

SUMMARY
Bacterial pneumonia is a global healthcare burden, and unwarranted inflammation is suggested as an important cause of mortality. Optimum levels of the anti-inflammatory cytokine IL-10 are essential to reduce inflammation and improve survival in pneumonia. Elevated levels of the mitochondrial-DAMP cardiolipin (CL), reported in tracheal aspirates of pneumonia patients, have been shown to block IL-10 production from lung MDSCs. Although CL-mediated K107 SUMOylation of PPARγ has been suggested to impair this IL-10 production, the mechanism remains elusive. We identify PIAS2 to be the specific E3-SUMOligase responsible for this SUMOylation. Moreover, we identify a concomitant CL-mediated PPARγ S112 phosphorylation, mediated by JNK-MAPK, to be essential for PIAS2 recruitment. Furthermore, using a clinically tested peptide inhibitor targeting JNK-MAPK, we blocked these post-translational modifications (PTMs) of PPARγ and rescued IL-10 expression, improving survival in murine pneumonia models. Thus, we explore the mechanism of mito-DAMP-mediated impaired lung inflammation resolution and propose a therapeutic strategy targeting PPARγ PTMs.
Graphical Abstract
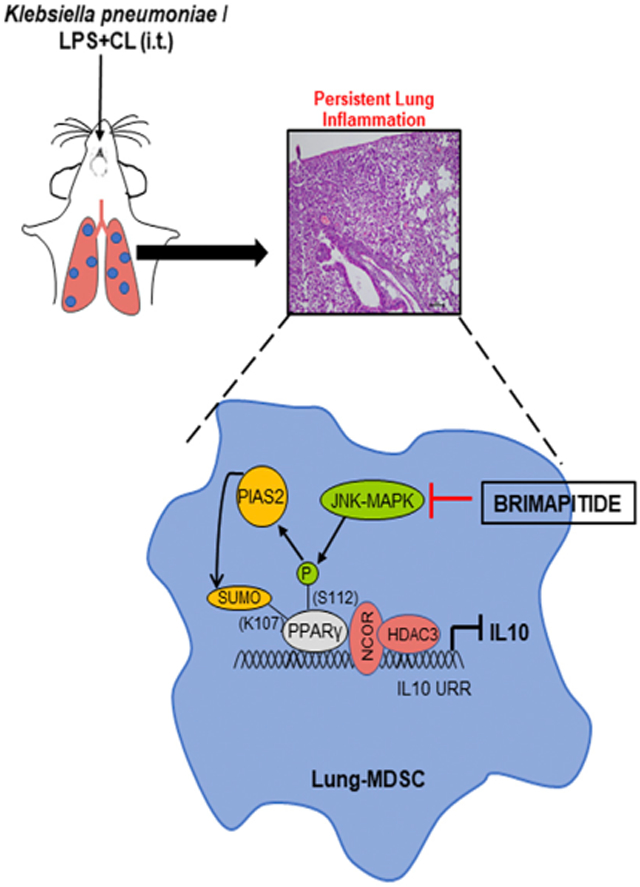
In Brief
Garg et al. identify the role of post-translational modifications of PPARγ in causing cardiolipin-mediated IL-10 suppression from lung myeloid-derived suppressor cells, thus increasing pneumonia severity. They identify multiple targets to reverse this suppression and improve IL-10 expression and lung inflammation resolution in bacterial pneumonia.
INTRODUCTION
Despite significant improvements in health care, bacterial pneumonia remains a leading cause of morbidity and mortality around the globe (McAllister et al., 2019; Mizgerd, 2008). Inflammation, in response to bacterial infection, is essential for pathogen clearance, and alveolar macrophages and neutrophils play a crucial role in this process. Alveolar macrophages initially recognize pathogenic bacteria and secrete mediators to recruit neutrophils to the infection site. Neutrophils kill bacteria by releasing superoxides and catabolizing enzymes, including proteases and lipases. Subsequently, with the recruitment of additional immune cells, there is a surge of inflammatory mediators in the form of cytokines and chemokines, triggering an inflammatory cascade. However, unchecked inflammation is in itself detrimental to the host and leads to collateral tissue damage, causing organ failure (Bhattacharya and Matthay, 2013; Levy and Serhan, 2014; Nathan and Ding, 2010; Robb et al., 2016). A well-known complication of pneumonia is the precipitation of respiratory failure in the form of acute lung injury (ALI) or acute respiratory distress syndrome (ARDS), in which aggravated inflammation drives disease progression (Johnson and Matthay, 2010; Rubenfeld et al., 2005).
To reduce the extent of this inflammation-mediated collateral damage, the host orchestrates sequential events for the timely resolution of inflammation. This process triggers an array of signaling pathways, cytokines, chemokines, and lipid mediators, among which interleukin-10 (IL-10) plays a central role (Ogawa et al., 2008; Ouyang and O’Garra, 2019). CD11b+F480+Ly6Gint Ly6Clo lung cells, known as myeloid-derived suppressor cells (lung MDSCs), have been identified as a major source of IL-10 by multiple studies (Arora et al., 2010; Poe et al., 2013). IL-10 from lung MDSCs directly suppresses proinflammatory cytokine production and also promotes neutrophil clearance (efferocytosis), to prevent persistent lung inflammation. The immunosuppressive role of MDSCs has been explored in various disease states, including those of infectious etiology (Gabrilovich and Nagaraj, 2009; Ostrand-Rosenberg and Sinha, 2009; Ray et al., 2013).
An important feature of diseases with aggravated inflammation such as ARDS is uncontrolled cell death (Matthay et al., 2019; Sauler et al., 2019). High cell death tends to release cellular components called DAMPs (damage-associated molecular patterns) into the extracellular compartment. Mitochondrial DAMPs are endogenous mitochondrial components that act as inflammatory triggers, leading to a vicious inflammatory cycle and disease progression (Cloonan and Choi, 2016; Krysko et al., 2011). Elevated levels of a mitochondrial DAMP, cardiolipin (CL), have already been shown to be present in the tracheal aspirates of patients suffering from severe pneumonia (Ray et al., 2010). CL is a phospholipid component of the mitochondrial inner membrane and its role in various diseases has been reported in the literature (Paradies et al., 2019). Moreover, oxidized CL has been shown to play an important role in apoptosis, further reflecting its potential for disease progression (Kagan et al., 2005). Earlier reports suggest that CL mediates SUMOylation of peroxisome proliferator-activated receptor γ (PPARγ) at its K107 residue, limits its transcriptional activity, and causes IL-10 suppression in bacterial pneumonia (Chakraborty et al., 2017). However, the detailed underlying mechanism remained elusive. In the present study, we identify protein inhibitor of activated STAT2 (PIAS2) to be the specific E3-SUMO (small ubiquitin-like modifier) ligase responsible for CL-mediated PPARγ K107 SUMOylation. Moreover, we find that a concomitant CL-induced S112 phosphorylation of PPARγ, mediated by the upstream c-Jun N-terminal kinase-mitogen-activated protein kinase (JNK-MAPK), is essential for PPARγ-PIAS2 interaction. Intriguingly, blocking JNK-MAPK in a sterile CL-mediated acute lung injury model as well as in a Klebsiella pneumoniae (KP)-mediated murine pneumonia model rescued IL-10 production and significantly reduced lung injury severity.
Thus, our study identifies how crosstalk between the post-translation modifications of PPARγ fine-tunes IL-10 expression under the influence of CL, and affects the resolution of lung inflammation during pneumonia. In addition, this study provides a therapeutic strategy to promote inflammation resolution by using a specific peptide inhibitor of JNK-MAPK. Interestingly, this particular JNK-MAPK inhibitor (Brimapitide/AM-111) is already being used in various clinical conditions, and repurposing this drug as an adjunct therapy in pneumonia may significantly reduce the mortality and morbidity associated with this dreaded disease.
RESULTS
PIAS2-E3-SUMOligase is critical for CL-mediated PPARγ SUMOylation and IL-10 suppression in lung MDSCs
It has been demonstrated that CL leads to persistent lung inflammation by impeding IL-10 expression and lung inflammation resolution (Chakraborty et al., 2017). CL-mediated PPARγ SUMOylation at its K107 residue was suggested to recruit the co-repressors histone deacetylase 3 (HDAC3) and NCOR to the IL-10 promoter, thus suppressing its expression. However, the specific E3-SUMO ligase crucial for this process remains unexplored. The PIAS family of proteins is reported to be the major E3-SUMOligases in many cell types, and 4 PIAS genes (PIAS1–4) with tissue-specific expression have been identified so far (Shuai, 2006). To explore their expression in lung MDSCs, we isolated these cells, as described earlier (Figure S1A). Our qPCR experiments showed that all four PIAS genes are abundantly present in lung MDSCs, and PIAS1 has the highest expression among them (Figure S1D). To identify whether one of these PIAS genes was responsible for the CL-mediated PPARγ SUMOylation and IL-10 suppression, we took a gene knockdown approach. Lung MDSCs were transfected with either scrambled small interfering RNA (siRNA) or siRNA targeting individual PIAS genes. The efficiency of each siRNA was confirmed by qPCR (Figures S1E-S1H). Cells with wild-type (WT) or silenced PIAS expression were treated with lipopolysaccharide (LPS) ± CL, and the IL-10 level in the culture supernatants was measured after 6 h. Interestingly, PIAS2 silencing rescued the IL-10 suppression caused by LPS + CL treatment. This effect was specific as it was not observed by silencing the other PIAS genes (Figure 1A).
Figure 1. PIAS2-E3-SUMOligase is critical for CL-mediated PPARγ SUMOylation and IL-10 suppression in lung MDSCs.

Lung MDSCs were enriched from C57BL/6 mice of 8–10 weeks of age and seeded in appropriate density (A–D); 106 lung MDSCs were transfected with scrambled or indicated PIAS siRNAs. At 24 h post-transfection, cells were stimulated with LPS (1 μg/mL) ± CL (10 μg/mL).
(A) IL-10 in culture supernatant measured by ELISA after stimulation for 6 h.
(B) PPARγ was immunoprecipitated followed by western blot analysis with SUMO1 antibody (left panel) along with its quantitative densitometry analysis (right panel).
(C and D) ChIP assay performed with histone deacetylase 3 (HDAC3) (C) and (D) NCOR antibody to investigate their recruitment to the IL-10 promoter. All ex vivo experiments were conducted with 2–3 replicates. The data shown are representative of 2 independent experiments.
(E–L) C57BL/6 mice 8–10 weeks old were administered LPS (1 mg/kg) ± CL (5 mg/kg) intratracheally (i.t.). 24 h later they were administered either scrambled or PIAS2 targeting siRNA mixed with in vivo transfection reagent JetPEI by tail vein injection. At 48 h post-transfection, mice were harvested. In vivo experiments were conducted with 4 mice in each of the LPS and LPS + CL-administered groups except where otherwise mentioned. The data shown are representative of 2 independent experiments.
(E) Diagrammatic representation of the experimental design for the in vivo experiments.
(F) Change in percentage of body weight as a time course. Significance and the respective p value mentioned in parentheses are indicative of the comparative analysis between scrm siRNA and PIAS2 siRNA-administered mice in the LPS + CL group.
(G) Lung pathology studied by H&E staining of lung tissue (left panel). Lung injury score was calculated according to American Thoracic Society (ATS) guidelines (right panel). A minimum of 20 random high-power fields were analyzed in each mouse; 40× images for representation were acquired from the left lower field of the respective 10× image in all groups, as indicated by the dotted box.
(H) Infiltrating cells in the BAL fluid counted manually using a cell counting chamber.
(I) Albumin levels in the BAL fluid measured using ELISA; 5 mice were kept in each of the LPS + CL-treated groups.
(J) IL-10:TNF-α ratio in lung tissue lysate measured by ELISA.
(K and L) Lung MDSCs were isolated from the LPS + CL-administered mice after administration of the scrambled or PIAS2 siRNA on day 3; 3 mice were considered in each group.
(K) PPARγ was immunoprecipitated from these cells, and SUMO1 expression in these groups of mice was checked by western blot analysis (left panel) along with quantitative densitometry analysis (right panel).
(L) ChIP assay performed to compare HDAC recruitment on the IL-10 promoter. The data shown are means ± SDs and representative of 2 independent experiments. The hinges in the box and whisker graphs represent the interquartile distance and the whiskers represent the range. Each dot on the plots represents a biological replicate.
