

Abstract
A major challenge in modern biology is understanding how the effects of short-term biological responses influence long-term evolutionary adaptation, defined as a genetically determined increase in fitness to novel environments. This is particularly important in globally important microbes experiencing rapid global change, due to their influence on food webs, biogeochemical cycles, and climate. Epigenetic modifications like methylation have been demonstrated to influence short-term plastic responses, which ultimately impact long-term adaptive responses to environmental change. However, there remains a paucity of empirical research examining long-term methylation dynamics during environmental adaptation in nonmodel, ecologically important microbes. Here, we show the first empirical evidence in a marine prokaryote for long-term m5C methylome modifications correlated with phenotypic adaptation to CO2, using a 7-year evolution experiment (1,000+ generations) with the biogeochemically important marine cyanobacterium Trichodesmium. We identify m5C methylated sites that rapidly changed in response to high (750 µatm) CO2 exposure and were maintained for at least 4.5 years of CO2 selection. After 7 years of CO2 selection, however, m5C methylation levels that initially responded to high-CO2 returned to ancestral, ambient CO2 levels. Concurrently, high-CO2 adapted growth and N2 fixation rates remained significantly higher than those of ambient CO2 adapted cell lines irrespective of CO2 concentration, a trend consistent with genetic assimilation theory. These data demonstrate the maintenance of CO2-responsive m5C methylation for 4.5 years alongside phenotypic adaptation before returning to ancestral methylation levels. These observations in a globally distributed marine prokaryote provide critical evolutionary insights into biogeochemically important traits under global change.
Keywords: microbial evolution, global change, ocean acidification, methylation, marine microbiology, nitrogen fixation
Introduction
Epigenetic modifications like methylation have been shown to regulate myriad transcriptional, translational, and phenotypic processes without a change in the underlying DNA sequence (Kronholm et al. 2017). Epigenetic regulation can mediate phenotypic plasticity in response to environmental change (West-Eberhard 2005; Nishikawa and Kinjo 2018). Phenotypic plasticity can be defined as a short-term change in phenotype in response to a perturbation without a change in the underlying genotype. In contrast, evolutionary adaptation results from a selective change in phenotype derived from an underlying change to the genetic (allelic) composition of a population as a result of natural selection (Collins et al. 2013; Lenski 2017). Environmentally induced epigenetic modifications of nucleotides or peptides that drive differentiated plastic phenotypes can serve as molecular proxies for adaptation through different evolutionary processes (Kronholm and Collins 2015; Schaum et al. 2016; Kronholm et al. 2017; Nishikawa and Kinjo 2018). This is especially important because the two processes can be linked, where epigenetics can “lead the way” for genetic adaptation to occur. For example, an environmentally induced, nongenetic change in phenotype can become genetically encoded during the course of selection, accompanied by a loss of environmental sensitivity in the adapted phenotype (Stajic et al. 2019). This evolutionary mechanism is known as genetic assimilation (Ehrenreich and Pfennig 2015; Danchin et al. 2019).
Regardless of mechanism, the selective fixation of beneficial traits in microbial populations is heavily influenced by the strength of selective pressure in a new environment, the exposure time relative to generation time, and the standing genetic variation. Many large-scale environmental sequencing studies examining marine microbial diversity have demonstrated local selection by environmental and ecological gradients driven by ocean circulation among globally distributed populations (Whittaker and Rynearson 2017; Delmont et al. 2019; Salazar et al. 2019). Understanding how timescales of physicochemical gradients drive evolution is critical to be able to predict how global biogeography will evolve as global change factors increase in speed and frequency (Hutchins and Boyd 2016; Boyd et al. 2018). Although these studies paint an extensive picture of biogeographic patterns relative to the global seascape, they offer little insight into specific evolutionary mechanisms that give rise to these patterns over time. As marine microbial populations can be exposed to one or many selection events through factors like ocean upwelling and/or advection (Doblin and van Sebille 2016), evolutionary processes have the potential to respond either as a relatively isolated incident, or in succession. Furthermore, several long-term evolution studies have revealed that short and long-term microbial responses to global change factors can change in magnitude and sign (Schluter et al. 2016; Schaum et al. 2018). Hence, it is important to study the molecular underpinnings connecting short- and long-term environmental responses to help explain large-scale biogeographic patterns shaped by local environmental gradients and how they may evolve due to global change.
Several recent empirical studies in model organisms such as yeast have demonstrated direct evidence for epigenetically mediated adaptive responses (Danchin et al. 2019; Stajic et al. 2019), whereas a collection of genomic studies have shown widespread epigenetic potential across diverse microbial phyla including cyanobacteria (Blow et al. 2016; Walworth et al. 2017; Hu et al. 2018; Gärtner et al. 2019). To address the lack of long-term studies examining marine microbial evolutionary epigenetics, we investigated m5C methylation dynamics during CO2 adaptation in a 7-year experimental evolution study of the biogeochemically critical cyanobacterial diazotroph Trichodesmium erythraeum IMS101 (hereafter IMS101) under future carbon dioxide (CO2) concentrations (Hutchins et al. 2013; Walworth et al. 2015; Walworth, Lee, et al. 2018). Trichodesmium is globally distributed and among the most important contributors to bioavailable nitrogen in the oligotrophic oceans, with some estimates suggesting it makes up as much as half of total N2 fixation in the subtropical gyre regions (Hutchins et al. 2015). It excretes organic metabolites and is a source of the relatively scarce organic coenzyme, cobalamin (Frischkorn et al. 2018; Walworth, Lee, et al. 2018). It can form extensive, recurring blooms and serves as a substrate for colonizing microbial consortia representing a substantial source of fixed carbon and nitrogen for new and recycled production (Walworth et al. 2015; Lee et al. 2017). Strain IMS101 was originally isolated from the Gulf Stream off of North Carolina and has since been maintained in laboratory cultures (Walworth et al. 2015). It remains a challenge to maintain in culture due to its inability to survive cryopreservation, thereby requiring numerous backup cultures to be maintained per biological replicate of each experimental treatment. IMS101, like other Trichodesmium isolates, has a large genome relative to other cyanobacteria at 7.75 Mb with only 60% of it coding for proteins (Walworth et al. 2015). It has no detectable plasmids, and large intergenic spaces (∼500-bp median length). IMS101 has over 5,000 protein coding genes and is littered with genome-wide transposons, introns, and noncoding RNAs (Pfreundt et al. 2014).
