

Abstract
Cellular energy production requires coordinated interactions between genetic components from the nuclear and mitochondrial genomes. This coordination results in coadaptation of interacting elements within populations. Interbreeding between divergent gene pools can disrupt coadapted loci and result in hybrid fitness breakdown. While specific incompatible loci have been detected in multiple eukaryotic taxa, the extent of the nuclear genome that is influenced by mitonuclear coadaptation is not clear in any species. Here, we used F2 hybrids between two divergent populations of the copepod Tigriopus californicus to examine mitonuclear coadaptation across the nuclear genome. Using developmental rate as a measure of fitness, we found that fast-developing copepods had higher ATP synthesis capacity than slow developers, suggesting variation in developmental rates is at least partly associated with mitochondrial dysfunction. Using Pool-seq, we detected strong biases for maternal alleles across 7 (of 12) chromosomes in both reciprocal crosses in high-fitness hybrids, whereas low-fitness hybrids showed shifts toward the paternal population. Comparison with previous results on a different hybrid cross revealed largely different patterns of strong mitonuclear coadaptation associated with developmental rate. Our findings suggest that functional coadaptation between interacting nuclear and mitochondrial components is reflected in strong polygenic effects on this life-history phenotype, and reveal that molecular coadaptation follows independent evolutionary trajectories among isolated populations.
Keywords: copepods, pool-seq, ATP synthesis, hybridization, postzygotic isolation
Significance
Production of cellular energy within mitochondria involves the interaction of proteins encoded in the nucleus as well as in the organelle itself. Because of this coordination, genes in the nuclear chromosomes are expected to be influenced by changes in the mitochondrial genome. Using a pair of hybrid crosses in an intertidal copepod, here, we show that by changing the mitochondrial background in hybrid individuals, the frequencies of nuclear genes change substantially in one generation in the direction of the same source population as the mitochondrial genome. This bias is strongest in the fastest-developing individuals, suggesting that high levels of fitness are achieved when nuclear and mitochondrial genes are inherited from the same parental populations because these genes coevolved to function in harmony.
Introduction
Cellular energy production via oxidative phosphorylation (OXPHOS) occurs inside the mitochondria, but relies on physical interactions between proteins encoded by both the mitochondrial (∼13 proteins) and nuclear genomes (∼60–80 proteins; Bar-Yaacov et al. 2012). Although OXPHOS is the ultimate generator of ATP, mtDNA replication, transcription, and gene translation also require coordination between the two genomes. Because of these intimate interactions, natural selection within an isolated population is expected to favor allele combinations between the two genomes that optimize physiological function. In many taxa, this process of coadaptation often involves the evolution of compensatory substitutions in nuclear genes as these respond to rapidly evolving mitochondrial genomes (Rand et al. 2004; Ballard and Pichaud 2014; Sloan et al. 2018).
Given sufficient divergence, secondary contact and hybridization is known to break up previously coevolved mitonuclear components, forcing nuclear-encoded proteins to function with a “novel” mitochondrial genome. Experiments in multiple systems have demonstrated reduced fitness of hybrids, termed hybrid breakdown, associated with functional “mismatches” between mitonuclear pathway components (Bolnick et al. 2008; Ellison et al. 2008; Chou et al. 2010; Meiklejohn et al. 2013). Mitochondrial dysfunction resulting from such incompatibilities is now appreciated as a possible driver of reproductive isolation, as well as possibly underpinning certain human cellular and physiological impairments (Burton et al. 2013; Reinhardt et al. 2013; Wolff et al. 2014; Zaidi and Makova 2019). Despite many examples of specific mitonuclear incompatibilities, it is unclear whether certain regions are more likely than others to be involved in mitonuclear incompatibilities, and whether hybrid breakdown observed in different population crosses is due to the same incompatible regions. Moreover, how does intensity of selection and number of incompatible regions change with increasing divergence?
The copepod Tigriopus californicus has been a model for examining hybrid breakdown for decades (Burton 1990a, 1990b). This species inhabits shallow pools on the supralittoral zone (“splash pools”) along the west coast of North America. Gene flow between adjacent populations is nearly zero, with many population pairs showing mtDNA sequence divergence in excess of 20% (Burton and Lee 1994; Burton et al. 2007; Willett and Ladner 2009). Divergence at nuclear genes is also high (Barreto et al. 2011). Nevertheless, laboratory crosses generate hybrids with highly fit F1 individuals, but starting at the F2 generation, hybrid populations suffer hybrid breakdown, in which fitness is markedly decreased, affecting several phenotypes, such as reduced female fecundity, slower developmental time to maturity, and lower viability (Burton 1990a; Edmands 1999; Ellison and Burton 2006; Ellison and Burton 2008; Foley et al. 2013). Although hybrid fitness is on average lower than that of parental populations, a major feature of this pattern of hybrid breakdown is a wider distribution of fitness levels among recombinant hybrids, with new extreme levels of low fitness reached in some individuals, whereas other recombinant individuals retain parental-level fitness (Edmands 1999; Ellison and Burton 2008; Barreto and Burton 2013). This pattern suggests that hybrid breakdown is caused by certain genetic incompatibilities, and does not occur due to a generalized genomic instability. Breakdown has also been shown to occur in mitochondrial function, with recombinant hybrids exhibiting, on average, reduced capacity for ATP synthesis (Ellison and Burton 2006; Ellison and Burton 2008). Hybrid breakdown has been traced to mitonuclear incompatibilities, and some studies have found association of specific mitonuclear genotypes and mitochondrial function. For example, mitonuclear subunits of the cytochrome c oxidase enzyme complex (complex IV of OXPHOS) that are “matched” (i.e., come from the same population) result in higher enzyme activity in vitro (Edmands and Burton 1999; Rawson and Burton 2002). Also, hybrids with matched mitochondrial RNA polymerase (mtRPOL) and mtDNA also have higher mitochondrial gene transcription (Ellison and Burton 2008). Although these target gene studies have identified loci at least partly involved in mitonuclear incompatibilities, genome-wide analyses have predicted that strong coding sequence coadaptation has occurred across many functional pathways (Barreto et al. 2018). Therefore, selective pressure imposed by the mitochondrial genome is likely manifested at multiple functional pathways.