Statistical significance was calculated using 2-way ANOVA with Tukey’s multiple comparisons test for (F), unpaired t test for (K) and (L), and 1-way ANOVA with Tukey’s multiple comparisons test for the rest of the panels. The numbers in parentheses indicate the respective p values for statistical significance between the marked groups. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; NS, not significant. See also Figure S1.
Furthermore, we validated our findings by checking the direct effect of PIAS2 silencing on PPARγ SUMOylation and found that silencing PIAS2 abrogated the increased-PPARγ SUMOylation found in LPS + CL-treated cells, as compared to that observed with LPS stimulation alone. Silencing PIAS1 was not effective in producing the same results (Figure 1B). Comparably, the increased HDAC3 recruitment to the IL-10 promoter in LPS + CL-treated cells was significantly reduced with PIAS2 silencing but not with PIAS1 knockdown (Figure 1C). Similar results were observed for NCOR recruitment to the IL-10 promoter upon PIAS2 silencing (Figure 1D). Collectively, these observations suggest that PIAS2 is critical for CL-mediated PPARγ SUMOylation, leading to co-repressor recruitment (HDAC3, NCOR) and IL-10 suppression in lung MDSCs.
Next, to investigate the role of PIAS2 in impairing the resolution of lung inflammation, we performed in vivo gene silencing using siRNA along with an in vivo transfection reagent (jetPEI). Mice were randomly distributed and administered LPS + CL intratracheally (i.t.), followed by tail vein injection of siRNA targeting PIAS2. Mice that received either PBS, CL only, LPS only, or LPS + CL were treated with scrambled siRNA and served as controls (Figure 1E). PIAS2 knockdown in lung MDSCs was confirmed using qPCR and was found to be maximum at 48 h post-transfection (data not shown). Although LPS-administered mice lost body weight initially, they regained weight by day 3. In contrast, mice that received LPS + CL and scrambled siRNA showed a marked loss of body weight and minimal regain up to day 3. LPS + CL-administered mice with PIAS2 knockdown regained weight markedly better (Figure 1F). Histopathological analysis also showed improved lung pathology in PIAS 2-silenced mice (Figure 1G). This was again reflected in reduced cell counts seen in bronchoalveolar lavage (BAL) (Figure 1H), as well as the lower BAL-albumin levels (Figure 1I), suggesting reduced severity of lung injury. As expected, mice that were administered only CL did not show any signs of inflammation. Furthermore, to investigate whether the improved lung pathology in PIAS2-silenced mice was a consequence of better inflammation resolution, we measured IL-10 levels in their lung tissue lysates. Following LPS + CL administration, IL-10 levels were significantly higher in PIAS2-silenced mice as compared to that in mice treated with scrambled siRNA (Figure S1I). This was not associated with a decrease in tumor necrosis factor-α (TNF-α) levels (Figure S1J), and a higher IL-10/TNF-α ratio was seen in PIAS2-silenced mice (Figure 1J). Finally, to investigate whether this rescue of the inflammation resolution process is through a reduced PPARγ SUMOylation, we enriched lung MDSCs from the two groups of LPS + CL-administered mice and performed immunoprecipitation (IP) and chromatin IP (ChIP) studies. IP studies showed that PIAS2 silencing markedly reduced CL-mediated PPARγ SUMOylation (Figure 1K), and this was validated by reduced HDAC3 recruitment to the IL-10 promoter post-PIAS2 silencing (Figure 1L). These observations suggest that PIAS2 gene silencing allows the resolution of CL-mediated persistent lung inflammation, likely through the rescue of IL-10 production and resolution of inflammation.
CL-mediated PPARγ K107 SUMOylation is controlled by a concomitant S112 phosphorylation
It has been shown that CL induces PPARγ SUMOylation specifically at the K107 residue, sparing the K395 SUMOylation site (Chakraborty et al., 2017). However, the underlying mechanism ensuring such specificity remains largely unexplored. To investigate this, we analyzed the amino acid sequence of PPARγ around its K107 SUMOylation site. A putative phosphorylation site at the serine 112 position was found in close proximity (Figure 2A). This indicates the presence of a phosphorylation-dependent SUMOylation motif (PDSM) where phosphorylation at the serine residue is suggested to influence SUMOylation of the neighboring lysine residue (Hietakangas et al., 2006). Therefore, we wondered whether CL may induce a concomitant phosphorylation of PPARγ at its S112 residue. Our western blot data showed that LPS + CL administration in mice caused a marked increase in PPARγ S112 phosphorylation as compared with that observed with only LPS administration (Figure 2B). As this increase in PPARγ S112 phosphorylation with CL treatment could reflect the effect from a number of cell types, we decided to enrich lung MDSCs from LPS ± CL-treated mice and check for PPARγ S112 phosphorylation. A similar trend was observed in enriched lung MDSCs using immunofluorescence microscopy (Figure 2C) to confirm that CL administration in vivo did cause PPARγ S112 phosphorylation in lung MDSCs. Furthermore, to validate whether CL exerts a direct effect on lung MDSCs to cause this phosphorylation, we stimulated them ex vivo with LPS ± CL and observed similar results (Figures 2D and 2E). To explore whether this phosphorylation was specific to CL and not an effect of LPS dose, we used a substantially higher dose of LPS (10 μg/mL) and found that this did not lead to increased PPARγ S112 phosphorylation or IL-10 suppression (Figures S2A and S2B).
Figure 2. CL-mediated PPARγ K107 SUMOylation is controlled by a concomitant S112 phosphorylation.

(A) Amino acid sequence of PPARγ showing a phosphorylation-dependent SUMOylation motif at the 112 residue and sequence homology between Homo sapiens and Mus musculus.
(B and C) LPS ± CL was administered i.t. in 8- to 10-week-old C57BL/6 mice. On day 6, mice were sacrificed for outcome measures.
(B) PPARγ S112 phosphorylation was detected in whole lung tissue lysate by western blot (left panel) along with its quantitative densitometry analysis (right panel).
(C) Representative immunofluorescence images showing expression of S112 phosphorylated PPARγ in enriched lung MDSCs. The nuclear stain DAPI is represented by blue and phospho-S112 PPARγ is represented by green pseudo-colors. Quantification of the mean fluorescence normalized to total-PPARγ is shown alongside. A minimum of 100 cells from 3 mice in each group was considered for quantitation.
(D and E) Lung MDSCs were enriched from 8- to 10-week-old C57BL/6 mice and stimulated ex vivo with LPS ± CL for 3h.
(D) PPARγ S112 phosphorylation detected in these cells by western blot, represented alongside its quantitative densitometry analysis.
(E) S112 phosphorylation of PPARγ detected by immunofluorescence along with quantitation of its mean fluorescence normalized to total PPARγ. A minimum of 200 cells from 2 biological replicates in each group was analyzed for quantitation. DAPI is represented by blue and phospho-S112 PPARγ is represented by green pseudo-colors.
(F–I) 106 RAW 264.7 cells were transfected with either wild-type PPARγ (FLAG-tagged) overexpression construct or PPARγ S112A mutant as shown. Cells were stimulated with LPS (1 μg/mL) ± CL (10 μg/mL) for 3 h. (F) PPARγ was immunoprecipitated with anti-FLAG antibody followed by western blot analysis with SUMO1 antibody (top panel) as well as PIAS2 antibody (center panel). Densitometric analysis of each blot is represented alongside as indicated. ChIP analysis was performed to investigate the recruitment of (G) HDAC and (H) NCOR to the IL-10 promoter. (I) IL-10 in culture supernatant was measured by ELISA after 6 h of LPS ± CL treatment.
All ex vivo and in vitro experiments were conducted with 2 replicates in each group. The data shown are means ± SDs and representative of 2 independent experiments. The hinges in the box and whisker graphs represent the interquartile distance and the whiskers represent the range. Each dot on the plots represents a biological replicate. Statistical significance was calculated using unpaired t test for (B)–(E) and 1-way ANOVA with Tukey’s multiple comparisons test for (F)–(I). The numbers in parentheses indicate the respective p values for statistical significance between the marked groups. *p < 0.05, **p < 0.01, ****p < 0.0001; NS, not significant. See also Figure S2.
Next, we wished to explore whether PPARγ S112 phosphorylation truly influences SUMOylation at the K107 residue. RAW264.7 macrophages are cells known to have undetectable levels of endogenous PPARγ. Exploiting this knowledge, we overexpressed WT PPARγ (FLAG-tagged) or phosphorylation-deficient (S112A) mutant PPARγ (FLAG-tagged) in these cells. On LPS + CL treatment, cells expressing WT PPARγ showed a marked increase in PPARγ SUMOylation as compared to that detected with LPS treatment alone. However, this was not seen in cells expressing phosphorylation-deficient PPARγ mutant (S112A) (Figure 2F, upper panel). Next, we studied the association of PIAS2 with PPARγ in these cells by IP. A marked increase in this association was observed following LPS + CL treatment of cells expressing WT PPARγ cells, but not mutant PPARγ (S112A)-expressing cells (Figure 2F, center panel). A similar trend was observed with respect to HDAC3 (Figure 2G) and NCOR recruitment (Figure 2H) to the IL-10 promoter. As anticipated, IL-10 levels were also significantly suppressed upon LPS + CL treatment in the culture supernatants of WT PPARγ-expressing cells but not in those of cells expressing the mutant construct (Figure 2I). These experiments show that CL induces the phosphorylation of PPARγ at the S112 residue, which influences its K107 SUMOylation and blockade of IL-10 expression.
JNK is required for CL-mediated PPARγ S112 phosphorylation
LPS-triggered Toll-like receptor 4 (TLR4) activation is known to be vital in the pathogenesis of bacterial pneumonia (Fitzgerald and Kagan, 2020; Triantafilou and Triantafilou, 2002). In addition, bacterial pneumonia has been shown to be associated with elevated levels of CL (Chakraborty et al., 2017; Ray et al., 2010). Thus, we decided to investigate the possible role of TLR4-activated MAPKs in regulating CL-mediated S112 phosphorylation. To this end, enriched lung MDSCs were treated with LPS ± CL in combination with specific inhibitors of either of the 3 MAPKs (U0126 against Erk, SB203580 against P38, and Brimapitide against JNK). Although LPS administration led to a slight increase in PPARγ S112 phosphorylation over untreated, LPS + CL treatment augmented the phosphorylation severalfold, which was abrogated upon pre-treatment with the JNK-MAPK inhibitor (Figure 3A). This reduction was not seen on treatment with other MAPK inhibitors. Interestingly, the marked suppression of IL-10 observed in LPS + CL-treated cells was also largely rescued upon treatment with the JNK-MAPK inhibitor (Figure 3B). As CDK-9 is a known upstream kinase causing PPARγ phosphorylation, and CDK9 expression in MDSCs was appreciated earlier, we decided to explore the effects of CDK-9 inhibition on CL-mediated PPARγ phosphorylation. We found that the CDK-9 inhibitor SNS-032 failed to reduce CL-mediated PPARγ S112 phosphorylation or rescue IL-10 suppression (Figures S3A and S3B). These data suggest that JNK-MAPK is a major upstream kinase responsible for CL-mediated PPARγ S112 phosphorylation in lung MDSCs. To validate our findings, we checked PPARγ SUMOylation (Figure 3C, upper panel) and PIAS2-PPARγ association (Figure 3C, center panel) with JNK inhibition and found a trend similar to that observed with PPARγ phosphorylation. Similarly, HDAC3 and NCOR recruitment to the IL-10 promoter was clearly reduced with the JNK inhibitor (JNKi) pre-treatment (Figures 3D and 3E). Finally, we used a genetic approach with the help of the DN.JNK plasmid transfected into RAW264.7 cells along with the full-length WT PPARγ construct. We found that the overexpression of DN.JNK significantly reduced CL-mediated PPARγ S112 phosphorylation and rescued IL-10 suppression (Figures S3C and S3D). To summarize, our data show that CL, in the presence of LPS, causes JNK-MAPK-mediated phosphorylation of the S112 residue of PPARγ. This leads to PIAS2-mediated SUMOylation of its K107 residue, recruiting the co-repressors HDAC3 and NCOR to the IL-10 promoter, suppressing IL-10 production and leading to impaired inflammation resolution (Figure 3F).
Figure 3. JNK is required for CL-mediated PPARγ S112 phosphorylation.