Due to the effects of epigenetically regulated phenotypic variation in response to environmental change in eukaryotes coupled to the widespread conservation of m5C methylation in prokaryotes (Blow et al. 2016), we hypothesized that m5C methylation may play a role in adaptation to a novel environment such as elevated CO2. Indeed, a few short-term studies have pointed to a role for multiple epigenetic controls in the acclimation of model cyanobacteria and diatoms to nitrogen stress and high-CO2, respectively (Hu et al. 2018; Huang et al. 2019). Furthermore, we and others recently demonstrated correlative evidence of m5C methylation potentially impacting transcription in IMS101 (Walworth et al. 2017) and the marine diatom Phaeodactylum tricornutum (Veluchamy et al. 2013). However, long-term adaptation and methylation has not yet been addressed. Other recent studies have demonstrated transcriptional and physiological evidence for adaptation (e.g., genetic assimilation) in multiple filamentous cyanobacteria (Walworth, Lee, et al. 2016; Koch et al. 2017). Adaptation is typically inferred through the evolutionary shift of reaction norms that underlie phenotypic expression (Ehrenreich and Pfennig 2015), where reaction norms are defined as short-term trait responses of a given genotype (e.g., N2 fixation and growth) as a function of two or more environments (Walworth, Lee, et al. 2016). Previously, we showed phenotypic evidence of genetic assimilation following high CO2 selection in Trichodesmium through evolutionary shifts in reaction norms (Walworth, Lee, et al. 2016), even after the high-CO2 adapted cell lines were grown in the ambient CO2 (ambient) environment for >2 years. Here, we build on this work by investigating the m5C methylome under long-term selective pressure to help elucidate m5C methylation dynamics to long-term high CO2 in the biogeochemically important diazotroph Trichodesmium. Theoretical and empirical studies examining epigenetic dynamics underlying adaptation have shown epigenetic modifications including m5C methylation to be influential early in the adaptive process, followed a later reduction in fitness effects (Schmitz et al. 2011; Kronholm and Collins 2015; Stajic et al. 2019; Walworth et al. 2020). Through our previous studies demonstrating phenotypic adaptation in IMS101, we hypothesized that epigenetic modifications would exhibit greater variation earlier in the adaptive process. Although various types of epigenetic modifications could influence CO2 adaptation, bisulfite sequencing was the only method that enabled us to examine epigenome-wide methylation dynamics with sufficient sequencing depth using minimal starting material (nanograms of DNA) given the culturing constraints outlined above. Since we demonstrated both robust physiological and transcriptional patterns consistent with genetic assimilation to high CO2, we leveraged bisulfite sequencing to examine if m5C methylation patterns are consistent with these trends including exhibiting more epigenetic activity early in an adaptive walk. We identified differentially methylated (DM) sites that rapidly changed in response to high-CO2 exposure during a short-term, plastic response, which were then maintained for at least 4.5 years. During this time, adaptive shifts in reaction norms to high-CO2 were observed through the fixation of multiple traits in the high-CO2 adapted lines, even after being returned to ambient CO2 environment for 2 years. Importantly, numerous DM sites maintained their high-CO2 induced methylation levels even after high-CO2 cell lines were returned to ambient CO2, demonstrating methylation levels to be specific to the phenotype and not just changes in carbonate chemistry. Although we note that adaptive shifts (i.e., adaptation) could have occurred prior to this 4.5-year time point, this timescale is in line with other studies demonstrating phenotypic adaptation in marine microbes after a few hundred generations of environmental change (Collins and Bell 2004; Lohbeck et al. 2012, 2013, 2014; Schaum and Collins 2014). Due to the challenges of culturing IMS101 outlined above, we conducted multiple assays to confirm adaptation prior to large-scale molecular, biogeochemical, and phenotypic assays. The vast majority of these DM sites were intragenic and overrepresented in select core metabolic pathways. At the 7-year mark, no DM sites were observed between ambient and high-CO2 adapted cell lines despite significant differences in CO2-responsive traits, indicating that methylation levels of initially responsive DM sites had returned to their ancestral levels. The fixation of multiple traits irrespective of CO2 condition alongside the return of high-CO2 responsive methylated sites to ambient levels provide evidence for an evolutionary pathway of biogeochemical significance under ocean acidification in a globally important microbe.
Results and Discussion
Long-Term Methylation Dynamics
As discussed above, we previously demonstrated physiological and transcriptional evidence of genetic assimilation following long-term high-CO2 exposure (∼850 generations) in Trichodesmium IMS101 (Hutchins et al. 2015; Walworth, Lee, et al. 2016). We originally started with a single population of IMS101 and split it into an ambient-selected (380 µatm; Amb) and high-selected (750 µatm; High) experimental CO2 treatment with six biological replicates each (six flasks per treatment). At the time of the start of the experiment, 380 µatm CO2 represented the present day (i.e., ambient) concentration in Earth’s atmosphere, whereas 750 µatm CO2 represented the future predicted atmospheric CO2 concentration in the year 2100 under business-as-usual conditions (Hutchins et al. 2007). A diagram of the experimental design can be viewed in supplementary figure S1, Supplementary Material online. Increased growth and N2-fixation rates induced after several weeks of exposure to 750 µatm CO2 (i.e., plastic response; Ambient-High or Amb-High) were maintained 4.5 years later (fig. 1a). When high-CO2 lines were returned to the 380 µatm CO2 condition at this time-point, their increased growth and N2-fixation rates were retained, demonstrating a loss of CO2 sensitivity (fig. 1a; High-Ambient or High-Amb). We then sampled the original experimental cell lines maintained in semicontinuous batch culture under 380 and 750 µatm CO2 at a final ∼7-year time point (∼1,000+ generations), which showed high-CO2 growth and N2-fixation rates to still be significantly greater than those of ambient cell lines (P < 0.05, Student’s t-test, n = 6; fig. 1b; no reciprocal transplant experiment was carried out at this time point due to loss of the cultures) (Walworth, Fu, et al. 2016, 2018; Lee et al. 2018). Furthermore, fold changes in trait values between the amb-CO2 and high-CO2 conditions remained conserved across 7 years of selection (supplementary fig. S2, Supplementary Material online).
Fig. 1.
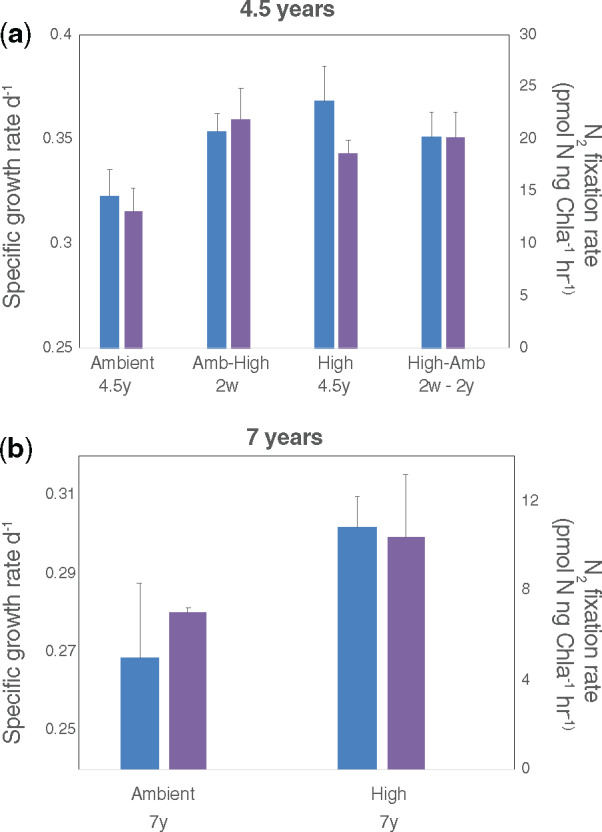
Trichodesmium N2 fixation and growth rates for 4.5- and 7-year time points. (a) The cell-specific growth rates (blue) and N2 fixation rates (purple) sampled at 4.5 years. The short-term (Amb-High) and long-term (High and High-Amb) responses are significantly higher than the Ambient condition. Below each sample name is the time of sampling after being transferred to the respective CO2 environment. For example, Ambient (4.5yr) means cell lines had been growing at ambient CO2 for 4.5 years. Amb-High means ambient cell lines were transferred to high CO2 and sampled after 2 weeks of growth. High-Amb means high cell lines were transferred to ambient CO2 and sampled periodically starting from 2 weeks of growth with the final time point being 2 years of growth. Bars and error bars for both plots are means and standard deviations of six biological replicates. (b) Cell-specific rates of the same cell lines and treatments shown in panel (a). Shown are the specific growth rates (blue) and N2 fixation rates (purple) sampled at 7 years for the Ambient and High cell lines.
In a prior study, we used bisulfite sequencing to characterize the genome-wide m5C methylation landscape of IMS101 and its biogeographic conservation across spatiotemporally separated isolates (Walworth et al. 2017). This method is specifically designed to investigate a type of modification known as m5C cytosine methylation, defined as the addition of a methyl group to the C5 carbon residue. Trichodesmium and other cyanobacteria contain the genomic capacity for other epigenetic modifications (Zehr et al. 1991; Blow et al. 2016), but this method does not capture those dynamics. To examine if genome-wide, site-specific methylation levels (Smet; number of reads with a methylated cytosine/total number of reads mapping to that cytosine) significantly changed in response to both short- (Amb-High; several weeks) and long-term (High; 4.5 years) CO2 exposure, we compared genome-wide methylation changes among experimental treatments using a modified version of the RADMeth algorithm with conservative thresholds (see Materials and Methods) (Dolzhenko and Smith 2014). From this, 967 and 1,136 DM sites were identified in Amb-High and High-CO2 treatments, respectively, relative to those maintained under constant 380 µatm CO2. We also bisulfite-sequenced the original population from which all cultures originated (Amb-ancestral) to examine if methylation levels of environmentally or growth rate-responsive DM cytosines were more similar to those of ambient or high-CO2 conditions at the 4.5-year mark. We note that at this step, we simply performed pairwise differential methylation analysis to detect DM sites but had no knowledge if site-specific methylation levels had increased or decreased in high-CO2 conditions relative to ambient CO2. All DM cytosines resided within the CpG motif with 44% DM on one strand and 56% on both strands (supplementary file 1, Supplementary Material online). Of the DM sites methylated on one strand (n = 800), we did not observe a strand preference as 49% and 51% were methylated on the sense and antisense strand, respectively. Overall, methylated cytosines have an average Smet of 74% (median = 84%), and overall DM sites show widespread reductions in Smet levels with an average of 53% (median = 57%).
