β producing cells in the beginning of inflammation, or an increase in apoptosis of T cells which are known to create an immunosuppressive environment (134). This could lead to a pathologic immunosuppression at the onset of inflammation. Lastly, the role of TGF-β may differ in sepsis and sterile trauma. Therefore, future studies should also address differences in non-sterile and sterile settings to evaluate appropriate therapeutic action in the different diseases.
Effects and Drivers of Pathological Release in Trauma and Sepsis
Assuming an adequate response to an infection, we postulate the role of TGF-β as follows: First, it acts as a chemoattractant for myeloid cells in the very beginning of infection to promote initial innate immune response. This underlying premise is based upon that TGF-β acts in a stimulatory manner on resting monocytes (48). Further, TGF-β impairs the activation of naïve T cells but can be overridden by CD28 co-stimulation in addition to low IL-2 doses (48). The team around Li et al. argues that this circumstance serves as a threshold to divide between self-antigen driven steady state inflammation, which is hindered by TGF-β, and a pathological condition in which the above mentioned stimuli override TGF-β inhibition to guarantee a proper immune response to the infection (48). As stated above, TGF-β curbs macrophages in inflammation which might serve to restore hemostasis and prevent excessive inflammation.
The data suggest that these regulated dynamics are severely impaired in sepsis. In the study from Hiraki et al., a murine abdominal sepsis model was utilized with a TGF-β depleting antibody administered 6 h after the intervention, which lead to improved survival of the mice (103). The group also showed a positive correlation between the percentage of Tregs of all CD4 T cells and the serum TGF-β levels (103). It is known from human clinical studies that an increased percentage of Tregs is correlated with worse outcome (45, 135). The group around Hiraki et al. therefore postulates that the disruption of TGF-β leads to a decrease of Treg abundance which might be harmful in the development of sepsis (103). It is also possible that TGF-β itself plays a detrimental role by depressing T cell functions and disrupting the positive feedback loop that promotes Treg proliferation (50, 115). The study did not examine which cell type provides the major source of TGF-β. It is assumed that the initial release of auto- and or alloantigens released when infection occurs leads to such a massive TGF-β release that homeostasis as described above can no longer be restored. Currently, it is not clear whether this is driven by DAMPs or PAMPs. In the study, the administration of the depleting antibody led to the restoration of this homeostasis. Of note is that a single administration was enough. A current study from Zeng et al. supports these findings (136). An anti-TGF-β antibody was administered, which led to increased IL-2 and IFN-γ levels. This was considered beneficial for the outcome. The authors, however, neglected to provide the timepoint of the anti-TGF-β-antibody administration (136).
Altogether, these studies contribute to the understanding of the role of TGF-β and Tregs in the initial phase of sepsis. However, it is not possible to draw a definite conclusion from what is currently known, given the fact that the effects of TGF-β differ a lot depending on location, timing and the activation status of the cells it affects as described in the section above.
Potential Therapeutic Modulation of TGF-β
Currently, TGF-β dynamics in human sepsis studies need to be expanded before a therapeutic modulation of TGF-β or its receptors should be applied. The focus of studies should be on the dynamics and the origin of sepsis (organ/tissue). In current clinical trials, a number of drugs are tested that might prove useful in the future. An overview of current TGF-β modulating drugs is provided in Table 2 . These include direct anti-TGF-β antibodies (e.g. Fresolimumab: Human monoclonal anti-TGFβ-antibody (Non-small cell lung carcinoma), Phase 1/2 NCT02581787) and receptor blockage (Galunisertib (LY2157299): small molecule inhibitor of the kinase domain of Type 1 TGFβ receptor (solid tumor, non-small cell lung cancer, hepatocellular carcinoma), Phase 1/2 NCT02423343). Interestingly, an anti- programmed cell death 1 ligand 1 (PD-L1)/TGF-β fusion protein is also being tested (e.g. SHR-1701: anti-PD-L1/TGFβ fusion protein (advanced solid tumors), Phase 1 NCT04324814). If TGF-β blockage proves beneficial in trauma or sepsis, application of this drug would be a very interesting approach as the upregulation of PD-L1 is associated with monocyte dysfunction and upregulation of its ligand programmed cell death protein 1 (PD-1) in lymphoid cells is a detrimental hallmark of post-traumatic and septic immune dysfunction (32). A first anti-PD-1 antibody (nivolumab) has already been tested for safety in septic patients (Phase 1b NCT02960854) (137). The potential therapeutic modulations of TGF-β are outlined in Figure 2 .
Table 2.
Clinical Trials targeting TGF-ß or its receptor.
| Cytokine | Drug | Sponsor | Description | Indication | Clinical trial |
|---|---|---|---|---|---|
| TGF-β | Juvista (Avotermin) | Renovo | TGF-β3 | Wound healing | Phase 2 NCT00629811 |
| TGFβ-resistant LMP-specific cytotoxic T lymphocytes | Baylor College of Medicine | TGFβ-resistant LMP-specific cytotoxic T lymphocytes | Lymphoma | Phase 1 NCT00368082 |
|
| LY3200882 | The Netherlands Cancer Institute | TGFβRI small molecule inhibitor | Colorectal metastatic cancer | Phase 1/2 NCT04031872 |
|
| TGFβ-resistant HER2/EBV-CTLs | Baylor College of Medicine | TGFβ-resistant HER2/EBV-Cytotoxic T cells | Treatment of Her2 positive malignancy | Phase 1 NCT00889954 |
|
| AVID200 | Formation Biologics | TGFβ-receptor ectodomain-IgG Fc fusion protein inhibitor of TGFβ | Malignant solid tumor | Phase 1 NCT03834662 |
|
| TGFβ2 Antisense-GMCSF Gene Modified Autologous Tumor Cell (TAG) Vaccine | Mary Crowley Medical Research Center | TGFβ2 Antisense-GMCSF Gene Modified Autologous Tumor Cell (TAG) Vaccine | Advanced metastatic carcinoma | Phase 1 NCT00684294 |
|
| AP 12009 | Isarna Therapeutics GmbH | Phosphorothioate antisense oligodeoxynucleotide specific for the mRNA of human TGFβ2 | Pancreatic neoplasms, melanoma, colorectal neoplasms | Phase 1 NCT00844064 |
|
| GPC3 and/or TGFβ targeting CAR-T cells | Second Affiliated Hospital of Guangzhou Medical University | Third/fourth generation of CAR-T cells that target GPC3 (GPC3-CART cell) and/or soluble TGFβ (GPC3/TGFβ-CART) | Hepatocellular carcinoma, lung cancer |
Phase 1 NCT03198546 |
|
| P144 cream | ISDIN | TGFβ1-inhibitor, designed to block interaction between TGFβ1 and TGFβ1 type III receptor | Healthy (tolerability and bioavailability) | Phase 1 NCT00656825 |
|
| GC 1008 | National Cancer Institute (NCI) | Human anti-TGFβ- monoclonal antibody | Renal cell carcinoma | Phase 1 NCT00923169 |
|
| M7824 | National Cancer Institute (NCI) | Bifunctional anti-PD-L1/TGFβ Trap fusion protein | Head and neck cancer | Phase 1/2 NCT04247282 |
|
| Fresolimumab | Maximilian Diehn, Stanford University | Human monoclonal anti-TGFβ-antibody | Non-small cell lung carcinoma | Phase 1/2 NCT02581787 |
|
| Galunisertib (LY2157299) | Eli Lilly and Company | Small molecule inhibitor of the kinase domain of Type 1 TGFβ receptor | Solid tumor, non-small cell lung cancer, hepatocellular carcinoma | Phase 1/2 NCT02423343 |
|
| Vactosertib (TEW-7197) | MedPacto, Inc. | Inhibitor of the serine/threonine kinase of the TGFBR1 | Recurrent advanced urothelial carcinoma | Phase 2 NCT04064190 |
|
| GS-1423 | Gilead Sciences | Anti-CD73- TGFβ-Trap bifunctional antibody | Advanced solid tumors | Phase 1 NCT03954704 |
|
| PF-03446962 | Pfizer | Monoclonal antibody against Activin Receptor-Like Kinase-1 (ALK1), a type I subclass of the TGFβ receptor | Advanced solid tumors | Phase 1 NCT00557856 |
|
| SHR-1701 | Atridia Pty Ltd. | Anti-PD-L1/TGFβ fusion protein | Advanced solid tumor | Phase 1 NCT04324814 |
|
| SRK-181 | Scholar Rock, Inc. | Inhibitor of the activation of latent TGFβ1 | Locally advanced or metastatic solid tumors | Phase 1 NCT04291079 |
|
| NIS793 | Novartis Pharmaceuticals | Anti-TGFβ-antibody | Breast-, lung-, hepatocellular-, colorectal-, pancreatic-, renal-cancer | Phase 1 NCT02947165 |
TSLP
How Does TSLP Affect Immune Cells?
