

Abstract
The full set of T cell receptors (TCRs) in an individual is known as his or her TCR repertoire. Defining TCR repertoires under physiological conditions and in response to a disease or vaccine may lead to a better understanding of adaptive immunity and thus has great biological and clinical value. In the past decade, several high-throughput sequencing-based tools have been developed to assign TCRs to germline genes and to extract complementarity-determining region 3 (CDR3) sequences using different algorithms. Although these tools claim to be able to perform the full range of fundamental TCR repertoire analyses, there is no clear consensus of which tool is best suited to particular projects. Here, we present a systematic analysis of 12 available TCR repertoire analysis tools using simulated data, with an emphasis on fundamental analysis functions. Our results shed light on the detailed functions of TCR repertoire analysis tools and may therefore help researchers in the field to choose the right tools for their particular experimental design.
Keywords: T-cell receptor repertoire, high-throughput sequencing, tools benchmarking, immunology, in silico simulation
Introduction
The set of all T cell receptors in an individual is known as his or her TCR repertoire. The TCR repertoire is characterized by incredible diversity, because each TCR is generated through consecutive biological processes consisting of somatic rearrangement, non-template insertion and deletion, and heterogeneous chain pairing. Theoretically, the number of distinct TCRs in an individual is estimated to be as high as 1013~1015 [1]. This diversity underlies the immune system’s ability to raise specific responses against a vast array of antigens, including pathogens, auto-antigens, toxins, allergens, and tumor neoantigens. Thus, the TCR repertoire plays a critical role in adaptive immunity, and analysis of TCR repertoires stands to improve our understanding of immune responses and may have broad implications for health and well-being. However, studies of the TCR repertoire are complicated by the number of molecules involved, because traditional methods, such as spectratyping, Sanger sequencing, and flow cytometry, can only characterize a limited number of TCRs.
High-throughput sequencing (HTS) technology can capture hundreds to thousands of millions of sequencing reads and thus enables researchers to characterize TCR repertoires with unprecedented depth. Indeed, high-throughput TCR repertoire sequencing (TCR Rep-Seq) and profiling has emerged as an important tool in fundamental research and clinical applications, such as vaccine design and monitoring therapeutic responses. To date, this versatile approach has been applied for studies of cancer [2], inflammation [3], autoimmune disease [4], hematopoietic stem cell transplantation [5], infection [6], and rare diseases [7, 8]. TCR Rep-Seq may also have the potential to trace an individual’s immune history and evaluate his or her ability to resist distinct pathogens [9, 10].
However, while capturing millions of distinct TCRs via HTS technology is straightforward, accurately and effectively extrapolating biological and/or clinical information from these data represents a significant challenge. TCR Rep-Seq analyses can be classified as either low-level or high-level analyses [11]. Low-level analyses investigate raw data processing, error correction, V (D) J assignments, and third complementary determining region (CDR3) extraction. High-level analyses examine repertoire diversity, shared and private clones, and antigen specificity. Several tools have been developed to unravel the complex information contained within TCR repertoires [12–26]. While the availability of these tools is helpful, there is no clear consensus of which one yields better results during analyses.
Afzal et al. reported a systematic comparison of ten TCR Rep-Seq tools [27]. In addition to the general properties such as ease of usage, customizability, Linux installation, and dependency on external tools, their study focused on comparisons of clonotype detection (i.e., the identification of unique V(D) J combinations), CDR3 identification, and error correction accuracy. While a thorough investigation of these high-level analyses is helpful for the community, the authors did not explicitly compare the performance of these tools in low-level or fundamental analyses. V(D) J assignment decodes the fundamental information for somatic recombination, and the CDR3 sequence determines the binding specificity for a particular TCR. The accuracy of these results is essential for high-level studies and for the subsequent qualitative and quantitative analysis of TCR Rep-Seq data. Thus, a comprehensive comparison of tools for these fundamental analyses is worthwhile.
In this study, we compared the fundamental performance of 12 tools for TCR Rep-Seq data analysis, focusing on read assignment rate, gene segment assignment accuracy, clone recall rate, and accuracy. In combination with prior reviews and comparative studies, these results provide a full-spectrum characterization of the available tools for TCR repertoire analysis. This work will be valuable in helping scientists select a method for their particular experimental design.
Results
Generic feature comparisons of the TCR Rep-Seq analysis tools
Table 1 shows the major features of twelve freely available TCR Rep-Seq analysis tools, ordered by year of publication. Though these tools were developed in different programming languages, all but IMGT/HighV-QUEST provide a stand-alone version for the ease of local implementation. Eight of these twelve tools can also process B-cell receptor sequencing (Ig-Seq) data. To deal with the challenge imposed by the huge volume of HTS data, IgBLAST, MiXCR, IMmunogenetic SEQuence analysis (IMSEQ), TRIg, and RTCR have implemented multi-thread modules to improve their efficiency.
Table 1.