We also checked for genetic changes shared by the high-CO2 adapted cell lines (High at 4.5 years, High-Amb at 4.5 years, and High at 7 years) using the BS-SNPer pipeline for bisulfite-seq data (Gao et al. 2015) and found 52 mutations (supplementary file 1, Supplementary Material online). Of the 52 mutations, 40 occurred in only 1 high-CO2 adapted sample, 7 occurred in 2 high-CO2 adapted samples, 4 in 3 high-CO2 adapted samples, and 1 in 4 high-CO2 adapted samples. None occurred across all high-CO2 samples (n = 9) nor did any occur in the genes or promoter regions of the five identified DNA-cytosine methyltransferases (Walworth et al. 2017). Additionally, no changes in expression levels were observed across treatments for any MTase genes (supplementary file 2, Supplementary Material online). Due to the limitations of short-read, bisulfite-seq data, and the numerous transposable elements found genome-wide in Trichodesmium (Walworth et al. 2015), other larger genomic changes may have occurred in the high-CO2 selected cell lines but were undetected. Additionally, high-CO2 selected cell lines may exhibit convergent phenotypes derived from different underlying genetic changes as previously observed in other microbial experimental evolution studies (Papadopoulos et al. 1999; Fong 2005). Here, we focus on long-term m5C methylome dynamics as they relate to phenotypic and transcriptomic changes.
We wanted to first ensure that comparative analysis among treatments was conducted on those methylation sites that exhibited significant pairwise differences between a high-CO2 treatment and the ambient reference condition (see Materials and Methods). We identified 686 DM cytosines that met this criterion among the high-CO2 treatments (Amb-High and High) and extracted the corresponding methylation levels from the ambient and 380-ancestral conditions. Bray−Curtis dissimilarities were calculated across all experimental treatments and hierarchical clustering with multiscale bootstrap resampling (replicates = 1,000) was performed to explore the possibility of treatment-specific methylation patterns. The 380-ancestral and ambient cultures formed 1 high-confidence cluster that significantly segregated (Approximately Unbiased P-value [AUP] > 0.95) (Suzuki and Shimodaira 2006) from the other cluster comprised the high-CO2 phenotypic lines (Amb-High and High replicates) (fig. 2). These analyses provide robust evidence that methylation changes of these DM sites are significantly correlated to changes in CO2 conditions, considering that the ambient and 380-ancestral cell lines were sampled 4.5 years apart. The formation of these clusters using this clustering method rejects the hypothesis that these clusters do not exist at the 0.05 significance level (i.e., AUP >0.95). Thus, this analysis provides strong statistical support that specific m5C methylation levels exhibit a significant short-term response (Amb-High) followed by long-term maintenance (High) to either CO2, growth rate, or both, consistent with previously identified phenotypic and transcriptional patterns (Walworth, Lee, et al. 2016).
Fig. 2.
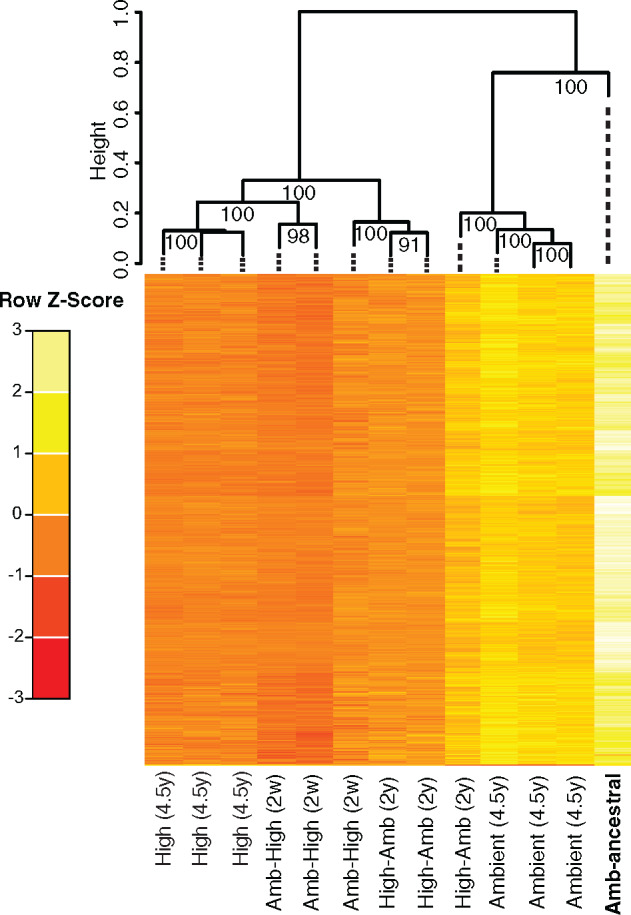
Hierarchical clustering (HC) with multiscale bootstrap resampling of methylation levels at identified shared DM sites at the 4.5-year time point. HC of Bray−Curtis dissimilarities calculated from shared (n = 686) DM methylation levels. Values at nodes are AUPs. The ancestral population is labeled in bold. Scale bar and colors depict standardized methylation levels. Both the plastic (Amb-High) and adaptive (High) high-CO2 conditions significantly segregate from the ancestral (Amb-ancestral) and ambient conditions, forming two high-confidence clusters separated by CO2 concentration. The replicates of the high-selected treatment transplanted back to ambient conditions (High-Amb) showed an interesting clustering pattern. Two of the High-Amb replicates cluster with the high-CO2 conditions, whereas one replicate clusters with the ambient conditions.
Interestingly, methylation levels in the plastic (Amb-High) and adaptive (High) treatments mostly exhibited reductions in the high-CO2 environment (fig. 2; supplementary file 1, Supplementary Material online). We tested the probability of sharing these DM sites between the Amb-High and High treatments by chance through a pairwise hypergeometric test between DM pools. We observed a very low probability (P < 10−4) that these DM sites were shared by chance alone, suggesting them to be nonrandomly associated with both the plastic and adaptive responses to high-CO2.
Next, we analyzed these shared DM sites’ methylation levels in steady-state High cell lines transplanted back to 380 µatm CO2 levels for 2 years (High-Amb). Interestingly, two of the High-Amb biological replicates significantly grouped with the high-CO2 conditions (Amb-High and High) whereas one replicate clustered with ambient treatments (Ambient and Amb-ancestral; fig. 2). Given that we sampled all selection treatments for methylation after CO2 adaptation (Hutchins et al. 2015; Walworth, Lee, et al. 2016), significant segregation between the High-Amb replicates suggests that exposing High cell lines to their ancestral CO2 conditions may have induced the return of CO2 responsive methylation modifications to their ancestral methylation levels, albeit at different rates. These differing rates may be due to differences in underlying genetic factors interacting with the ambient environment (e.g., genetic drift and/or genetic divergence) after 4+ years of selection at high-CO2, but future studies are needed to confirm this hypothesis.
This separation may also be due to our time of sampling. The exact timing of adaptation in the High cell lines before our 4+-year sampling period is unknown. If adaptation occurred much earlier than our sampling time point, then methylation levels may have been receding for some time at differing rates. Had we performed the High-Amb transplantation experiment just after high-CO2 adaptation (assuming it occurred at a similar time in all replicates), the High-Amb biological replicates may have still had similar enough methylation levels to cluster together mirroring their High counterparts, as they are technically the same cell lines. Regardless, if environmentally induced methylation changes are primarily influential early in an adaptive walk, and then regress following adaptation as evidenced here and in other systems (Schmitz et al. 2011; Kronholm and Collins 2015; Danchin et al. 2019; Draghi 2019; Stajic et al. 2019), then methylation levels should eventually return to ancestral conditions. This should occur irrespective of the ambient or high-CO2 environment (i.e., either in High-Amb or High), with the former occurring at a faster rate, as we observed (fig. 2).