Thymic stromal lymphopoietin (TSLP) is a member of the IL-2 cytokine family and is closely related to IL-7 (138). It appears in two variants, a long form of TSLP (lfTSLP) and a short form (sfTSLP). These two forms do not derive from alternative splicing but are promoted by different putative promoter regions (139). Their effects on immune cells differ widely. Of note, many articles do not differentiate between the long and the short form of TSLP (139). In a review of Bjerkan et al., the authors summarized that the in absence of infection the short form is steadily expressed and the long form is absent (139). The short form seems to provide homeostasis especially on healthy barrier tissue like in the skin and gut. However inflammation can induce the expression of the long form and the short form becomes downregulated (139).
The diseases TSLP are best studied in are asthma, inflammatory bowel diseases and cancer, in which a Th2 dominated inflammation occurs or Th2 cells promote cancer cell survival (138). Keratinocytes, epithelial cells, mast cells, smooth muscle cells, fibroblasts and dendritic cells are producers of TSLP (140). However, it can also be produced by mast cells, human monocytes, macrophages and granulocytes, murine basophils and cancer cells (141). Unlike cells producing TSLP, which are comparably few, many more cells become activated by TSLP. Cells known to become activated by TSLP, are dendritic cells, innate lymphoid cells 2 (ILC2), CD4 T and Th2 cells, natural killer T cells, CD8 T cells and B cells, regulatory T cells, eosinophils, neutrophils, murine (but not human) basophils, monocytes, mast cells, macrophages, platelets, and sensory neurons (141).
Mouse and human TSLP bind to the combination of the IL-7 receptor α chain (IL-7Rα) and the TSLPR (TSLP receptor) chain, but only rarely to TSLPR alone (142). An extensive review about the intracellular pathways of TSLPR signaling would exceed the scope of this review, but was compiled by Zhong et al. and Yu et al. (143, 144). The lfTSLP receptor is expressed on several immune cells such as dendritic cells, T cells, B cells, natural killer cells, monocytes, basophils, eosinophils, and epithelial cells (139). The TSLPR was also found on tissue in heart, skeletal muscle, kidney, and liver (142). On CD4 T cells the TSLPR is upregulated upon TCR stimulation assuming a regulative role in inflammation (142). It is believed to portray an endothelial reaction to endogenous and exogenic triggers and drive a Th2-dominated inflammatory reaction mediated by dendritic cells. However, these Th2 cells also produce TNF-α, but not IL-10 (145).
It has been demonstrated that TSLP can exert potent antimicrobial effects. It could be shown that TSLP inhibits the growth of Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, Staphylococcus epidermidis and fungal species (139). Furthermore, TSLP augments ROS production in neutrophils by triggering the complement C5 system which enhances killing of S. aureus in human and mice (146).
Dynamics in Trauma and Sepsis
TSLP has not been studied extensively in sepsis to date. Several studies provide conflicting results concerning the effect of TSLP in animal sepsis models or human studies (44, 47, 147, 148). These findings are summarized in Table 3 and visualized in Figure 3 . Undisputed findings of the mentioned murine and human studies are that TSLP serum levels increase within the initial phase of the inflammation. Depending on the injury model or clinical circumstances, this initial peak ranges from 6 (147) to 24 h (44) and seems to decrease with the course of the inflammation.
Table 3.
The effect of TSLP in sepsis.
| Sepsis Model/Intervention | Intervention/Mouse model | Murine or human | Mortality | Cytokine expression | Other parameters | Study |
|---|---|---|---|---|---|---|
| CLP | Anti TSLP-Ab | M | ↓ | TNF-a, IL-17 (ip) ↓ | Neutrophil no (ip) ↑ Neutrophil ox. burst ↑ CFU ↓ |
Kuethe et al. (147) |
| CLP | Anti TSLP-Ab | M | Drop in body temp ↑ CFU ↔ |
Piliponsky et al. (44) | ||
| CLP | TSLP administration | M | TNF, IL-17A, IL-6, KC (plasma and ip) ↓ | Drop in body temp ↓ Neutrophil no (ip) ↓ CFU (ip) ↔ (blood) ↑ |
||
| CLP | TLSPR-/- | M | ↑ | TNF, IL-17A, IL-6, KC (plasma and ip) ↑ | Drop in body temp ↑ Neutrophil/Macrophage no (ip) ↑ CFU (ip and blood) ↓ |
|
| CLP | Lys-Cre+; Tslpr fl/fl (reduced TSLPR expression) | M | ↑ | TNF, IL-17A, IL-6, KC (plasma) ↑ | Neutrophil/Macrophage no (ip) ↑ CFU (ip and blood) ↓ IL-6 (within Neutrophils and Macrophages) ↑ |
|
| LPS stim (in vitro: Neutrophils) |
TSLP administration | H | IL-6 (within Neutrophils) ↓ | |||
| Sepsis | LPS stim (in vitro) |
H | IL-6, TNF-α, IL-1b (non-classical and intermediate Monocytes) (in patients above av. TSLP serum levels vs lower TSLP Serum levels) ↑ | Yu et al. (47) | ||
| Sepsis | PMA (in vitro) | H | Neutrophil ox burst and phagocytosis ↔ (in patients above av. TSLP serum levels vs lower TSLP Serum levels) ↑ |
|||
| Sepsis | Cell stim (in vitro) |
H | TNF-a, IFN-y (CD4 T cells) (in patients above av. TSLP serum levels vs lower TSLP Serum levels) ↑ | |||
| LPS injection (ip) | TSLP siRNA injection (reduces TSLP levels) Or TSLP-/- |
M | IL-6, VEGF, ICAM-1, and MIP2, TNF-a (serum, liver)↓ | Han et al. (148) | ||
| LPS injection (ip) | TSLP administration | M | IL-6 (serum) ↑ | |||
| LPS stim (in vitro: Monocytes (RAW264.7, WT and TSLP-/-)) |
TSLP siRNA transfection: RAW264.7 cells (reduces TSLP levels). WT: Anti-TSLP-Ab TSLP-/-: no intervention |
M | IL-6, ICAM-1, MIP-2, TNF-a, NO production in all types on Monocytes ↓ | |||
| CLP | – | M | TSLP (serum) ↑ | Kuethe et al. (147) | ||
| CLP | – | M | TSLP (plasma, ip) ↑ | Neutrophil TSLPR expression ↑ | Piliponsky et al. (44) | |
| Severe Sepsis | – | H | TSLP (plasma) ↑ | |||
| Sepsis | – | H | TSLP (serum) ↑ (in patients with hyperleukocytosis (HL) and high neutrophil ratio (HNR) vs without HL and HNR) |
Mortality ↑ (in patients above average TSLP serum levels vs lower TSLP serum levels) | Yu et al. (47) | |
| Sepsis | H | TSLP (serum) ↑ | Han et al. (148) | |||
| LPS, E.coli injection (ip) | M | TSLP (plasma, ip) ↑ |
Figure 3.

TSLP expression in murine and human sepsis and its experimental modification. Murine and human sepsis acutely lead to increase of systemic TSLP blood levels. Murine studies blocking TSLP provided contradictive results, which might be explained by differing methodology. (A) Several murine sepsis (cecal ligation and puncture and intraperitoneal injection of live bacteria) and endotoxemia (intraperitoneal injection of lipopolysaccharide (LPS)) models, as well as human observational trials in sepsis and severe sepsis revealed that TSLP levels are elevated in the blood within 24 h (44, 147, 148). (B) Three murine studies (44, 147, 148) assessed the effect of TSLP blockage and provided contradictive results that are highlighted in this figure. An explanation for these differences might be found in the different methods that were used. The figure contains adapted graphics from Les Laboratoirs Servier - Medical Art under the terms of the Creative Commons Attribution License (CC BY) for non-commercial use. The use, distribution or reproduction in other forums is permitted: https://creativecommons.org/licenses/by/3.0/legalcode, last accessed July 30th, 2020.
Up to now, none of the studies included a long-term analysis of TSLP levels and none of the studies conducted in sepsis differentiated between the long and the short TSLP form. As the long form is considered to become upregulated in inflammatory settings (139), it is likely that the long form is what was found to be elevated in the serum and peritoneum. This is of note as it might provide an explanation for the conflicting results found in the TSLP/sepsis studies concerning inflammatory and immunosuppressive actions of TSLP.