In order to quantify the role of mitonuclear coadaptation on nuclear genome evolution, a recent study by Healy and Burton (2020) compared nuclear allele frequencies between fast-developing (high fitness) F2 hybrids from reciprocal crosses of two T. californicus populations (San Diego [SD] and Santa Cruz [SC], California). They found strong selection favoring the respective maternal alleles along chromosomes 1–5 (out of 12), demonstrating that maintaining compatibility between nuclear and mitochondrial alleles at multiple loci is important for individual development and mitochondrial function. A major advantage of the T. californicus system is the presence of a population divergence continuum across its geographic range (Edmands 1999, 2001; Barreto et al. 2018), permitting inquiries about the repeatability and accumulation of functional variants. Lima et al. (2019) took advantage of this framework and compared allele frequency shifts associated with viability to adulthood in reciprocal crosses of three different interpopulation hybrid F2 samples. Like Healy and Burton (2020), they found evidence for strong mitonuclear effects in several chromosomes in each cross. However, only chromosome 10 was similarly involved in all crosses, and each cross had at least two uniquely affected chromosomes.
Here, we extend this line of investigation by quantifying nuclear allele frequency changes in reciprocal hybrid crosses, with individuals differing in developmental rate and ATP synthesis rates. We use a divergent interpopulation cross (SD and Strawberry Hill [SH], Oregon) and examine the evolution of mitonuclear coadaptation across the nuclear genome, identifying single nucleotide polymorphisms (SNPs) via whole-genome sequencing. In addition, by comparing our results to those of Healy and Burton (2020) (same phenotypes and approach, different crosses), we ask if the same nuclear regions are involved, or whether SD and SH have accumulated different or more coadapted loci.
Materials and Methods
Copepod Culturing and Scoring of Developmental Rates
Copepods were collected from high intertidal pools in San Diego, CA (henceforth SD: 32.45, –117.25) and Strawberry Hill Wayside, OR (SH: 44.25, –124.11) and maintained in large cultures in 400-ml beakers containing artificial seawater (ASW; Instant Ocean, Blacksburg, VA) at 35 parts per thousand salinity. The beakers were kept in constant-temperature incubators at 20 °C and 12 h:12 h light:dark cycles. Cultures were fed a mixture of live microalgae (Isochrysis galbana and Nannochloropsis oculata) and dry commercial fish food (ground Spirulina and TetraMin flakes). Once a month, partial water changes and food additions were performed, and beakers mixed haphazardly to promote outbreeding. Stock cultures were maintained in this manner for a minimum of two months (approximately two generations) before individuals were used in any of the experiments below.
Interpopulation crosses (SD♀×SH♂ and its reciprocal SH♀×SD♂) were initiated by combining, in 100 mm × 15 mm Petri dishes, virgin females (separated from mate-guarded pairs) with males from the other population. Approximately 100 pairs of each cross were started, and maintained in multiple dishes (∼20 pairs per dish) in the same conditions as stock cultures. After mating occurred, males were removed and the females monitored. Females were transferred to a new Petri dish once F1 larvae were observed, and larvae from multiple plates were pooled in a beaker to maximize outbreeding. The F1 were allowed to mature and form breeding pairs, which were then transferred to new Petri dishes. After mating occurred, males were removed and females monitored for egg development. Egg sacs that were well-developed were carefully removed from gravid females using a fine syringe needle and transferred to a new Petri dish. Clutches manipulated in this manner generally hatch within a few hours, but we removed any eggs that had not hatched after 18 h. Only five egg sacs were hatched in each plate to control for density, and food amount was standardized across all dishes.
Since all F2 clutches in a given dish hatched within 18 h, they were assigned the same hatch date and then monitored daily for metamorphosis into copepodids (CI stage). Development time for each individual was scored as the number of days from hatch to CI. Each scored copepodid was then isolated into a well of a 24-well plate, assigned a unique ID number, and allowed to grow to maturity to increase DNA yield for genetic analysis. Isolated adult copepods were starved for 24 h to clear their gut prior to being saved in 1.5-ml tubes at –80 °C until DNA isolation. Hatch dishes were monitored daily until no individuals were seen. We hatched enough egg sacs until at least 2,000 individuals were scored per reciprocal cross.
To obtain parental-level fitness distributions, we scored development time for pure SD and SH individuals in the same manner. Developed egg sacs were haphazardly picked from gravid females found in healthy stock cultures, and clutches hatched and monitored as above. A minimum of ∼1,200 hatched individuals were phenotyped per parental population.
Sequence Divergence
The overall genomic divergence between SD and SH has never been reported. Here, we calculated raw percent sequence divergence between SD and SH and between SD and SC by performing reciprocal BlastN alignments between the annotated SD genes (Barreto et al. 2018) and a published SH transcriptome assembly (NCBI accession GHUE00000000; Graham and Barreto 2019), and published SC transcriptome (Pereira et al. 2016; Dryad accession dryad.23s61).
Genome Sequencing and Analyses
For Pool-seq (Schlötterer et al. 2014), we prepared four pools of copepods, each comprising the 10% fastest- or slowest-growing copepods from each reciprocal cross (fig. 1). Pools of individuals were placed in a 1.5-ml tube containing isolation buffer and DNA was isolated using phenol: chloroform (Sambrook and Russell 2010). Possible contaminant RNA was removed with RNase A incorporated in the protocol. For each pool, we constructed a whole-genome DNA library using the Nextera DNA Library Preparation Kit (Illumina, San Diego, CA). Each pool received unique dual barcodes during PCR enrichment of 10 cycles. Post-PCR samples were then size-selected from a 2.5% agarose gel in the range 350–550 bp, and gel slices were purified with MinElute Gel Extraction kit (Qiagen, Hilden, Germany). After quantification (Qubit fluorescence, Thermo Fisher, Waltham, MA), purified samples were pooled in equimolar amounts and sequenced as 150-bp paired-end reads across 1.5 lanes of the Illumina HiSeq3000 platform at the Center for Genome Research and Biocomputing (Oregon State University, Corvallis, Oregon).
Fig. 1.