Moreover, the 380-ancestral and ambient cell lines are significantly more similar to each other than those of the short- (several weeks; Amb-High) and long-term (4.5 years; High) high-CO2 treatments. Taken together with the maintenance of specific High-Amb methylation profiles, these data provide evidence for long-term m5C methylation during adaptation in this cyanobacterium, similar to previous epigenetic dynamics during adaptation in eukaryotic model systems (Danchin et al. 2019; Stajic et al. 2019). Numerous High-Amb DM sites also remained differential methylated even after ambient CO2 exposure (fig. 2) for several hundred generations, further supporting the notion that DM sites are specific to the phenotype rather than to mere changes in carbonate chemistry. However, more research is needed to determine if methylation plays a causative role in mediating the CO2 adaptive response in Trichodesmium.
The vast majority of the 686 shared DM sites between the Amb-High and High lines (supplementary file 1, Supplementary Material online) were distributed among 196 genes containing varying amounts of DM sites (1 − 11 DM sites per gene) in both treatments (fig. 3a;supplementary file 2, Supplementary Material online). To examine if gene-bounded, shared-DM sites were overrepresented in any particular metabolic pathways, hypergeometric tests with Benjamini and Hochberg’s FDR correction (Benjamini and Hochberg 1995) (FDR <0.05; see Materials and Methods) were conducted with Gene Ontology (GO) categories (The Gene Ontology Consortium 2018) as previously described (Walworth et al. 2017). Significantly enriched GO categories included pathways involved in select core metabolic processes including cell signaling, protein metabolism, membrane processes, and cobalamin metabolism (fig. 3a). Trichodesmium was recently demonstrated to be a potential important source for the often limiting coenzyme cobalamin (vitamin B12) in the open oceans (Frischkorn et al. 2018; Walworth, Lee, et al. 2018). Although methylation changes are enriched in these central cellular processes, more targeted studies are needed to investigate the mechanistic roles of methylation changes within genes in these pathways.
Fig. 3.

Overrepresented GO pathways and transcription of genes containing DM sites in high-CO2. (a) The Venn diagram displays the distribution of genes containing DM sites between high-CO2 plastic (Amb-High) and adapted (High) treatments. Significantly GO-enriched pathways shared between these treatments representing the 196 genes are displayed below. (b) Expression levels of all 196 shared genes containing DM sites as a function of CO2 condition. No significant differences among treatment-specific expression pools were observed. (c) Normalized expression levels of the 196 shared genes as a function of Rmet levels across all high-CO2 conditions. Above each boxplot are four metrics and from top to bottom are: no. of genes in that box, mean Rmet value, minimum Rmet value, and maximum Rmet value. We observed a gradual expression reduction with increasing methylation density, with bin 5 exhibiting a significantly reduced median expression level (FDR < 0.05) and the highest Rmet levels (Rmet ≥0.01) relative to all other bins, suggesting a nonrandom population-wide transcriptional reduction for this pool. (d) Length-normalized expression of upregulated genes as a function of treatment in bin 5.
Correlating Methylation to Transcription
Next, we examined the dynamics between methylation changes and gene expression to identify potential methylated sites correlated to transcriptional changes. We must note here that our data below between methylation and expression levels are strictly correlative and solely identify interesting targets for future study. Although m5C methylation is generally widespread throughout microbial genomes, some evidence suggests that it may only indirectly or directly influence a small fraction of transcription in cyanobacteria, and therefore must be involved in alternative roles like restriction−modification systems (Blow et al. 2016) or those that have yet to be characterized (Walworth et al. 2017; Hu et al. 2018; Gärtner et al. 2019). In a previous study, we observed significantly reduced transcript levels in genes exhibiting relatively higher methylation density (Rmet; number of methylated cytosines/total base pairs per gene) within the gene body at Rmet levels approaching 0.01 (Walworth et al. 2017). We also conducted this analysis for cytosine-specific methylation levels (Rc; number of methylated cytosines/number of per-gene cytosines), which yielded the same results as Rmet and Rc values show strong correlation (R2 = 0.89). This is generally consistent with observations in eukaryotic algae, where transcriptional repression was observed in genes with higher Rmet (Veluchamy et al. 2013). To first scan all shared intragenic DM sites, we plotted the length- and depth-normalized mRNA expression levels (see Materials and Methods) of all 196 shared genes containing DM sites as a function of CO2 condition, which yielded no significant difference between the expression pools (fig. 3b; Kruskal−Wallis test; P > 0.05)—supporting prior evidence that cyanobacterial methylation may only influence a small fraction of gene expression (Walworth et al. 2017; Hu et al. 2018; Gärtner et al. 2019).
Next, we searched for DM sites within genome-wide promoters (n = 6,080) that were experimentally verified in IMS101 (Pfreundt et al. 2014) and found five promoters containing DM sites (supplementary file 3, Supplementary Material online). Every promoter only had one DM site except for the promoter of the Tery_4419 gene annotated as a hypothetical protein, which had two DM sites in the promoter region and one DM site in the gene body. Four out of the five sites within these promoters including those of Tery_4419 were DM in both Amb-High and High-selected conditions (supplementary file 3, Supplementary Material online). However, Tery_4419 was the only one gene that also exhibited differential expression across both of the high-CO2 conditions (fig. 4a). Similar to increasing methylation density correlated with transcriptional changes (e.g., Rmet), IMS101 genes may need to contain a certain density of m5C DM sites across the promoter and/or the gene body (see below) to influence transcription. More targeted research is needed in this area.
Fig. 4.

Gene-specific transcript profiles containing DM sites in high-CO2. Methylation levels (Smet; no. of reads with a methylated cytosine/total no. of reads mapping to that cytosine) of intragenic/promoter DM sites (left axis) and expression level of the corresponding gene (right axis). (a) Methylation (boxplot) and transcriptional (bar plot) profiles of a gene (Tery_4419) containing 2 DM sites in its promoter and 1DM site in the gene body while also exhibiting differential expression across the high-CO2 conditions. (b) An intragenic DM site exhibiting significantly reduced methylation levels (Smet) across high-CO2 phenotypes (yellow bar; left y-axis) in concert with significantly increased transcript levels of the gene Tery_2760 (blue bars, right y-axis). (c) Methylation (boxplot) and transcriptional profiles (bar plot) of a gene (Tery_2481) containing DM sites and upregulated expression. This gene contains 19 intragenic DM sites and the clear boxplots show the distribution of Smet methylation levels of all sites across experimental conditions demonstrating reduced methylation in high-CO2 phenotypes (left y-axis). The bar plots show the corresponding gene expression level (right y-axis) exhibiting highest expression in high-CO2 phenotypes. (d) This plot is the same as in (b) except now there are two intragenic DM sites (light blue and orange bars) plotted adjacent to the corresponding transcript levels of that gene (dark blue bars) *In transcript plots (a−d), no transcript data are available for the 380-ancestral. Bars and error bars in bar plots are means and standard deviations of three biological replicates for methylation levels and two biological replicates for RNA-Seq. Genes inside of overrepresented GO pathways are denoted above the plots.
To investigate increasing gene−body methylation, we binned the 196 shared genes by their Rmet levels across both ambient and high-CO2 treatments, respectively, and plotted their length-normalized transcription as a function of Rmet (fig. 3c). We experimented with different numbers of bins until a specific bin (bin 5) with a certain Rmet range showed significantly different median expression versus all other bins (Kruskal−Wallis, post hoc Dunn test with Benjamini–Hochberg correction; FDR <0.05). Since it is unknown which of these 196 genes’ expression may at least be partially influenced by methylation, this method allowed us to use a sliding window of methylation density to search for a set of genes that exhibited consistently different expression changes associated with significantly higher methylation density. Consistent with previous findings (Veluchamy et al. 2013; Walworth et al. 2017), we observed gradual transcriptional reduction with increasing methylation density. The significantly reduced median expression level in the final bin (bin 5) with the highest Rmet levels (Rmet ≥0.01) relative to all other bins suggests a nonrandom population-wide transcriptional reduction for this pool (fig. 3c). Although bin 5 exhibits the strongest statistical evidence for at least partial transcriptional reduction under increased methylation density (n = 39; supplementary file 2, Supplementary Material online), it is worth noting that genes in other bins (e.g., 2, 3, and 4) also display a similar although nonsignificant trend (fig. 3c). Here, we focus on representative genes in bin 5 as candidates that demonstrate consistently different expression changes associated with higher methylation density and leave genes in other bins for future studies. Although the significantly increased methylation levels in these genes are significantly correlated to corresponding reductions in their expression, it is of course possible that methylation may play alternative or indirect roles in addition to, or rather than, directly impacting transcription.