The first study that provided an examination of the effect of TSLP in sepsis was performed by Kuethe et al. (147). This group found that TSLP blockade decreased mortality and dampened the production of inflammatory cytokines (147). A similar study was conducted by Piliponsky et al. in 2016 (44). In contrast to the study of Kuethe et al. the administration of anti-TSLP-Ab in a murine abdominal sepsis model lead to an increase in morbidity and did not improve bacterial clearance (44). The authors argue that this may be explained by different injection times and inflammation dynamics. Otherwise, they confirmed the immunosuppressive role of the TSLP-TSLPR interaction as TSLPR-/- mice displayed increased intraperitoneal levels of TNF-a, IL-6, IL-17A and KC after onset of abdominal sepsis (44). However, TSLPR-/- mice had impaired bacterial clearance leading the authors to compare the effect of TSLPR with the effect that can be seen in IL-10 deficient mice succumbing to sepsis due to impaired bacterial clearance (44). Piliponsky et al. depicted that TSLP mainly dampens myeloid cytokine production, which then contributes to reduced morbidity by limiting inflammation (44). Overall, they conclude that the upregulation of TSLP in sepsis aims to restore immune hemostasis by dampening inflammation (44). In contrast, Yu et al. demonstrated a negative correlation between serum TSLP levels in patients with high ratio of neutrophils and increased mortality in a septic patient cohort (47). Moreover, they showed TSLP induces inflammation in neutrophils and T lymphocytes and certain monocyte subtypes (intermediate and non-classical) leading to increased production of e.g. IL-6, TNF-α and IFN-γ (47). They therefore conclude that TSLP has a pro-inflammatory effect on monocytes and lymphocytes but does not improve antibacterial clearance of neutrophils, as phagocytosis and respiratory burst were unchanged (47). In line with these findings is a study by Han et al. They revealed that TSLP-deficient mice show lower levels of IL-6, VEGF, ICAM-1, and MIP2 in serum in LPS challenged mice (148). They also demonstrated TSLP upregulates macrophage-mediated inflammation as TSLP neutralization and TSLP siRNA silencing reduced the production of IL-6, TNF-α and NO after LPS stimulation. Moreover, they showed LPS or E. coli stimulated macrophages via TLR4 produce TSLP (148). The finding of decreased proinflammatory cytokines in TSLP-deficient mice contradicts the findings of Piliponsky et al. and Kuethe et al., although Han et al. in part used the same experimental approach (Anti-TSLP-Ab) (44, 147, 148). The team of Han et al. argues that the different septic models (LPS vs cecal ligation and puncture (CLP)) may provide an explanation for this. They summarize that TSLP triggers proinflammatory reactions especially of macrophages and might lead to organ dysfunction in sepsis (148).
Taken together, these studies provide surprisingly conflicting data about the effect of TSLP in sepsis. It will be critical to consider the differences between the models to explain these results. The CLP model conducted by Kuethe et al. and Piliponsky et al. constitutes a physiologically more relevant model as it includes pathogen related tissue damage, phagocytic uptake of pathogens by neutrophils and macrophages, as well as the release of other pathogenic antigens compared to the administration of LPS alone, which lacks all these important characteristics. Moreover, Han et al. used relatively low LPS doses (148) which may not induce an inflammatory reaction as strong as in the CLP model. This may indicate a different role of TSLP depending on the injury severity and inflammatory milieu. However, until further direct experimental comparisons, this remains speculative.
The following points should be addressed in further studies to clarify the role of TSLP in sepsis. First, a discrimination between the short and the long form should be conducted in future studies as their effects are considered to differ (139). Secondly, studies should differentiate between the severity of sepsis, as Piliponsky as well as Yu et al. reported differences in the effect of TSLP regarding the severity of the disease (44, 47). Thirdly, none of the studies so far address the release of TSLP by endothelial cells, which are considered the main producers (140). The effect of TSLP on macrophages and endothelial cells seem most important as they might steer potential pro- or anti-inflammatory effects. Lastly, the dynamics of the septic disease should be examined, as this may also be a potential cause of the current conflicts in the data.
Effects and Drivers of Pathological Release in Trauma and Sepsis
The production of TSLP is triggered by allergens, pro-inflammatory cytokines, viruses, bacteria, fungi and tryptases (141). With respect to a septic setting, the challenge of epithelial cells with bacteria is likely the most relevant. This poses the important question of whether the stimulation occurs directly by microbes or in an indirect manner mediated by consecutively upregulated pro-inflammatory cytokines. In a study performed by Lan et al. it is demonstrated that Staphylococcus aureus directly induces epithelial cell-derived TSLP release via TLR-2-binding (149). Although not in a septic model, this might explain the upregulation of TSLP in sepsis (149). If TSLP is considered to restore immune hemostasis in sepsis, which still has to be proven due to the conflicting results past studies provided, then overwhelming microbial challenge might lead to detrimentally high release of TSLP. This could provide an explanation for conflicting data as lower levels might restore homeostasis, but by surpassing a certain threshold, TSLP may trigger inflammation and therefore worsen outcome. However, this currently remains a matter of speculation.
Potential Therapeutic Modulation of TSLP
As discussed in the previous section more research is needed to characterize the effect of TSLP in trauma or sepsis. However, antibodies directed against TSLP are currently tested for safety in clinical trials aiming to treat allergic diseases (e.g. MEDI9929: human monoclonal antibody immunoglobulin IgG2λ directed against TSLP (asthma), Phase 2 NCT02698501). An overview of current TSLP modulating drugs is provided in Table 4 . These might prove useful in clinical trials when dynamics and functions of TSLP in trauma and sepsis have been elucidated.
Table 4.
Clinical Trials targeting TSLP.
| Cytokine | Drug | Sponsor | Description | Indication | Clinical trial |
|---|---|---|---|---|---|
| TSLP | MEDI9929 | Celeste Porsbjerg | Human monoclonal antibody immunoglobulin IgG2λ directed against TSLP | Asthma | Phase 2 NCT02698501 |
| AMG 157 (MEDI9929) | National Institute of Allergy and Infectious Diseases (NIAID) | Anti-TSLP antibody | Cat Allergy | Phase 1/2 NCT02237196 |
Conclusion
Sepsis is responsible for one out of three in-hospital deaths (150) while trauma is still the leading course of death for people under the age of 46 in the United States (151). The following discusses the two most pressing challenges for these pathophysiologies:
1) The development of methods to stratify patients, to evaluate which of three clinical trajectories the respective patient will follow: early death, rapid recovery or chronic critical illness (19). Promising strategies are phenotyping innate and adaptive immune cells, using flow cytometry (32), assessing genetic or transcriptional changes within innate and adaptive immune cells (152), evaluating cytokine levels and their ratio’s (31) and most importantly use functional assessment of immune cell behavior (46, 153). The use of functional assays has proven useful to examine the capability of cells to produce cytokines in response to stimuli and thus enables the assessment of the current immune status (46). Moreover, patient endotypes within the sepsis and trauma populations should be defined (10). These endotypes should be studied to determine if they are associated with good or bad clinical outcomes. Knowing the endotype associated with the immune status should allow for a more personalized and effective therapeutic strategy. All of the clinical, phenotypic and functional assays should be analyzed in combination with the standard clinical data on the ICU’s. We believe that dynamic immune monitoring prospectively should become a standard in intensive care medicine.
2) If proper stratification of patients is successful the next step would be to therapeutically address immune perturbation. Currently the use of three cytokines is being tested to improve immune function in sepsis and infection: IFN-γ to treat sepsis-induced immunoparalysis (Phase 3, NCT01649921), granulocyte-macrophage colony-stimulating factor (GM-CSF) to decrease ICU acquired infections (Phase 3, NCT02361528), IL-7 to restore lymphocyte counts in sepsis patients (Phase 2, NCT02640807). Beside cytokine modulation the blockage of the programmed cell death protein 1 (PD-1), a checkpoint inhibitor with elevated expressed in sepsis patients (154), might prove beneficial, regarding its ability to hinder T cell apoptosis in murine sepsis model and in vitro human sepsis studies (155–157). Moreover, the blockage of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), another immune checkpoint receptor overexpressed in sepsis patients (155, 158) that improved T cell survival in mice (159), might provide a therapeutic approach.
Other than the above mentioned, there are no current approaches to clinically test modulation of immunosuppressive cytokines, such as IL-10, TGF-β and TSLP in sepsis or trauma. Immunosuppression is responsible for significant numbers of deaths in sepsis and trauma patient populations (13). It is therefore key to further examine how immunosuppressive cytokines affect immune hemostasis and how they can be beneficially modulated.