Miscellaneous features of the TCR repertoire analysis tools
| Tools | Published year | Citations* | Implementation | Accessibility | Aligner/Algorithm | PCR/Sequencing error correction | Multi-thread supported | In silico simulation as benchmark | BCR-supported | Authors |
|---|---|---|---|---|---|---|---|---|---|---|
| IMGT/HighV-QUEST | 2012 | 515 | Java | WS | GPA (NW) | No | No | No | Yes | Alamyar et al. |
| MiTCR | 2013 | 132 | Java | SA | N-mers | Yes | No | Yes | No | Bolotin et al. |
| Decombinator | 2013 | 80 | Python | SA | Half tag | No* | No | Yes | No | Thomas et al. |
| IgBLAST | 2013 | 316 | C++ | WS/SA | BLAST | No | Yes | No | Yes | Ye et al. |
| Vidjil | 2014 | 41 | C++ | WS/SA | K-word | Yes | No | No | Yes | Giraud et al. |
| MiXCR | 2015 | 248 | Java | SA | Kaligner | Yes | Yes | Yes | Yes | Bolotin et al. |
| TCRklass | 2015 | 27 | C++, Perl | SA | K-string | Yes | No | Yes | No | Yang et al. |
| LymAnalyzer | 2015 | 30 | Java | SA | Fast-tag | Yes | No | Yes | Yes | Yu et al. |
| IMonitor | 2015 | 41 | C++, Perl, R | SA | BLAST | Yes | No | Yes | Yes | Zhang et al. |
| IMSEQ | 2015 | 48 | C++ | SA | SCF matching | Yes | Yes | Yes | Yes | Kuchenbecker et al. |
| RTCR | 2016 | 20 | C, Python | SA | Bowtie2 | Yes | Yes | Yes | No | Gerritsen et al. |
| TRIg | 2016 | 3 | Perl | SA | Nucmer | No | Yes | No | Yes | Hung et al. |
Note: Citation statistics was obtained from Google Scholar before Mar 20, 2019 (https://scholar.google.com/). WS and SA in column Availability represent Web Service and Stand Alone, respectively. The new release of Decombinator has incorporated a barcode-based error-correction algorithm. Attributes assigned here for each tool refers to stand alone version of it providing a stand alone version is available for this tool.
Tools that do not follow standard TCR analysis procedure or that were designed for specific purposes were excluded. For instance, Molecular Identifier Groups-based Error Correction (MIGEC) was excluded from the set because it requires a unique molecular identifier (UMI) in the sequencing reads. To facilitate the selection of tools based on both experimental design and analysis requirements, we have provided a decision flowchart (Figure 1).
Figure 1.
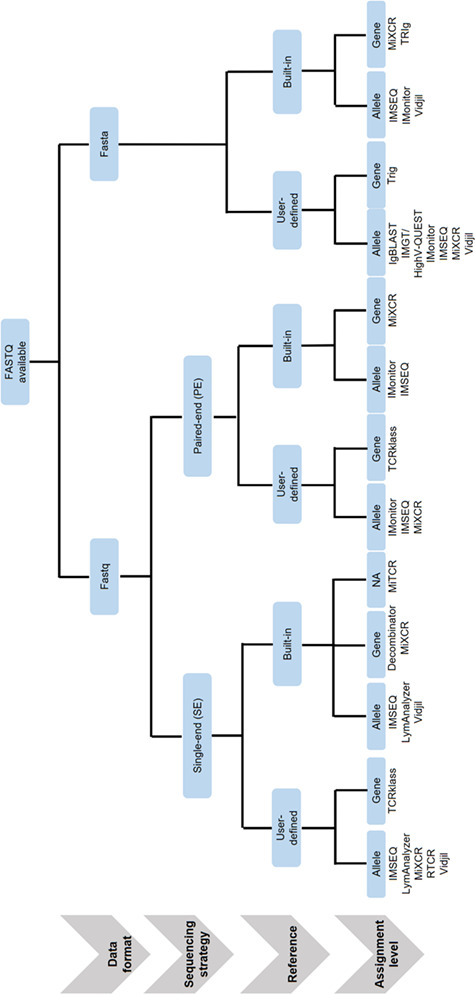
Flowchart showing the guidelines for TCR Rep-Seq tool selection. This flowchart is a step-by-step guideline showing how to select a tool for given input file formats, different sequencing strategies, customization of reference germ line databases, and expected output details.
Identifying V(D) J gene segments, one of the key processes in analyzing TCR Rep-Seq datasets, is carried out by different heuristic algorithms. Most of the tools calculate the frequencies of k-mer strings in the reference gene/allele set and store their positions in an indexed database. During assignment, the k-mer sequences from the query reads are compared to the ones in the database, and a full alignment is performed by sequential extension. For example, MiXCR manipulates alignment by introducing a modified k-mer chaining algorithm that randomly picks seeds from query sequences to align against a pre-calculated index, which in turn stores the positions of all seeds in germline reference sequences for TCR/BCR targeted repertoire sequencing data. RTCR, on the other hand, benefits from a seeded-alignment by taking advantage of the fast alignment of the Bowtie tool. IMGT/HighV-QUEST uses global pairwise alignment to identify gene segments. All tools except MiTCR can give assignment details for query sequences, but only a subset of them can distinguish alleles (see Supplementary Table 1).
To extract CDR3, the critical component of a TCR, all tools utilize the conserved cysteine (Cys or C) at the 104th position and the FGXG motif, albeit with slightly different approaches. TCRklass uses a six reading-frame translation for the query sequence to compare with predefined germline reference k-string profile to locate the positions of conserved residues. LymAnalyzer identifies the nucleotides that encode FGXG and subsequently searches upstream nucleotides that encode the C within the same reading frame. All other tools rely on the assignment of germline gene segments for CDR3 identification. These tools have varying tolerances for out-of-frame errors, internal stop codons, and mutations within the conserved C and FGXG (see Supplementary Table 1).