Lung MDSCs were enriched from C57BL/6 mice of 8–10 weeks of age, seeded at appropriate densities, and stimulated with LPS (1 μg/mL) ± CL (10 μg/mL) for 3 h, in combination with different MAPK inhibitors, as mentioned.
(A and B) PPARγ S112 phosphorylation studied by western blot analysis after 3 h of stimulation (A). Quantitative densitometry is shown alongside (B) IL-10 in culture supernatant measured by ELISA after stimulation of cells for 6 h.
(C–E) Cells were stimulated for 3 h with LPS ± CL in combination with JNK MAPK-inhibitor.
(C) PPARγ SUMOylation studied by immunoprecipitation followed by western blot analysis using anti- SUMO1 and anti-PIAS2 antibody, respectively. Densitometry analysis is plotted in the right panel.
(D) HDAC recruitment to IL-10 promoter investigated by ChIP assay.
(E) NCOR recruitment to the IL-10 promoter investigated using ChIP assay.
(F) Diagrammatic representation showing the possible mechanism of CL-mediated IL-10 suppression, based on the results of this study.
Experiments were conducted with 2–3 replicates in each group. The data shown are means ± SDs and representative of 2 independent experiments. Statistical significance was calculated using 1-way ANOVA with Tukey’s multiple comparisons test. Each dot on the plots represents a biological replicate. The numbers in parentheses indicate the respective p values for statistical significance between the marked groups. *p < 0.05, ***p < 0.001, ****p < 0.0001; NS, not significant. See also Figure S3.
Blocking JNK resolves CL-mediated persistent lung inflammation by rescuing IL-10
Subsequently, we wondered whether inhibiting JNK by using a clinically tested specific peptide inhibitor could promote lung inflammation resolution and reduce lung injury severity by rescuing IL-10 production. To this end, we administered mice LPS + CL, as described earlier, to induce persistent lung inflammation. After 48 h, mice were divided randomly into three groups. The first group was kept as a vehicle control, while the second was treated with the JNKi via tail vein injection. To confirm whether any change in lung inflammation severity is through IL-10 rescue, we treated a third group with JNKi in combination with an IL-10 receptor-blocking antibody. The first two groups received the respective isotype controls (Figure 4A).
Figure 4. Blocking JNK resolves CL-mediated persistent lung inflammation by rescuing IL-10.

C57BL/6 mice of 8–10 weeks of age were treated i.t. with LPS + CL. After 48 h, mice were divided into 3 groups. The first group received appropriate isotype and vehicle (veh) controls, the second group was treated with JNK inhibitor (JNKi) (3 doses) + isotype control, and the third group received IL-10receptor-blocking antibody (6 doses) in conjugation with JNKi (3 doses) through tail vein injection. Experiments were conducted with 10 mice in the first group, 14 mice inthe second group, and 4 mice in the third group, except where mentioned. The data shown are representative of 2 independent experiments, except (B)–(E).
(A) Diagrammatic representation of the experimental design, including dosage schedule.
(B) Change in percentage of body weight monitored over 5 days. Significance and the respective p value mentioned in parentheses is indicative of the comparative analysis between vehicle + isotype control and JNKi + isotype control-administered mice.
(C) Lung pathology studied by H&E staining of lung tissue section (left panel). Lung injury score was calculated as described earlier (right panel). Four mice were considered in each group for quantification.
(D) Counts of infiltrating cells in BAL fluid.
(E) Albumin measured in BAL fluid by ELISA.
(F) Ratio of IL-10 and TNF-α expression level in lung tissue homogenate measured by ELISA. Five mice in the first group and 6 mice in the second group were considered for the analysis.
(G and H) Lung MDSCs were enriched from 3 mice in each group, and the ChIP assaywas performed on the IL-10 promoter to study the (G) HDAC3 and (H) NCOR recruitment.
(B)–(E) presents data pooled from 2 experiments, and (F)–(H) are data representative of 2 independent experiments. Data shown are means ± SDs. The hinges in the box and whisker graphs represent the interquartile distance and the whiskers represent the range. Each dot on the plots represents a biological replicate. Statistical significance was calculated using 2-way ANOVA with Tukey’s multiple comparisons test for (B), 1-way ANOVA with Tukey’s multiple comparisonstest for (C)–(E), and unpaired t test for (F)–(H). The numbers in parentheses indicate the respective p values for statistical significance between the marked groups. *p < 0.05, **p < 0.01, ****p < 0.0001; NS, not significant. See also Figure S4.
We noticed that although all three groups of mice showed an initial loss of body weight, the JNKi + isotype control-treated mice displayed better regain of weight as compared to the other two groups (Figure 4B). On day 6, lung inflammation was studied by multiple parameters. The JNKi + isotype control-treated group presented improved lung histology with negligible lung tissue damage as compared with the vehicle-administered group, as observed on H&E-stained sections. However, this was absent when the anti-IL-10-receptor antibody was administered in addition to the JNKi (Figure 4C). Similarly, total cell counts (Figure 4D) and albumin levels (Figure 4E) in BAL were significantly reduced in the JNKi + isotype control-treated group but not in the anti-IL-10-receptor antibody-treated group as compared to the vehicle-treated mice. Interestingly, administration of the JNKi also led to a decrease in the neutrophil fraction in the BAL, suggesting a reduction in inflammation and improvement in lung injury severity (Figure S4A).
To investigate whether this improvement in lung histology is due to a better anti-inflammatory response, we decided to check the IL-10:TNF-α ratio in the lung homogenates. We found that the JNKi-treated group had a higher IL-10:TNF-α ratio as compared to the vehicle-administered mice (Figure 4F). IL-10 levels alone were also found to be elevated in the JNKi-treated group, which was not associated with a decrease in the TNF-α levels (Figures S4B and S4C). In agreement, recruitment of HDAC3 (Figure 4G) and NCOR (Figure 4H) to the IL-10 promoter was markedly reduced in lung MDSCs enriched from these mice. These results suggest that JNK-MAPK inhibition could improve lung-inflammation resolution, possibly by rescuing IL-10 production in lung MDSCs.
JNK inhibition improves bacterial pneumonia-induced inflammation through IL-10 restoration
Since our findings in the present study suggested a potential for JNK inhibition to improve the resolution of lung inflammation, we asked whether this strategy of JNK inhibition could be combined with antibiotic therapy to reduce unwarranted inflammation and mortality due to bacterial pneumonia. KP is considered one of the major pathogens associated with nosocomial pneumonia, and elevated levels of CL in the BAL of mice infected with KP was shown in previous reports (Chakraborty et al., 2017). Therefore, we chose a KP-mediated murine pneumonia model to check the protective efficacy of JNKi as an adjuvant therapy. A dose of 1,000 CFU of KP inoculum is reported as the approximate median lethal dose (LD50) for mice at 72 h post-infection (Poe et al., 2013) and was chosen for the experiment. At 24 h post-intratracheal administration of KP, mice were divided into different groups. While one group received antibiotic + vehicle control, the other group was treated with the JNKi in combination with antibiotic administered through tail vein injection for 2 consecutive days. Vehicle-administered groups infected with KP, treated with or without JNKi were kept as controls (Figure 5A). To investigate whether JNK inhibition can improve survival in these KP-infected mice, we monitored them until day 5. We noticed that while antibiotic treatment improved survival to almost 60%, an adjuvant therapy of JNKi with the antibiotic further increased survival to >90% (Figure 5B). Since the difference in mortality between these two groups was highest on day 3, we used the same time point to inspect the extent of inflammation, assuming an obvious difference in inflammation at this time. We noted that on day 3, although the antibiotic + vehicle control group regained weight better than the vehicle controls, the addition of JNKi to antibiotic treatment further improved weight gain significantly (Figure 5C). Moreover, lung histology of antibiotic+ JNKi-treated mice also showed minimal tissue injury and was markedly better than the antibiotic + vehicle group (Figure 5D). Interestingly, the administration of only JNKi was also not detrimental as it did not increase the severity of inflammation as compared to vehicle control. We next decided to investigate the bacterial proliferation in the lungs and bacterial dissemination in the bloodstream. We found that administration of antibiotic along with JNKi did not increase the bacterial proliferation as compared to antibiotic + vehicle-treated group (Figures 5E and 5F). Bacterial counts in both of these groups were significantly lower than that in the vehicle controls. In addition, we found that the administration of only JNKi was not detrimental as it did not increase the bacterial counts as compared to vehicle control.
Figure 5. JNK inhibition improves bacterial pneumonia-induced inflammation through IL-10 restoration.

(A–F) 8-to 10-week-old C57BL/6 mice were infected i.t. with 103 CFU of Klebsiella pneumoniae; 24 h after infection, mice were randomly distributed into 4 groups. One group was treated only with antibiotic doses (2 doses), while the other group received JNKi in addition to the antibiotic. A third group was kept as a vehicle control and the fourth group was administered JNKi with vehicle. Experiments were conducted with 6 mice in the antibiotic group and 9 mice in the antibiotic + JNKi group, except where mentioned. Mice were either harvested on day 3 or observed until day 5 for survival analysis. The data shown are representative of 2 independent experiments, except in (B).
(A) Diagrammatic representation of the experimental design, including the dosage schedule.
(B) Percentage of survival of mice in the 3 groups until day 5, represented by Kaplan-Meier survival curves; 21 mice were observed in the antibiotic and the antibiotic + JNKi groups.
(C) Change in body weight monitored over 3 days (presented as percentage change from initial body weight). Significance and the respective p value mentioned in parentheses in (B) and (C) are indicative of the comparative analysis between antibiotic and antibiotic + JNKi-treated mice.
(D) Representative images of the H&E sections of the lung tissue studied for assessing lung pathology. Lung injury score was calculated as per the ATS guidelines, as mentioned.
(E) Bacterial CFU in lung normalized to the lung weights.
(F) Bacterial CFU/mL blood. Six mice were considered in the antibiotic + vehicle and antibiotic + JNKi groups each for (E) and (F).
(G) Albumin measured in BAL fluid by ELISA.
(H) Ratio of IL-10 and TNF-α expression level in lung tissue homogenate measured by ELISA.
(I) Western blot analysis for studying phospho-S112 PPARγ expression in the lung tissue lysates from 3 mice in each group, accompanied by its densitometric analysis.
(J) Lung MDSCs were enriched from 3 mice in the first group and 4 mice in the second, and PPARγ was immunoprecipitated. SUMO expression checked by western blot is represented accompanied by its densitometric analysis.
(K) Lung MDSCs were enriched from 3 mice in each group, and ChIP assay was conducted to assess HDAC3 recruitment on the IL-10 promoter.
(C)–(K) shows data representative of 2 independent experiments. (B) shows cumulative data pooled from 2 experiments. The data shown are means ± SDs. The hinges in the box and whisker graphs represent the interquartile distance and the whiskers represent the range. Each dot on the plots represents a biological replicate. Statistical significance was calculated using 2-way ANOVA with Tukey’s multiple comparisons test for (C), 1-way ANOVA for (D), and unpaired t test for (E)–(H). The numbers in parentheses indicate the respective p values for statistical significance between the marked groups. *p < 0.05, **p < 0.01, ***p< 0.001; NS, not significant. See also Figure S5.
We next measured the BAL albumin levels and found significantly lower levels in the antibiotic + JNKi group compared to that in antibiotic + vehicle-treated mice (Figure 5G). Interestingly, the IL-10:TNF-α ratio in the antibiotic + JNKi-treated mice was significantly higher, suggesting an improved anti-inflammatory response in this group of mice (Figure 5H). IL-10 levels alone were also found to be higher in this group, which was not associated with a decrease in the TNF-α levels. (Figures S5A and S5B). To validate whether the observed improvement was due to a PPARγ-mediated pathway, we investigated S112 phosphorylation of PPARγ in these groups of mice. We found a higher PPARγ S112 phosphorylation in the antibiotic + vehicle group as compared to that in the antibiotic + inhibitor-treated mice (Figure 5I). This was also reflected by the IP assays and ChIP analysis in enriched lung-MDSCs, which showed higher PPARγ SUMOylation and HDAC3 recruitment to the IL-10 promoter in the antibiotic + vehicle group (Figures 5J and 5K). qPCR analysis of these cells also reflected a higher IL-10 expression in the antibiotic + JNKi-treated group (Figure S5C). These observations show that the administration of a JNKi as an adjunct to antibiotic therapy can significantly improve survival in murine KP-induced pneumonia models, possibly through reversal of CL-mediated IL-10 suppression and promotion of inflammation resolution.