As highlighted in this review the effect of the potent anti-inflammatory cytokines IL-10, TGF-β, and TSLP depends on dynamics, dose and receptive cell type. Thus, the following prerequisites for these specific cytokines should be met to approach pharmaceutical therapeutic modulation of IL-10, TGF-β, and TSLP: 1) Future pre-clinical or clinical studies should focus on temporal expression of these cytokines. 2) The collective literature shows improved predictive power when a combination of cytokines was assessed to predict outcome (31, 160, 161). Therefore, they should be evaluated relatively to each other and other pro-inflammatory cytokines. 3) Future research should focus on which cells are the main contributors to the systemic release and functionally assess which stimuli drive the release of the respective cytokine, so the specific cell function can be therapeutically modulated.
If these criteria are met and a clear characterization of cytokine behavior emerges, we suggest translating results and immune modulating therapeutics from other pathophysiologies, such as autoimmune diseases and cancer, to sepsis and trauma. These promising treatments that are being tested in these diseases potentially share common mechanisms. Thus, these FDA approved drugs for cancer and autoimmunity could potentially be trialed in the setting of sepsis or trauma to address pathological cytokine release of IL-10, TGF-β, and TSLP leading to disbalanced immunosuppression.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
CB and CC outlined the review. CB drafted the review, tables, and figures. CB and NB designed the figures. CC, NB, CS, MH, and PC reviewed the drafted review. The latter three especially providing critical clinical input concerning therapeutic approaches. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by funding from the Deutsche Forschungsgemeinschaft (German Research Foundation) (BE 7016/1-1) (CB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1. Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol (2001) 13:114–9. 10.1016/S0952-7915(00)00191-6 [DOI] [PubMed] [Google Scholar]
- 2. Raymond SL, Holden DC, Mira JC, Stortz JA, Loftus TJ, Mohr AM, et al. Microbial recognition and danger signals in sepsis and trauma. Biochim Biophys Acta (2017) 1863(10):2564–73. 10.1016/j.bbadis.2017.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ (2003) 10:45–65. 10.1038/sj.cdd.4401189 [DOI] [PubMed] [Google Scholar]
- 4. Schulte W, Bernhagen J, Bucala R. Cytokines in sepsis: potent immunoregulators and potential therapeutic targets–an updated view. Mediators Inflammation (2013) 2013:165974. 10.1155/2013/165974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cohen J. The immunopathogenesis of sepsis. Nature (2002) 420:885–91. 10.1038/nature01326 [DOI] [PubMed] [Google Scholar]
- 6. Eskilsson A, Mirrasekhian E, Dufour S, Schwaninger M, Engblom D, Blomqvist A. Immune-induced fever is mediated by IL-6 receptors on brain endothelial cells coupled to STAT3-dependent induction of brain endothelial prostaglandin synthesis. J Neurosci (2014) 34:15957–61. 10.1523/JNEUROSCI.3520-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Q, Fisher DT, Clancy KA, Gauguet JM, Wang WC, Unger E, et al. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat Immunol (2006) 7:1299–308. 10.1038/ni1406 [DOI] [PubMed] [Google Scholar]
- 8. Chen Q, Wang WC, Bruce R, Li H, Schleider DM, Mulbury MJ, et al. Central role of IL-6 receptor signal-transducing chain gp130 in activation of L-selectin adhesion by fever-range thermal stress. Immunity (2004) 20:59–70. 10.1016/S1074-7613(03)00358-3 [DOI] [PubMed] [Google Scholar]
- 9. Andrades ME, Morina A, Spasic S, Spasojevic I. Bench-to-bedside review: sepsis - from the redox point of view. Crit Care (2011) 15:230. 10.1186/cc10334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seymour CW, Kennedy JN, Wang S, Chang CH, Elliott CF, Xu Z, et al. Derivation, Validation, and Potential Treatment Implications of Novel Clinical Phenotypes for Sepsis. JAMA (2019) 321:2003–17. 10.1001/jama.2019.5791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis-associated disseminated intravascular coagulation and thromboembolic disease. Mediterr J Hematol Infect Dis (2010) 2:e2010024. 10.4084/mjhid.2010.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dewar DC, Mackay P, Balogh Z. Epidemiology of post-injury multiple organ failure in an Australian trauma system. ANZ J Surg (2009) 79:431–6. 10.1111/j.1445-2197.2009.04968.x [DOI] [PubMed] [Google Scholar]
- 13. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers (2016) 2:16045. 10.1038/nrdp.2016.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE, Jr., Hui JJ, Chang KC, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol (2001) 166:6952–63. 10.4049/jimmunol.166.11.6952 [DOI] [PubMed] [Google Scholar]
- 15. Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, et al. Increased percentage of CD4+CD25+ regulatory T cells during septic shock is due to the decrease of CD4+CD25- lymphocytes. Crit Care Med (2004) 32:2329–31. 10.1097/01.CCM.0000145999.42971.4B [DOI] [PubMed] [Google Scholar]
- 16. Hollen MK, Stortz JA, Darden D, Dirain ML, Nacionales DC, Hawkins RB, et al. Myeloid-derived suppressor cell function and epigenetic expression evolves over time after surgical sepsis. Crit Care (2019) 23:355. 10.1186/s13054-019-2628-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Drewry AM, Ablordeppey EA, Murray ET, Beiter ER, Walton AH, Hall MW, et al. Comparison of monocyte human leukocyte antigen-DR expression and stimulated tumor necrosis factor alpha production as outcome predictors in severe sepsis: a prospective observational study. Crit Care (2016) 20:334. 10.1186/s13054-016-1505-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH, Osborne DF, Wagner TH, et al. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol (2002) 168:2493–500. 10.4049/jimmunol.168.5.2493 [DOI] [PubMed] [Google Scholar]
- 19. Stortz JA, Mira JC, Raymond SL, Loftus TJ, Ozrazgat-Baslanti T, Wang Z, et al. Benchmarking clinical outcomes and the immunocatabolic phenotype of chronic critical illness after sepsis in surgical intensive care unit patients. J Trauma Acute Care Surg (2018) 84:342–9. 10.1097/TA.0000000000001758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosenthal MD, Moore FA. Persistent Inflammation, Immunosuppression, and Catabolism: Evolution of Multiple Organ Dysfunction. Surg Infections (2016) 17:167–72. 10.1089/sur.2015.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Inoue S, Suzuki K, Komori Y, Morishita Y, Suzuki-Utsunomiya K, Hozumi K, et al. Persistent inflammation and T cell exhaustion in severe sepsis in the elderly. Crit Care (2014) 18:R130. 10.1186/cc13941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, et al. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg (2012) 72:1491–501. 10.1097/TA.0b013e318256e000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hawkins RB, Raymond SL, Stortz JA, Horiguchi H, Brakenridge SC, Gardner A, et al. Chronic Critical Illness and the Persistent Inflammation, Immunosuppression, and Catabolism Syndrome. Front Immunol (2018) 9:1511. 10.3389/fimmu.2018.01511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McBride MA, Owen AM, Stothers CL, Hernandez A, Luan L, Burelbach KR, et al. The Metabolic Basis of Immune Dysfunction Following Sepsis and Trauma. Front Immunol (2020) 11:1043. 10.3389/fimmu.2020.01043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rubio I, Osuchowski MF, Shankar-Hari M, Skirecki T, Winkler MS, Lachmann G, et al. Current gaps in sepsis immunology: new opportunities for translational research. Lancet Infect Dis (2019) 19:e422–36. 10.1016/S1473-3099(19)30567-5 [DOI] [PubMed] [Google Scholar]