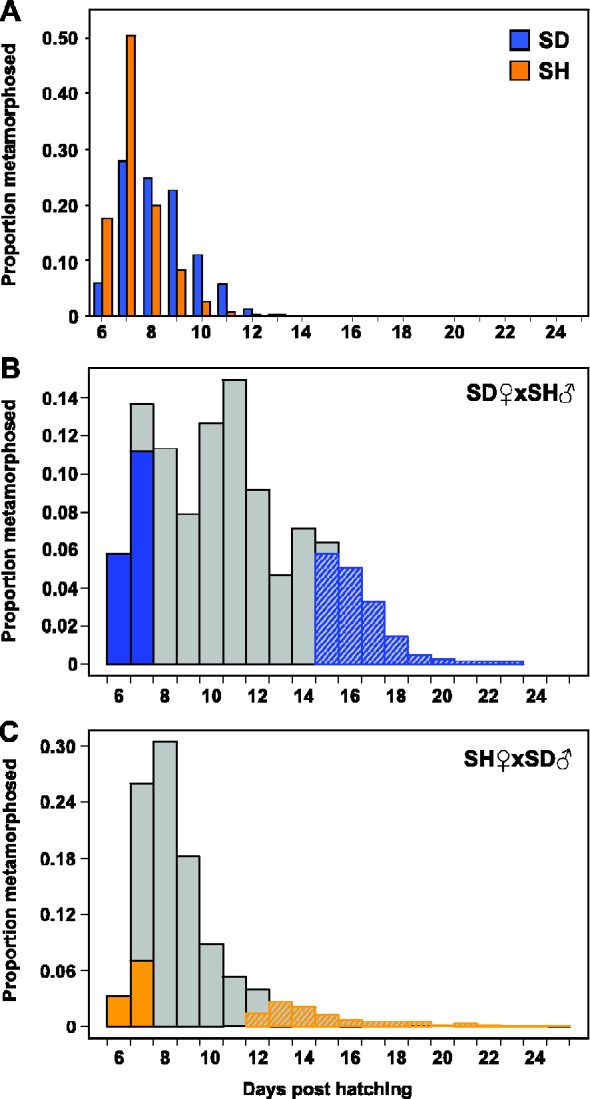
Frequency distributions of developmental time to metamorphosis in Tigriopus californicus. Individuals were scored as the number of days from hatching to reaching copepodid I stage. (A) Outbred parental populations, San Diego (SD, n = 1,368) and Strawberry Hill (SH, n = 1,416); (B) SD♀×SH♂ (n = 2,097); (C) SH♀×SD♂ (n = 3,093). Shaded areas in B and C represent the subset of individuals (n = 300) pooled from each phenotypic range.
Mapping of reads and quantification of allele frequencies followed the pipeline designed by Lima and Willett (2018), and used in other studies (Lima et al. 2019; Healy and Burton 2020). Before allele frequency analyses, we generated a reference genome assembly for the SH population. A single whole-genome Nextera sample was prepared from a pool of 20 full-sib adult copepods (i.e., from the same clutch). This sample was sequenced as 150-bp paired-end reads on half of one lane of an Illumina HiSeq3000 instrument. Reads were trimmed for base quality, and adaptor contamination using “cutadapt” (Martin 2011), and only reads >50 bp were retained. To generate the assembly, we followed the approach in Barreto et al. (2018) which uses the de novo assembly for the SD population in reference-guided assemblies of other populations. In brief, SH reads were mapped to the SD assembly using bwa-mem (Li and Durbin 2009) with more permissive parameters (-k 15 -B 3 -O 5 -E 0), and only uniquely mapped reads were retained with samtools (Li et al. 2009). We then used samtools and bcftools to extract the consensus sequence from the alignments. This process retains the scaffolding of the SD assembly while replacing the sequence with that from the SH reads. Because of the divergence between these two populations, large gaps are expected, especially in intergenic regions. Nevertheless, the coordinates are maintained between SD and SH. The two assemblies were then “equalized” with respect to all gaps (“N” positions) so that differences in assembly quality did not influence mapping. In addition, a catalog of SNP positions that are fixed between SD and SH were identified by mapping and variant calling of population-specific reads (the SH reads obtained here, and SD reads from Barreto et al. 2018, accession SRX469409) to the other’s genome assembly.
Reads from the four hybrid pools were quality-filtered as above, and then mapped in parallel to the SD and SH assemblies using bwa-mem. Reads with MAPQ scores lower than 20 were removed. PoPoolation2 (Kofler et al. 2011) was used to determine allele counts mapped to each population genome, with minimum coverage of 50, maximum coverage of 500, and minimum allele read count of 4. We cross-checked the SNP positions between a pool’s mapping to SD and SH, and retained only sites that were common in both mappings and in common with the catalog of fixed SNPs between populations. In each pool, each SNP site’s read counts were averaged between the mapping to the two references to obtain final read count per allele. For demonstration of this method's accuracy, see Lima and Willett (2018) and Lima et al. (2019).
For statistical analyses, we retained loci positions that were common across the four hybrid pools and that had a minimum read coverage of 80×. To quantify change in allele frequencies between certain pairs of hybrid pools (e.g., fast developers between crosses), we calculated the Z-statistic developed by Huang et al. (2012) and applied a false-discovery rate (FDR) correction (Benjamini and Hochberg 1995) at α = 0.01. The Z-statistic for each SNP locus is a direct measure of the difference in allele frequency between pools, while taking into account read coverage and pool size.
ATP Synthesis Rates
A distinct set of crosses were generated, at a smaller scale (35–40 parental pairs per reciprocal cross), to quantify mitochondrial function in fast and slow developers. F2 hatching, monitoring, and scoring were done as above. Metamorphosed larvae were cultured in individual wells of 24-well plates. At day 32 post hatching, when most individuals had already reached adulthood, 6–7 pools of eight copepods for each group (fast and slow developers for each reciprocal cross) were used in ATP assays following the method in Harada et al. (2019). Briefly, live samples were transferred to a 1.5-ml tube and rinsed once with 200 µl of ice-cold homogenization buffer (HB; 400 mM sucrose, 100 mM KCl, 6 mM EGTA, 3 mM EDTA, 70 mM HEPES, 1% BSA, pH 7.6). Each sample was homogenized in 800 μl cold HB using a 2-ml glass tissue grinder (Sigma–Aldrich), and transferred to new 1.5-ml tubes. These were centrifuged for 5 min at 1,000 × g at 4°C. The supernatant was transferred to new tubes and centrifuged for 10 min at 11,000 × g (4 °C). The supernatant was pipetted out and discarded, and the mitochondrial pellet gently resuspended in 55 μl of assay buffer (AB; 560 mM sucrose, 100 mM KCl, 10 mM KH2PO4, 70 mM HEPES, pH 7.6) and kept on ice until assay (up to 1 h).