Both PCR- and HTS-based approaches may introduce erroneous bases and chimeric sequences [28, 29]. Thus, the TCR repertoire analysis tools use different error correction strategies to remove these sequence artifacts. LymAnalyzer and TCRklass remove reads with low quality and/or low frequency using specific thresholds. MiTCR, MiXCR, and IMonitor use a cluster-based error correction method to remove errors through a quality-based correction algorithm. IMSEQ and RTCR use a complex error correction scheme in which each base’s quality is considered.
During their development, most of these tools were evaluated by in silico benchmark tests (Table 1). Therefore, simulated data with known V(D) J recombination and CDR3s represents a powerful and effective method for comparing the TCR analysis tools.
Generation of in silico datasets
TCR repertoire data is characterized by differential germline gene expression, preferential D-J recombination [30], random insertions and deletions (indels) at the V-D and D-J junctions, and artifacts caused by PCR and HTS. There are many tools that can simulate TCR Rep-Seq data. Safonova et al. developed IgSimulator, which is suitable for antibody repertoire simulation but neglects the specific features mentioned above [31]. IGoR is capable of learning the empirical features from a training dataset, but does not account for artifacts introduced in the PCR process [32]. We constructed a simulation pipeline that incorporates known recombination features and artifacts aforementioned (Figure 2).
Figure 2.
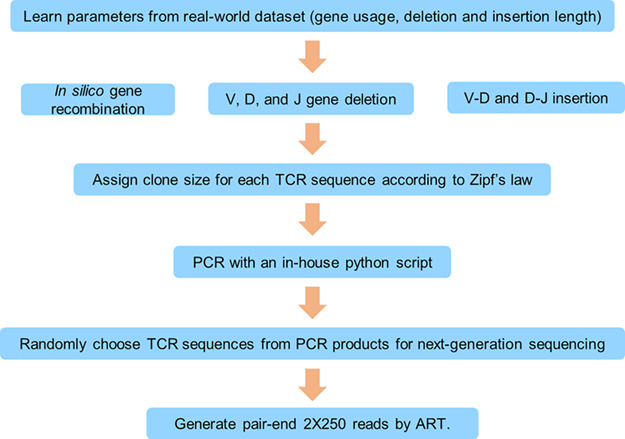
Simulation pipeline for benchmark data. Simulated data were modeled after real-world data from a deep sequencing dataset. This model consists of statistics for gene usage, gene deletion and gene insertion. These parameters were considered to generate original individual clonotypes, sizes for which were assigned based on Zipf’s law. Subsequent PCR process and NGS were implemented using an in-house python script and a published sequencing simulator (ART), respectively. The amplified sequences were then randomly selected for subsequent HTS, which yielded in silico datasets resembling a real-world TCR repertoire dataset.
To mimic the real TCR repertoire data as much as possible, we calculated the properties of TCR repertoires, including the usage of V(D) J gene segments, indels in the V-D and D-J junctions, and the distribution of CDR3 length from real-world datasets [33]. A combination of in-house python scripts and A next-generation sequencing Read simulaTor (ART) was used to incorporate these properties accordingly. To evaluate the performance of the TCR Rep-Seq tools, we generated two different sets of data. Both datasets were generated based on the properties learned from two different real-world data (see Materials and Methods). The first dataset (hereafter referred to Dataset A) contains 200,000 clones with a shallower sequencing depth, more singleton TCRs, and less indels in the junctional region. The other data set (hereafter referred to Dataset B) contains 15,000 clones with a deeper sequencing depth (no singletons) and more indels in the junctions. We compared the simulated data to real-world data to ensure quality (see Supplementary Figure 1). Our subsequent analyses focused on Dataset A, and any necessary comparisons to Dataset B were performed when different results occurred between them.
Comparisons of germline gene segment assignment
As discussed above, germline gene assignment is the foundation for novel germline gene prediction, CDR3 extraction, and subsequent evaluation of repertoire diversity and evenness. Therefore, accurate assignment of reads to gene segments is critical in TCR Rep-Seq data processing.