- 26. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol (2010) 11:373–84. 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 27. Robertson CM, Coopersmith CM. The systemic inflammatory response syndrome. Microbes Infection (2006) 8:1382–9. 10.1016/j.micinf.2005.12.016 [DOI] [PubMed] [Google Scholar]
- 28. Islam MN, Bradley BA, Ceredig R. Sterile post-traumatic immunosuppression. Clin Trans Immunol (2016) 5:e77. 10.1038/cti.2016.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Neunaber C, Zeckey C, Andruszkow H, Frink M, Mommsen P, Krettek C, et al. Immunomodulation in polytrauma and polymicrobial sepsis - where do we stand? Recent Patents Inflammation Allergy Drug Discovery (2011) 5:17–25. 10.2174/187221311794474892 [DOI] [PubMed] [Google Scholar]
- 30. Jawa RS, Anillo S, Huntoon K, Baumann H, Kulaylat M. Interleukin-6 in surgery, trauma, and critical care part II: clinical implications. J Intensive Care Med (2011) 26:73–87. 10.1177/0885066610395679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sapan HB, Paturusi I, Jusuf I, Patellongi I, Massi MN, Pusponegoro AD, et al. Pattern of cytokine (IL-6 and IL-10) level as inflammation and anti-inflammation mediator of multiple organ dysfunction syndrome (MODS) in polytrauma. Int J Burns Trauma (2016) 6:37–43. [PMC free article] [PubMed] [Google Scholar]
- 32. Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol (2017) 14:121. 10.1038/nrneph.2017.165 [DOI] [PubMed] [Google Scholar]
- 33. Hotchkiss RS, Osmon SB, Chang KC, Wagner TH, Coopersmith CM, Karl IE. Accelerated lymphocyte death in sepsis occurs by both the death receptor and mitochondrial pathways. J Immunol (2005) 174:5110–8. 10.4049/jimmunol.174.8.5110 [DOI] [PubMed] [Google Scholar]
- 34. Bergmann CB, Hefele F, Unger M, Huber-Wagner S, Biberthaler P, van Griensven M, et al. Platelets modulate the immune response following trauma by interaction with CD4+ T regulatory cells in a mouse model. Immunol Res (2016) 64:508–17. 10.1007/s12026-015-8726-1 [DOI] [PubMed] [Google Scholar]
- 35. Monneret G, Debard AL, Venet F, Bohe J, Hequet O, Bienvenu J, et al. Marked elevation of human circulating CD4+CD25+ regulatory T cells in sepsis-induced immunoparalysis. Crit Care Med (2003) 31:2068–71. 10.1097/01.CCM.0000069345.78884.0F [DOI] [PubMed] [Google Scholar]
- 36. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. Jama (2011) 306:2594–605. 10.1001/jama.2011.1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pugh AM, Auteri NJ, Goetzman HS, Caldwell CC, Nomellini V. A Murine Model of Persistent Inflammation, Immune Suppression, and Catabolism Syndrome. Int J Mol Sci (2017) 18. 10.3390/ijms18081741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu YC, Zou XB, Chai YF, Yao YM. Macrophage polarization in inflammatory diseases. Int J Biol Sci (2014) 10:520–9. 10.7150/ijbs.8879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Venet F, Lepape A, Monneret G. Clinical review: flow cytometry perspectives in the ICU - from diagnosis of infection to monitoring of injury-induced immune dysfunctions. Crit Care (2011) 15:231. 10.1186/cc10333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest (2016) 126:23–31. 10.1172/JCI82224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med (2007) 204:1463–74. 10.1084/jem.20062602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ouyang W, O’Garra A. IL-10 Family Cytokines IL-10 and IL-22: from Basic Science to Clinical Translation. Immunity (2019) 50:871–91. 10.1016/j.immuni.2019.03.020 [DOI] [PubMed] [Google Scholar]
- 43. Chen W, Ten Dijke P. Immunoregulation by members of the TGFbeta superfamily. Nat Rev Immunol (2016) 16:723–40. 10.1038/nri.2016.112 [DOI] [PubMed] [Google Scholar]
- 44. Piliponsky AM, Lahiri A, Truong P, Clauson M, Shubin NJ, Han H, et al. Thymic Stromal Lymphopoietin Improves Survival and Reduces Inflammation in Sepsis. Am J Respir Cell Mol Biol (2016) 55:264–74. 10.1165/rcmb.2015-0380OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang LF, Yao YM, Dong N, Yu Y, He LX, Sheng ZY. Association between regulatory T cell activity and sepsis and outcome of severely burned patients: a prospective, observational study. Crit Care (2010) 14:R3. 10.1186/cc8232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mazer M, Unsinger J, Drewry A, Walton A, Osborne D, Blood T, et al. IL-10 Has Differential Effects on the Innate and Adaptive Immune Systems of Septic Patients. J Immunol (2019) 203:2088–99. 10.4049/jimmunol.1900637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yu Q, Li Y, Wang H, Xiong H. TSLP induces a proinflammatory phenotype in circulating innate cells and predicts prognosis in sepsis patients. FEBS Open Bio (2019) 9:2137–48. 10.1002/2211-5463.12746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol (2006) 24:99–146. 10.1146/annurev.immunol.24.021605.090737 [DOI] [PubMed] [Google Scholar]
- 49. Ng TH, Britton GJ, Hill EV, Verhagen J, Burton BR, Wraith DC. Regulation of adaptive immunity; the role of interleukin-10. Front Immunol (2013) 4:129. 10.3389/fimmu.2013.00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sabat R, Grutz G, Warszawska K, Kirsch S, Witte E, Wolk K, et al. Biology of interleukin-10. Cytokine Growth Factor Rev (2010) 21:331–44. 10.1016/j.cytogfr.2010.09.002 [DOI] [PubMed] [Google Scholar]
- 51. Mannino MH, Zhu Z, Xiao H, Bai Q, Wakefield MR, Fang Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett (2015) 367:103–7. 10.1016/j.canlet.2015.07.009 [DOI] [PubMed] [Google Scholar]
- 52. Trifunovic J, Miller L, Debeljak Z, Horvat V. Pathologic patterns of interleukin 10 expression–a review. Biochem Med (Zagreb) (2015) 25:36–48. 10.11613/BM.2015.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saxena A, Khosraviani S, Noel S, Mohan D, Donner T, Hamad AR. Interleukin-10 paradox: A potent immunoregulatory cytokine that has been difficult to harness for immunotherapy. Cytokine (2015) 74:27–34. 10.1016/j.cyto.2014.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Penaloza HF, Schultz BM, Nieto PA, Salazar GA, Suazo I, Gonzalez PA, et al. Opposing roles of IL-10 in acute bacterial infection. Cytokine Growth Factor Rev (2016) 32:17–30. 10.1016/j.cytogfr.2016.07.003 [DOI] [PubMed] [Google Scholar]
- 55. Mittal SK, Roche PA. Suppression of antigen presentation by IL-10. Curr Opin Immunol (2015) 34:22–7. 10.1016/j.coi.2014.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kany S, Vollrath JT, Relja B. Cytokines in Inflammatory Disease. Int J Mol Sci (2019) 20. 10.3390/ijms20236008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest (2000) 117:1162–72. 10.1378/chest.117.4.1162 [DOI] [PubMed] [Google Scholar]
- 58. Petitjean G, Chevalier MF, Tibaoui F, Didier C, Manea ME, Liovat AS, et al. Level of double negative T cells, which produce TGF-beta and IL-10, predicts CD8 T-cell activation in primary HIV-1 infection. AIDS (London England) (2012) 26:139–48. 10.1097/QAD.0b013e32834e1484 [DOI] [PubMed] [Google Scholar]
- 59. de Waal Malefyt R, Haanen J, Spits H, Roncarolo MG, te Velde A, Figdor C, et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med (1991) 174:915–24. 10.1084/jem.174.4.915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Creery WD, Diaz-Mitoma F, Filion L, Kumar A. Differential modulation of B7-1 and B7-2 isoform expression on human monocytes by cytokines which influence the development of T helper cell phenotype. Eur J Immunol (1996) 26:1273–7. 10.1002/eji.1830260614 [DOI] [PubMed] [Google Scholar]
- 61. Bazzoni F, Tamassia N, Rossato M, Cassatella MA. Understanding the molecular mechanisms of the multifaceted IL-10-mediated anti-inflammatory response: lessons from neutrophils. Eur J Immunol (2010) 40:2360–8. 10.1002/eji.200940294 [DOI] [PubMed] [Google Scholar]
- 62. Lichte P, Pfeifer R, Kobbe P, Tohidnezhad M, Pufe T, Almahmoud K, et al. Inhalative IL-10 treatment after bilateral femoral fractures affect pulmonary inflammation in mice. Ann Anat (2015) 200:73–8. 10.1016/j.aanat.2015.02.005 [DOI] [PubMed] [Google Scholar]