ATP synthesis measurements were performed with a luciferase luminescence protocol using CellTiter-Glo reagent (Promega, Madison, WI). From each sample, 5 μl of mitochondrial resuspension were saved (–80 °C) for protein quantification. Samples were then split into two aliquots of 25 μl and placed in PCR tubes. To each aliquot, 5 μl of complex I substrate were added and mixed by pipetting, with final substrate concentration of 5 mM pyruvate, 2 mM malate and 1 mM ADP. To one set of aliquots, 25 μl of CellTiter-Glo was added immediately following substrate mixing; this stops ATP synthesis and was used as initial (time “0”) ATP levels. The other set of aliquots was incubated in a thermal cycler at 20 °C for 10 min, to allow for ATP synthesis, and then CellTiter-Glo was added to stop the reactions. Previous experiments using this protocol showed that between 5 min and 3 h, ATP synthesis in vitro is linear, and can hence be quantified within that time period (Ellison and Burton 2006; Ellison and Burton 2008). We used a 10-min synthesis incubation for consistency with more recent work in this species (Harada et al. 2019; Healy and Burton 2020). After addition of CellTiter-Glo, all samples were then incubated in the dark for 10 min, following manufacturer's protocol, to allow luminescence signal to stabilize, and then transferred to a white half-area plate along with ATP standards. Luminescence was measured in a Tecan Spark plate reader (Männedorf, Switzerland), and an absolute ATP synthesis rate for each sample was calculated by subtracting initial from final luminescence values and comparing these to the ATP standard curve run on the same plate. Sample rates were normalized to protein content that was obtained from the saved mitochondrial aliquot using the NanoOrange Protein Quantitation Kit (Thermo Fisher, Waltham, WA). After verification that data were normally distributed (Shapiro–Wilk test: W = 0.969, P = 0.603), ATP synthesis rates between fast and slow developers in each reciprocal cross were compared with t-tests.
Comparison to SD x SC Cross
To more directly compare our observed allele frequency shifts to the previously studied cross SD×SC (Healy and Burton 2020), we obtained the Z-scores they observed for the comparison between fast-developer pools, available from their supplementary materials, and compared their distribution to ours, per chromosome. Because the number of SNPs obtained in each study differed, we downsampled both sets by randomly sampling 1,000 SNP sites per chromosome in each data set. Distributions of Z-scores (absolute values) were then compared with a t-test per chromosome and adjusted for multiple comparisons with FDR correction.
Results
Developmental Rate Variation
Pattern of time-to-metamorphosis was similar between the pure SD and SH larvae. Out of 1,368 and 1,416 phenotyped individuals in SD and SH, respectively, over 50% reached the CI stage in 8 or fewer days from hatch date (fig. 1A). The slowest developers reached CI in 13 days in both populations. The distribution of rates among F2 larvae was substantially different than that of parentals, particularly with regards to the range. In both reciprocal crosses, hybrid breakdown is reflected by the presence of much slower rates of development, with some individuals taking 23 days or more to reach CI stage (fig. 1B and C). The cross with SD mitochondrial background (SD♀ × SH♂) was more strongly affected, with over 25% of larvae taking longer than 13 days (i.e., beyond parental range) to reach CI (fig. 1B), whereas only 6% of larvae in the reciprocal cross were in this slow range (fig. 1C). In total, 2,097 individuals were phenotyped from SD♀×SH♂ and 3,093 from SH♀× SD♂.
For estimating allele frequencies in high- and low-fitness groups, we pooled ∼300 fast-developing and ∼300 slow-developing copepods within each cross (shading in fig. 1B and C). Based on the distributions of developmental rates, fast developers metamorphosed in days 6 and 7 posthatching in both crosses. Pools of slow developers included individuals with CI days ≥12 in SH♀×SD♂ and ≥ 15 in SD♀× SH♂.
ATP Synthesis Rates
Mitochondrial function was assessed as rates of ATP synthesis in vitro from organelles isolated from fast- and slow-developing pools of copepods from each cross. For each developmental group, we quantified Complex I-fueled ATP synthesis rates in seven replicates in the SD♀ × SH♂ cross, and in six replicates in the SH♀ × SD♂ cross. ATP synthesis rates were on average ∼60% higher in the fast developers compared with slow developers in both crosses (SD♀ × SH♂: t = 2.49, P = 0.029; SH♀ × SD♂: t = 2.42, P = 0.038; fig. 2).
Fig. 2.

Rate of ATP synthesis in fast- and slow-developing F2 hybrid copepods. Developmental rates were quantified as the number of days from hatching until metamorphosis into copepodid I stage (fast: 6 days; slow: 15+ days). Measurements are of complex I-fueled ATP production in isolated mitochondria. Rates between fast and slow groups within each hybrid cross were significantly different (t-tests, P < 0.05).
Sequencing and SNP Calling Summary
A total of 253 million paired-reads were obtained from the single SH sample and used in the reference-based assembly of that population’s genome. After equalization, the assemblies contained ∼17% gapped regions. Across hybrid pools, 461 million paired-reads were obtained (range: 49–152 million), and after mapping, average coverage per base pair was 146× for SD♀×SH♂-fast, 155× for SD♀×SH♂-slow, 172× for SH♀×SD♂-fast, and 60× for SH♀×SD♂-slow. At our final minimum coverage threshold (>80X), the first three pools above had between 2.2 and 2.7 million SNPs, whereas SH♀×SD♂-slow had 146,507 SNP sites. Nevertheless, the four pools had a total of 106,929 SNP positions in common, and a mean of 8,910 SNPs per chromosome (range: 6,289–10,225; table 1), which were used in our main analyses.
Table 1.
Number and Density of SNP Loci Examined in Common across the Four Pooled Samples (SD♀×SH♂ Fast- and Slow-Developers, and SH♀×SD♂ Fast- and Slow-Developers)
| Chromosome | Total SNPs | SNPs/Mbp |
|---|---|---|
| 1 | 9,915 | 601 |
| 2 | 8,275 | 542 |
| 3 | 8,179 | 552 |
| 4 | 8,241 | 616 |
| 5 | 9,796 | 594 |
| 6 | 7,420 | 486 |
| 7 | 9,518 | 578 |
| 8 | 10,225 | 652 |
| 9 | 9,818 | 621 |
| 10 | 9,997 | 613 |
| 11 | 9,256 | 586 |
| 12 | 6,289 | 348 |
Allele Frequency Deviations
High-fitness (fast-developing) copepods of the reciprocal crosses exhibited widespread shifts in nuclear allele frequencies across the genome. All chromosomes had multiple SNPs with significantly different frequencies between the crosses. Four chromosomes (2, 4, 9, and 10) in both crosses had large regions which showed shifts in the direction of their respective mitochondrial background (i.e., mitonuclear match; fig. 3), and three (5, 8, and 11) with smaller regions showing this pattern. Chromosome 1 had strong frequency differences, but only one cross followed a pattern of mitochondrial matching. A pattern of mitonuclear mismatch (increase in allele frequency from paternal population) was present, albeit weakly, in a portion of chromosome 7. Comparisons of allele frequencies between slow developers also had significant deviations across large regions of all chromosomes, but patterns varied widely among chromosomes (fig. 4). Chromosomes 4, 6, and 10 showed strong mitonuclear mismatching (i.e., alleles matching mitochondrial background decreased in frequency), but matching was observed on chromosome 1 and weakly on chromosome 11.