Focusing on genes in bin 5, all intragenic methylation levels exhibited significant reductions going from ambient to high CO2. Hence, we next examined if the pooled transcription of these genes displayed an average increase going from ambient to high CO2. Upon plotting the normalized expression of upregulated genes as a function of treatment within bin 5 (supplementary file 2, Supplementary Material online), we observe a gradual increase in expression going from the ambient environment to the plastic (Amb-High) and then the adapted cell lines (High) (fig. 3d). The High-Amb lines are the High cell lines moved to the ambient environment. Indeed, the High-Amb expression pool exhibited slightly lower average expression values relative to the High treatment (fig. 3d). This is possibly due to divergence of the High-Amb biological replicates at the molecular level, particularly the replicate that grouped with the ambient CO2 cell lines (fig. 2). Despite this, the Amb-High, High, and High-Amb treatments share statistically indistinguishable phenotypes (i.e., growth and N2 fixation; fig. 1) (Walworth, Lee, et al. 2016). Interestingly, High average transcription levels are typically highest for these 196 genes (fig. 3d) and lowest for intragenic DM sites relative to other treatments (e.g., fig. 4). However, with population-wide transcriptomics alone, only correlation and not causation can be inferred to link these methylation and transcriptional changes.
Accordingly, our primary aim was to identify gene targets and enriched pathways exhibiting significant transcriptional shifts that robustly correlate to significant intragenic methylation shifts for further targeted studies. To do this, we looked for significant differences in both expression and methylation through independent pairwise analyses. We screened for genes in bin 5 exhibiting significant upregulated expression (see Materials and Methods) in at least one high-CO2 condition (Amb-High or High) while also retaining the same sign in the other corresponding high-CO2 condition relative to the ambient treatment. We also ensured that selected genes harbored both significantly DM sites and Rmet ≥ 0.01. From these criteria, 17 candidates emerged (supplementary table S2 and file 2, Supplementary Material online). Roughly half of these (n = 8; supplementary table S2, Supplementary Material online) appeared within overrepresented GO pathways containing significant methylation shifts (see above) including protein binding (GO: 0005515), membrane processes (GO: 0016021), and proteolysis (GO: 0006508). These genes, and particularly the eight in GO-enriched pathways, are thus strong candidates for more targeted studies as significant molecular activity was detected from independent methods (i.e., transcriptomics and methylomics) and several independent statistical tests (supplementary file 2, Supplementary Material online). Figure 4 shows four of these genes exhibiting strikingly consistent methylation and transcriptional patterns. For example, we observed a gene annotated as a tetratricopeptide repeat (TPR) that harbored 19 DM sites with reduced methylation levels and upregulated mRNA expression (fig. 4c; supplementary files 1 and 2, Supplementary Material online). TPR domains are found across all domains of life and participate in a variety of functions, ranging from cell division to RNA metabolism to protein transport. They have garnered recent interest for the discovery of their role in photosynthetic metabolism, particularly in the assembly and stability of photosystems I and II (Klinkert et al. 2004). In the cyanobacterium Synechocystis, a membrane-bound TPR protein was demonstrated to be involved in the processing of the D1 reaction center (Klinkert et al. 2004) and additionally was subsequently found to play a role in photosynthetic performance and light-dependent chlorophyll synthesis (Schottkowski et al. 2009). Other genes involved in oxidative stress and photosynthetic maintenance displayed similar profiles (fig. 4b and d; see supplementary note S1, Supplementary Material online). These results are generally supportive of prior observations in Trichodesmium under both short- and long-term CO2 exposure that showed significant shifts in PSI:PSII ratios and photosynthetic electron transport transcripts (Levitan et al. 2007; Walworth, Lee, et al. 2016). These data may show molecular evidence for the association of redox changes and protein turnover that potentially result in previously observed changes in PSI:PSII ratios under high-CO2, which may in turn liberate iron (Fe) from the Fe−heavy PSI complex for use in N2 fixation.
Return of high-CO2 Induced Methylation to Ancestral Levels
At the 7-year mark (1,000+ generations) of this evolution experiment, we again bisulfite-sequenced the ambient and High cell lines (hereafter *ambient and *High, respectively) and compared methylation levels with those of the Amb-ancestral and CO2-selected conditions at the 4.5-year mark. Upon hierarchical clustering of Bray−Curtis dissimilarities calculated from methylation levels from the same 686 shared genes identified above across 7 years of selection, the 7-year *ambient replicates clustered with the Amb-ancestral and 4.5-year ambient conditions as expected. However, the 7-year *High replicates also clustered with all ambient treatments, indicating the return of methylation levels in initially responsive DM sites back to ambient ancestral levels (fig. 5).
Fig. 5.

Hierarchical clustering (HC) with multiscale bootstrap resampling of DM sites. HC of Bray−Curtis dissimilarities calculated from shared (n = 686) DM methylation levels in figure 2. Values at nodes are AUPs. The ancestral population is labeled in bold and the 7-year time points are labeled in green. Both the plastic (Amb-High) and adaptive (High) high-CO2 conditions at the 4.5-year mark maintain segregation from the ancestral (Amb-ancestral) and ambient conditions as in plots in figure 2. Also consistent with plots in figure 2, two of the High-Amb replicates still cluster with the 4.5-year high-CO2 conditions, whereas one replicate clusters with the ambient conditions. This demonstrates CO2-responsive methylation levels to be returning to ancestral conditions once placed back into the ancestral environment. Upon adding *ambient and *High selected conditions at the 7-year time point (green), the *ambient replicates cluster with the Amb-ancestral and 4.5-year ambient conditions as expected, but the *High replicates also cluster with all ambient conditions, demonstrating the return of methylation levels originally triggered by high-CO2 back to their ancestral ambient levels.
These results are in line with prior modeling and empirical studies examining genetic assimilation theory where phenotypic fixation is observed irrespective of treatment while initial, environmentally induced nongenetic changes return to ancestral levels prior to selection (Madlung et al. 2002; Pigliucci et al. 2006; Schlichting and Wund 2014; Ehrenreich and Pfennig 2015; Kronholm and Collins 2015). The two overarching clusters are strongly statistically supported (AUP >0.95), which rejects the null hypothesis that these clusters do not exist at the 0.05 significance level. Furthermore, no significantly DM sites were detected between the *ambient and *High conditions, which serves as robust evidence for statistically analogous global methylation states at the 7-year mark. This contrasts strongly with observations of significantly different methylated sites at 4.5 years of selection (fig. 2), and with the significant phenotypic differences in growth and N2-fixation rates still observed after 7 years between the *ambient and *High treatments (fig. 1). Hence, at this 7-year time point any previous phenotypic differences that may have been associated with nongenetic changes are presumably now decoupled from them, potentially implicating a genetic change.
Conclusion
In summary, we identified m5C methylated sites that rapidly changed in response to high-CO2 exposure during a short-term, plastic response (Amb-High) relative to their levels in both the ancestral population (Amb-ancestral) and ambient selected conditions. High-CO2 methylation levels at these sites were then maintained for at least 4.5 years, during which time adaptive shifts in reaction norms were observed (High cell lines). This is evidenced by the fixation of multiple traits (i.e., growth and N2 fixation) that were retained in the High lines after being returned to a ambient environment (i.e., High-Amb). The vast majority of these DM sites were intragenic and overrepresented in select core metabolic pathways, thereby implicating m5C methylation changes to be significantly associated with these genes during high-CO2 selection. Additionally, we identified specific genes that both harbored DM sites and exhibited differential mRNA expression as candidates for potential mechanistic studies under long-term high-CO2. Their annotated functions suggest central roles in cellular redox changes and protein turnover potentially associated with photosynthetic activity. Multiple lines of evidence from other independent analyses examining Trichodesmium metabolism under high-CO2 have also indicated significant cellular investment toward energetic and photosystem rearrangement, which seem to facilitate increased growth and N2 fixation. Placing the high-CO2 cell lines back in the ambient environment (High-Amb) appears to influence the return of high-CO2 methylation levels back to their ancestral state with different rates among the High-Amb replicates. Upon resampling the ambient and High cell lines at the 7-year mark, no DM sites were observed between conditions, indicating that methylation levels of initially responsive DM sites had returned to their ancestral levels. This is strongly supported by the clustering of all sampled replicates from across 7 years of selection. Though more work needs to be done to determine if methylation played a causative role in the phenotype, these long-term dynamics are consistent with genetic assimilation theory wherein the adapted phenotype is maintained through the loss of environmental sensitivity while initial molecular changes return to ancestral levels in the selection environment. Further study examining methylation during adaptation in marine photosynthetic microbes will help to elucidate the importance of short-term microevolutionary processes on long-term evolutionary divergence to interacting ocean and global change dynamics. Here, we provide a first step to investigating long-term m5C methylation in conjunction with high-CO2 selection for a globally distributed marine microbe. Importantly, the long-term dynamics of these methylated sites were highly consistent with both prior observations in other systems, and those predicted by genetic assimilation theory. This work presents a framework for examining long-term roles of epigenomes as potential mechanisms in biogeochemically important trait evolution under global change.