DISCUSSION
Non-resolving bacterial pneumonia with its obscure pathophysiology remains a leading cause of mortality and morbidity worldwide (McAllister et al., 2019). Besides microbial invasion, unwarranted inflammation and collateral tissue damage form a critical component of its pathogenesis (Bhattacharya and Matthay, 2013; Levy and Serhan, 2014; Nathan and Ding, 2010; Robb et al., 2016). Mitochondrial DAMPs such as cardiolipin (CL) are endogenous inflammatory triggers that are being increasingly recognized as key components of disease progression (Cloonan and Choi, 2016; Krysko et al., 2011). Elevated levels of the mito-DAMP CL have been reported in the lung fluids of patients suffering from severe pneumonia as well as in the murine pneumonia models (Chakraborty et al., 2017; Ray et al., 2010). Several previous studies (Arora et al., 2010; Chakraborty et al., 2017; Peñaloza et al., 2019; Poe et al., 2013) and our present study have identified lung MDSCs as a major source of IL-10, necessary for lung inflammation resolution (Figures S1B and S1C). Although previous work showed that CL blocks IL-10 expression through PPARγ K107 SUMOylation in lung MDSCs, the precise mechanism remained unexplored (Chakraborty et al., 2017). Notably, this site is distinct from the PPARγ K395 SUMOylation known to be mediated by the ligand rosiglitazone. In the present study, we identify PIAS2 as the major E3 SUMO ligase responsible for this CL-mediated PPARγ K107 SUMOylation (Figure 1). In addition, we recognize a concomitant JNKmediated PPARγ S112 phosphorylation caused by CL as being essential for its K107 SUMOylation and IL-10 suppression (Figures 2 and 3). Finally, using a specific peptide inhibitor against JNK-MAPK, we identify a therapeutic approach to reduce the severity of both CL-mediated sterile lung injury and KP-induced murine pneumonia (Figures 4 and 5).
SUMOylation is a sequence of steps involving multiple enzymes, which adds a SUMO protein to an acceptor-lysine residue of a substrate. SUMOylation is known to alter function, subcellular localization, and stability of a target protein (Flotho and Melchior, 2013). E3-SUMO-ligase is a critical enzyme that catalyzes the final step of the SUMOylation pathway, and is therefore considered a major drug target. The PIAS family of proteins is reported to be the major E3-SUMO ligases, and four PIAS genes have been identified thus far (Shuai, 2006). However, there is limited knowledge regarding their expression in immune cells, especially lung MDSCs, which have an important role in the resolution of inflammation. Our data show that all four PIAS genes (PIAS1–4) are highly expressed in lung MDSCs (Figure S1D).
PIAS1 has been reported to play an important role in the ligand-dependent K395 SUMOylation of PPARγ (Liu and Shuai, 2008; Pascual et al., 2005). However, the E3-SUMO ligase critical for SUMOylation of the K107 site remains unexplored. Our study demonstrates that PIAS2, but not PIAS1, is required for CL-mediated K107 SUMOylation (Figure 1B), leading to suppressed IL-10 expression in lung MDSCs (Figure 1A). Moreover, using a siRNAmediated gene-silencing approach, we show that PIAS2 knockdown rescues IL-10 production and improves the resolution of lung inflammation (Figures 1F-1J). We assessed the inflammatory parameters in mice sacrificed 48 h post-siRNA administration, a time point that coincides with maximum siRNA efficiency.
The literature is increasingly appreciative of the pathophysiological importance of PIAS2 as an E3-SUMO-ligase. A recent study has shown that PIAS2 promotes SUMOylation of α-synuclein and leads to its accumulation, aggravating the severity of Parkinson’s disease (Rott et al., 2017). Another study has demonstrated PIAS2-mediated SUMOylation of Oct4, controlling Nanog expression, a pivotal transcription factor essential for maintaining the pluripotency of embryonic stem cells (Wu et al., 2012). In contrast, our study demonstrates an adverse role of PIAS2 in immune cells, mediated through PPARγ SUMOylation and IL-10 suppression. Although we focused on lung MDSCs, the underlying mechanism may be applicable to other immune cells, particularly macrophages, in which PPARγ plays a central role in differentiation and polarization toward an inflammatory or regulatory phenotype.
SUMOylation generally occurs in specific lysine residues situated within the consensus tetra peptide ΨKXE, (Ψ, large hydrophobic residue; x, any amino acid) sequence. However, reports suggest that SUMOylation could be influenced by other post-translational modifications (PTMs), particularly phosphorylation, which occur mostly at a serine residue situated four C-terminal amino acids apart from the acceptor lysine residue. Such a motif (ΨKXEXXSP) is known as a phosphorylation-dependent SUMOylation motif (PDSM) (Flotho and Melchior, 2013; Geiss-Friedlander and Melchior, 2007). PDSM is highly conserved and is seen in several transcription regulators, including heat shock factor 1 (HSF1), GATA1, c-Myb, and ATF6, suggesting its importance in regulating their activity (Hietakangas et al., 2006). Interestingly, the K107 SUMOylation motif of PPARγ resembles such PDSMs (Figure 2A). Through our experiments, we demonstrate that CL mediates a concomitant PPARγ phosphorylation at its S112 residue, which is critical for its K107 SUMOylation and IL-10 suppression (Figure 2). Our ex vivo studies also show this S112 phosphorylation to be a direct influence of CL on lung MDSCs (Figures 2D and 2E). It is possible that CL could also be causing this phosphorylation in other lung cells; however, exploring it is beyond the scope of the present study.
Previous reports show that a variety of stimuli, including epidermal growth factor (EGF), platelet-derived growth factor (PDGF), transforming growth factor β (TGF-β), insulin, prostaglandin F2α (PGF2α), and stressors such as UVcan trigger PPARγ S112 phosphorylation (Adams et al., 1997; Brunmeir and Xu, 2018; Hu et al., 1996; Reginato et al., 1998). Although mostly carried out in cell line-based in vitro systems, these studies associated PPARγ S112 phosphorylation with its impaired transcriptional activity. The physiological significance of this was highlighted by a study demonstrating that PPARγ S112A mutation protects mice from obesity-induced insulin resistance, which is known to be influenced by PPARγ (Rangwala et al., 2003). A meta-analysis of genome-wide association studies (GWAS) confirmed the correlation between S112A allele occurrence and reduced type 2 diabetes mellitus risk (Gouda et al., 2010). In addition, the association between reduced IL-10 levels and type 2 diabetes mellitus/metabolic syndrome was identified earlier (Esposito et al., 2003; van Exel et al., 2002). These studies reinforce the association between PPARγ S112 phosphorylation and its compromised activity. However, the underlying mechanism of this association remains incompletely understood.
Although an earlier study suggested the interaction of PPARγ with PIAS2 following rosiglitazone administration in an overexpressed HEK293T-based system (Ohshima et al., 2004), its physiological role was not examined. Using IP experiments, we provide direct evidence, in both an overexpressed system (Figure 2F) and primary immune cells (Figure 3C), that PPARγ S112 phosphorylation promotes its interaction with PIAS2 and plays a critical role in suppressing the production of the key anti-inflammatory cytokine IL-10.
Several in vitro studies demonstrate the role of multiple kinases, including MAPKs, in triggering PPARγ S112 phosphorylation under diverse conditions. However, some contrasting studies have reported increased PPARγ activity following S112 phosphorylation by the cyclin-dependent kinases cdk7 and cdk9 (Compe et al., 2005; Helenius et al., 2009; Iankova et al., 2006). This suggests the importance of the cellular context and the upstream kinase in determining the consequence of S112 phosphorylation on PPARγ activity. Therefore, we decided to explore the upstream kinase in lung MDSCs, responsible for CL-mediated PPARγ S112 phosphorylation, and identified JNK-MAPK to play a major role (Figure 3A). Furthermore, our data confirm that JNK inhibition blocks PPARγ S112 phosphorylation, inhibits its interaction with PIAS2 to prevent its K107 SUMOylation, and in turn rescues IL-10 production in lung MDSCs (Figures 3 and S3). This provides crucial mechanistic insight linking MAPK-mediated S112 phosphorylation with compromised PPARγ activity.
Earlier, the CL metabolite cyclic phosphatidic acid (cPA) was shown to impair PPARγ activity by inducing its K107 SUMOylation (Chakraborty et al., 2017). However, the direct effect of cPA on PPARγ S112 phosphorylation has not been explored. Since a cPA-dependent or -independent PPARγ phosphorylation would have limited influence over downstream events, it was not examined in this study.
Having dissected the critical role of PPARγ PTMs in suppressing IL-10 production in lung MDSCs, we decided to validate the effect of short-term systemic JNK inhibition on CL-mediated ALI and KP-induced murine pneumonia. For the sterile model, treatment with the JNKi was started on the 2nd day post-LPS + CL, to coincide with MDSC recruitment to the lung (Poe et al., 2013). An IL-10 receptor-blocking antibody was used to verify that the protection offered by JNK inhibition indeed occurs through IL-10 rescue. As the literature suggests that IL-10 secretion and inflammation resolution starts almost simultaneously with injury progression (Tamayo et al., 2011), we administered six doses of the anti-IL-10R antibody starting on the day of LPS + CL administration. KP, one of the major etiological agents in hospital-acquired pneumonias (Agaba et al., 2017; Gaynes et al., 2005), was used to generate the murine pneumonia model. Survivorship bias in this experiment was avoided by identifying the 3rd day post-infection as the time point that demonstrated the maximum difference in mortality (Figure 5B). All of the other parameters of inflammation were assessed at this time point. Interestingly, JNK inhibition significantly minimized lung inflammation in both sterile and KP-mediated ALI and increased the levels of the critical regulatory cytokine IL-10 (Figures 4B-4F and 5B-5H).
The central role of IL-10 in promoting inflammation resolution through its anti-inflammatory, anti-apoptotic, and regenerative properties is well appreciated in the literature (Ogawa et al., 2008; Ouyang and O’Garra, 2019). Apart from several animal studies, in which IL-10 administration attenuated airway inflammatory diseases (Arai et al., 2000; Inoue, 2000; Inoue et al., 2001; Zhao et al., 2014), IL-10 therapy is in clinical trials for various inflammatory diseases such as inflammatory bowel disease (IBD), psoriasis, and rheumatoid arthritis (RA) (Ouyang and O’Garra, 2019). However, for diseases with infectious etiologies such as pneumonia, the administration of exogenous IL-10 is a risky venture, considering its tendency to reduce microbial clearance. In fact, some animal studies have even proposed the use of IL-10-blocking antibodies as a therapeutic strategy for pneumonia (Greenberger et al., 1995; van der Poll et al., 1996; van der Sluijs et al., 2004). Although this benefits microbial clearance, it triggers unchecked inflammatory damage, which possibly accounts for its limited translational benefit. It is possible that with the standardization of the correct dose, correct time of administration, and correlation with microbial load, this approach may be beneficial in pneumonia. However, further complications, posed as the wide heterogeneity in IL-10 secretion seen in populations (Brockmann et al., 2018; Poli et al., 2002), makes this standardization an improbable task. A similar predicament is seen with corticosteroid therapy for reducing inflammation in ALI and other intensive care unit (ICU) disorders, their usage remaining controversial (Britt et al., 2006; Hough, 2014; Long and Koyfman, 2017; Monteverde-Fernández et al., 2019). Although corticosteroid administration showed promising results in animal models of ALI, clinical trials achieved mixed results, with some showing no clinical benefit or even increased mortality, subject to the time of administration (Peter et al., 2008; The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome [ARDS] Clinical Trials Network, 2006). Moreover, the adverse effects of steroids seen in the form of salt retention, alterations in glycemic status, myopathies, and neurological abnormalities have critical consequences in the ICU. Cumulatively, these limitations are reflected in the guidelines formulated by the Scandinavian Society of Anaesthesiology and Intensive Care Medicine (SSAI) as well as the 2018 Faculty of Intensive Care Medicine (FICM)/Intensive Care Society (ICS), which recommend against the use of steroids in ALI/ARDS, citing the lack of evidence (Claesson et al., 2016; Monteverde-Fernández et al., 2019). In this regard, by identifying the mechanism of CL-mediated IL-10 suppression, we provide an alternate therapeutic strategy for reducing lung inflammation in pneumonia. The use of a clinically tested specific JNKi would promote homeostatic stability, instead of blindly tipping inflammatory balance toward excessive or inadequate inflammation. Permitting endogenous inflammation resolution alongside antibiotic therapy can be expected to reduce tissue damage without overtly compromising microbial clearance. This is evident from our study in which JNK inhibition was not detrimental as far as bacterial proliferation was concerned (Figures 5E and 5F), and is possibly responsible for the reduction in mortality (Figure 5B).