Fig. 3.

Pool-seq analysis of allele frequencies in fast-developing F2 hybrid copepods. (A) Allele frequencies for each reciprocal cross along the 12 autosomes. Each point represents a mean of frequencies across SNPs in 200-kb windows. Legend on right side of panel aids in interpretation of mitonuclear matching/mismatching; (B) Results of Z-statistics (Huang et al. 2012) comparing allele frequencies between crosses at individual SNP loci (n = 106,929). Red dashed line marks the false-discovery rate (FDR) threshold of 0.01.
Fig. 4.

Pool-seq analysis of allele frequencies in slow-developing F2 hybrid copepods. (A) Allele frequencies for each reciprocal cross along the 12 autosomes. Each point represents a mean of frequencies across SNPs in 200-kb windows. Legend on right side of panel aids in interpretation of mitonuclear matching/mismatching; (B) Results of Z-statistics (Huang et al. 2012) comparing allele frequencies between crosses at individual SNP loci (n = 106,929). Red dashed line marks the false-discovery rate (FDR) threshold of 0.01.
The distributions of allele frequency changes among SD×SH fast developers were compared with the ones quantified for another cross, SD×SC, from Healy and Burton (2020). Here we compared absolute values of Z-statistics (1,000 randomly sampled SNPs) from both studies for each chromosome. Consistent with broad patterns reported for these crosses, the SD×SC cross had stronger frequency deviations (i.e., higher Z-scores) than SD×SH on chromosomes 2, 3, and 5, whereas SD×SH showed higher deviations on chromosomes 8–11. Chromosomes 1 and 4, which had very significant deviations within each study, showed no difference in distribution of Z-scores between studies (fig. 5).
Fig. 5.

Comparison of allele frequencies deviations between crosses across chromosomes. Plotted are boxplot distributions of Z-statistics quantifying the difference in frequencies between fast-developers from reciprocal crosses in SD×SH (this study), and in SD×SC (SC = Santa Cruz, Healy and Burton 2020). Each boxplot comprises Z-statistics of 1,000 randomly sampled SNP loci. P-values are from Wilcoxon tests comparing Z-statistics between crosses, and underlined chromosomes are those showing significant differences.
Genomic Divergence among Populations
We estimated genetic divergence between SD and SH based on raw percent difference at nuclear and mtDNA gene sequences. MtDNA protein-coding genes (n = 13) ranged from 11.1% to 35.2%, with highest values occurring in genes from OXPHOS complex I. Among nuclear genes (n = 14,485), median differentiation was 2.8%, with maximum of 22.8%. In comparison, nuclear gene sequences between SD and SC (n = 13,704) showed a median and maximum differentiation of 2.5% and 22.3%, respectively. The overall distributions of nuclear gene differences are similar, but SD-SH have evolved higher levels of divergence and in more genes than SD-SC (Wilcoxon test: W = 86,503,875, P < 10−15; fig. 6). Differences in mtDNA genes are similarly very high, as is common in this system, but some genes in complex I are nearly 7% more divergent between SD-SH than SD-SC (table 2).
Fig. 6.
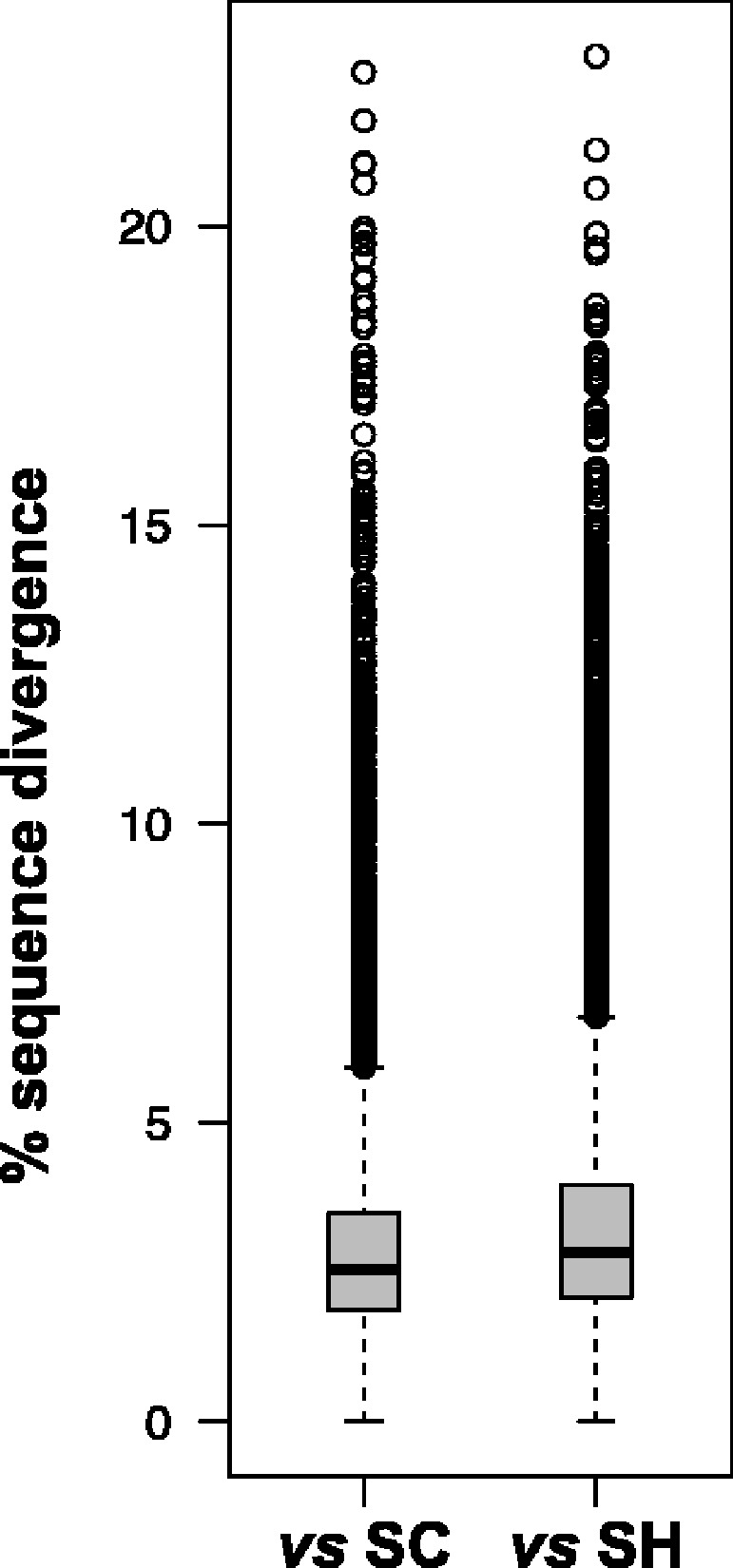
DNA sequence differentiation between SD and two populations across nuclear protein-coding genes. SD versus SC has 13,704 gene alignments, and SD versus SH has 14,485.