Materials and Methods
Culturing and Physiology
Detailed culturing and physiology methods and results can be found in previously published studies (Hutchins et al. 2015; Walworth, Lee, et al. 2016; Lee et al. 2018). Briefly, stock cultures of Trichodesmium strain IMS101 were maintained at the University of Southern California (Los Angeles, CA) in a modified Aquil medium devoid of added combined nitrogen with standard vitamins and trace metals, which included 500 nM iron and 20 μM phosphate. Cultures were grown at 26°C under a light–dark cycle of 12:12 (L:D) and a light intensity of 120 mmol photons per m2 s−1 incident irradiance. A diagram of the experimental design can be viewed in supplementary figure S1, Supplementary Material online. At the beginning of the experiment, we started with a single population of IMS101 and split it into an ambient-selected (380 µatm; Amb) and high-selected (750 µatm; High) experimental CO2 treatment. Six biological replicates per treatment were examined using semi-continuous culturing methods where each replicate was diluted individually based on the growth rate calculated for the respective replicate. For all phenotypic assays, six biological replicates were used to calculate means and standard deviations. Each replicate bottle was kept optically thin to avoid self-shading, nutrient limitation, and perturbations to targeted CO2 levels. Cultures were constantly bubbled with prepared air/CO2 mixtures (Praxair, Los Angeles, CA) to sustain stable CO2 concentrations of 380 (ambient) and 750 (high-CO2) μatm CO2 for a total of 7 years. Growth and N2 fixation rates reported in this study were measured during the middle of the photoperiod at 4.5 and 7 years, respectively, as previously described (Walworth, Fu, et al. 2016; Walworth, Lee, et al. 2016). To measure the CO2 plastic response following 4.5 years of growth at 380 µatm CO2, subcultures of ambient cell lines were transferred to 750 µatm CO2 for 2 weeks (Amb-High) and growth and N2-fixation rates were measured during the middle of the photoperiod. The correlated response (High-Amb) was measured in the same manner, except that High cell lines were transferred to 380 µatm CO2 for 2 weeks before assaying and were then maintained under the changed CO2 conditions for over 2 years. The phenotypic trends of these published experiments are presented for readers in figure 1 and supplementary figure S1, Supplementary Material online, in order to facilitate interpretation of the epigenetic results presented here.
Nucleic Acid Sampling and Isolation for Illumina Sequencing
Three independent biological replicates per treatment (three randomly selected separate flasks out of six total flasks per treatment) were gently filtered during the middle of the photoperiod onto 5 mm polycarbonate filters (Sigma Aldrich, St. Louis, MO), immediately flash frozen, and stored in liquid nitrogen until DNA extraction as previously described (Walworth et al. 2017). DNA was extracted from frozen filters using the FastDNA Spin Kit for Soil (MP Biomedicals, Santa Ana, CA) according to the manufacturer’s protocol. Extracted DNA was then sent to the USC Genomics and Cytometry Core for library construction using the NEXTflex Bisulfite-Seq library preparation kit (PerkinElmer, Waltham, MA) followed by sequencing on the Illumina HiSeq.
Two corresponding biological replicates for RNA extraction (cells sampled from the same flasks as the bisulfite samples) were taken concomitantly with the above bisulfite samples during the middle of the photoperiod, as described in Walworth, Fu, et al. (2016). Briefly, cells were swiftly and gently filtered at 11 AM with 5-µm polycarbonate filters (Whatman), immediately flash frozen, and stored in liquid nitrogen until RNA extraction. RNA was extracted as previously described (Walworth et al. 2017). Briefly, the Ambion MirVana miRNA Isolation Kit (Thermo Fisher Scientific, Waltham, MA) was used in an RNAse free environment according to the manufacturer’s instructions followed by two incubations with Ambion’s Turbo DNA-free kit to degrade trace amounts of DNA. Extracted RNA was then sent to the UC San Diego IGM Genomics Center for library construction and Illumina sequencing. Briefly, rRNA was removed from total RNA using the Ribo-Zero rRNA Removal Kit (Illumina, San Diego, CA), and libraries were constructed with the TruSeq Stranded mRNA Library Prep Kit (Illumina) followed by 50 base pair, single end sequencing with the Illumina HiSeq. All WGBS and RNA sampling occurred simultaneously. Due to the well-known microbiome community associated with Trichodesmium cells (Lee et al. 2017), we conducted deep sequencing to obtain high coverage of IMS101 (e.g., >60×) for both methylation and RNA analyses for robust statistical support of observed trends. Raw reads were quality checked using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
Methylation Bioinformatics
Raw reads were trimmed and quality processed using Trimmomatic (v0.32) (Bolger et al. 2014) using the following settings: LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:35. Reads were then mapped onto the IMS101 genome followed by methylation identification and site-specific estimation of methylation levels using the MethPiPe pipeline (Song et al. 2013) with default settings. A cytosine was flagged as methylated if the residue had a combined coverage of ≥5 reads and if methylation was detected in all biological replicates with at least 20% of total reads being methylated in at least one experimental condition (Veluchamy et al. 2013; Walworth et al. 2017). Methylation statistics can be found in Supplemental File 1. Differential methylation analysis was conducted with a modified version of RADMeth (Dolzhenko and Smith 2014) that skips the step where significance of each site is adjusted based on the significance of its neighbors. This step was originally designed to enable detection of regional changes in mammalian epigenomes at sites with low coverage. The coverages in our samples are high enough to enable detection of methylation changes without this adjustment. A DM cytosine was considered significant if the change in the cytosine-specific (Smet) methylation level (no. of reads with a methylated cytosine/total no. of reads mapping to that cytosine) was >1/3 relative to the corresponding cytosine in the stated reference treatment and if it retained a Benjamini−Hochberg false discovery rate (FDR) <0.001 (Benjamini and Hochberg 1995). A flowchart of the analysis pipeline can be found in supplementary figure S2, Supplementary Material online. Methylation levels for all detected m5C residues can be found in supplementary file 1, Supplementary Material online, along with methylation and RNA sequencing statistics. Methylation and RNA sequencing statistics can also be found in supplementary table S1, Supplementary Material online.
SNP Analysis
Quality processed reads (see above) were first aligned to the T. erythraeum IMS101 reference genome with Bismark software (Krueger and Andrews 2011). SNP analysis was conducted using the BS-SNPer pipeline with default settings. BEDTools was used for genome-wide comparative analysis of genetic features (Quinlan and Hall 2010) including experimentally confirmed promoter regions from Pfreundt et al. (2014).
Differential Expression Analysis
Transcript normalization and differential expression was conducted as previously described (Walworth et al. 2017). Briefly, Trimmomatic version 0.35 (Bolger et al. 2014) was used to quality trim and filter with the following settings: SE -threads 35 -phred33 LEADING: 3 TRAILING: 3 SLIDINGWINDOW: 4:15 MINLEN: 35. Processed fastq files were next mapped onto IMS101, IMG-called genes (Chen et al. 2019) using Bowtie2 v2.2.5 (Ben Langmead and Salzberg 2012) with default settings. Genes containing an average of <10 counts across libraries were removed. Because we compared the distribution of bins containing numerous genes, transcripts were first normalized by gene length via a “Loess regression” in the EDASeq package (Risso et al. 2011) followed by library-depth normalization using calculated size factors per library in the DESeq2 package. Finally, differential expression analysis was conducted using DESeq2 (Love et al. 2014). Genes with a Benjamin and Hochberg’s FDR <0.05 were deemed differentially expressed. Normalized expression levels for all detected genes can be found in supplementary file 2, Supplementary Material online. A flowchart of the analysis pipeline can be found in supplementary figure S3, Supplementary Material online.