ARDS is a frequently encountered condition in ICUs, marked by aggravated lung inflammation and a predisposition to nosocomial pneumonias (Bellani et al., 2016; Markowicz et al., 2000; Matthay et al., 2012; Pham and Rubenfeld, 2017). It is plagued by the lack of specific treatment options. Considering the extensive cell death seen in ARDS (Matthay et al., 2019; Sauler et al., 2019), it is possible that mito-DAMPs such as CL may play a major role in driving the pathogenesis of this disease by suppressing inflammation resolution. It is noteworthy that the elevated CL levels found in pneumonia have been specifically identified as host CL and not bacterial in origin (Chakraborty et al., 2017). Validating the findings of the present study in ARDS could lead to the introduction of JNK inhibition as a therapeutic strategy for this disease state.
The JNK signaling pathway is an important cellular process that plays a crucial role in various physiological processes (Cui et al., 2007; Davies and Tournier, 2012). Although the JNKi we used (Brimapitide, AM-111) is a specific peptide inhibitor, blocking this vital cellular process could have unintended consequences. Interestingly, a Phase I dose escalation study found the intravenous use of this drug to be safe in healthy volunteers (Deloche et al., 2014). Moreover, this drug has been granted fast track status in Phase II and Phase III clinical trials for sensorineural hearing loss (SNHL) and ocular diseases (e.g., NCT: NCT02561091, NCT00802425, NCT02235272, NCT02508337, NCT02809118, NCT01570205). Several preclinical studies have reported benefits of this drug in animal models of chronic colitis, neuropathic pain, and chronic brain disorders (Kersting et al., 2013; Wang et al., 2017; Zhao et al., 2012). However, to the best of our knowledge, no prior reports for use of this drug in acute lung diseases are available, and its safety efficacy in the context of ALI needs to be elucidated. In addition, although our study shows benefits of this drug as an adjuvant to antibiotics in pneumonia, without an increase in bacterial proliferation, any other adverse effects need to be explored for better safety profiling. This is important in today’s scenario, with the emergence of microbial antibiotic resistance, which warrants proper antibiotic sensitivity testing before use of this inhibitor.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for protocols, resources and reagents should be directed to the lead contact Dr. Krishnendu Chakraborty (krishnendu.c@igib.res.in, krishnenduind@gmail.com)
Materials availability
The plasmid generated in the study is available on request, directed to the lead contact and subject to institute’s material transfer policy.
Data and code Availability
Original data have been deposited to Mendeley Data:https://doi.org/10.17632/xdgghg2jpt.1
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animals
All experiments were conducted after prior approval from the Institutional Animal Ethics Committee at the Institute of genomics and Integrative Biology, Delhi, India (IGIB/IAEC/26/28/2020; IGIB/IAEC/5/28/2020; IGIB/IAEC/JUL2019/16). 8-10 weeks old male C57BL/6 mice were used for all experiments. Mice were housed and bred under pathogen-free conditions in individually ventilated cages with temperature and humidity control (20-23°C; 40%–50% humidity). Each cage housed a maximum of 5 mice. A standard 12-hour photoperiod was maintained in the animal house. The mice were given access to chow diet and water ad libitum. For in vivo experiments, littermates were randomly assigned into different experimental groups and at least four mice were used in each group unless otherwise stated. Specific sample sizes are mentioned in the legends. During experiments, moribund animals with a weight loss of more than 30% of their weight were euthanized. Wherever feasible, the experimenters were blinded to the mice groups, especially while administering treatment regimen or assessing outcomes. Intratracheal administration was done using the oropharyngeal intratracheal administration technique as described by Allen (2014). For intravenous administrations, either of the lateral tail veins were used. Mice with failed intravenous or intratracheal administrations were excluded from the study. All experiments were typically performed at least twice as independent experiments, unless specified.
Primary cells
Primary lung MDSCs were isolated from 8-10week old male C57BL/6 mice using MACS enrichment and Flow Cytometry Sorting. Purity of the sorted cells was found to be 90%–95% by flow cytometry. The total number of cells were counted, seeded in appropriate density and maintained in DMEM supplemented with 10% FBS and Antibiotic-antimycotic cocktail (Sigma-Aldrich, Cat#A5955). The cells were cultured at 37°C in cell culture incubators (Eppendorf) with 5%CO2.
In vitro experiments
In vitro experiments were performed with RAW 264.7 cell line obtained from ATCC (ATCC TIB-71). Cells were discarded after 15 passages. The cells were maintained in DMEM supplemented with 10% FBS and Antimycotic-Antibacterial cocktail. Cells were maintained in the cell culture incubators at 37°C with 5% CO2. Cells were sub cultured by scrapping; a sub cultivation ratio of 1:4 was maintained and media was changed every 2 days.
METHOD DETAILS
Animal experiments
A murine Acute Lung Injury model was created as described earlier (Chakraborty et al., 2017). Briefly, C57BL/6 mice of 8 to 10 weeks age were divided in two groups. While one group of mice was administered LPS (100 μL dose of 1mg/kg, intra-tracheally), the other received CL (100 μL dose of 5 mg/kg, i.t.) in combination with LPS. Inflammation was assessed by tracking weight change, quantifying lung injury on H&E sections, counting cell numbers and measuring Albumin leak in BAL as well as assay of pro- and anti-inflammatory cytokines in the lung lysate.
For validating the effect of PIAS2 gene knockdown on lung injury severity, pilot experiments were conducted using siRNA targeting PIAS2, administered in vivo. A single dose of 40 μg of siRNA combined with the non-viral in vivo transfection reagent JetPEI® (Polyplus transfection) was administered to each mouse through tail-vein injection. A N/P ratio of 8 in a total volume of 400 μL of 5% Dextrose was maintained for each injection, following the manufacturer’s protocol. The maximum efficiency of the siRNA was found to be at 48 hours which was selected for the final experiments as indicated in the figures. Scrambled siRNA was administered as control.
To investigate the efficacy of the JNK-inhibitor (Brimapitide/AM-111, Cat # HY-P0069, MedChem Express) in promoting lung-inflammation resolution, mice were administered (i.t) with LPS + CL. After random distribution, one group of mice was treated with the water-soluble cell-permeable peptide-inhibitor; specifically targeting JNK-MAPK (0.3mg/kg each dose, intravenous) for three consecutive days, starting 48 hours post LPS+CL administration, while the other group received vehicle control. The third group was administered an IL-10 receptor blocking antibody (25mg/kg each dose, intravenous, Cat # BE0050, clone 1B1.3A, BioXcell) along with the JNK inhibitor to assess the role of IL-10 in improving lung inflammation. The other 2 groups were administered the respective isotype control (Cat # BE0088, BioXcell). A total of 6 consecutive doses of the IL-10R blocking antibody were administered starting on the day of LPS+CL administration. On day 6, mice were sacrificed and lung-inflammation was assessed.
For establishing the murine pneumonia model, mice were infected intratracheally with 1000cfu of Klebsiella pneumoniae (ATCC 43816). 24 hours post-infection mice were divided in two groups. While one group of mice was treated with Gentamicin (Cat#15750078, GIBCO, 10mg/kg, intravenous), the other group was treated with the same dose of Gentamicin along with the JNK inhibitor (0.3mg/kg each dose, intravenous). The mice were given 2 consecutive doses of the antibiotic ± inhibitor, 24 hours apart. Mice were either observed until the 5th day for survival analysis or sacrificed on the 3rd day for assessment of inflammation.
Ex vivo experiments
Experiments with enriched lung-MDSCs were performed as described previously (Chakraborty et al., 2017). Briefly, lung-MDSCs were cultured in DMEM with 10% FBS and stimulated with LPS (1 μg/ml). After one hour of LPS treatment, CL (10 μg/ml) was added to culture medium. For cytokine assay by ELISA, culture supernatant was collected after stimulation for 6 hours. For western blot analysis, immunoprecipitation and ChIP experiments cells were stimulated for 3 hours. Cells were either lysed with appropriate lysis-buffer for IP and western blot experiments, or fixed with 1% formaldehyde for ChIP assay. For immunofluorescence studies, the experiments were done on Poly-L-Lysine coated glass coverslips. The cells were then fixed with 4% Formaldehyde before staining.
In vitro experiments
RAW264.7 cells were transfected with FLAG tagged overexpression construct of either wild-type PPARγ or phosphorylation deficient PPARγ(S112A), as described. 24 hours post transfection, cells were stimulated with LPS(1 μg/ml) and later with cardiolipin (CL)(10 μg/ml). For ELISA, cell supernatants were collected after 6hours of stimulation while for IP and ChIP studies, cells were collected in the appropriate buffer after 3 hours.
Cell isolation and sorting
MDSCs were isolated as described earlier (Chakraborty et al., 2017). Briefly, lungs were dissociated in collagenase-DNase suspension (Collagenase from Clostridium histolyticum, Cat # 10103586001, Roche and DNAase I from Bovine Pancreas, Cat # 11284932001, Roche) using a gentle MACS Dissociator (Miltenyi Biotec) according to the manufacturer’s protocol. Single-cell suspensions were obtained by passing the dissociated tissue through a 70 μm cell strainer (BD Biosciences) and washed with PBS containing 2% FBS. CD11b cells were enriched by labeling them with PE and using anti-PE antibody-coated magnetic beads (Cat # 130048801, Miltenyi Biotec). Live cells were gated from these enriched cells using a cell viability dye (SYTOX Green Ready Flow Reagent, Invitrogen, Cat# R37168) and then sorted to 90%–95% purity by flow cytometry (BD FACS Melody) using fluorochromelabeled antibodies to sort CD11b+ F4/80+ Ly6Gint Ly6Clo cells.
Flow cytometry
CD11b positive cells were enriched using labeling with PE and using anti-PE antibody coated magnetic beads. These cells were then stained using antibodies against Ly6G (PE-Cy5 conjugated, clone RB6-8C5, Biolegend, Cat # 108409), Ly6C (PE-Cy7 conjugated, clone HK1.4, Biolegend, Cat#128017) and F4/80(PerCP-Cy5.5 conjugated, Clone BM8, Biolegend, Cat#123127). The cells were then fixed and permeabilised using a fixation permeabilization solution (Cytofix/Cytoperm, BD, Cat#554722) and then stained with antibody against IL-10(FITC conjugated, clone JES5-16E3, Biolegend, Cat#505005). Flow cytometry was then used to assess the IL-10 production in CD11b+ cells. The data was then analyzed using FlowJo.
Site-directed mutagenesis
S112 phosphorylation sites in PPARγ were mutated to alanine using QuikChange II XL Site-Directed Mutagenesis Kit (Cat # 200521) from Agilent Technologies following manufacturer’s instructions. Briefly, PPARγ overexpression construct (purchased from Addgene; Cat # 8895) was used as template in a PCR reaction containing the primers harboring the desired mutations. Following PCR amplification, the methylated template strands were removed by digestion with DpnI and the undigested constructs containing the desired mutations were used for transforming transformation-competent E. coli cells. The plasmids were purified and used for transfecting RAW 264.7 cells (ATCC, Cat # ATCC TIB-71).
Transfection
Lung-MDSCs were transfected ex vivo with siRNA or pLpA-DN.JNK1 plasmid(Addgene, Cat#51942), while RAW 264.7 cells were transfected with FLAG-tagged overexpression construct of either wild-type PPARγ or phosphorylation-deficient PPARγ (S112A), using the Amaxa Mouse Macrophage Nucleofector Kit from Lonza (Cat # VPA-1009), following the manufacturer’s protocol with minor modifications. In brief, 106 cells were resuspended in 100 μL of nucleofection solution and mixed with 2 μg of siRNA or plasmid DNA (overexpression constructs). Cells were then transferred to an electroporation cuvette and Nucleofector 2b (Lonza)-electroporator was used to give appropriate electrical pulse according to a pre-set program. 500 μL of pre-warmed (37°C) culture medium was added immediately, and cells were transferred to a 30mm culture dish containing 2ml of pre-warmed culture medium. 24 hours post transfection gene silencing was verified by qPCR while exogenous FLAG-tagged PPARγ expression was verified by western blot analysis.