To ensure a fair comparison,we used the same germline reference sequences for all analyses (see Materials and Methods). For Decombinator, we modified the tag file after replacing the germline reference files. Figure 3a and b show the percent of V and J gene assignment and the corresponding assignment accuracy for all tools with Dataset A. All tools except MiTCR provide read assignment information. Ten of the other eleven tools assigned germline variable (V) or junctional (J) gene segments to nearly all reads for Dataset A (Figure 3a and b). IMSEQ distinguished itself by a slightly lower assignment rate (around 95%). It is worthwhile to mention that IMSEQ computes reverse complements of the V(D)J reads (Read 2) for input files by default (the blue dots in Figure 3a and b). The input reads were therefore standardized to a single direction to process reads with different orientations. The V and J gene assignment percentages of LymAnalyzer, IMSEQ and Decombinator were notably lower in Dataset B (Supplementary Figure 2a and b). A careful investigation of these incorrectly assigned reads revealed that these tools are susceptible to deletions occurring in the V and J gene segments. This effect is caused by the fact that during the assignment step, these tools heavily depend on preselected tags or substrings located at the 3’ end of the germline reference sequences. Long deletions tend to interfere with this initial match with the germline reference. As shown in Figure 3c and d, TCRklass, Decombinator, and TRIg do not report alleles for V and J alleles assignment. While the other tools performed well in J allele assignment, the assignment accuracy for the V alleles varied. Among the tested tools, IMonitor showed 20% incorrect V allele assignments. Most of these misassignments happened between alleles of the same genes. For example, 30.3%, 15.7%, and 13% reads between allele pairs TRBV20-1*01 and TRBV20-1*02, TRBV10-3*01 and TRBV10-3*02, and TRBV7-2*01 and TRBV7-2*04, respectively were missassigned. Two sequence alignments showed that the differences between each pair of sequences are minor: an insertion/deletion at the 3’ terminus and one nucleotide mismatch before the 87th nucleotide. Because the first 87 bases were cut off by IMonitor when building germline references and because indels occur frequently at the V-D junction, these pairs of alleles are indistinguishable with IMonitor (Supplementary Figure 3a, b, and c).
Figure 3.
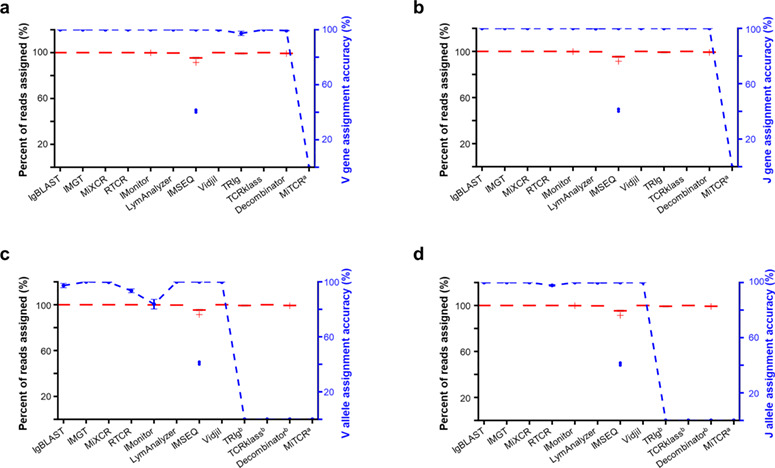
Statistics and comparisons of V and J gene segment assignments. (Left Y axis) The red boxplots show the percentage of reads assigned with V (a) and J (b) gene segments, V (c) and J (d) alleles. (Right Y axis) The blue dashed line indicates the accuracy of germline gene segment assignment. Note: aMiTCR does not report gene fragment assignments; bTCRklass, bDecombinator, bTRIg do not report allele information.
CDR3 extraction
The diversity and richness of a TCR repertoire are also essential data points, and confidently acquiring these measurements relies on the preciseness of CDR3 extraction and clonotype analysis. Though great efforts have been made in the past decade to standardize TCR repertoire data sharing and comparison, the field has not yet achieved a unanimous clonotype definition (Supplementary Table 1) [34, 35]. Nevertheless, the nucleotide CDR3 sequence is generally accepted as the identity of a TCR clone. We therefore used the nucleotide CDR3 sequence in the following analyses.
In most analyses, only productive reads are considered informative. However, the definitions of productivity vary among the tools used. In this study, we chose to retain reads with functional CDR3s – the ones that had no frame-shift mutations or internal stop codons and ran from the conserved 104th C to the FGXG. If a tool reported only amino acid CDR3s, the corresponding nucleotide sequences were extracted accordingly.
CDR3s identified by the tools were classified into three categories: true CDR3s, non-singleton false-positive CDR3s, and singleton false-positive CDR3s (Figure 4a). For Dataset A, eight tools (Decombinator, IMGT/HighV-QUEST, IMSEQ, IMonitor, IgBLAST, LymAnalyzer, TRIg, and Vidjil) successfully recovered nearly all genuine CDR3s, but still reported a considerable number of false positives, dominated by singletons, were also incorrectly identified. In all, 41.3 to 49.4 percent of the CDR3s reported by these eight tools did not exist in the simulated data. On the other hand, MiTCR, MiXCR, RTCR, and TCRklass reported negligible false positives ranging from 0.4 to 7.6 percent. However, MiTCR, MiXCR and TCRklass failed to identify 23 to 41 percent of true CDR3s. As shown in Figure 4b, RTCR surpassed all other tools in both recall and accuracy of CDR3 extraction.
Figure 4.
