

Abstract
The origin and deep history of retroviruses remain mysterious and contentious, largely because the diversity of retroviruses is incompletely understood. Here, we report the discovery of lokiretroviruses, a novel major lineage of retroviruses, within the genomes of a wide range of vertebrates (at least 137 species), including lampreys, ray-finned fishes, lobe-finned fishes, amphibians, and reptiles. Lokiretroviruses share a similar genome architecture with known retroviruses, but display some unique features. Interestingly, lokiretrovirus Env proteins share detectable similarity with fusion glycoproteins of viruses within the Mononegavirales order, blurring the boundary between retroviruses and negative sense single-stranded RNA viruses. Phylogenetic analyses based on reverse transcriptase demonstrate that lokiretroviruses are sister to all the retroviruses sampled to date, providing a crucial nexus for studying the deep history of retroviruses. Comparing congruence between host and virus phylogenies suggests lokiretroviruses mainly underwent cross-species transmission. Moreover, we find that retroviruses replaced their ribonuclease H and integrase domains multiple times during their evolutionary course, revealing the importance of domain shuffling in the evolution of retroviruses. Overall, our findings greatly expand our views of the diversity of retroviruses, and provide novel insights into the origin and complex evolutionary history of retroviruses.
Keywords: retroviruses, paleovirology, phylogenetics
Introduction
Retroviruses infect a wide range of vertebrates (Gifford and Tristem 2003; Hayward et al. 2015; Xu et al. 2018), and cause various diseases in human populations, severely threatening global public health. The replication of retroviruses requires reverse transcription of viral RNA into double-stranded DNA (dsDNA) and integration of viral dsDNA into host genomes (Stoye 2012; Johnson 2019). Retroviruses occasionally infect the host germ line, and can become vertically inherited as endogenous retroviruses (ERVs) (Stoye 2012; Johnson 2019). ERVs accumulated in the host genomes partially document retroviral infections over time, and thus provide “molecular fossils” for studying the deep history and macroevolution of retroviruses as well as the evolution of ancient host–retrovirus interactions (Stoye 2012; Johnson 2019).
The diversity of retroviruses has been extensively studied, and nearly 70 exogenous retroviruses and thousands of ERVs have been identified (Herniou et al. 1998; Hayward et al. 2015; Xu et al. 2018; Walker et al. 2019). Currently, exogenous retroviruses are classified into two subfamilies, Orthoretrovirinae and Spumaretrovirinae (Walker et al. 2019). Orthoretrovirinae includes six genera (Alpharetrovirus, Betaretrovirus, Gammaretrovirus, Deltaretrovirus, Epsilonretrovirus, and Lentivirus), whereas Spumaretrovirinae (also known as foamy virus) includes five genera (Bovispumavirus, Equispumavirus, Felispumavirus, Prosimiispumavirus, and Simiispumavirus) (Walker et al. 2019). Traditionally, ERVs have been grouped into classes I, II, and III based on their close relatedness to Gammaretrovirus, Betaretrovirus, and Spumaretrovirus (Gifford and Tristem 2003; Gifford et al. 2018). However, the classification systems between exogenous and endogenous retroviruses are incompatible, and some ERVs cannot be readily classified (Gifford et al. 2018; Xu et al. 2018).
Retroviruses share similarity with Metaviridae (the Ty3/Gypsy retrotransposon), Pseudoviridae (the Ty1/Copia retrotransposon), Belpaoviridae (the Bel/Pao retrotransposon), and Caulimoviridae in terms of domain architecture and sequence homology (Xiong and Eickbush 1990; Krupovic et al. 2018). These five virus families are thought to originate from a common viral ancestor, and have been unified into a single virus order, Ortervirales (Krupovic et al. 2018). Phylogenetic analyses based on reverse transcriptase (RT) suggest retroviruses are closely related to metaviruses (Doolittle et al. 1989; Xiong and Eickbush 1990; Eickbush and Jamburuthugoda 2008; Krupovic et al. 2018). Retroviruses encode a dual ribonuclease H (RH) domain: retroviruses acquired a new RH domain, and the preexisting RH domain degenerated to become the tether domain (Malik and Eickbush 2001; Smyshlyaev et al. 2013; Ustyantsev et al. 2015). Sporadic phylogenetic analyses of RH suggest that retroviral RH is not monophyletic, and different retroviruses might have acquired RH from different sources (Malik and Eickbush 2001; Smyshlyaev et al. 2013; Ustyantsev et al. 2015; Gong and Han 2018). However, much remains obscure about the origin and deep history of retroviruses (Hayward 2017).