Statistical Analysis
Hypergeometric tests were conducted as previously described (Walworth, Lee, et al. 2016; Walworth, Fu, et al. 2018). GO annotations for Trichodesmium were downloaded from the Genome2D web server (http://genome2d.molgenrug.nl). The phyper function in R was used to test for significance and P values were corrected by the Benjamini−Hochberg false discovery method (Benjamini and Hochberg 1995) with the p.adjust function (P < 0.05).
Bray−Curtis dissimilarities were calculated from growth- and depth-normalized methylation levels with the vegan package (Oksanen et al. 2013) and hierarchical clustering with multiscale bootstrapping (n = 1,000) was carried out with the pvclust package (Suzuki and Shimodaira 2006). Heatmaps were generated using the Heatplus package (Ploner 2015).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by the US National Science Foundation (Grant Nos. OCE 1260490, OCE 1657757, and OCE 1851222). We thank Naomi M. Levine, Ian Ehrenreich, and Sinead Collins for insightful discussions.
Data Availability
Raw read files for Whole-genome bisulfite-sequenced libraries (WGBS) have been deposited in the NCBI’s Gene Expression Omnibus (Edgar et al. 2002) and are accessible through GEO Series accession number GSE138176. The ambient an Amb-High RNA raw reads can be found in another GEO project with accession number GSE86992. Within the GSE86992, the specific samples for the ambient and Amb-High have sample accession numbers of GSM2318497, GSM2318498, GSM2318499, and GSM2318500. The rest of the RNA-Seq libraries for the high-CO2 and High-Amb can be found in GSE138176.
References
- Langmead B, Salzberg SL.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y.. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57(1):289–300. [Google Scholar]
- Blow MJ, Clark TA, Daum CG, Deutschbauer AM, Fomenkov A, Fries R, Froula J, Kang DD, Malmstrom RR, Morgan RD, et al. 2016. The epigenomic landscape of prokaryotes. PLoS Genet. 12(2):e1005854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd PW, Collins S, Dupont S, Fabricius K, Gattuso J-P, Havenhand J, Hutchins DA, Riebesell U, Rintoul MS, Vichi M, et al. 2018. Experimental strategies to assess the biological ramifications of multiple drivers of global ocean change—a review. Glob Change Biol. 24(6):2239–2261. [DOI] [PubMed] [Google Scholar]
- Chen I, Chu K, Palaniappan K, Pillay M, Ratner A, Huang J, Huntemann M, Varghese N, White JR, Seshadri R, et al. 2019. IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 47(D1):D666–D677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S, Bell G.. 2004. Phenotypic consequences of 1,000 generations of selection at elevated CO2 in a green alga. Nature 431(7008):566–569. [DOI] [PubMed] [Google Scholar]
- Collins S, Rost B, Rynearson TA.. 2013. Evolutionary potential of marine phytoplankton under ocean acidification. Evol Appl. 7(1):140–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danchin E, Pocheville A, Rey O, Pujol B, Blanchet S.. 2019. Epigenetically facilitated mutational assimilation: epigenetics as a hub within the inclusive evolutionary synthesis. Biol Rev. 94(1):259–282. [Google Scholar]
- Delmont TO, Kiefl E, Kilinc O, Esen OC, Uysal I, Rappé MS, Giovannoni S, Eren AM.. 2019. Single-amino acid variants reveal evolutionary processes that shape the biogeography of a global SAR11 subclade. Elife 8:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doblin MA, van Sebille E.. 2016. Drift in ocean currents impacts intergenerational microbial exposure to temperature. Proc Natl Acad Sci U S A. 113(20):5700–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolzhenko E, Smith AD.. 2014. Using beta-binomial regression for high-precision differential methylation analysis in multifactor whole-genome bisulfite sequencing experiments. BMC Bioinformatics 15(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghi J. 2019. Links between evolutionary processes and phenotypic robustness in microbes. Semin Cell Dev Biol. 88:46–53. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE.. 2002. Gene expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30(1):207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenreich IM, Pfennig DW.. 2015. Genetic assimilation: a review of its potential proximate causes and evolutionary consequences. Ann Bot. 117(5):769–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong SS. 2005. Parallel adaptive evolution cultures of Escherichia coli lead to convergent growth phenotypes with different gene expression states. Genome Res. 15(10):1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischkorn KR, Haley ST, Dyhrman ST.. 2018. Coordinated gene expression between Trichodesmium and its microbiome over day–night cycles in the North Pacific Subtropical Gyre. ISME J. 12(4):997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Zou D, Mao L, Liu H, Song P, Chen Y, Zhao S, Gao C, Li X, Gao Z, et al. 2015. BS-SNPer: SNP calling in bisulfite-seq data. Bioinformatics 31(24):4006–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gärtner K, Klähn S, Watanabe S, Mikkat S, Scholz I, Hess WR, Hagemann M.. 2019. Cytosine N4-methylation via M.Ssp6803II is involved in the regulation of transcription, fine-tuning of DNA replication and DNA repair in the cyanobacterium Synechocystis sp. PCC 6803. Front Microbiol. 10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Xiao P, Jiang Y, Dong M, Chen Z, Li H, Hu Z, Lei A, Wang J.. 2018. Transgenerational epigenetic inheritance under environmental stress by genome-wide DNA methylation profiling in cyanobacterium. Front Microbiol. 9:2943–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Ding J, Gao K, de Carvalho MHC, Tirichine L, Bowler C, Lin X.. 2019. A potential role for epigenetic processes in the acclimation response to elevated pCO2 in the model diatom Phaeodactylum tricornutum. Front Microbiol. 9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins DA, Boyd PW.. 2016. Marine phytoplankton and the changing ocean iron cycle. Nat Clim Change 6(12):1072–1079. [Google Scholar]
- Hutchins DA, Fu F, Zhang Y, Warner ME, Feng Y, Portune K, Bernhardt PW, Mulholland MR.. 2007. CO2 control of Trichodesmium N2 fixation, photosynthesis, growth rates, and elemental ratios. Limnol Oceanogr. 52(4):1293–1304. [Google Scholar]
- Hutchins DA, Fu F-X, Webb EA, Walworth N, Tagliabue A.. 2013. Taxon-specific response of marine nitrogen fixers to elevated carbon dioxide concentrations. Nat Geosci. 6:1–6. [Google Scholar]
- Hutchins DA, Walworth NG, Webb EA, Saito MA, Moran D, McIlvin MR, Gale J, Fu F-X.. 2015. Irreversibly increased nitrogen fixation in Trichodesmium experimentally adapted to elevated carbon dioxide. Nat Commun. 6: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinkert B, Ossenbühl F, Sikorski M, Berry S, Eichacker L, Nickelsen J.. 2004. PratA, a periplasmic tetratricopeptide repeat protein involved in biogenesis of photosystem II in Synechocystis sp. PCC 6803. J Biol Chem. 279(43):44639–44644. [DOI] [PubMed] [Google Scholar]
- Koch R, Kupczok A, Stucken K, Ilhan J, Hammerschmidt K, Dagan T.. 2017. Plasticity first: molecular signatures of a complex morphological trait in filamentous cyanobacteria. BMC Evol Biol. 17(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronholm I, Bassett A, Baulcombe D, Collins S.. 2017. Epigenetic and genetic contributions to adaptation in Chlamydomonas. Mol Biol Evol. 34(9):2285–2306. [DOI] [PubMed] [Google Scholar]
- Kronholm I, Collins S.. 2015. Epigenetic mutations can both help and hinder adaptive evolution. Mol Ecol. 25(8):1856–1868. [DOI] [PubMed] [Google Scholar]
- Krueger F, Andrews SR.. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27(11):1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MD, Walworth NG, McParland EL, Fu F-X, Mincer TJ, Levine NM, Hutchins DA, Webb EA.. 2017. The Trichodesmium consortium: conserved heterotrophic co-occurrence and genomic signatures of potential interactions. ISME J. 11(8):1813–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MD, Webb EA, Walworth NG, Fu F-X, Held NA, Saito MA, Hutchins DA.. 2018. Transcriptional activities of the microbial consortium living with the marine nitrogen-fixing cyanobacterium Trichodesmium reveal potential roles in community-level nitrogen cycling. Appl Environ Microbiol. 84:e02026–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenski RE. 2017. Convergence and divergence in a long-term experiment with bacteria. Am Nat. 190(S1):S57–S68. [DOI] [PubMed] [Google Scholar]
- Levitan O, Rosenberg G, Setlik I, Setlikova E, Grigel J, Klepetar J, Prasil O, Berman-frank I.. 2007. Elevated CO2 enhances nitrogen fixation and growth in the marine cyanobacterium Trichodesmium. Global Change Biol. 13(2):531–538. [Google Scholar]
- Lohbeck KT, Riebesell U, Collins S, Reusch TBH.. 2013. Functional genetic divergence in high CO2 adapted Emiliania huxleyi populations. Evolution 67(7):1892–1900. [DOI] [PubMed] [Google Scholar]
- Lohbeck KT, Riebesell U, Reusch TBH.. 2012. Adaptive evolution of a key phytoplankton species to ocean acidification. Nature Geosci. 5(5):346–351. [Google Scholar]
- Lohbeck KT, Riebesell U, Reusch TBH.. 2014. Gene expression changes in the coccolithophore Emiliania huxleyi after 500 generations of selection to ocean acidification. Proc R Soc B 281(1786):20140003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S.. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12), doi:10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madlung A, Masuelli RW, Watson B, Reynolds SH, Davison J, Comai L.. 2002. Remodeling of DNA methylation and phenotypic and transcriptional changes in synthetic Arabidopsis allotetraploids. Plant Physiol. 129(2):733–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K, Kinjo AR.. 2018. Mechanism of evolution by genetic assimilation. Biophys Rev. 10(2):667–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J, Blanchet FG, Kindt R.. 2013. Vegan: Community Ecology Package 2.0–3, https://cran.r-project.org/web/packages/vegan/index.html.