- 63. Keel M, Ungethum U, Steckholzer U, Niederer E, Hartung T, Trentz O, et al. Interleukin-10 counterregulates proinflammatory cytokine-induced inhibition of neutrophil apoptosis during severe sepsis. Blood (1997) 90:3356–63. 10.1182/blood.V90.9.3356 [DOI] [PubMed] [Google Scholar]
- 64. Cassatella MA, Meda L, Bonora S, Ceska M, Constantin G. Interleukin 10 (IL-10) inhibits the release of proinflammatory cytokines from human polymorphonuclear leukocytes. Evidence for an autocrine role of tumor necrosis factor and IL-1 beta in mediating the production of IL-8 triggered by lipopolysaccharide. J Exp Med (1993) 178:2207–11. 10.1084/jem.178.6.2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lewkowicz N, Mycko MP, Przygodzka P, Cwiklinska H, Cichalewska M, Matysiak M, et al. Induction of human IL-10-producing neutrophils by LPS-stimulated Treg cells and IL-10. Mucosal Immunol (2016) 9:364–78. 10.1038/mi.2015.66 [DOI] [PubMed] [Google Scholar]
- 66. Kasten KR, Muenzer JT, Caldwell CC. Neutrophils are significant producers of IL-10 during sepsis. Biochem Biophys Res Commun (2010) 393:28–31. 10.1016/j.bbrc.2010.01.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhao HQ, Li WM, Lu ZQ, Sheng ZY, Yao YM. The growing spectrum of anti-inflammatory interleukins and their potential roles in the development of sepsis. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res (2015) 35:242–51. 10.1089/jir.2014.0119 [DOI] [PubMed] [Google Scholar]
- 68. Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol (2009) 10:1178–84. 10.1038/ni.1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tschoeke SK, Oberholzer C, LaFace D, Hutchins B, Moldawer LL, Oberholzer A. Endogenous IL-10 regulates sepsis-induced thymic apoptosis and improves survival in septic IL-10 null mice. Scand J Immunol (2008) 68:565–71. 10.1111/j.1365-3083.2008.02176.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mumm JB, Emmerich J, Zhang X, Chan I, Wu L, Mauze S, et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell (2011) 20:781–96. 10.1016/j.ccr.2011.11.003 [DOI] [PubMed] [Google Scholar]
- 71. Naing A, Infante JR, Papadopoulos KP, Chan IH, Shen C, Ratti NP, et al. PEGylated IL-10 (Pegilodecakin) Induces Systemic Immune Activation, CD8(+) T Cell Invigoration and Polyclonal T Cell Expansion in Cancer Patients. Cancer Cell (2018) 34:775–791 e3. 10.1016/j.ccell.2018.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yao Y, Simard AR, Shi FD, Hao J. IL-10-producing lymphocytes in inflammatory disease. Int Rev Immunol (2013) 32:324–36. 10.3109/08830185.2012.762361 [DOI] [PubMed] [Google Scholar]
- 73. Kobbe P, Lichte P, Schreiber H, Reiss LK, Uhlig S, Pape HC, et al. Inhalative IL-10 attenuates pulmonary inflammation following hemorrhagic shock without major alterations of the systemic inflammatory response. Mediators Inflammation (2012) 2012:512974. 10.1155/2012/512974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cao C, Yu M, Chai Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis (2019) 10:782. 10.1038/s41419-019-2015-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Misra AK, Levy MM, Ward NS. Biomarkers of Immunosuppression. Crit Care Clin (2020) 36:167–76. 10.1016/j.ccc.2019.08.013 [DOI] [PubMed] [Google Scholar]
- 76. Xia BT, Beckmann N, Winer LK, Kim Y, Goetzman HS, Veile RE, et al. Amitriptyline Treatment Mitigates Sepsis-Induced Tumor Necrosis Factor Expression and Coagulopathy. Shock (2018) 51(3):356–63. 10.1097/SHK.0000000000001146 [DOI] [PubMed] [Google Scholar]
- 77. Latifi SQ, O’Riordan MA, Levine AD. Interleukin-10 controls the onset of irreversible septic shock. Infection Immun (2002) 70:4441–6. 10.1128/IAI.70.8.4441-4446.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Stensballe J, Christiansen M, Tonnesen E, Espersen K, Lippert FK, Rasmussen LS. The early IL-6 and IL-10 response in trauma is correlated with injury severity and mortality. Acta Anaesthesiol Scand (2009) 53:515–21. 10.1111/j.1399-6576.2008.01801.x [DOI] [PubMed] [Google Scholar]
- 79. Feng M, Sun T, Zhao Y, Zhang H. Detection of Serum Interleukin-6/10/18 Levels in Sepsis and Its Clinical Significance. J Clin Lab Anal (2016) 30:1037–43. 10.1002/jcla.21977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chen P, Stanojcic M, Jeschke MG. Septic predictor index: A novel platform to identify thermally injured patients susceptible to sepsis. Surgery (2018) 163:409–14. 10.1016/j.surg.2017.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gogos CA, Drosou E, Bassaris HP, Skoutelis A. Pro- versus anti-inflammatory cytokine profile in patients with severe sepsis: a marker for prognosis and future therapeutic options. J Infect Dis (2000) 181:176–80. 10.1086/315214 [DOI] [PubMed] [Google Scholar]
- 82. Chuang TY, Chang HT, Chung KP, Cheng HS, Liu CY, Liu YC, et al. High levels of serum macrophage migration inhibitory factor and interleukin 10 are associated with a rapidly fatal outcome in patients with severe sepsis. Int J Infect Dis (2014) 20:13–7. 10.1016/j.ijid.2013.12.006 [DOI] [PubMed] [Google Scholar]
- 83. Frencken JF, van Vught LA, Peelen LM, Ong DSY, Klein Klouwenberg PMC, Horn J, et al. An Unbalanced Inflammatory Cytokine Response Is Not Associated With Mortality Following Sepsis: A Prospective Cohort Study. Crit Care Med (2017) 45:e493–9. 10.1097/CCM.0000000000002292 [DOI] [PubMed] [Google Scholar]
- 84. van Vught LA, Klein Klouwenberg PM, Spitoni C, Scicluna BP, Wiewel MA, Horn J, et al. Incidence, Risk Factors, and Attributable Mortality of Secondary Infections in the Intensive Care Unit After Admission for Sepsis. JAMA (2016) 315:1469–79. 10.1001/jama.2016.2691 [DOI] [PubMed] [Google Scholar]
- 85. Ivanov P, Anderson P. Post-transcriptional regulatory networks in immunity. Immunol Rev (2013) 253:253–72. 10.1111/imr.12051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Easton R, Balogh ZJ. Peri-operative changes in serum immune markers after trauma: a systematic review. Injury (2014) 45:934–41. 10.1016/j.injury.2013.12.002 [DOI] [PubMed] [Google Scholar]
- 87. Li X, Xu Z, Pang X, Huang Y, Yang B, Yang Y, et al. Interleukin-10/lymphocyte ratio predicts mortality in severe septic patients. PloS One (2017) 12:e0179050. 10.1371/journal.pone.0179050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhao S, Wu D, Wu P, Wang Z, Huang J. Serum IL-10 Predicts Worse Outcome in Cancer Patients: A Meta-Analysis. PloS One (2015) 10:e0139598. 10.1371/journal.pone.0139598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gabrysova L, Howes A, Saraiva M, O’Garra A. The regulation of IL-10 expression. Curr Top Microbiol Immunol (2014) 380:157–90. 10.1007/978-3-662-43492-5_8 [DOI] [PubMed] [Google Scholar]
- 90. Tamassia N, Le Moigne V, Calzetti F, Donini M, Gasperini S, Ear T, et al. The MyD88-independent pathway is not mobilized in human neutrophils stimulated via TLR4. J Immunol (2007) 178:7344–56. 10.4049/jimmunol.178.11.7344 [DOI] [PubMed] [Google Scholar]
- 91. Lachmann G, von Haefen C, Kurth J, Yuerek F, Spies C. Innate immunity recovers earlier than acquired immunity during severe postoperative immunosuppression. Int J Med Sci (2018) 15:1–9. 10.7150/ijms.21433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ocuin LM, Bamboat ZM, Balachandran VP, Cavnar MJ, Obaid H, Plitas G, et al. Neutrophil IL-10 suppresses peritoneal inflammatory monocytes during polymicrobial sepsis. J Leukoc Biol (2011) 89:423–32. 10.1189/jlb.0810479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature (1997) 390:350–1. 10.1038/37022 [DOI] [PubMed] [Google Scholar]
- 94. Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest (2002) 109:41–50. 10.1172/JCI0211638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ortega-Gomez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med (2013) 5:661–74. 10.1002/emmm.201202382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lawrence SM, Corriden R, Nizet V. How Neutrophils Meet Their End. Trends Immunol (2020) 41:531–44. 10.1016/j.it.2020.03.008 [DOI] [PubMed] [Google Scholar]