In this study, we systematically screened the presence of ERVs within the deuterostome genomes and report the discovery of a novel major lineage of retroviruses, referred to as lokiretroviruses, in the genomes of a wide range of vertebrates. Lokiretroviruses exhibit a genome architecture similar to known retroviruses, but display a number of unique features. Evolutionary analyses of lokiretroviruses and retroviruses sampled previously provide novel insights into the diversity and the complex evolutionary history of retroviruses.
Results
The Discovery of Lokiretroviruses
Initially, we employed a similarity search and phylogenetic analysis combined approach to search for the genetic elements that are closely related to retroviruses within the deuterostome genomes. Interestingly, we identified a lineage of retrotransposons that form a sister group to all the known retroviruses within the genomes of species from five vertebrate classes, including Petromyzontida, Actinopterygii (at least 120 species within 36 orders), Sarcopterygii, Amphibian, and Reptile (fig. 1A and supplementary fig. S1, Supplementary Material online). This retrotransposon lineage is referred to as lokiretroviruses, following the name of Loki, the cunning trickster god in Norse mythology. To further characterize lokiretroviruses, we reconstructed lokiretrovirus consensus sequences for 24 representative vertebrate species (supplementary data set S1; fig. S1, Supplementary Material online).
Fig. 1.

Distribution and genome structure of lokiretrovirus. (A) Distribution of endogenous lokiretrovirus. Orange circles indicate the presence of endogenous lokiretroviruses. The empty orange circle indicates highly degraded endogenous lokiretroviruses in Testudines. The first column of numbers represents the number of genomes used to screen lokiretroviruses. The second column of numbers represents the number of genomes where lokiretroviruses were identified. (B) Consensus genome structure of lokiretrovirus. The lokiretrovirus genome encodes at least three ORFs (gag, pol, and env) flanked by two LTRs. Some lokiretrovirus genomes encode an additional smc gene. (C) Comparison of capsid (CA) secondary structure among Loki-Str (the lokiretrovirus from Salmo trutta), MuLV (PDB accession No.: 6HWW.A), and Ty3 (PDB accession No.: 6R24.D). The helices and arrows represent α-helices and β-strands, respectively. “T” and “S” indicate hydrogen bonded turn and bend, respectively. (D) Model of lokiretrovirus Env protein. Sp, fp, cc, and tm represent signal peptide, fusion peptide, coiled-coil motif, and transmembrane domain, respectively. RXKR is the cleavage site. The alignment between Loki-Str Env and the consensus sequence of fusion glycoprotein F0 (pfam No.: PF00523) was shown.
The consensus sequences comprise at least three predicted genes (gag, pol, and env) flanked by two long terminal repeats (LTRs) (fig. 1B). For the putative Gag protein, we found a large part of it shares significant structural similarity with murine leukemia virus (MuLV) capsid (CA) (confidence = 97.83%) and Ty3 (belonging to Metaviridae) CA (confidence = 95.23%) (fig. 1C). The lokiretrovirus pol gene encodes all the retroviral enzymes, including protease (PR), RT, RH, and integrase (IN). Like retroviruses, the lokiretrovirus genomes also encode a degraded RH domain known as the tether domain between RT and RH domains (fig. 1B). The lokiretrovirus Env proteins possess signal peptide, fusion peptide, coiled-coil (CC) motif, transmembrane (TM) domain, and a RXKR proteolytic cleavage site (fig. 1D). Unexpectedly, the lokiretrovirus Env proteins share detectable sequence similarity with the fusionglycoproteins (pfam No.: PF00523) of Paramyxoviridae and Pneumovirinae within the Mononegavirales order (e-value = 2.2 × 10−7; fig. 1D). Unlike retroviruses, some lokiretrovirus genomes encode a protein homologous to structural maintenance of chromosomes (SMC) proteins that bind DNA and function in many aspects of chromosome dynamics (Harvey et al. 2002) (supplementary fig. S2, Supplementary Material online). The smc gene appears to be acquired during the evolution of lokiretroviruses (supplementary fig. S2, Supplementary Material online). Taken together, our results demonstrate that lokiretroviruses share similar genome architecture with retroviruses, and display several unique features.