RNA isolation, cDNA synthesis and qPCR
Lung tissue was homogenized using a high-speed homogenizer and RNA was isolated using a RNeasy kit (Cat #74106, QIAGEN) following the manufacturer’s instructions. cDNA was synthesized using High-Capacity cDNA Reverse Transcription kit (Cat # 4368814, Applied Biosystems) according to the manufacturer’s instructions. Quantitative PCR was performed in a Roche lightcycler-480 using Kapa SYBRFast qPCR master-mix (Cat # KK4602, Roche) and primers designed by Primer3 online primerdesigning tool. Results were analyzed by measuring the relative mRNA expression using the 2−ΔCt method with hprt as an internal reference control.
Primer Sequences:
PIAS1: FP 5′ atcaggtagcgtcccacaac 3′/RP 5′ cgaggcttgatgaggaagac 3′
PIAS2: FP 5′ gactttgcttggcagagacc 3′/RP 5′ ttggcacagctgaagacaac 3′
PIAS3: FP 5′ ggacgtgtcctgtgtgtgac 3′/RP 5′ ctctgatgcctccttcttgg 3′
PIAS4: FP 5′ gcctggtgtggaacctaaga 3′/RP 5′ atagttgccccaggtgacag 3′
hprt: FP 5′ agtcccagcgtcgtgattag 3′/RP 5′ tgatggcctcccatctcctt 3′
Immunoprecipitation (IP)
Immunoprecipitation assay was performed using Pierce Crosslink Immunoprecipitation Kit (Cat # 26147) following supplier’s instructions. Briefly, desired antibodies (10 μg per IP) were adsorbed onto protein A/G agarose beads and crosslinked. Cells were washed with cold PBS and lysed. Cell lysates were cleared by centrifugation and supernatants were collected. Cell lysate was mixed with agarose slurry and incubated for 1 h on rocker at 4°C for pre-clearing. Agarose slurry beads were removed by centrifugation and the supernatant was transferred to fresh tubes. Protein concentration was determined by BCA protein estimation assay. 600 μg of total protein was incubated with 10 μg of antibody-crosslinked protein-A/G agarose and incubated overnight on a rocker at 4°C. After multiple washes, the antigen was eluted with 40 μL of elution buffer and analyzed on SDS–PAGE (polyacrylamide gel electrophoresis) gel.
ChIP
Chromatin immunoprecipitation experiments were performed using Chromaflash High Sensitivity ChIP kit (Cat # P-2027-48, Epigentek, USA) following the manufacturer’s instructions. Briefly, 106 lung-MDSCs were fixed using 1% Formaldehyde for 10 minutes at room temperature, followed by addition of Glycine for 5 minutes. Cells were washed with cold PBS and lysed with working lysis buffer. For proper lysis, cells were incubated for 10 minutes on ice with intermittent vortexing. Supernatant was removed and chromatin pellet was dissolved in 100 μL of ChIP-buffer. Chromatin was sheared in a bath sonicator (150 W and 50% power output) with cooling facility and 40 cycles of sonication was applied. Each cycle of sonication comprised of 15 s of pulse followed by 15 s of rest. Supernatant was collected after 10 minutes of centrifugation at 10000 rcf. at 4°Cand proper shearing was confirmed by running on an agarose gel. Next, supernatant was transferred to the wells, pre-coated with desired ChIP-grade antibody targeting NCOR or HDAC3 and incubated overnight at 4°C. Wells were washed with wash buffer twice, followed by the addition of 40 μL of DNA-release buffer containing Proteinase K and incubated for 45 minutes at 60°C. Proteinase K was inactivated, by heating samples at 95°C for 10 minutes. 2 μL of Immunoprecipitated genomic DNA and specific primers (FP 5′ tatcggacgttcaacccagg 3′/RP 5′ gccctcatctgtggattcc 3′) were used to amplify the region containing the PPRE at −349 bp of the IL-10 promoter. An IgG control was kept for assessing background enrichment.
Immunofluorescence
Cells were allowed to adhere to glass coverslips before being fixed with 4% Formaldehyde (Cat # 28908, ThermoFisher Scientific) in PBS for 10minutes at Room Temperature. Permeabilization was done in a buffer containing 0.2%Triton X-100, 0.2% BSA and 1mM Sodium Azide in PBS on ice for 10 minutes and cells were then blocked in a blocking buffer comprising of 0.02%Triton-x100, 3%BSA and 1mM Sodium Azide in PBS. Primary antibody incubation was done in the same buffer for 1 hour at room temperature followed by secondary antibody incubation in blocking buffer using fluorochrome tagged anti-rabbit antibodies. Cells were washed between steps using a buffer consisting of 0.02%Triton X-100, 1.5%BSA and 1mM Sodium Azide in PBS. The coverslips were then mounted on frosted glass slides (Cat # 2948, Corning) using DAPI mountant (Prolong Diamond Antifade Mountant with DAPI, Cat # P36966, Invitrogen) and allowed to dry before being sealed using a colorless nail polish paint.
Images were obtained on a Nikon Eclipse Ti2 Confocal Microscope and were analyzed on the Nikon Elements software.
Image intensity quantitation was done using ImageJ. Images were converted to 8-bit and a common threshold was applied all images to convert them into binary masks. Regions of Interest were then marked on the binary masks with a size cut-off of 8 μm to reduce noise. These marked regions were overlaid on the original unprocessed images and the mean Intensity of the images was calculated and compared.
Western blot analysis
Lung tissue was homogenized in Cytokine buffer containing NaCl, Tris and Tween-20 supplemented with Protease Inhibitor Cocktail (Cat # 04693159001, Roche) and Phosphatase Inhibitor Cocktail (Cat # 04906837001, Roche). Cell lysates were prepared by harvesting cells in Cell Lysis Buffer (Cat. # 9803, Cell Signaling Technologies) supplemented with Protease and Phosphatase Inhibitor. The lysates were cleared of debris by centrifugation and total protein estimation was performed on the collected supernatant by Bicinchoninic Acid (BCA) assay. Proteins were resolved by loading 15-20 μg of protein on a 10%SDS-PAGE Gel. The proteins were then transferred to a polyvinylidene difluoride (PVDF) membrane by using a wet transfer technique. Blots were probed with antibodies against the protein of interest overnight at 4°C, followed by HRP-conjugated Secondary Antibody (Cat # 111035003, Jackson ImmunoResearch Laboratories) for 1hr at RT. The blots were developed using Super Signal West Femto-chemiluminescence substrate (Cat # 34095, ThermoFisher Scientific) or Bio-Rad West Clarity Max substrate (Cat # 1705062) Quantification was done using ImageJ.
Histology
Lungs were perfused by injection of cold PBS through the pulmonary vasculature and the excised lungs were stored in 10%Formalin in PBS for 48 hr for fixation. The lungs were then paraffin embedded, mounted on glass slides and stained with Haemotoxylin and Eosin using the protocol mentioned by Zhou and Moore (2017). Briefly, 5 μm sections were cut from the paraffin blocks, mounted on glass slides and deparaffinised using Xylene. They were then hydrated using decreasing concentrations of Alcohol, washed with distilled water before staining with Haemotoxylin solution and counterstained with Eosin. The sections were then dehydrated with increasing alcohol concentrations, followed by clearing with Xylene and mounting on glass slides with DPX.
The lung injury scores were then computed by using the American Thoracic Society guidelines as described by Matute-Bello et al. (2011). A minimum of 20 random high-power fields (40X objective) were scored by an independent blinded pathologist. The inflammation score was based on 5 different parameters– neutrophils in the alveolar space, neutrophils in the interstitial space, presence of hyaline membranes, presence of proteinaceous membranes in the airspaces, and alveolar septal thickening. A score of 0-2 was given to each parameter for each field. A final score was computed using the individual score according to the equation given below.
Analysis of secreted cytokines
Lungs were homogenized in a buffer containing 50mM Tris-HCl, pH 7.4, 150mM NaCl, 0.02% Tween 20, and EDTA-free protease inhibitor cocktail (Roche). Debris-free supernatant was collected for cytokine measurement. IL-10 and TNFα levels in mouse lung extracts were measured, using DuoSet Elisa kit (R&D Systems, Cat # DY417 and Cat# DY410 respectively) according to the manufacturer’s protocol
Measurement of BAL-albumin
Amount of albumin leak in BAL was measured using mouse albumin-specific ELISA kit (Abcam, Cat # ab108792) following the manufacturer’s instructions.
Bacterial counts
Bacterial counts in the lung and blood were calculated by a single plate-serial dilution method. A small portion of the left lung were taken and weighed in pre-weighed microcentrifuge tubes. The tissue was homogenized in 1ml of sterile PBS and diluted serially in sterile PBS. 20 μL of each dilution was plated on TSA (Tryptone Soya Agar) plates and allowed to dry completely before leaving them overnight in a 37°C incubator. Bacterial colonies were counted and the CFU count was calculated, normalized against the lung weights. Blood was collected in microcentrifuge tubes precoated with EDTA by cardiac aspiration. The blood was serially diluted in sterile PBS and 20 μL of each dilution was plated on TSA plates and allowed to dry. The plates were left overnight in a 37°C incubator and the colonies were counted to calculate the CFU count/ml of blood.