- 97. Goren I, Allmann N, Yogev N, Schurmann C, Linke A, Holdener M, et al. A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am J Pathol (2009) 175:132–47. 10.2353/ajpath.2009.081002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Korner A, Schlegel M, Theurer J, Frohnmeyer H, Adolph M, Heijink M, et al. Resolution of inflammation and sepsis survival are improved by dietary Omega-3 fatty acids. Cell Death Differ (2018) 25:421–31. 10.1038/cdd.2017.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Walker J, Dichter E, Lacorte G, Kerner D, Spur B, Rodriguez A, et al. Lipoxin a4 increases survival by decreasing systemic inflammation and bacterial load in sepsis. Shock (2011) 36:410–6. 10.1097/SHK.0b013e31822798c1 [DOI] [PubMed] [Google Scholar]
- 100. Kang JW, Lee SM. Resolvin D1 protects the liver from ischemia/reperfusion injury by enhancing M2 macrophage polarization and efferocytosis. Biochim Biophys Acta (2016) 1861:1025–35. 10.1016/j.bbalip.2016.06.002 [DOI] [PubMed] [Google Scholar]
- 101. Schutters K, Kusters DH, Chatrou ML, Montero-Melendez T, Donners M, Deckers NM, et al. Cell surface-expressed phosphatidylserine as therapeutic target to enhance phagocytosis of apoptotic cells. Cell Death Differ (2013) 20:49–56. 10.1038/cdd.2012.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Oberholzer A, Oberholzer C, Moldawer LL. Interleukin-10: A complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med (2002) 30:S58–63. 10.1097/00003246-200201001-00008 [DOI] [PubMed] [Google Scholar]
- 103. Hiraki S, Ono S, Tsujimoto H, Kinoshita M, Takahata R, Miyazaki H, et al. Neutralization of interleukin-10 or transforming growth factor-beta decreases the percentages of CD4+ CD25+ Foxp3+ regulatory T cells in septic mice, thereby leading to an improved survival. Surgery (2012) 151:313–22. 10.1016/j.surg.2011.07.019 [DOI] [PubMed] [Google Scholar]
- 104. Pajkrt D, van der Poll T, Levi M, Cutler DL, Affrime MB, van den Ende A, et al. Interleukin-10 inhibits activation of coagulation and fibrinolysis during human endotoxemia. Blood (1997) 89:2701–5. 10.1182/blood.V89.8.2701 [DOI] [PubMed] [Google Scholar]
- 105. Pajkrt D, Camoglio L, Tiel-van Buul MC, de Bruin K, Cutler DL, Affrime MB, et al. Attenuation of proinflammatory response by recombinant human IL-10 in human endotoxemia: effect of timing of recombinant human IL-10 administration. J Immunol (1997) 158:3971–7. [PubMed] [Google Scholar]
- 106. Wong JJM, Leong JY, Lee JH, Albani S, Yeo JG. Insights into the immuno-pathogenesis of acute respiratory distress syndrome. Ann Transl Med (2019) 7:504. 10.21037/atm.2019.09.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. van der Poll T, Jansen PM, Montegut WJ, Braxton CC, Calvano SE, Stackpole SA, et al. Effects of IL-10 on systemic inflammatory responses during sublethal primate endotoxemia. J Immunol (1997) 158:1971–5. [PubMed] [Google Scholar]
- 108. Remick DG, Garg SJ, Newcomb DE, Wollenberg G, Huie TK, Bolgos GL. Exogenous interleukin-10 fails to decrease the mortality or morbidity of sepsis. Crit Care Med (1998) 26:895–904. 10.1097/00003246-199805000-00025 [DOI] [PubMed] [Google Scholar]
- 109. Sanjabi S, Oh SA, Li MO. Regulation of the Immune Response by TGF-beta: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol (2017) 9. 10.1101/cshperspect.a022236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Tran DQ. TGF-beta: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol (2012) 4:29–37. 10.1093/jmcb/mjr033 [DOI] [PubMed] [Google Scholar]
- 111. Cottrez F, Groux H. Regulation of TGF-beta response during T cell activation is modulated by IL-10. J Immunol (2001) 167:773–8. 10.4049/jimmunol.167.2.773 [DOI] [PubMed] [Google Scholar]
- 112. Curotto de Lafaille MA, Lino AC, Kutchukhidze N, Lafaille JJ. CD25- T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J Immunol (2004) 173:7259–68. 10.4049/jimmunol.173.12.7259 [DOI] [PubMed] [Google Scholar]
- 113. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. 10.1084/jem.20030152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kitani A, Fuss I, Nakamura K, Kumaki F, Usui T, Strober W. Transforming growth factor (TGF)-beta1-producing regulatory T cells induce Smad-mediated interleukin 10 secretion that facilitates coordinated immunoregulatory activity and amelioration of TGF-beta1-mediated fibrosis. J Exp Med (2003) 198:1179–88. 10.1084/jem.20030917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8:523–32. 10.1038/nri2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature (2007) 449:361–5. 10.1038/nature06110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Fogel-Petrovic M, Long JA, Misso NL, Foster PS, Bhoola KD, Thompson PJ. Physiological concentrations of transforming growth factor beta1 selectively inhibit human dendritic cell function. Int Immunopharmacol (2007) 7:1924–33. 10.1016/j.intimp.2007.07.003 [DOI] [PubMed] [Google Scholar]
- 118. Delvig AA, Lee JJ, Chrzanowska-Lightowlers ZM, Robinson JH. TGF-beta1 and IFN-gamma cross-regulate antigen presentation to CD4 T cells by macrophages. J Leukoc Biol (2002) 72:163–6. [PubMed] [Google Scholar]
- 119. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell (2009) 16:183–94. 10.1016/j.ccr.2009.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Balazovich KJ, Fernandez R, Hinkovska-Galcheva V, Suchard SJ, Boxer LA. Transforming growth factor-beta1 stimulates degranulation and oxidant release by adherent human neutrophils. J Leukoc Biol (1996) 60:772–7. 10.1002/jlb.60.6.772 [DOI] [PubMed] [Google Scholar]
- 121. Monneret G, Finck ME, Venet F, Debard AL, Bohe J, Bienvenu J, et al. The anti-inflammatory response dominates after septic shock: association of low monocyte HLA-DR expression and high interleukin-10 concentration. Immunol Lett (2004) 95:193–8. 10.1016/j.imlet.2004.07.009 [DOI] [PubMed] [Google Scholar]
- 122. Knapp S, Thalhammer F, Locker GJ, Laczika K, Hollenstein U, Frass M, et al. Prognostic value of MIP-1 alpha, TGF-beta 2, sELAM-1, and sVCAM-1 in patients with gram-positive sepsis. Clin Immunol Immunopathol (1998) 87:139–44. 10.1006/clin.1998.4523 [DOI] [PubMed] [Google Scholar]
- 123. Yeh FL, Shen HD, Fang RH. Deficient transforming growth factor beta and interleukin-10 responses contribute to the septic death of burned patients. Burns J Int Soc Burn Injuries (2002) 28:631–7. 10.1016/S0305-4179(02)00113-4 [DOI] [PubMed] [Google Scholar]
- 124. Gabriel VA. Transforming growth factor-beta and angiotensin in fibrosis and burn injuries. J Burn Care Res (2009) 30:471–81. 10.1097/BCR.0b013e3181a28ddb [DOI] [PubMed] [Google Scholar]
- 125. Marie C, Cavaillon JM, Losser MR. Elevated levels of circulating transforming growth factor-beta 1 in patients with the sepsis syndrome. Ann Intern Med (1996) 125:520–1. 10.7326/0003-4819-125-6-199609150-00034 [DOI] [PubMed] [Google Scholar]
- 126. de Pablo R, Monserrat J, Reyes E, Diaz D, Rodriguez-Zapata M, la Hera A, et al. Sepsis-induced acute respiratory distress syndrome with fatal outcome is associated to increased serum transforming growth factor beta-1 levels. Eur J Intern Med (2012) 23:358–62. 10.1016/j.ejim.2011.10.001 [DOI] [PubMed] [Google Scholar]
- 127. Ayala A, Knotts JB, Ertel W, Perrin MM, Morrison MH, Chaudry IH. Role of interleukin 6 and transforming growth factor-beta in the induction of depressed splenocyte responses following sepsis. Arch Surg (1993) 128:89–94; discussion 94-5. 10.1001/archsurg.1993.01420130101015 [DOI] [PubMed] [Google Scholar]
- 128. Nullens S, De Man J, Bridts C, Ebo D, Francque S, De Winter B. Identifying Therapeutic Targets for Sepsis Research: A Characterization Study of the Inflammatory Players in the Cecal Ligation and Puncture Model. Mediators Inflammation (2018) 2018:5130463. 10.1155/2018/5130463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ahmad S, Choudhry MA, Shankar R, Sayeed MM. Transforming growth factor-beta negatively modulates T-cell responses in sepsis. FEBS Lett (1997) 402:213–8. 10.1016/S0014-5793(96)01535-9 [DOI] [PubMed] [Google Scholar]