Table 2.
Raw DNA Sequence Differentiation of mtDNA-Encoded Genes between San Diego (SD) and Two Other Populations (SC, Santa Cruz; SH, Strawberry Hill)
| mtDNA Protein-Coding Genes(Number of Genes) | % Difference |
|
|---|---|---|
| SD Versus SC | SD Versus SH | |
| Complex I (7) | 24.8–27.8 | 22.2–35.2 |
| Complex III (1) | 21.1 | 21.7 |
| Complex IV (3) | 19.5–21.9 | 18.9–19.4 |
| Complex V (2) | 8.1–25.3 | 11.1–24.9 |
Discussion
Hybridization that produces offspring with reduced fitness, but that are otherwise still viable and fertile, represents an early stage of reproductive isolation between the parental lineages. The importance of incompatibilities between nuclear and cytoplasmic (chloroplast or mitochondrial) genomes in generating hybrid breakdown has long been appreciated, and may occur disproportionately in taxa that lack heteromorphic sex chromosomes (Bolnick et al. 2008; Burton et al. 2013; Bar-Yaacov et al. 2015; Sloan et al. 2017; Sunnucks et al. 2017; Hill et al. 2019). The strength and consequence of selection on nuclear genomes, as imposed by its coadaptation to the mitochondrial genome, is still poorly examined, especially in experimental settings (see Zaidi and Makova 2019 for an example in admixed populations). In this study, we demonstrate that even after a single generation of recombination, mitochondrial backgrounds strongly affect the direction of nuclear allele frequency changes, with high-fitness cohorts (with fast-development) exhibiting a biased allele frequency favoring the maternal allele, reciprocally, across small to wide swaths of seven chromosomes. Conversely, frequency shifts in the direction of the paternal population were minimal. Moreover, we found that F2 hybrids differing in developmental rates also differ in mitochondrial fitness, as measured by OXPHOS ATP synthesis capacity. This association between strong mitonuclear matching and these two phenotypes suggests that variation in developmental time to metamorphosis is at least partially influenced by efficiency of mitochondrial function. Our findings among low-fitness (slow-developing) copepod pools revealed less consistent patterns of nuclear frequency shifts, with mitonuclear mismatch occurring in only three chromosomes, and others even showing mitonuclear match. We argue that together these findings suggest that maintenance of parental-level developmental rates, as represented by our pools of fast developers, requires a polygenic mechanism of mitonuclear matching. However, disruption of even only a few coadapted loci may be sufficient for causing substantial negative effects in this phenotype.
In an effort to provide a comparative framework for learning about repeatability of mitonuclear coadaptation, we examined a new cross (SD×SH) but followed a similar experimental protocol as Healy and Burton (2020), which examined hybrids between SD and SC. In both crosses, developmental rate and mitochondrial ATP output were categorically associated in F2 hybrids, supporting previous evidence that disruption of mitochondrial function is likely the ultimate cause of slower-than-parental development. As with SD×SC, mitonuclear effects were also clear and pervasive across the genome of SD×SH hybrids. While allele frequency changes in the F2 pools (away from 0.5 in the F1) were numerically low (∼0.05–0.15), such shifts are actually a high proportion of the maximum change expected. Assuming “lethality” of a homozygous genotype (or in this case, its exclusion from the high-fitness pool), the maximum allele frequency change is 0.167 in an F2 cohort following an F1×F1 cross. A major contrast of patterns between these crosses was the identity of nuclear chromosomes with strongest effects. Although mitonuclear effects were observed in chromosomes 2, 4, and 5 in both crosses, SD×SH also showed clear involvement of loci in chromosomes 8, 9, 10, and 11 among fast-developing copepods. These latter chromosomes showed no tendency for mitonuclear matching in the SD×SC cross. Altogether, these comparisons reveal that functional coadaptation among interacting nuclear- and mtDNA-encoded molecules can follow very distinct evolutionary trajectories in isolated populations, and that no specific genes or functions are more likely than others to be affected in contributing to fast-development phenotypes. A recent study in three sets of reciprocal crosses, also using Pool-seq, similarly found largely different genomic architecture regarding mitonuclear coadaptation affecting a different phenotype, viability to adulthood (Lima et al. 2019). Such lack of parallelism is likely the result of natural demographic dynamics of T. californicus populations. Specifically, the nearly complete lack of gene flow among populations prevents adaptive polymorphism from being shared, and the very low effective population sizes (Pereira et al. 2016) allows drift to push allele frequencies in very unique trajectories among locations. Therefore, which nuclear compensatory variants become favored in a given population depends on the idiosyncrasy of which mitochondrial haplotypes are segregating.
In addition to mitonuclear effects, epistasis amongst nuclear genes has also been detected in T. californicus interpopulation hybrids. Allelic patterns of nuclear-nuclear effects on given phenotypes are evidenced by frequency shifts favoring the same parental population in both reciprocal crosses, because the interacting alleles can be inherited similarly in both crosses regardless of mtDNA background. Using a QTL approach for viability to adulthood in SD×SC, Foley et al. (2013) observed genotypic ratios consistent with nuclear-nuclear effects in some markers on chromosome 11. Nuclear-nuclear interactions affecting viability were also detected by Lima et al. (2019) across three different crosses; here, a total of four chromosomes were involved, but none overlapped among crosses. Nevertheless, nuclear-only effects were much weaker and less pervasive than mitonuclear effects in both of these past studies of viability. In our study with SD×SH, we detected only a weak signal of nuclear-nuclear interactions associated with fast development (on chromosome 3), with frequency deviations along the chromosome often not differing from ∼0 (fig. 3). A similarly weak pattern on a single chromosome (8) was found in SD×SC fast developers (Healy and Burton 2020). The regions with nuclear-nuclear patterns were substantially weaker than any mitonuclear patterns in both crosses. Overall, epistasis between mitonuclear components appear to be more important in T. californicus, but epistasis between nuclear genes also contribute to some of the patterns observed in hybrids and vary in strength depending on interpopulation divergence and phenotype measured.