Lokiretroviruses Are Sister to Known Retroviruses
To explore the relationship between lokiretroviruses, retroviruses, and other orterviruses, we performed phylogenetic analyses based on RT proteins, and found lokiretroviruses are sister group to all the known retroviruses (supplementary figs. S3, S4A, and data set S2, Supplementary Material online). Furthermore, we performed phylogenetic analyses of lokiretroviruses, representative retroviruses, and metaviruses based on RT proteins using two different alignment methods. Our phylogenetic analyses show that retroviruses and lokiretroviruses form two independent clades with robust supports (lokiretrovirus monophyly: ultrafast bootstrap approximation [UFBoot] = 100% for both alignments; retrovirus monophyly: UFBoot = 97% for both alignments) (fig. 2A, supplementary data sets S3 and S4, Supplementary Material online). Lokiretroviruses form a sister group to known retroviruses with robust supports (UFBoot = 100% for both alignments), but do not fall within the diversity of sampled retroviruses (fig. 2A, supplementary data sets S3 and S4; fig. S3 and data set S2, Supplementary Material online). Therefore, our phylogenetic analyses demonstrate that lokiretroviruses are sister to retroviruses sampled to date, and represents a novel major lineage of retroviruses.
Fig. 2.

Phylogenetic relationship of lokiretroviruses and retroviruses. (A) Phylogenetic relationship among lokiretroviruses, retroviruses, and metaviruses based on their RT protein sequences (supplementary data sets S3 and S4, Supplementary Material online). The support values for selected nodes are shown in the form of UFBoot for align-Ma/UFBoot for align-3D. Ma and 3D indicate sequences aligned using MAFFT and PROMALS3D, respectively. (B) Comparison of the phylogenies between lokiretroviruses and their hosts. The left is the phylogeny of lokiretroviruses based on the RT proteins, whereas the right is the host tree based on the fish tree of life (Rabosky et al. 2018).
Frequent Interspecies Jump by Lokiretroviruses
We observed that the lokiretrovirus phylogeny is generally incongruent with the host phylogeny (especially for ray-finned fishes), indicating that lokiretroviruses might have undergone frequent host switching (fig. 2B). For instance, different from vertebrates, the lokiretrovirus from Anguilla rostrata (a ray-finned fish) rather than lamprey lokiretroviruses branch earliest within the lokiretrovirus tree. Then, we quantitatively compared the phylogeny of lokiretroviruses with that of their vertebrate hosts using an event-based approach. We assembled two data sets: one includes all of the 24 consensus sequences of Petromyzontida (2), Actinopterygii (17), Sarcopterygii (1), and Amphibian (4), and the other includes 17 lokiretrovirus consensus sequences from Actinopterygii. We found no statistically significant congruence between virus and host phylogenies for both data sets with exception of one set of event costs (table 1). Taken together, these results suggest that lokiretroviruses evolved mainly through frequent cross-species transmission.
Table 1.
Host–Virus Phylogeny Congruence Test for Lokiretrovirus
| Data Sets (No. of Species Used) | Event Costsa | Total Cost | No. of Events |
P Valueb | ||||
|---|---|---|---|---|---|---|---|---|
| Cospeciation | Duplication | Duplication and Host Switching | Loss | Failure to Diverge | ||||
| Total (24) | 0, 1, 2, 1, 1 | 32 | 10 | 0 | 13 | 6 | 0 | p = 0.006 |
| Total (24) | 0, 1, 1, 2, 0 | 18 | 5 | 0 | 18 | 0 | 0 | p > 0.05 |
| Total (24) | –1, 0, 0, 0, 0 | –13 | 13 | 0 | 10 | 27 | 0 | p > 0.05 |
| Actinopterygii (17) | 0, 1, 2, 1, 1 | 26 | 5 | 0 | 11 | 4 | 0 | p > 0.05 |
| Actinopterygii (17) | 0, 1, 1, 2, 0 | 14 | 2 | 0 | 14 | 0 | 0 | p > 0.05 |
| Actinopterygii (17) | –1, 0, 0, 0, 0 | –8 | 8 | 0 | 8 | 22 | 0 | p > 0.05 |
Event cost schemes are for cospeciation, duplication, duplication with host switch, loss, and failure to diverge, respectively.
P-value represents statistical analyses results by using the method of random parasite tree with the sample size of 500.
Complex Evolution of Retrovirus RH Domain
Retrovirus genomes encode a dual RH domain (Malik and Eickbush 2001; Ustyantsev et al. 2015). They acquired a new RH domain during their evolution, and the preexisting RH domain degenerated into a short domain known as “tether” (Malik and Eickbush 2001; Ustyantsev et al. 2015). Interestingly, we found lokiretrovirus genomes also encode a dual RH domain (fig. 1B). The tether domain of lokiretrovirus shares detectable structure and sequence similarity with MuLV tether domain (confidence = 100%; sequence identity = 24%) and HIV-1 tether domain (confidence = 99%; sequence identity = 15%) (fig. 3A). Moreover, the putative tether domain of lokiretrovirus also share significant similarity with the RH domain of Saccharomyces cerevisiae Ty3 (metavirus) (confidence = 100%; sequence identity = 32%) (fig. 3A). It follows that lokiretroviruses and retroviruses are indeed closely related, and the degeneration of the preexisting RH domain that is closely related to metavirus RH occurred before the most recent common ancestor of lokiretroviruses and retroviruses.