QUANTIFICATION AND STATISTICAL ANALYSIS
Results shown are mean values ± s.d.(Standard Deviation). Sample sizes for each experiment is displayed on the figures as well as specified in the legends. Each point on the graph represents a biological replicate. All figures displayed are a representative of 2 independent experiments except the survival curve which displays pooled data. Unpaired t test was used for comparisons involving two groups, one-way or two-way ANOVA was used for multiple pairwise comparisons. Kaplan–Meier survival curves are derived from four groups for survival experiments. The P value was calculated using log-rank test. Differences between groups were considered significant when p ≤ 0.05. Statistically significant differences are represented as follows: * P value < 0.05; **P value < 0.01; ***P value < 0.001;****P value < 0.0001. All statistical analyses were performed using GraphPad Prism 8 software. Specific statistical details are as mentioned in the figures and their legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti- mouse IL-10R Neutralizing Antibody Isotype: Rat IgG1, κ |
Bioxcell | Cat# BE-0050; RRID: AB_1107611 Isotype Cat# BE-0088; RRID: AB_1107775 |
| Anti-PE microbeads | Miltenyi Biotec | Cat# 130-048-801; RRID: AB_244373 |
| Rabbit polyclonal anti-HDAC3 | Abcam | Cat# ab7030; RRID: AB_305708 |
| Rabbit Polyclonal anti- NCOR | Abcam | Cat# ab3482; RRID: AB_10860296 |
| Rabbit Polyclonal anti-DDDDK tag(FLAG sequence) | Abcam | Cat# ab1162; RRID: AB_298215 |
| Rabbit Polyclonal anti-PPARγ | Abcam/ SantaCruz Biotechnology | Cat# ab19481/sc7273; RRID: AB_444944/AB_628115 |
| Rabbit Polyclonal anti-phospo-PPARγ (Ser112) | Thermo | Cat# PA5-36763; RRID: AB_2553712 |
| Rabbit Polyclonal anti-SUMO-1 | Cell Signaling Technology | Cat# 4930; RRID: AB_10698887 |
| PE Rat Monoclonal anti-CD11b (clone:M1/70), Isotype: PE Rat IgG2b,κ | Biolegend | Cat# 101207; RRID: AB_312790 isotype Cat# 400607; RRID: AB_326551 |
| PerCP/Cy5.5 Rat Monoclonal anti-F4/80 (clone:BM8) Isotype: PerCP/Cy5.5 Rat IgG2a, κ | Biolegend | Cat# 123127; RRID: AB_893496 Isotype Cat# 400531; RRID: AB_2864286 |
| PE/Cyanine5 Rat Monoclonal anti-Ly6G(Gr-1) (Clone: RB6-8C5); Isotype: PE/Cyanine5 Rat IgG2b κ | Biolegend | Cat# 108409; RRID: AB_313374 Isotype Cat# 400609; RRID: AB_326553 |
| PE/Cyanine7 Rat Monoclonal anti-Ly6C(clone: HK1.4) Isotype: PE/Cyanine7 Rat IgG2c κ | Biolegend | Cat# 128017; RRID: AB_1732093 Isotype Cat# 400721 |
| FITC Rat monoclonal anti- IL-10(Clone: JES5-16E3) | Biolegend | Cat# 505005; RRID: AB_315359 |
| Isotype: Rat IgG2b, κ | Isotype Cat# 400633; RRID: AB_893678 | |
| Alexa Fluor 488 Donkey anti-Rabbit IgG (H+L) | Molecular Probes | Cat# A-21206; RRID: AB_2535792 |
| Peroxidase Goat Anti-Rabbit IgG(H+L) | Jackson ImmunoResearch Labs | Cat# 111-035-003; RRID: AB_2313567 |
| Bacterial and virus strains | ||
| Klebisella pneumoniae pneumoniae | ATCC | ATCC 43816 |
| Chemicals, peptides, and recombinant proteins | ||
| Non-Viral in vivo transfection reagent | Polyplus Transfection | in vivo – jetPEI® |
| D-JNK-1 Inhibitor (AM-111/XG-102/Brimapitide, 0.3mg/kg or final concentration 2 μM) | Medchem Express | Cat#HY-P0069 |
| Lipopolysaccharide from Escherichia coli O26:B6 | Sigma Aldrich | Cat# 2654 |
| Collagenase from Clostridium histolyticum | Roche | Cat#10103586001 |
| DNAaseI from Bovine Pancreas | Roche | Cat#11284932001 |
| KAPA SYBR FAST qPCR mastermix | Roche | Cat#KK4602 |
| Prolong™ anti-fade mountant with DAPI | Invitrogen | Cat# P36966 |
| cOmplete ™ EDTA-free Protease Inhibitor Cocktail | Roche | Cat#04693159001 |
| phosSTOP ™ Phosphatase Inhibitor | Roche | Cat#04906837001 |
| Cell Lysis Buffer (10X) | Cell Signaling Technology | Cat#9803 |
| Cardiolipin Sodium from Bovine Heart | Avanti Polar Lipids | Cat#840012C |
| MEK/Erk MAPK Inhibitor (U0126, Final Conc. 20 μM) | Cell Signaling Technology | Cat#9903S |
| P38 MAPK inhibitor (SB203580, Final Conc. 10 μM) | Sigma Aldrich | Cat#S8307 |
| CDK9 Inhibitor (SNS-032, Final Conc. 10nM) | MedChem Express | Cat#HY-10008 |
| Fixation/Permeabilization solution (Cytofix/ Cytoperm) | Becton, Dickinson and company | Cat#554722 |
| Viability Dye (SYTOX Green Ready Flow Reagent) | Invitrogen | Cat#R37168 |
| Gentamicin | GIBCO | Cat#15750078 |
| Critical commercial assays | ||
| Mouse IL-10 DuoSet ELISA kit | RnD systems | Cat#DY417 |
| Mouse TNFα DuoSet ELISA kit | RnD systems | Cat#DY410 |
| Mouse Albumin ELISA kit | Abcam | Cat#ab108792 |
| Mouse macrophage nucleofector kit | Lonza | Cat# VPA-1009 |
| Chromaflash high sensitivity ChIP kit | Epigentek | Cat# P-2027-48 |
| Pierce Crosslink Immunoprecipitation kit | Thermo Scientific | Cat# 26147 |
| RNeasy kit for RNA isolation | QIAGEN | Cat# 74106 |
| HighCapacity cDNA reverse transcription kit | Applied Biosystems | Cat# 4368814 |
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent | Cat#200521 |
| Deposited data | ||
| Mendeley Data | This paper | https://doi.org/10.17632/xdgghg2jpt.1 |
| Experimental models: cell lines | ||
| Murine macrophage cell line: RAW264.7 | ATCC | ATCC TIB-71; RRID:CVCL_0493 |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Oligonucleotides | ||
| esiRNA targeting mouse PIAS1 | Sigma-Aldrich | Cat# EMU076871 |
| esiRNA targeting mouse PIAS2 | Sigma-Aldrich | Cat# EMU090071 |
| esiRNA targeting mouse PIAS3 | Sigma-Aldrich | Cat# EMU026711 |
| esiRNA targeting mouse PIAS4 | Sigma-Aldrich | Cat# EMU062861 |
| Scrambled Control siRNA | SantaCruz Biotech | Cat# sc37007 |
| PCR primers for mouse PIAS1: FP atcaggtagcgtcccacaac/RP cgaggcttgatgaggaagac |
This paper (Synthesized on order by IDT) | N/A |
| PCR primers for mouse PIAS2: FP gactttgcttggcagagacc/RP ttggcacagctgaagacaac |
This paper (Synthesized on order by IDT) | N/A |
| PCR primers for mouse PIAS3: FP ggacgtgtcctgtgtgtgac /RP ctctgatgcctccttcttgg |
This paper (Synthesized on order by IDT) | N/A |
| PCR primers for mouse PIAS4: FP gcctggtgtggaacctaaga /RP atagttgccccaggtgacag |
This paper (Synthesized on order by IDT) | N/A |
| PCR primers for mouse hprt: FP agtcccagcgtcgtgattag /RP tgatggcctcccatctcctt |
This paper (Synthesized on order by IDT) | N/A |
| PCR primers for mouse IL-10 promoter: FP tatcggacgttcaacccagg/ RP ggccctcatctgtggattcc |
(Chakraborty et al., 2017) (Synthesized on order by IDT) | PMID: 28074841 |
| Recombinant DNA | ||
| pCDNA flag PPAR gamma plasmid | (Hauser et al., 2000) | Addgene plasmid#8895; RRID#Addgene_8895 |
| PPARγ S112A mutant | This paper | N/A |
| pLpA-DN.JNK1 | (Liang et al., 2003) | Addgene plasmid#51942 RRID:Addgene_51942 |
| Software and algorithms | ||
| ImageJ | (Schneider et al., 2012) | RRID:SCR_003070 |
| FlowJo ver 10.7.1 | Becton Dickinson & Co. | RRID:SCR_008520 |
| Prism 8 | GraphPad Software | RRID:SCR_002798 |
| NIS Elements | Nikon | RRID:SCR_014329 |
Highlights.
PPARγ S112 phosphorylation is crucial for its K107 SUMOylation and IL-10 suppression
PIAS2 is the specific E3 SUMOligase causing CL-mediated PPARγ K107 SUMOylation
JNK-MAPK is responsible for CL-mediated PPARγ S112 phosphorylation
Repurposing a clinically used JNK inhibitor can improve survival in pneumonia
ACKNOWLEDGMENTS
We thank Dr. Timothy Oriss, research associate professor, University of Pittsburgh, for his expert opinion regarding flow cytometry. This work was supported by the Department of Biotechnology, Government of India-Ramalingaswami fellowship grant (BT/RLF/Re-entry/26/2016), Department of Biotechnology(DBT), Government of India-extramural grant (BT/PR32560/MED/30/2212/2020), and a Science and Engineering Board, Ministry of Science and Technology, Government of India(SERB)- Core Research Grant (CRG/2019/000691), to K.C., and a National Institutes of Heath, United States Department of Health and Human Resource (NIH)-PO1 (HL114453), to P.R. M.G. acknowledges fellowship and contingency support from the Indian Council of Medical Research (ICMR) as a Nurturing Clinical Scientist fellow (HRD/Head-NCS-10-2018). S.J. received a junior research fellowship from the Council of Scientific and Industrial Research (CSIR), Government of India. S.S. received a senior research fellowship from ICMR.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.Org/10.1016/j.celrep.2021.108736.
REFERENCES
- Adams M, Reginato MJ, Shao D, Lazar MA, and Chatterjee VK (1997). Transcriptional activation by peroxisome proliferator-activated receptor γ is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J. Biol. Chem 272, 5128–5132. [DOI] [PubMed] [Google Scholar]
- Agaba P, Tumukunde J, Tindimwebwa JVB, and Kwizera A (2017). Nosocomial bacterial infections and their antimicrobial susceptibility patterns among patients in Ugandan intensive care units: a cross sectional study. BMC Res. Notes 10, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen IC (2014). The Utilization of Oropharyngeal Intratracheal PAMP Administration and Bronchoalveolar Lavage to Evaluate the Host Immune Response in Mice. J. Vis. Exp (86), 51391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai T, Abe K, Matsuoka H, Yoshida M, Mori M, Goya S, Kida H, Nishino K, Osaki T, Tachibana I, et al. (2000). Introduction of the interleukin-10 gene into mice inhibited bleomycin-induced lung injury in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol 278, L914–L922. [DOI] [PubMed] [Google Scholar]
- Arora M, Poe SL, Oriss TB, Krishnamoorthy N, Yarlagadda M, Wenzel SE, Billiar TR, Ray A, and Ray P (2010). TLR4/MyD88-induced CD11b+Gr-1 int F4/80+ non-migratory myeloid cells suppress Th2 effector function in the lung. Mucosal Immunol. 3, 578–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, Gattinoni L, van Haren F, Larsson A, McAuley DF, et al. ; LUNG SAFE Investigators; ESICM Trials Group (2016). Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 315, 788–800. [DOI] [PubMed] [Google Scholar]
- Bhattacharya J, and Matthay MA (2013). Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu. Rev. Physiol 75, 593–615. [DOI] [PubMed] [Google Scholar]
- Britt RC, Devine A, Swallen KC, Weireter LJ, Collins JN, Cole FJ, and Britt LD (2006). Corticosteroid use in the intensive care unit: at what cost? Arch. Surg 141, 145–149. [DOI] [PubMed] [Google Scholar]
- Brockmann L, Soukou S, Steglich B, Czarnewski P, Zhao L, Wende S, Bedke T, Ergen C, Manthey C, Agalioti T, et al. (2018). Molecular and functional heterogeneity of IL-10-producing CD4+ T cells. Nat. Commun 9, 5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunmeir R, and Xu F (2018). Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci 19, 1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty K, Raundhal M, Chen BB, Morse C, Tyurina YY, Khare A, Oriss TB, Huff R, Lee JS, StCroix CM, et al. (2017). The mito-DAMP cardiolipin blocks IL-10 production causing persistent inflammation during bacterial pneumonia. Nat. Commun 8, 13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson J, Freundlich M, Gunnarsson I, Laake JH, Møller MH, Vandvik PO, Varpula T, and Aasmundstad TA (2016). Scandinavian clinical practice guideline on fluid and drug therapy in adults with acute respiratory distress syndrome. Acta Anaesthesiol. Scand 60, 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloonan SM, and Choi AMK (2016). Mitochondria in lung disease. J. Clin. Invest 126, 809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compe E, Drané P, Laurent C, Diderich K, Braun C, Hoeijmakers JHJ, and Egly J-M (2005). Dysregulation of the peroxisome proliferator-activated receptor target genes by XPD mutations. Mol. Cell. Biol 25, 6065–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Zhang M, Zhang YQ, and Xu ZH (2007). JNK pathway: diseases and therapeutic potential. Acta Pharmacol. Sin 28, 601–608. [DOI] [PubMed] [Google Scholar]
- Davies C, and Tournier C (2012). Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem. Soc. Trans 40, 85–89. [DOI] [PubMed] [Google Scholar]
- Deloche C, Lopez-Lazaro L, Mouz S, Perino J, Abadie C, and Combette J-M (2014). XG-102 administered to healthy male volunteers as a single intravenous infusion: a randomized, double-blind, placebo-controlled, dose-escalating study. Pharmacol. Res. Perspect 2, e00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito K, Pontillo A, Giugliano F, Giugliano G, Marfella R, Nicoletti G, and Giugliano D (2003).Association of low inter leukin-10 levels with the metabolic syndrome in obese women. J. Clin. Endocrinol. Metab 88, 1055–1058. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, and Kagan JC (2020). Toll-like Receptors and the Control of Immunity. Cell 180, 1044–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotho A, and Melchior F (2013). Sumoylation: a regulatory protein modification in health and disease. Annu. Rev. Biochem 82, 357–385. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, and Nagaraj S (2009). Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol 9, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaynes R, and Edwards JR; National Nosocomial Infections Surveillance System (2005). Overview of nosocomial infections caused by gram-negative bacilli. Clin. Infect. Dis 41, 848–854. [DOI] [PubMed] [Google Scholar]