- 130. Xu F, Lin SH, Yang YZ, Guo R, Cao J, Liu Q. The effect of curcumin on sepsis-induced acute lung injury in a rat model through the inhibition of the TGF-beta1/SMAD3 pathway. Int Immunopharmacol (2013) 16:1–6. 10.1016/j.intimp.2013.03.014 [DOI] [PubMed] [Google Scholar]
- 131. Garcia-Lazaro JF, Thieringer F, Luth S, Czochra P, Meyer E, Renteria IB, et al. Hepatic over-expression of TGF-beta1 promotes LPS-induced inflammatory cytokine secretion by liver cells and endotoxemic shock. Immunol Lett (2005) 101:217–22. 10.1016/j.imlet.2005.06.003 [DOI] [PubMed] [Google Scholar]
- 132. Kumar A, Kumar A, Paladugu B, Mensing J, Parrillo JE. Transforming growth factor-beta1 blocks in vitro cardiac myocyte depression induced by tumor necrosis factor-alpha, interleukin-1beta, and human septic shock serum. Crit Care Med (2007) 35:358–64. 10.1097/01.CCM.0000254341.87098.A4 [DOI] [PubMed] [Google Scholar]
- 133. Ganeshan K, Johnston LK, Bryce PJ. TGF-beta1 limits the onset of innate lung inflammation by promoting mast cell-derived IL-6. J Immunol (2013) 190:5731–8. 10.4049/jimmunol.1203362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Chen W, Frank ME, Jin W, Wahl SM. TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity (2001) 14:715–25. 10.1016/S1074-7613(01)00147-9 [DOI] [PubMed] [Google Scholar]
- 135. Conway Morris A, Datta D, Shankar-Hari M, Stephen J, Weir CJ, Rennie J, et al. Cell-surface signatures of immune dysfunction risk-stratify critically ill patients: INFECT study. Intensive Care Med (2018) 44:627–35. 10.1007/s00134-018-5247-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Jianqiong Zeng HX, Zhu G, Kuang C, Wu Y. Changes of cytokines levels after decreasing regulatory T cells activity during sepsis. Int J Clin Exp Pathol (2017) 10(3):2911–6. [Google Scholar]
- 137. Hotchkiss RS, Colston E, Yende S, Crouser ED, Martin GS, Albertson T, et al. Immune checkpoint inhibition in sepsis: a Phase 1b randomized study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of nivolumab. Intensive Care Med (2019) 45:1360–71. 10.1007/s00134-019-05704-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ziegler SF, Roan F, Bell BD, Stoklasek TA, Kitajima M, Han H. The biology of thymic stromal lymphopoietin (TSLP). Adv Pharmacol (2013) 66:129–55. 10.1016/B978-0-12-404717-4.00004-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Bjerkan L, Sonesson A, Schenck K. Multiple Functions of the New Cytokine-Based Antimicrobial Peptide Thymic Stromal Lymphopoietin (TSLP). Pharm (Basel) (2016) 9. 10.3390/ph9030041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Takai T. TSLP expression: cellular sources, triggers, and regulatory mechanisms. Allergol Int (2012) 61:3–17. 10.2332/allergolint.11-RAI-0395 [DOI] [PubMed] [Google Scholar]
- 141. Varricchi G, Pecoraro A, Marone G, Criscuolo G, Spadaro G, Genovese A, et al. Thymic Stromal Lymphopoietin Isoforms, Inflammatory Disorders, and Cancer. Front Immunol (2018) 9:1595. 10.3389/fimmu.2018.01595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. He R, Geha RS. Thymic stromal lymphopoietin. Ann N Y Acad Sci (2010) 1183:13–24. 10.1111/j.1749-6632.2009.05128.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Zhong J, Sharma J, Raju R, Palapetta SM, Prasad TS, Huang TC, et al. TSLP signaling pathway map: a platform for analysis of TSLP-mediated signaling. Database (Oxford) (2014) 2014:bau007. 10.1093/database/bau007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Yu X, Li H, Ren X. Signaling cascades initiated by TSLP-mediated signals in different cell types. Cell Immunol (2012) 279:174–9. 10.1016/j.cellimm.2012.10.005 [DOI] [PubMed] [Google Scholar]
- 145. Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol (2002) 3:673–80. 10.1038/ni805 [DOI] [PubMed] [Google Scholar]
- 146. West EE, Spolski R, Kazemian M, Yu ZX, Kemper C, Leonard WJ. A TSLP-complement axis mediates neutrophil killing of methicillin-resistant Staphylococcus aureus. Sci Immunol (2016) 1. 10.1126/sciimmunol.aaf8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kuethe JW, Prakash PS, Midura EF, Johnson BL,3, Kasten KR, Caldwell CC. Thymic stromal lymphopoietin mediates the host response and increases mortality during sepsis. J Surg Res (2014) 191:19–24. 10.1016/j.jss.2014.05.024 [DOI] [PubMed] [Google Scholar]
- 148. Han NR, Moon PD, Kim HM, Jeong HJ. TSLP Exacerbates Septic Inflammation via Murine Double Minute 2 (MDM2) Signaling Pathway. J Clin Med (2019) 8. 10.3390/jcm8091350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lan F, Zhang N, Holtappels G, De Ruyck N, Krysko O, Van Crombruggen K, et al. Staphylococcus aureus Induces a Mucosal Type 2 Immune Response via Epithelial Cell-derived Cytokines. Am J Respir Crit Care Med (2018) 198:452–63. 10.1164/rccm.201710-2112OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Hajj J, Blaine N, Salavaci J, Jacoby D. The “Centrality of Sepsis”: A Review on Incidence, Mortality, and Cost of Care. Healthc (Basel) (2018) 6. 10.3390/healthcare6030090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Rhee P, Joseph B, Pandit V, Aziz H, Vercruysse G, Kulvatunyou N, et al. Increasing trauma deaths in the United States. Ann Surg (2014) 260:13–21. 10.1097/SLA.0000000000000600 [DOI] [PubMed] [Google Scholar]
- 152. Mira JC, Szpila BE, Nacionales DC, Lopez MC, Gentile LF, Mathias BJ, et al. Patterns of gene expression among murine models of hemorrhagic shock/trauma and sepsis. Physiol Genomics (2016) 48:135–44. 10.1152/physiolgenomics.00072.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Smedman C, Gardlund B, Nihlmark K, Gille-Johnson P, Andersson J, Paulie S. ELISpot analysis of LPS-stimulated leukocytes: human granulocytes selectively secrete IL-8, MIP-1beta and TNF-alpha. J Immunol Methods (2009) 346:1–8. 10.1016/j.jim.2009.04.001 [DOI] [PubMed] [Google Scholar]
- 154. Guignant C, Lepape A, Huang X, Kherouf H, Denis L, Poitevin F, et al. Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care (2011) 15:R99. 10.1186/cc10112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Hutchins NA, Unsinger J, Hotchkiss RS, Ayala A. The new normal: immunomodulatory agents against sepsis immune suppression. Trends Mol Med (2014) 20:224–33. 10.1016/j.molmed.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Zhang Y, Li J, Lou J, Zhou Y, Bo L, Zhu J, et al. Upregulation of programmed death-1 on T cells and programmed death ligand-1 on monocytes in septic shock patients. Crit Care (2011) 15:R70. 10.1186/cc10059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Chang K, Svabek C, Vazquez-Guillamet C, Sato B, Rasche D, Wilson S, et al. Targeting the programmed cell death 1: programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Crit Care (2014) 18:R3. 10.1186/cc13176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Shipkova M, Wieland E. Surface markers of lymphocyte activation and markers of cell proliferation. Clin Chim Acta (2012) 413:1338–49. 10.1016/j.cca.2011.11.006 [DOI] [PubMed] [Google Scholar]
- 159. Chang KC, Burnham CA, Compton SM, Rasche DP, Mazuski RJ, McDonough JS, et al. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care (2013) 17:R85. 10.1186/cc12711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Reinhart K, Bauer M, Riedemann NC, Hartog CS. New approaches to sepsis: molecular diagnostics and biomarkers. Clin Microbiol Rev (2012) 25:609–34. 10.1128/CMR.00016-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Taneja I, Reddy B, Damhorst G, Dave Zhao S, Hassan U, Price Z, et al. Combining Biomarkers with EMR Data to Identify Patients in Different Phases of Sepsis. Sci Rep (2017) 7:10800. 10.1038/s41598-017-09766-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.