Organellar-nuclear (or cytonuclear) genetic interactions have received strong experimental support in many systems, and have been shown to be particularly clear in plant systems, where chloroplast-nuclear incompatibilities can significantly affect germination and survival (Fishman and Willis 2006; Barnard-Kubow et al. 2016). Investigating cytonuclear coadaptation is particularly attractive because of the greatly reduced set of possible cellular functions and molecular interactions involved, allowing for more targeted investigation of genetic mechanisms. In T. californicus, Barreto et al. (2018) annotated 605 nuclear-encoded proteins predicted to function exclusively within mitochondria. Of these, only 146 participate in cellular functions that require interaction with mtDNA directly (e.g., replication, n = 3 proteins; transcription, n = 3) or with its encoded elements (OXPHOS, n = 57; ribosomal assembly, n = 67; and protein translation, n = 16). Our study joins a few recent others showing that selective pressure imposed by mtDNA on the nuclear genome results in complex patterns. With only one generation of recombination, exacerbated by the rarity of recombination in T. californicus females (Burton et al. 1981), our experiment lacks the power to identify smaller regions within affected chromosomes. The four chromosomes most clearly showing mitonuclear matching in SD×SH fast developers (2, 4, 9, and 10) harbor a total of 69 genes encoding mitochondrial proteins that work across nearly all cellular functions mentioned above (supplementary table S1, Supplementary Material online), with no function being overrepresented when compared with the remaining 77 genes (Fisher's exact test, P > 0.35 for each function). A study using later-generation or recombinant inbred lines and coarse mapping would provide sufficient resolution to further narrow the set of loci.
While identifying specific interactions may be valuable for investigations of molecular function and may be more experimentally tractable, we argue that the T. californicus system can provide excellent opportunities for testing more general hypotheses regarding the repeatability and accumulation of incompatibilities. Studies of mitonuclear coadaptation generally start by acknowledging nucleotide divergences in mtDNA, regardless of what processes have contributed to them. Recent physiological work in T. californicus (Harada et al. 2019), and other systems (Camus et al. 2017; Greimann et al. 2020), have demonstrated strong environmental effects on patterns of mtDNA variation. Integrating these evolutionary processes into studies of mitonuclear function will be key in understanding differences in reproductive isolation among populations.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank M. Dasenko and K. Carter for help in genome data sequencing, and T. Lima, T. Healy, and R. Burton for assistance with computational analyses and feedback on earlier versions of the results. We also thank A. Graham for comments on the manuscript. This research was supported by a National Science Foundation award [DEB #1556455 to F.S.B.].
Data Availability
Raw reads from Pool-seq Illumina sequencing are deposited in NCBI Sequence Read Archive (SRA; accessions SRR9019159–SRR9019162 for the four hybrid pools, and SRR9019163 for the pure SH pool). Tables of mapped read counts and estimated allele frequencies are deposited in Dryad (accession doi.org/10.5061/dryad.9w0vt4bdm).
Literature Cited
- Ballard JWO, Pichaud N.. 2014. Mitochondrial DNA: more than an evolutionary bystander. Funct Ecol. 28(1):218–231. [Google Scholar]
- Barnard-Kubow KB, So N, Galloway LF.. 2016. Cytonuclear incompatibility contributes to the early stages of speciation. Evolution 70(12):2752–2766. [DOI] [PubMed] [Google Scholar]
- Barreto F, Moy G, Burton R.. 2011. Interpopulation patterns of divergence and selection across the transcriptome of the copepod Tigriopus californicus. Mol Ecol. 20(3):560–572. [DOI] [PubMed] [Google Scholar]
- Barreto FS, et al. 2018. Genomic signatures of mitonuclear coevolution across populations of Tigriopus californicus. Nat Ecol Evol. 2(8):1250–1257. [DOI] [PubMed] [Google Scholar]
- Barreto FS, Burton RS.. 2013. Elevated oxidative damage is correlated with reduced fitness in interpopulation hybrids of a marine copepod. Proc R Soc B. 280(1767):20131521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Yaacov D, et al. 2015. Mitochondrial involvement in vertebrate speciation? The case of mito-nuclear genetic divergence in chameleons. Genome Biol Evol. 7(12):3322–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Yaacov D, Blumberg A, Mishmar D.. 2012. Mitochondrial-nuclear co-evolution and its effects on OXPHOS activity and regulation. BBA – Gene Regul Mech. 1819(9–10):1107–1111. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y.. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 57(1):289–300. [Google Scholar]
- Bolnick DI, Turelli M, López-Fernández H, Wainwright PC, Near TJ.. 2008. Accelerated mitochondrial evolution and ‘Darwin’s corollary’: asymmetric viability of reciprocal F1 hybrids in centrarchid fishes. Genetics 178(2):1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton RS. 1990a. Hybrid breakdown in developmental time in the copepod Tigriopus californicus. Evolution 44(7):1814–1822. [DOI] [PubMed] [Google Scholar]
- Burton RS. 1990b. Hybrid breakdown in physiological response: a mechanistic approach. Evolution 44(7):1806–1813. [DOI] [PubMed] [Google Scholar]
- Burton RS, Byrne R, Rawson P.. 2007. Three divergent mitochondrial genomes from California populations of the copepod Tigriopus californicus. Gene 403(1–2):53–59. [DOI] [PubMed] [Google Scholar]
- Burton RS, Feldman M, Swisher S.. 1981. Linkage relationships among five enzyme-coding gene loci in the copepod Tigriopus californicus: a genetic confirmation of achiasmatic meiosis. Biochem Genet. 19(11–12):1237–1245. [DOI] [PubMed] [Google Scholar]
- Burton RS, Lee B.. 1994. Nuclear and mitochondrial gene genealogies and allozyme polymorphism across a major phylogeographic break in the copepod Tigriopus californicus. Proc Natl Acad Sci USA. 91(11):5197–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton RS, Pereira RJ, Barreto FS.. 2013. Cytonuclear genomic interactions and hybrid breakdown. Annu Rev Ecol Evol Syst. 44(1):281–302. [Google Scholar]
- Camus MF, Wolff JN, Sgrò CM, Dowling DK.. 2017. Experimental support that natural selection has shaped the latitudinal distribution of mitochondrial haplotypes in Australian Drosophila melanogaster. Mol Biol Evol. 34(10):2600–2612. [DOI] [PubMed] [Google Scholar]