Fig. 3.

Structural analyses and phylogenetic trees of tether and RH domain. (A) Comparison of secondary structure among Loki-Str tether, MuLV tether (PDB accession No.: 1RW3.A), HIV-1 tether (PDB accession No.: 2ZD1.B), and Ty3 RH (PDB accession No.: 4OL8.A). The helices and arrows represent α-helices and β-strands, respectively. “T,” “S,” and “G” indicate hydrogen bonded turn, bend, and 3-turn helix, respectively. (B) Phylogenetic trees of the RH domain (supplementary data sets S5 and S6, Supplementary Material online). The support values for selected nodes are shown in the form of UFBoot for align-Ma/UFBoot for align-3D. Ma and 3 D indicate sequences aligned using MAFFT and PROMALS3D, respectively.
To explore how retrovirus RH originated and evolved, we performed phylogenetic analyses of the RH domain. We found that retroviruses do not form a monophyletic group, but cluster into at least three groups with strong supports (fig. 3B, supplementary data sets S5 and S6, Supplementary Material online), which largely agrees with previous studies (for example, Ustyantsev et al. 2015). Lokiretroviruses and foamy viruses form group I with strong supports (UFBoot = 96% for the alignment generated by Mafft [align-Ma]). Lentiviruses form group II (UFBoot = 100% for align-Ma; UFBoot = 99% for alignment generated by PROMALS3D [align-3D]) and cluster with non-LTR retrotransposons with lower supports (UFBoot = 71% for align-Ma; UFBoot = 68% for align-3D). The other retroviruses form group III (UFBoot = 93% for align-Ma; UFBoot = 91% for align-3D), and cluster with RH from eukaryotes and bacteria with medium support (UFBoot = 75% for align-Ma). Therefore, these results suggest that the RH domains of retroviruses replaced their RH domain multiple times during their evolutionary course.
Complex Evolution of Retrovirus IN Domain
To explore how the IN domains of retroviruses evolved, we performed phylogenetic analyses and found that the IN domain of retrovirus and lokiretrovirus are not monophyletic. Lokiretrovirus IN is more closely related to CGIN1 genes with strong supports (UFBoot = 97% for both alignments) (fig. 4, supplementary data sets S7 and S8, Supplementary Material online). Retroviruses excluding lokiretroviruses cluster into at least four groups: group I includes gamma-retroviruses and epsilon-retroviruses (UFBoot = 100% for both alignments), group II includes tetrapod foamy viruses and coelacanth endogenous foamy virus (UFBoot = 100% for both alignments), group III includes fish endogenous foamy viruses (UFBoot = 92% for align-Ma), and group IV includes the other retroviruses (UFBoot = 84% for align-Ma; UFBoot = 75% for align-3D). Group IV appears to be a sister group to metavirus IN, which is consistent with the analyses of RT domains (fig. 4, supplementary data sets S7 and S8, Supplementary Material online). Therefore, our study indicates that retrovirus IN domains do not share a single origin either, and they replaced their IN domain multiple times during their evolutionary course.
Fig. 4.
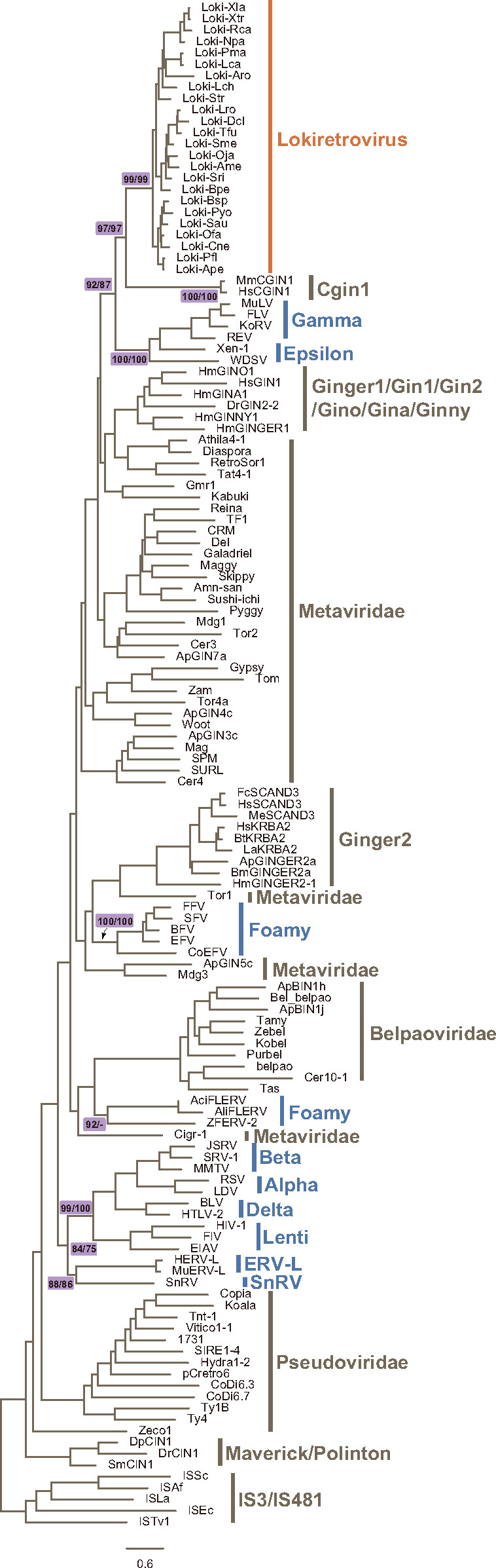
Phylogenetic trees of the IN domain (supplementary data sets S7 and S8, Supplementary Material online). The support values for selected nodes are shown in the form of UFBoot for align-Ma/UFBoot for align-3D., where Ma and 3D indicate sequences aligned using MAFFT and PROMALS3D, respectively.