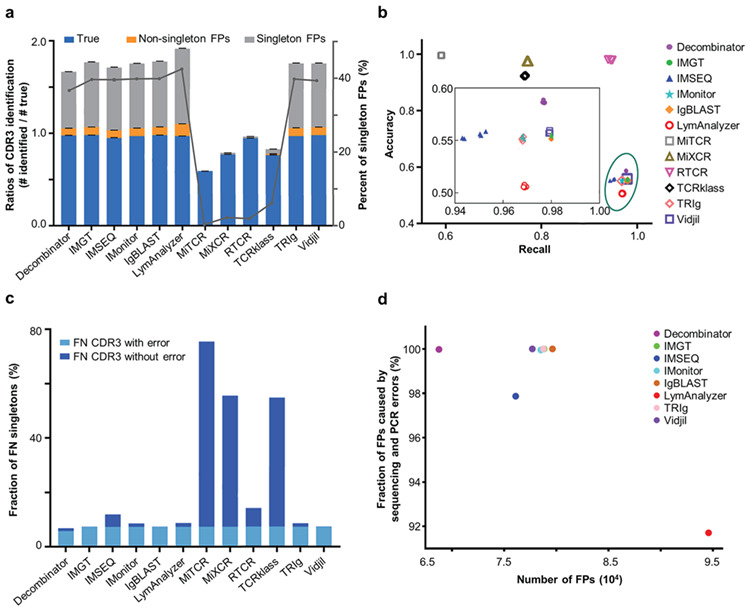
CDR3 identification results. a. The bar graph shows the ratio of the reported number of unique CDR3 nucleotide sequences to the “True” number (Left Y axis). The ratio of CDR3 identification is calculated as the number of reported CDR3s divided by the number of true CDR3s. The sections of blue, light orange, and light grey indicate the proportion of “True” CDR3s, non-singleton false positives, and singleton false positives, respectively. The error bars indicate the standard deviations. The grey line shows the percentage of singleton false positives identified by each tool (Right Y axis). MiTCR, MiXCR, RTCR and TCRklass reported the fewest false-positive CDR3s. b. Recall and accuracy of the resulting repertoires generated by twelve tools for five replicates. Recall (X axis) is defined as the fraction of simulated CDR3s that were correctly identified. Accuracy is defined as the fraction of simulated CDR3s in the total identified ones (Y axis). c. The fraction of singletons among the false-negative CDR3s. The light blue bars at the bottom indicate the fraction of singleton CDR3s with either PCR or sequencing errors, and the darker blue bars stand for those singletons without errors. d. The fraction of false positives that were caused by PCR or HTS errors. The X axis indicates the number of false positives identified by different tools, and the Y axis shows the fraction of error-containing false positives.
We then focused on the causes of the incorrectly identified CDR3s. We first examined the clone size distribution of the false-negative CDR3s. Most of the CDR3s that were not identified were from smaller clones and especially singletons (Supplementary Figure 4a). More than 60% of the false negatives identified by MiXCR and TCRklass are singletons in the simulated data. Singleton CDR3s also constituted 47% of the false negatives identified by MiTCR. Indeed, all the tested tools missed a range of singleton CDR3s in the in silico data (Figure 4c). Singleton CDR3s with sequencing errors were frequently missed by all tools. MiTCR, MiXCR, and TCRklass also failed to identify a considerable number of error-free CDR3s. We then examined the false-positive CDR3s. Most of the false-positive CDR3s shown in Figure 4a were caused by base errors generated during PCR or HTS (Figure 4d).
Since base errors caused by PCR and high-throughput sequencing are inevitable, we went on to characterize the false discovery rates in CDR3 identification step with error-free reads. As shown in Table 2, only LymAnalyzer generated high percent of false positive CDR3s. Even the tools without error correction, such as IgBLAST and TRIg performed very well. MiTCR, MiXCR, and TCRklass failed to identify 24% to 43% of the CDR3s in Dataset A, which mimics the lower depth and more clones. However, their false negative rates were acceptable with Dataset B. This indicates that higher sequencing depth is important for accurately identifying CDR3s for these three tools. Moreover, more indels in the junctional regions in Dataset B caused higher false positives in general (Supplementary Figure 6). To summarize these tools’ performance with error-free reads, LymAnalyzer suffered from significant false positive; MiTCR, MiXCR, and TCRklass may fail to identify singleton CDR3s; and all other tools performed well. Combining with the previous result, one can tell that the base errors introduced during sample preparation and sequencing are the major problems for CDR3 calling. In other words, most of the tools faithfully reported CDR3s with base errors caused by sample amplification and sequencing. However, these errors are intrinsic to the TCR Rep-seq. Since the errors are not avoidable, increasing sequencing depth and choosing the tools with better error correction performance seem to be the solution for the time being.
Table 2.
The false discovery rates for these 12 tools with error-free reads
| Software | Dataset A | Dataset B | ||
|---|---|---|---|---|
| FP (%) | FN (%) | FP (%) | FN (%) | |
| Decombinator | 0.0427 | 0.6391 | 0.3248 | 0.4637 |
| IgBLAST | 0.0000 | 0.0085 | 0.1303 | 0.2029 |
| IMGT/HighV-QUEST | 0.0009 | 0.0063 | 0.1736 | 0.1596 |
| IMonitor | 0.0000 | 1.1637 | 0.1765 | 1.6920 |
| IMSEQ | 0.0000 | 3.0483 | 0.0000 | 0.8741 |
| LymAnalyzer | 7.0614 | 0.9908 | 59.5584 | 1.3454 |
| MiTCR | 0.0000 | 43.2870 | 0.0159 | 9.0488 |
| MiXCR | 0.0000 | 24.0104 | 0.1612 | 1.7092 |
| RTCR | 0.0336 | 2.4030 | 0.1159 | 0.3545 |
| TCRklass | 0.1378 | 24.5734 | 2.9391 | 4.4167 |
| TRIg | 0.0000 | 1.2274 | 0.0295 | 1.7520 |
| Vidjil | 0.0002 | 0.0375 | 0.2457 | 0.2029 |
Note: FP: false positive, FN: False negative. The first column listed all the tools used in this analysis.