- Geiss-Friedlander R, and Melchior F (2007). Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol 8, 947–956. [DOI] [PubMed] [Google Scholar]
- Gouda HN, Sagoo GS, Harding A-H, Yates J, Sandhu MS, and Hig-gins JPT (2010). The association between the peroxisome proliferator-activated receptor-γ2 (PPARG2) Pro12Ala gene variant and type 2 diabetes mellitus: a HuGE review and meta-analysis. Am. J. Epidemiol 171, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberger MJ, Strieter RM, Kunkel SL, Danforth JM, Goodman RE, and Standiford TJ (1995). Neutralization of IL-10 increases survival in a murine model of Klebsiella pneumonia. J. Immunol 155, 722–729. [PubMed] [Google Scholar]
- Hauser S, Adelmant G, Sarraf P, Wright HM, Mueller E, and Spiegelman BM (2000). Degradation of the peroxisome proliferator-activated receptor γ is linked to ligand-dependent activation. J. Biol. Chem 275, 18527–18533. [DOI] [PubMed] [Google Scholar]
- Helenius K, Yang Y, Alasaari J, and Mäkelä TP (2009). Mat1 inhibits peroxisome proliferator-activated receptor γ-mediated adipocyte differentiation. Mol. Cell. Biol 29, 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietakangas V, Anckar J, Blomster HA, Fujimoto M, Palvimo JJ, Nakai A, and Sistonen L (2006). PDSM, a motif for phosphorylation-dependent SUMO modification. Proc. Natl. Acad. Sci. USA 103, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough CL (2014). Steroids for acute respiratory distress syndrome? Clin. Chest Med 35, 781–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E, Kim JB, Sarraf P, and Spiegelman BM (1996). Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 274,2100–2103. [DOI] [PubMed] [Google Scholar]
- Iankova I, Petersen RK, Annicotte J-S, Chavey C, Hansen JB, Kratchmarova I, Sarruf D, Benkirane M, Kristiansen K, and Fajas L (2006). Peroxisome proliferator-activated receptor γ recruits the positive transcription elongation factor b complex to activate transcription and promote adipogenesis. Mol. Endocrinol 20, 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue G (2000). Effect of interleukin-10 (IL-10) on experimental LPS-induced acute lung injury. J. Infect. Chemother 6, 51–60. [DOI] [PubMed] [Google Scholar]
- Inoue S, Suzuki M, Nagashima Y, Suzuki S, Hashiba T, Tsuburai T, Ikehara K, Matsuse T, and Ishigatsubo Y (2001). Transfer of heme oxygenase 1 cDNA by a replication-deficient adenovirus enhances interleukin 10 production from alveolar macrophages that attenuates lipopolysaccharide-induced acute lung injury in mice. Hum. Gene Ther 12, 967–979. [DOI] [PubMed] [Google Scholar]
- Johnson ER, and Matthay MA (2010). Acute lung injury: epidemiology, pathogenesis, and treatment. J. Aerosol Med. Pulm. Drug Deliv 23, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, et al. (2005). Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol 1, 223–232. [DOI] [PubMed] [Google Scholar]
- Kersting S, Behrendt V, Kersting J, Reinecke K, Hilgert C, Stricker I, Herdegen T, Janot MS, Uhl W, and Chromik AM (2013). The impact of JNK inhibitor D-JNKI-1 in a murine model of chronic colitis induced by dextran sulfate sodium. J. Inflamm. Res 6, 71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, and Vandenabeele P (2011). Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 32, 157–164. [DOI] [PubMed] [Google Scholar]
- Levy BD, and Serhan CN (2014). Resolution of acute inflammation in the lung. Annu. Rev. Physiol 76, 467–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Bueno OF, Wilkins BJ, Kuan CY, Xia Y, and Molkentin JD (2003). c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. EMBO J. 22, 5079–5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, and Shuai K (2008). Targeting the PIAS1 SUMO ligase pathway to control inflammation. Trends Pharmacol. Sci 29, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long B, and Koyfman A (2017). Controversies in Corticosteroid Use for Sepsis. J. Emerg. Med 53, 653–661. [DOI] [PubMed] [Google Scholar]
- Markowicz P, Wolff M, Djedaïni K, Cohen Y, Chastre J, Delclaux C, Merrer J, Herman B, Veber B, Fontaine A, and Dreyfuss D; ARDS Study Group (2000). Multicenter prospective study of ventilator-associated pneumonia during acute respiratory distress syndrome. Incidence, prognosis, and risk factors. Am. J. Respir. Crit. Care Med 161, 1942–1948. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Ware LB, and Zimmerman GA (2012). The acute respiratory distress syndrome. J. Clin. Invest 122, 2731–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A, Herridge M, Randolph AG, and Calfee CS (2019). Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 5, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, and Kuebler WM; Acute Lung Injury in Animals Study Group (2011). An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol 44, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister DA, Liu L, Shi T, Chu Y, Reed C, Burrows J, Adeloye D, Rudan I, Black RE, Campbell H, and Nair H (2019). Global, regional, and national estimates of pneumonia morbidity and mortality in children younger than 5 years between 2000 and 2015: a systematic analysis. Lancet Glob. Health 7, e47–e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizgerd JP (2008). Acute lower respiratory tract infection. N. Engl. J. Med 358,716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteverde-Fernández N, Cristiani F, McArthur J, and González-Dambrauskas S (2019). Steroids in pediatric acute respiratory distress syndrome. Ann. Transl. Med 7, 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C, and Ding A (2010). Nonresolving inflammation. Cell 140, 871–882. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Duru EA, and Ameredes BT (2008). Role of IL-10 in the resolution of airway inflammation. Curr. Mol. Med 8, 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima T, Koga H, and Shimotohno K (2004). Transcriptional activity of peroxisome proliferator-activated receptor γ is modulated by SUMO-1 modification. J. Biol. Chem 279, 29551–29557. [DOI] [PubMed] [Google Scholar]
- Ostrand-Rosenberg S, and Sinha P (2009). Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol 182, 4499–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, and O’Garra A (2019). IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 50, 871–891. [DOI] [PubMed] [Google Scholar]
- Paradies G, Paradies V, Ruggiero FM, and Petrosillo G (2019). Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 8, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, and Glass CK (2005). A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 437, 759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehaloza HF, Noguera LP, Ahn D, Vallejos OP, Castellanos RM, Vaz-quez Y, Salazar-Echegarai FJ, González L, Suazo I, Pardo-Roa C, et al. (2019). Interleukin-10 Produced by Myeloid-Derived Suppressor Cells Provides Protection to Carbapenem-Resistant Klebsiella pneumoniae Sequence Type 258 by Enhancing Its Clearance in the Airways. Infect. Immun 87, e00665–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter JV, John P, Graham PL, Moran JL, George IA, and Bersten A (2008). Corticosteroids in the prevention and treatment of acute respiratory distress syndrome (ARDS) in adults: meta-analysis. BMJ 336, 1006–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham T, and Rubenfeld GD (2017). Fifty Years of Research in ARDS. The Epidemiology of Acute Respiratory Distress Syndrome. A 50th Birthday Review. Am. J. Respir. Crit. Care Med 195, 860–870. [DOI] [PubMed] [Google Scholar]
- Poe SL, Arora M, Oriss TB, Yarlagadda M, Isse K, Khare A, Levy DE, Lee JS, Mallampalli RK, Chan YR, et al. (2013). STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal Immunol. 6, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli F, Nocco A, Berra S, Scalamogna M, Taioli E, Longhi E, and Sirchia G (2002). Allele frequencies of polymorphisms of TNFA, IL-6, IL-10 and IFNG in an Italian Caucasian population. Eur. J. Immunogenet 29, 237–240. [DOI] [PubMed] [Google Scholar]
- Rangwala SM, Rhoades B, Shapiro JS, Rich AS, Kim JK, Shulman GI, Kaestner KH, and Lazar MA (2003). Genetic modulation of PPARgamma phosphorylation regulates insulin sensitivity. Dev. Cell 5, 657–663. [DOI] [PubMed] [Google Scholar]
- Ray NB, Durairaj L, Chen BB, McVerry BJ, Ryan AJ, Donahoe M, Waltenbaugh AK, O’Donnell CP, Henderson FC, Etscheidt CA, et al. (2010). Dynamic regulation of cardiolipin bythe lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat. Med 16, 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Chakraborty K, and Ray P (2013). Immunosuppressive MDSCs induced by TLR signaling during infection and role in resolution of inflammation. Front. Cell. Infect. Microbiol 3, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginato MJ, Krakow SL, Bailey ST, and Lazar MA (1998). Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor γ. J. Biol. Chem 273, 1855–1858. [DOI] [PubMed] [Google Scholar]
- Robb CT, Regan KH, Dorward DA, and Rossi AG (2016). Key mechanisms governing resolution of lung inflammation. Semin. Immunopathol 38, 425–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rott R, Szargel R, Shani V, Hamza H, Savyon M, Abd Elghani F, Bandopadhyay R, and Engelender S (2017). SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. USA 114, 13176–13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, and Hudson LD (2005). Incidence and outcomes of acute lung injury. N. Engl. J. Med 353, 1685–1693. [DOI] [PubMed] [Google Scholar]
- Sauler M, Bazan IS, and Lee PJ (2019). Cell Death in the Lung: The Apoptosis-Necroptosis Axis. Annu. Rev. Physiol 81, 375–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuai K (2006). Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 16, 196–202. [DOI] [PubMed] [Google Scholar]
- Tamayo E, Fernández A, Almansa R, Carrasco E, Heredia M, Lajo C, Goncalves L, Gómez-Herreras JI, de Lejarazu RO, and Bermejo-Martin JF (2011). Pro- and anti-inflammatory responses are regulated simultaneously from the first moments of septic shock. Eur. Cytokine Netw 22,82–87. [DOI] [PubMed] [Google Scholar]
- The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network (2006). Efficacy and Safety of Corticosteroids for Persistent Acute Respiratory Distress Syndrome. N. Engl. J. Med 354, 1671–1684. [DOI] [PubMed] [Google Scholar]
- Triantafilou M, and Triantafilou K (2002). Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol. 23, 301–304. [DOI] [PubMed] [Google Scholar]
- van der Poll T, Marchant A, Keogh CV, Goldman M, and Lowry SF (1996). Interleukin-10 impairs host defense in murine pneumococcal pneumonia. J. Infect. Dis 174, 994–1000. [DOI] [PubMed] [Google Scholar]
- van derSluijs KF, van Elden LJR, Nijhuis M, Schuurman R, Pater JM, Florquin S, Goldman M, Jansen HM, Lutter R, and van derPoll T (2004). IL-10isanimportantmediatoroftheenhancedsusceptibilitytopneumococcal pneumonia after influenza infection. J. Immunol 172, 7603–7609. [DOI] [PubMed] [Google Scholar]
- van Exel E, Gussekloo J, de Craen AJM, Frölich M, Bootsma-Van Der Wiel A, and Westendorp RGJ; Leiden 85 Plus Study (2002). Low production capacity of interleukin-10 associates with the metabolic syndrome and type 2 diabetes: the Leiden 85-Plus Study. Diabetes 51, 1088–1092. [DOI] [PubMed] [Google Scholar]
- Wang C, Kong X, Zhu C, Liu C, Sun D, Xu Q, Mao Z, Qin Q, Su H, Wang D, et al. (2017). Wu-tou decoction attenuates neuropathic pain via suppressing spinal astrocytic IL-1R1/TRAF6/JNK signaling. Oncotarget 8, 92864–92879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Guo Z, Wu H, Wang X, Yang L, Shi X, Du J, Tang B, Li W, Yang L, and Zhang Y (2012). SUMOylation represses Nanog expression via modulating transcription factors Oct4 and Sox2. PLoS ONE 7, e39606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Spigolon G, Bonny C, Culman J, Vercelli A, and Herdegen T (2012). The JNK inhibitor D-JNKI-1 blocks apoptotic JNK signaling in brain mitochondria. Mol. Cell. Neurosci 49, 300–310. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Xiong Z, Lechner EJ, Klenotic PA, Hamburg BJ, Hulver M, Khare A, Oriss T, Mangalmurti N, Chan Y, et al. (2014). Thrombospondin-1 triggers macrophage IL-10 production and promotes resolution of experimental lung injury. Mucosal Immunol. 7, 440–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, and Moore BB (2017). Lung Section Staining and Microscopy. Bio Protoc. 7, e2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data have been deposited to Mendeley Data:https://doi.org/10.17632/xdgghg2jpt.1