Discussion
In this study, we screened the presence of ERVs within the genome of deuterostomes, and report the discovery of a novel major retrovirus lineage, lokiretroviruses. A single virus from this lineage has been reported in the sea lamprey genome through mining a large number of vertebrates previously, but phylogenetic analyses show that it falls within known retroviruses with an abnormal long branch (Hayward et al. 2015; Xu et al. 2018). In this study, we found lokiretroviruses were/are widely distributed in the genomes of vertebrates, at least including lampreys, ray-finned fishes, lobe-finned fishes, amphibians, and reptiles (reptile lokiretroviruses are highly degraded; supplementary fig. S4, Supplementary Material online). Within ray-finned fishes, endogenous lokiretrovirus elements were identified at least 120 species from 36 orders. Some complete endogenous lokiretrovirus elements share identical or nearly identical LTRs, and the integrations were estimated to occur from 0 to 36.82 Ma (supplementary table S1, Supplementary Material online), suggesting exogenous lokiretroviruses might have long been circulating in vertebrates. The lokiretrovirus genomes share similar structure with the retrovirus genomes in general, but display several unique features, including the presence of the smc gene, a novel domain not present in sampled retroviruses, in some lokiretroviruses and an env gene similar to the fusion glycoproteins of some negative sense single-stranded RNA viruses.
After several decades of extensive phylogenetic analyses and virus discovery, the understanding of the diversity and evolution of retroviruses had been thought to be largely completed. To date, seven genera of exogenous retroviruses (Alpha-, Beta-, Gamma-, Delta-, Epsilonretrovirus, Lentivirus, and Spumavirus) and three class of ERVs (classes I, II, and III) have been described. To our great surprise, we identified a novel major lineage of retroviruses, lokiretroviruses, which are distantly related to all the sampled retroviruses. Our phylogenetic analyses of the RT domain provide strong evidence that lokiretroviruses are sister to the retroviruses sampled to date. Thus, the discovery of lokiretroviruses provides an important nexus for studying the evolution of retroviruses. Together with the similarity between retroviruses and lokiretroviruses, we propose that lokiretroviruses might represent a novel subfamily within the family Retroviridae.
Unexpectedly, we found that lokiretrovirus Env proteins share detectable sequence similarity with the fusion glycoproteins of viruses from Paramyxoviridae and Pneumovirinae within the Mononegavirales order, indicating that lokiretrovirus Env and Paramyxoviridae and Pneumovirinae fusion glycoproteins derived from a common viral ancestor (hereafter referred to as virus X). However, we did not find significant overall sequence similarity between lokiretrovirus Env and retrovirus Env. Four different scenarios could be conceived (fig. 5A): 1) The common ancestor of retroviruses and lokiretroviruses acquired Env from virus X. Env evolved rapidly along the evolution of retroviruses, resulting in high sequence divergence (thus low similarity) between retroviruses and lokiretroviruses; 2) The common ancestor of retroviruses and lokiretroviruses acquired Env from unknown viral source, and lokiretroviruses replaced its Env with Env from virus X; 3) The common ancestor of retroviruses and lokiretroviruses acquired Env from virus X, and retroviruses replaced its Env with Env from unknown viral source; 4) Retroviruses and lokiretroviruses independently acquired Env from unknown viral source and from virus X. Currently, we cannot formally exclude any of these four possibilities. However, it has long been recognized that retrovirus Env and paramyxovirus fusion glycoproteins share similar sequence motifs, such as signal peptide, fusion peptide, CC motif, TM domain, and proteolytic cleavage site ( Colman and Lawrence 2003; Lamb and Jardetzky 2007). These proteins might be ultimately derived from a common viral ancestor. Previous studies found filoviruses appear to acquire Env protein horizontally from retroviruses (Bénit et al. 2001). Our discovery of similarity among lokiretrovirus Env and Paramyxoviridae and Pneumovirinae fusion glycoproteins represent an independent case that blurs the deep boundary between retroviruses and negative sense single-stranded RNA viruses and provide novel evolutionary framework to understand the origin and evolution of Env in retroviruses.