In addition, we also identified differences in the performance of the tools tested. While most of the identified false-positive CDR3s share a uniform length distribution, LymAnalyzer also reported a set of CDR3s of longer than 200 nucleotides (Supplementary Figure 4b). The algorithms used to identify CDR3s make it susceptible to substitution error in conserved locus, for LymAnalyzer identifies the nucleotides that encode FGXG and subsequently searches upstream nucleotides which encode the C within the same reading frame. In contrast to LymAnalyzer, IMGT/HighV-Quest, IgBLAST can accurately extract CDR3s (Supplementary Figure 6). The false positives aforementioned were caused by sequencing error. When analyzing Dataset B, which is characterized by a greater sequencing depth and no singletons, MiXCR identified nearly all CDR3s. Taken together, the intrinsic sequencing errors in TCR Rep-seq data are the major roadblock for CDR3 extraction. One can choose tools without error-correction functions such as IMGT/HighV-QUEST, IgBLAST, or TRIg if these errors are removed or mitigated beforehand via specific tools [36–38]. To use raw sequencing reads, MiXCR and RTCR should be the tools of choice.
Clonality and runtime efficiency analysis
TCR clones that specifically recognize antigens expand to become the major clones in an individual’s TCR repertoire. Thus, the clones that represent more T cells and subsequently more reads in the repertoire sequencing dataset are of particular interest in the study of adaptive immunity. We therefore evaluated the performance of these tools in clonality analyses. We first examined the recovery of individual clones as a function of their frequency. For the top 100 ranked clones, all tools performed well, with correlation coefficients greater than 0.9 compared with the true ranked clones (Figure 5a, see Materials and Methods). Eight tools achieved near identical coefficients. For the top 1000 clones and all non-singleton clones, the overall performance of all tools improved, but certain variabilities exist. Overall, the performance of MiXCR and RTCR remains stably the best (Supplementary Figure 7). A careful examination of the clonotype data showed that several tools (excluding IMGT/HighV-QUEST, IgBLAST, MiXCR, vidjil and RTCR) failed to identify certain major clones (Supplementary Figure 7c). Thus, IMGT/HighV-QUEST, IgBLAST, MiXCR, vidjil and RTCR represent the best choices for clonotype analyses.
Figure 5.
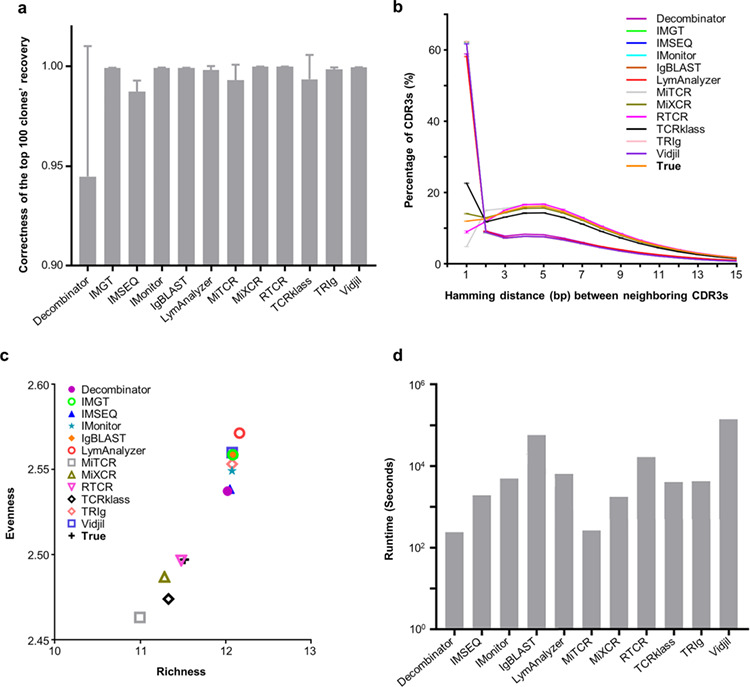
Clonality and runtime efficiency analyses results. a. Rank consistency of the top 100 clones between the true set and each tool’s recovery set. The grey bars indicate the average Spearman rank correlation coefficient based on 5 replicates, and the error bars indicate standard deviations. Higher bars indicate better recovery for the top 100 clones. b. Distribution of Hamming distance between nearest neighbor CDR3s. Each CDR3 was compared to all CDR3s with the same length, and the closest match is defined as its nearest neighbor. The Hamming distance was then calculated accordingly (X axis). The Y axis indicates the percent of CDR3s having specific distance to their nearest neighbors. Distributions closer to the True distribution are better than others. c. The distributions of repertoire richness and evenness. The richness and evenness were calculated according to Renyi entropies (see Materials and Methods). d. Runtime comparisons among tools. The bars indicate how many seconds does a tool needed to finish the calculation with same memory and CPU unit. The faster tools are indicated by the lower bars.
The Hamming distance between the nearest clones and the clonal plane has been proposed to be a good measurement of TCR repertoire analyses [39]. MiXCR and RTCR showed the closest Hamming distance to the actual value (Figure 5b). RTCR was also more faithful in clonal plane analyses of repertoire evenness and richness distributions (Figure 5c).