- Chou J, Hung Y, Lin K, Lee H, Leu J.. 2010. Multiple molecular mechanisms cause reproductive isolation between three yeast species. PLoS Biol. 8(7):e1000432–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmands S. 1999. Heterosis and outbreeding depression in interpopulation crosses spanning a wide range of divergence. Evolution 53(6):1757–1768. [DOI] [PubMed] [Google Scholar]
- Edmands S. 2001. Phylogeography of the intertidal copepod Tigriopus californicus reveals substantially reduced population differentiation at northern latitudes. Mol Ecol. 10(7):1743–1750. [DOI] [PubMed] [Google Scholar]
- Edmands S, Burton RS.. 1999. Cytochrome c oxidase activity in interpopulation hybrids of a marine copepod: a test for nuclear-nuclear or nuclear-cytoplasmic coadaptation. Evolution 53(6):1972–1978. [DOI] [PubMed] [Google Scholar]
- Ellison CK, Burton RS.. 2006. Disruption of mitochondrial function in interpopulation hybrids of Tigriopus californicus. Evolution 60(7):1382–1391. [PubMed] [Google Scholar]
- Ellison CK, Burton RS.. 2008. Genotype-dependent variation of mitochondrial transcriptional profiles in interpopulation hybrids. Proc Natl Acad Sci USA. 105(41):15831–15836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison CK, Burton RS.. 2008. Interpopulation hybrid breakdown maps to the mitochondrial genome. Evolution 62(3):631–638. [DOI] [PubMed] [Google Scholar]
- Ellison CS, Niehuis O, Gadau J.. 2008. Hybrid breakdown and mitochondrial dysfunction in hybrids of Nasonia parasitoid wasps. J Evol Biol. 21(6):1844–1851. [DOI] [PubMed] [Google Scholar]
- Fishman L, Willis JH.. 2006. A cytonuclear incompatibility causes anther sterility in Mimulus hybrids. Evolution 60(7):1372–1381. [DOI] [PubMed] [Google Scholar]
- Foley BR, Rose CG, Rundle DE, Leong W, Edmands S.. 2013. Postzygotic isolation involves strong mitochondrial and sex-specific effects in Tigriopus californicus, a species lacking heteromorphic sex chromosomes. Heredity 111(5):391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham AM, Barreto FS.. 2019. Loss of the HIF pathway in a widely distributed intertidal crustacean, the copepod Tigriopus californicus. Proc Natl Acad Sci USA. 116(26):12913–12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greimann ES, et al. 2020. Phenotypic variation in mitochondria-related performance traits across New Zealand snail populations. Integr Comp Biol. 60(2):275–287. [DOI] [PubMed] [Google Scholar]
- Harada AE, Healy TM, Burton RS.. 2019. Variation in thermal tolerance and Its relationship to mitochondrial function across populations of Tigriopus californicus. Front Physiol. 10:729–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy TM, Burton RS.. 2020. Strong selective effects of mitochondrial DNA on the nuclear genome. Proc Natl Acad Sci USA. 117(12):6616–6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill GE, et al. 2019. Assessing the fitness consequences of mitonuclear interactions in natural populations. Biol Rev. 94(3):1089–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, et al. 2012. Epistasis dominates the genetic architecture of Drosophila quantitative traits. Proc Natl Acad Sci USA. 109(39):15553–15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler R, Pandey RV, Schlötterer C.. 2011. PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 27(24):3435–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima TG, Burton RS, Willett CS.. 2019. Genomic scans reveal multiple mito-nuclear incompatibilities in population crosses of the copepod Tigriopus californicus. Evolution 73(3):609–620. [DOI] [PubMed] [Google Scholar]
- Lima TG, Willett CS.. 2018. Using Pool-seq to search for genomic regions affected by hybrid inviability in the copepod T. californicus. J Hered. 109(4):469–476. [DOI] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 17(1):10–12. [Google Scholar]
- Meiklejohn CD, et al. 2013. An incompatibility between a mitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLoS Genet. 9(1):e1003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira RJ, Barreto FS, Pierce NT, Carneiro M, Burton RS.. 2016. Transcriptome-wide patterns of divergence during allopatric evolution. Mol Ecol. 25(7):1478–1493. [DOI] [PubMed] [Google Scholar]
- Rand D, Haney R, Fry A.. 2004. Cytonuclear coevolution: the genomics of cooperation. Trends Ecol Evol. 19(12):645–653. [DOI] [PubMed] [Google Scholar]
- Rawson P, Burton R.. 2002. Functional coadaptation between cytochrome c and cytochrome c oxidase within allopatric populations of a marine copepod. Proc Natl Acad Sci USA. 99(20):12955–12958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt K, Dowling DK, Morrow EH.. 2013. Mitochondrial replacement, evolution, and the clinic. Science 341(6152):1345–1346. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW.. 2010. Purification of nucleic acids by extraction with phenol:chloroform. Cold Spring Harb Protoc. 2006(1):pdb.prot4455. [DOI] [PubMed] [Google Scholar]
- Schlötterer C, Tobler R, Kofler R, Nolte V.. 2014. Sequencing pools of individuals—mining genome-wide polymorphism data without big funding. Nat Rev Genet. 15(11):749–763. [DOI] [PubMed] [Google Scholar]
- Sloan DB, et al. 2018. Cytonuclear integration and co-evolution. Nat Rev Genet. 19:635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Havird JC, Sharbrough J.. 2017. The on-again, off-again relationship between mitochondrial genomes and species boundaries. Mol Ecol. 26(8):2212–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunnucks P, Morales HE, Lamb AM, Pavlova A, Greening C.. 2017. Integrative approaches for studying mitochondrial and nuclear genome co-evolution in oxidative phosphorylation. Front Genet. 8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett CS, Ladner J.. 2009. Investigations of fine-scale phylogeography in Tigriopus californicus reveal historical patterns of population divergence. BMC Evol Biol. 9(1):139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff JN, Ladoukakis ED, Enríquez JA, Dowling DK.. 2014. Mitonuclear interactions: evolutionary consequences over multiple biological scales. Phil Trans R Soc B. 369(1646):20130443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi AA, Makova KD.. 2019. Investigating mitonuclear interactions in human admixed populations. Nat Ecol Evol. 3(2):213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw reads from Pool-seq Illumina sequencing are deposited in NCBI Sequence Read Archive (SRA; accessions SRR9019159–SRR9019162 for the four hybrid pools, and SRR9019163 for the pure SH pool). Tables of mapped read counts and estimated allele frequencies are deposited in Dryad (accession doi.org/10.5061/dryad.9w0vt4bdm).