Fig. 5.

Complex evolutionary history of Env and enzyme domains of retroviruses and lokiretroviruses. (A) Four possible scenarios for the origin of lokiretrovirus and retrovirus env gene. F represents the fusion glycoprotein. (B) Complex evolutionary history of RH and IN domains of lokiretroviruses and retroviruses. RH and IN domains in different colors indicate they derived from different sources.
Our phylogenetic analyses of both RH and IN domains show that retroviruses form multiple distinct groups, suggesting that recurrent replacements of RH and IN domains occurred during their evolution. It follows that domain shuffling might shape the complexity of the retrovirus genomes. Based on the RT and RH phylogenies, we infer that the RH proteins of lokiretroviruses and foamy viruses might represent the most ancient RH lineage of retroviruses. The common ancestor of retroviruses acquired a new RH domain possibly from eukaryote hosts. The preexisting RH domain degraded into the tether domain, which explains the significant similarity between the tether domain of retroviruses and the RH domain of metaviruses. Like retroviruses, lokiretrovirus genome encodes a dual RH (tether and RH) domain, suggesting that the degradation of preexisting RH domain occurred before the most recent common ancestor of lokiretroviruses and retroviruses. After diverging from foamy viruses, retroviruses replaced the RH domain with a RH domain possibly also from eukaryote hosts, and lentiviruses replaced its RH domain by a RH possibly from non-LTR retrotransposons (fig. 5B). Reconciling the RT and IN phylogenies, we infer group IV IN of retroviruses might represent the original IN, because it is sister to metavirus IN (fig. 4, supplementary data sets S7 and S8, Supplementary Material online). Fish foamy viruses, tetrapod/coelacanth foamy viruses, lokiretroviruses, and the common ancestor of gamma- and epsilon-retroviruses replaced their IN domains independently with IN domains from different sources (fig. 5B). However, these evolutionary scenarios should be taken with cautions, because the phylogenies of RH and IN domains are notoriously difficult to reconstruct, and some nodes of our RH and IN phylogenies are only weakly supported.
In conclusion, the discovery of lokiretroviruses greatly expands the diversity of retroviruses, and provides a crucial novel retroviral group to illuminate the complex evolutionary history of retroviruses. Our findings illustrate the value of discovering novel viral lineages to understand the ancient evolution of viruses and the diversity of the viral world.
Materials and Methods
The Discovery of Lokiretroviruses
We used a similarity search and phylogenetic analysis combined approach to mine the genetic elements that are closely related to retroviruses. The TBlastN algorithm was used to screen all the available genomes of Echinodermata (14), Hemichordata (2), Urochordata (14), Cephalochordata (6), and Cyclostomata (4) (supplementary table S2, Supplementary Material online) with representative retrovirus RT proteins (supplementary table S3, Supplementary Material online) as the queries and an e cut-off value of 10−5. The significant hits and representative RT protein sequences of LTR retrotransposons (Llorens et al. 2011) were aligned using MAFFT 7.402 (Katoh and Standley 2013). Initial phylogenetic analyses were carried out using an approximate maximum-likelihood (ML) method implemented in FastTree 2.1.10 (Price et al. 2010). We found some hits (lokiretroviruses) from the genome of Petromyzon marinus cluster together to form a sister group to known retroviruses. To further explore the distribution of lokivirueses, all the available vertebrate genomes (supplementary table S2, Supplementary Material online) were further mined for lokiretroviruses using the TBlastN algorithm with the RT protein of the P. marinus lokiretrovirus as the query and an e cut-off value of 10−5. Phylogenetic analyses were also performed using FastTree 2.1.10 (Price et al. 2010). Lokiretrovirus-like hits from 24 representative species (supplementary fig. S1, Supplementary Material online) that cover the major diversity of lokiretroviruses were retrieved for consensus sequence reconstruction.