Finally, we compared the runtime efficiencies for all tools with 2,472,403 raw reads (SRR8733525). IMGT/HighV-QUEST is running online and thus was omitted from this comparison. Of the other tools, Decombinator and MiTCR are the fastest, and Vidjil and IgBLAST are the slowest (Figure 5d; running environment and conditions provided in Supplementary Information). If high performance computing is available to the researchers, the runtime should not be an issue for any of the tested tools.
Conclusions and discussion
The TCR repertoire is an essential constituent of adaptive immunity. Perturbation of an individual’s TCR repertoire leads to vulnerability to infections and diseases, and infections can alter a person’s TCR repertoire. Physiological changes have also been proved to associate with fluctuations of the repertoire [40, 41]. Therefore, the accurate characterization and delineation the TCR repertoire in cross-sectional and longitudinal studies are important for fundamental studies and for clinical applications related to adaptive immunology. The immense diversity of the TCR repertoire has been a major roadblock for the researchers in the field. However, the advent of HTS technology has made it possible to investigate the TCR repertoire as well as its constituent TCR molecules.
Here we report a systematic comparison of 12 TCR repertoire tools, with an emphasis on fundamental analyses. These results may help researchers in the field choose the optimal tool for their analyses. If an experimental design requires annotating germline genes, researchers should avoid MiTCR, which does not report germlines. In addition, IMSEQ requires that the input reads be provided in a single strand and performed less ideally. Moreover, if distinguishing alleles are desired, researchers should avoid TCRklass, Decombinator, and TRIg, which do not report allele assignments.
While these tools can also extract CDR3 sequences, most of the tools are limited by false-positive CDR3s. And the intrinsic base error introduced during PCR and sequencing are the reason for high false discovery rates for many tools. Once these inherent sequencing errors are removed beforehand, IMGT/HighV-QUEST, IgBLAST, and Vidjil would perform well. If raw reads are to be fed to the tools, RTCR is the best choices. Moreover, MiXCR would also suffice if the sequencing depth is deep enough.
Aside from germline reference customization, all comparisons were conducted with the default or recommended parameters, and thus the accuracy of some tools may be sub-optimal. Unique molecular barcodes (UMIs) have been used for better qualitative and quantitative analyses of immune repertoires [42, 43]. However, because majority of the tested tools do not incorporate the related algorithms, the performance of these datasets with UMIs was not evaluated. We believe that future studies will be aided by more advanced tools and comprehensive analysis modules to complement TCR repertoire sequencing.
Materials and Methods
Inclusion criteria for the TCR analysis tools
To process the huge size of high-throughput sequencing data conveniently, most of the tools selected for this study must have a standalone version that can be implemented within a high performance computing (HPC) environment. We further required the tools to incorporate standard TCR analysis procedures, including germline gene segment assignment, CDR3 extraction, etc. IMGT/HighV-QUEST was also included as it was developed first and represents the most cited tool for this work.
In silico data simulation
In order to generate in silico datasets with TCR repertoire-specific features, we downloaded germline database version 3.1.18 from IMGT (http://www.imgt.org) on April 20, 2018. Based on a previous report, the selections of D and J are dependent upon each other [30]. Thus, the usage frequencies of the V and D-J gene segments were calculated from real-world data from the PBMC from male 1 at day 1 (SRR060699-SRR060725) and a dataset generated by our laboratory [33]. For each germline gene segment, we performed a statistical analysis of the 3’ end deletion in V, the 5’ end deletion in J, and the 5’ and 3’ end deletions in D from a real-world dataset generated by our lab as well as the published dataset. The insertion size and frequency between V-D and D-J were also calculated. Accordingly, an initial number of unique TCR beta chain sequences were generated, each of which represents a clone. The clone sizes of these initial sequences were then assigned following Zipf’s law [44]. To incorporate the amplification efficiency and nucleotide substitution (frequency: 5.0e-5 [45]), we generated more than one billion in silico PCR products using an in-house Python script. We then used the sequencing read simulator ART to generate raw sequencing data that incorporated the variations in sequencing errors and base qualities [46].
Germline reference unification across tools
All tools but IgBLAST and TCRklass were installed with a built-in TRB reference set. However, diversified spectrums of germline references have been observed for those tools, making a comparison of performance across assignments difficult. We therefore selected 146 alleles corresponding to sixty-six TRBV genes, 3 alleles corresponding to two TRBD genes and 16 alleles corresponding to fourteen TRBJ genes as the standard germline reference set for all tools (with MiTCR excluded) (Supplementary Table 2). Germline references for IMGT/HighV-QUEST could not be customized as flexibly as were the other standalone tools, so we selected the ‘F+ORF+in-frame’ for IMGT/HighV-QUEST reference directory set, and all genes within the predefined standard germline reference set were included in this directory, enabling no intrinsic error due to germline reference incompleteness. Germline reference normalization for IgBLAST and TCRklass was as easy as following germline reference building guidelines provided within their manual. For the rest of the tools, more effort should be made to manage customization.