- Papadopoulos D, Schneider D, Meier-Eiss J, Arber W, Lenski RE, Blot M.. 1999. Genomic evolution during a 10,000-generation experiment with bacteria. Proc Natl Acad Sci U S A. 96(7):3807–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfreundt U, Kopf M, Belkin N, Berman-Frank I, Hess WR.. 2014. The primary transcriptome of the marine diazotroph Trichodesmium erythraeum IMS101. Sci Rep. 4:6187–6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigliucci M, Murren CJ, Schlichting CD.. 2006. Phenotypic plasticity and evolution by genetic assimilation. J Exp Biol. 209(12):2362–2367. [DOI] [PubMed] [Google Scholar]
- Ploner A. 2015. Heatplus: heatmaps with row and/or column covariates and colored clusters. R Package Version 2.18.0, https://github.com/alexploner/Heatplus.
- Quinlan AR, Hall IM.. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6):841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Schwartz K, Sherlock G, Dudoit S.. 2011. GC-content normalization for RNA-Seq data. BMC Bioinformatics. 12(1):480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar G, Paoli L, Alberti A, Huerta-Cepas J, Ruscheweyh H-J, Cuenca M, Field CM, Coelho LP, Cruaud C, Engelen S, et al. 2019. Gene expression changes and community turnover differentially shape the global ocean metatranscriptome. Cell 179(5):1068–1083.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaum C-E, Buckling A, Smirnoff N, Studholme DJ, Yvon-Durocher G.. 2018. Environmental fluctuations accelerate molecular evolution of thermal tolerance in a marine diatom. Nat Commun. 9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaum C-E, Rost BOR, Collins SEA.. 2016. Environmental stability affects phenotypic evolution in a globally distributed marine picoplankton. ISME J. 10(1):75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaum CE, Collins S.. 2014. Plasticity predicts evolution in a marine alga. Proc R Soc B 281(1793):20141486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schottkowski M, Ratke J, Oster U, Nowaczyk M, Nickelsen J.. 2009. Pitt, a novel tetratricopeptide repeat protein involved in light-dependent chlorophyll biosynthesis and thylakoid membrane biogenesis in Synechocystis sp. PCC 6803. Mol Plant. 2(6):1289–1297. [DOI] [PubMed] [Google Scholar]
- Schlichting CD, Wund MA.. 2014. Phenotypic plasticity and epigenetic marking: an assessment of evidence for genetic accommodation. Evolution 68(3):656–672. [DOI] [PubMed] [Google Scholar]
- Schluter L, Lohbeck KT, Gro ger JP, Riebesell U, Reusch TBH.. 2016. Long-term dynamics of adaptive evolution in a globally important phytoplankton species to ocean acidification. Sci Adv. 2(7):e1501660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, Schultz MD, Lewsey MG, O'Malley RC, Urich MA, Libiger O, Schork NJ, Ecker JR.. 2011. Transgenerational epigenetic instability is a source of novel methylation variants. Science 334(6054):369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Q, Decato B, Hong EE, Zhou M, Fang F, Qu J, Garvin T, Kessler M, Zhou J, Smith AD.. 2013. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS ONE. 8(12):e81148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajic D, Perfeito L, Jansen LET.. 2019. Epigenetic gene silencing alters the mechanisms and rate of evolutionary adaptation. Nat Ecol Evol. 3(3):491–498. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Shimodaira H.. 2006. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22(12):1540–1542. [DOI] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. 2018. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47:D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veluchamy A, Lin X, Maumus F, Rivarola M, Bhavsar J, Creasy T, O’Brien K, Sengamalay NA, Tallon LJ, Smith AD.. 2013. Insights into the role of DNA methylation in diatoms by genome-wide profiling in Phaeodactylum tricornutum. Nat. Commun. 4:1–10. [DOI] [PubMed] [Google Scholar]
- Walworth N, Pfreundt U, Nelson WC, Mincer T, Heidelberg JF, Fu F, Waterbury JB, Glavina del Rio T, Goodwin L, Kyrpides NC, et al. 2015. Trichodesmium genome maintains abundant, widespread noncoding DNA in situ, despite oligotrophic lifestyle. Proc Natl Acad Sci U S A. 112(14):4251–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth NG, Fu F-X, Webb EA, Saito MA, Moran D, Mcllvin MR, Lee MD, Hutchins DA.. 2016. Mechanisms of increased Trichodesmium fitness under iron and phosphorus co-limitation in the present and future ocean. Nat Commun. 7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth NG, Fu F-X, Lee MD, Cai X, Saito MA, Webb EA, Hutchins DA.. 2018. Nutrient-colimited Trichodesmium as a nitrogen source or sink in a future ocean. Appl Environ Microbiol. 84:e02137-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth NG, Hutchins DA, Dolzhenko E, Lee MD, Fu F, Smith AD, Webb EA.. 2017. Biogeographic conservation of the cytosine epigenome in the globally important marine, nitrogen-fixing cyanobacterium Trichodesmium. Environ Microbiol. 19(11):4700–4713. [DOI] [PubMed] [Google Scholar]
- Walworth NG, Lee MD, Fu F-X, Hutchins DA, Webb EA.. 2016. Molecular and physiological evidence of genetic assimilation to high CO2 in the marine nitrogen fixer Trichodesmium. Proc Natl Acad Sci U S A. 113(47):E7367–E7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth NG, Lee MD, Suffridge C, Qu P, Fu F-X, Saito MA, Webb EA, Sañudo-Wilhelmy SA, Hutchins DA.. 2018. Functional genomics and phylogenetic evidence suggest genus-wide cobalamin production by the globally distributed marine nitrogen fixer Trichodesmium. Front Microbiol. 9:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth NG, Zakem EJ, Dunne JP, Collins S, Levine NM.. 2020. Microbial evolutionary strategies in a dynamic ocean. Proc Natl Acad Sci U S A. 117(11):5943–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West-Eberhard MJ. 2005. Developmental plasticity and the origin of species differences. Proc Natl Acad Sci U S A. 102(Suppl 1):6543–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker KA, Rynearson TA.. 2017. Evidence for environmental and ecological selection in a microbe with no geographic limits to gene flow. Proc Natl Acad Sci U S A. 114(10):2651–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehr JP, Ohki K, Fujita Y, Landry D.. 1991. Unique modification of adenine in genomic DNA of the marine cyanobacterium Trichodesmium sp. strain NIBB 1067. J Bacteriol. 173(21):7059–7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw read files for Whole-genome bisulfite-sequenced libraries (WGBS) have been deposited in the NCBI’s Gene Expression Omnibus (Edgar et al. 2002) and are accessible through GEO Series accession number GSE138176. The ambient an Amb-High RNA raw reads can be found in another GEO project with accession number GSE86992. Within the GSE86992, the specific samples for the ambient and Amb-High have sample accession numbers of GSM2318497, GSM2318498, GSM2318499, and GSM2318500. The rest of the RNA-Seq libraries for the high-CO2 and High-Amb can be found in GSE138176.