Consensus Sequence Reconstruction
The retrieved lokiretrovirus hits were bidirectionally extended to identify typical domains of retroviruses using conserved domain (CD) search with the default parameters (Marchler-Bauer et al. 2017). The LTRs were identified using LTR_Finder (Xu and Wang 2007) or BlastN. For each species, the longest sequence was retrieved and was then used as the query to search for its homologs using BlastN with an e cut-off value of 10−5, an identity cut-off value of 80% and a length cut-off value of 600 nt. The specific hits were aligned using MAFFT 7.402 (Katoh and Standley 2013). The consensus sequences were reconstructed and ORFs were predicted using Geneious (Kearse et al. 2012). Domains and motifs were annotated using phmmer (Potter et al. 2018), CD search (Marchler-Bauer et al. 2017), and SignalP 3.0 (Bendtsen et al. 2004).
Phylogenetic Analyses and Secondary Structure Prediction
Lokiretrovirus sequences from reptiles were excluded in the phylogenetic analyses due to their highly degraded nature (supplementary fig. S4, Supplementary Material online). All protein sequences (supplementary table S4, Supplementary Material online) were aligned using two methods, MAFFT 7.402 with the L-INS-I strategy (Katoh and Standley 2013) and PROMALS3D with the default parameters (Pei et al. 2008). The alignments were manually trimmed to remove ambiguous regions. Phylogenetic trees were reconstructed using a ML approach implemented in IQ-tree 2 (Minh et al. 2020). The best-fit model for each tree was estimated using Model Finder implemented in IQ-tree 2 (Kalyaanamoorthy et al. 2017). The ultrafast bootstrap approximation support was estimated with 1,000 replications (Hoang et al. 2018). The Phyre2 web server (Kelley et al. 2015) was used to compare the secondary structure of CA and tether/RH proteins of the lokiretrovirus from Salmo trutta (Loki-Str), MuLV, HIV-1, and S. cerevisiae Ty3.
Dating Analyses
The divergence between 5′ LTR and 3′ LTR can be used to estimate the minimum time of the ERV integration. The time can be estimated through: t = d/2 μ, where d represents the genetic distance between 5′ LTR and 3′ LTR of an ERV, and μ represents the host neutral evolutionary rate. Due to no neutral evolutionary rate available for fishes, we used a neutral rate estimated for mammals, ∼2.2 × 10−9 substitutions/site/year (Kumar and Subramanian 2002), to calculate the insertion time. The distance between 5′ and 3′ LTRs of a complete endogenous lokiretrovirus was calculated with the Kimura 2-parameter substitution model and four Gamma rate categories.
Host–Virus Phylogeny Congruence Analysis
The phylogeny of lokiretroviruses was compared with that of their hosts using Jane 4 (Conow et al. 2010). Different sets of cost values for five types of events (for cospeciation, duplication, duplication with host switch, loss, and failure to diverge: 0, 1, 2, 1, 1; –1, 0, 0, 0, 0; and 0, 1, 1, 2, 0) were examined (Xu et al. 2018). The statistical analyses were performed using the method of random parasite tree with the sample size of 500. Species tree used in this analysis was based on the fish tree of life (Rabosky et al. 2018).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Data Availability
The data underlying this article are available in NCBI (https://www.ncbi.nlm.nih.gov/) database. The accession numbers of genomes used, the consensus sequences of lokiretroviruses, and the alignments are available in supplementary table S2, supplementary data set 1, and supplementary data sets 2–8, Supplementary Material online.
Supplementary Material
Acknowledgments
This study was supported by National Natural Science Foundation of China (31922001 and 31701091) and Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.
References
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S.. 2004. Improved prediction of signal peptides: signalP 3.0. J Mol Biol. 340(4):783–795. [DOI] [PubMed] [Google Scholar]
- Bénit L, Dessen P, Heidmann T.. 2001. Identification, phylogeny, and evolution of retroviral elements based on their envelope genes. J Virol. 75(23):11709–11719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman PM, Lawrence MC.. 2003. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol. 4(4):309–319. [DOI] [PubMed] [Google Scholar]
- Conow C, Fielder D, Ovadia Y, Libeskind-Hadas R.. 2010. Jane: a new tool for the cophylogeny reconstruction problem. Algorithms Mol Biol. 5(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle RF, Feng DF, Johnson MS, McClure MA.. 1989. Origins and evolutionary relationships of retroviruses. Q Rev Biol. 64(1):1–30. [DOI] [PubMed] [Google Scholar]
- Eickbush TH, Jamburuthugoda VK.. 2008. The diversity of retrotransposons and the properties of their reverse transcriptases. Virus Res. 134(1–2):221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford R, Tristem M.. 2003. The evolution, distribution and diversity of endogenous retroviruses. Virus Genes. 26(3):291–315. [DOI] [PubMed] [Google Scholar]
- Gifford RJ, Blomberg J, Coffin JM, Fan H, Heidmann T, Mayer J, Stoye J, Tristem M, Johnson WE.. 2018. Nomenclature for endogenous retrovirus (ERV) loci. Retrovirology 15(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Han GZ.. 2018. Insect retroelements provide novel insights into the origin of hepatitis B viruses. Mol Biol Evol. 35(9):2254–2259. [DOI] [PubMed] [Google Scholar]
- Harvey SH, Krien MJ, O'Connell MJ.. 2002. Structural maintenance of chromosomes (SMC) proteins, a family of conserved ATPases. Genome Biol. 3(2):REVIEWS3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward A, Cornwallis CK, Jern P.. 2015. Pan-vertebrate comparative genomics unmasks retrovirus macroevolution. Proc Natl Acad Sci USA. 112(2):464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward A. 2017. Origin of the retroviruses: when, where, and how? Curr Opin Virol. 25:23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herniou E, Martin J, Miller K, Cook J, Wilkinson M, Tristem M.. 1998. Retroviral diversity and distribution in vertebrates. J Virol. 72(7):5955–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS.. 2018. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 35(2):518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE. 2019. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat Rev Microbiol. 17(6):355–370. [DOI] [PubMed] [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS.. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ.. 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 10(6):845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupovic M, Blomberg J, Coffin JM, Dasgupta I, Fan H, Geering AD, Gifford R, Harrach B, Hull R, Johnson W, et al. 2018. Ortervirales: new virus order unifying five families of reverse-transcribing viruses. J Virol. 92:e00515–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Subramanian S.. 2002. Mutation rates in mammalian genomes. Proc Natl Acad Sci USA. 99(2):803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb RA, Jardetzky TS.. 2007. Structural basis of viral invasion: lessons from paramyxovirus F. Curr Opin Struct Biol. 17(4):427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens C, Futami R, Covelli L, Domínguez-Escribá L, Viu JM, Tamarit D, Aguilar-Rodríguez J, Vicente-Ripolles M, Fuster G, Bernet GP, et al. 2011. The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res. 39(Database):D70–D74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik HS, Eickbush TH.. 2001. Phylogenetic analysis of ribonuclease H domains suggests a late, chimeric origin of LTR retrotransposable elements and retroviruses. Genome Res. 11(7):1187–1197. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, et al. 2017. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45(D1):D200–D203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R.. 2020. IQ-TREE 2: new Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol. 37(5):1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J, Kim BH, Grishin NV.. 2008. PROMALS3D: a tool for multiple sequence and structure alignment. Nucleic Acids Res. 36(7):2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, Finn RD.. 2018. HMMER web server: 2018 update. Nucleic Acids Res. 46(W1):W200–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP.. 2010. Arkin, FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5(3):e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabosky DL, Chang J, Title PO, Cowman PF, Sallan L, Friedman M, Kaschner K, Garilao C, Near TJ, Coll M, et al. 2018. An inverse latitudinal gradient in speciation rate for marine fishes. Nature 559(7714):392–395. [DOI] [PubMed] [Google Scholar]
- Smyshlyaev G, Voigt F, Blinov A, Barabas O, Novikova O.. 2013. Acquisition of an Archaea-like ribonuclease H domain by plant L1 retrotransposons supports modular evolution. Proc Natl Acad Sci USA. 110(50):20140–20145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoye JP. 2012. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol. 10(6):395–406. [DOI] [PubMed] [Google Scholar]
- Ustyantsev K, Novikova O, Blinov A, Smyshlyaev G.. 2015. Convergent evolution of ribonuclease h in LTR retrotransposons and retroviruses. Mol Biol Evol. 32(5):1197–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker PJ, Siddell SG, Lefkowitz EJ, Mushegian AR, Dempsey DM, Dutilh BE, Harrach B, Harrison RL, Hendrickson RC, Junglen S, et al. 2019. Changes to virus taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2019). Arch Virol. 164(9):2417–2429. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Eickbush TH.. 1990. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 9(10):3353–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zhao H, Gong Z, Han GZ.. 2018. Endogenous retroviruses of non-avian/mammalian vertebrates illuminate diversity and deep history of retroviruses. PLoS Pathog. 14(6):e1007072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Wang H.. 2007. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35(Web Server):W265–W268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in NCBI (https://www.ncbi.nlm.nih.gov/) database. The accession numbers of genomes used, the consensus sequences of lokiretroviruses, and the alignments are available in supplementary table S2, supplementary data set 1, and supplementary data sets 2–8, Supplementary Material online.