Germline references for LymAnalyzer were included in the compressed.jar file. Thus, the germline reference substitution took place after the.jar file was decompressed. Later recompression created a functional.jar file again. A tool named repseqio (Version v1.2.12) was employed for germline reference normalization for MiXCR. Decombinator required a predefined tag list, each of which uniquely identified a gene. An in-house python script was used to extract tags for supplement genes. Importantly, two gene pairs, TRBV6-2/ TRBV6-3 and TRBV24-1/TRBV24/OR9-2, were identical in sequence. For each pair, only one of them was selected for representation. Intergenic assignments within the two pairs were considered accurate gene recoveries. IMonitor provided a shell script named run.sh for germline reference customization. As for IMSEQ, RTCR, TRIg and Vidjil, formatted germline reference sequences were carefully prepared and were used to replace old ones. The germline reference within TRIg’s built-in directory is characterized by a complete long nucleotide sequence extracted from human chromosome 7 that covers all TRBV, TRBD, and TRBJ gene locations. Due to the inclusion of orphan genes (i.e., TRBV), we extracted nucleotide sequences from chromosome 9 (hg19) to include those gene locations (location information based on hg19 was obtained from the NCBI Gene Database), and an additional length of 10 kb both upstream and downstream were extracted together.
Assignment accuracy calculation
The exact V, D, and J gene segments is known for each of the simulated reads. After each tool finished its assignment of these reads, we extracted the assigned germline gene segment for each read. We defined a correct assignment as a match between the simulated gene with the assigned gene. The accuracy was calculated as the percentage of the total correctly assigned reads relative to the total number of assigned reads.
Recall and accuracy
Because the data were simulated, we know precisely the correct CDR3 for every read. We refer to the correct CDR3 for each read and each dataset and refer them as the true CDR3. We then calculated two indices, recall and accuracy to evaluate the performance of tools. We defined recall as the CDR3s correctly identified by each tool divided by the true CDR3s. We defined accuracy as corrected CDR3s divided by the CDR3s found by the software.
Clone rank consistency measurement
Spearman rank correlation coefficients (ρ) were used to measure clone rank consistency between the true clone set and each tool’s recovery set. Specifically, for each true clone, we could obtain two ranks, a true rank known from a simulated model and a recovered rank that was reported by each tool. For this analysis, missing clones are simply not taken into consideration. In this way, for each tool, we could get a series of rank pairs, based on which ρ can be derived. We utilized the cor function in R (v3.2.1) to implement this analysis, with parameter “use” is set as “complete.obs”.
Richness and evenness
The Rényi entropy of order α, where α≥ 0 and α≠ 1, is defined as:
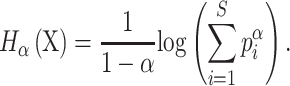 |
The richness is defined when α= 0 as:
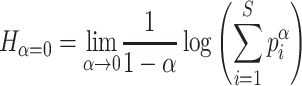 |
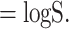 |
The evenness is defined as when α
 as:
as:
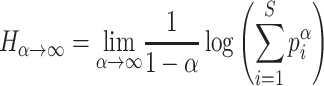 |
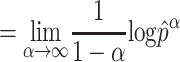 |
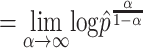 |
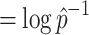 |
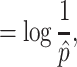 |
Where 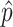 is the maximal frequency among all the clones.
is the maximal frequency among all the clones.
Runtime calculation
To make a fair comparison of the runtime of these tools, we set reasonable parameters and provided each with equal computational resources. All tools that incorporated a multi-thread programing module were run on a single thread. Memory sizes for MiTCR and LymAnalyzer were set as 16 GB. IMGT/HighV-QUEST is a web-based analysis tool and was not included in our runtime analysis. The command lines for all tools are provided in the supplementary files.
Data availability
The two simulated datasets were submitted to the NCBI Sequence Read Archive. The accession numbers for Dataset A are from SRR8733522 to SRR8733526. The accession numbers for Dataset B are from SRR8755318 to SRR8755322.
Key Points:
Compares the performance of TCR Rep-Seq analysis tools carrying out fundamental analysis
Reveals significant differences in gene assignment and error correction performance among these tools
Provides additional guidelines for TCR Rep-Seq analysis tool selection
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) (31771479, 81822036, and 31770931), the Science Fund for Creative Research Groups of the NSFC (81521003), NSFC Projects of International Cooperation and Exchanges of NSFC (61661146004), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01S131), Municipal Planning Projects of Scientific Technology of Guangdong (201804020083), the Science and Technology Program of Guangzhou (201400000004), the National Natural Science Foundation of Guangdong (2015B050501006), the Team Program of Guangdong Natural Science Foundation (2014A030312002), the Thousand Talent Plan of China, and the Guangdong Natural Science Funds for Distinguished Young Scholar (2017A030306030).
Author Contributions
Y.Z., X.Y., Y.Z., Y.Z., Y.Z., M.W., J.X.O., H.Z., J.W. analyzed and interpreted the data. C.L. coordinated the project. Z.Z. designed the research. Y.Z., X.Y., Y.Z., H.Z., W.Y., and Z.Z. wrote and edited the manuscript with input from the co-authors.
Supplementary Material
Zhenhai Zhang is a professor and dean of the Bioinformatics Department at the Southern Medical University. His work focuses on the development and applications of immune repertoire sequencing. Zhang has published in Science, Cell, and other journals.
Yanfang Zhang, Yanxia Zhang, and Yan Zhu are Ph.D. candidates majored in Bioinformatics at Southern Medical University (SMU).
Minhui Wang is an M.D. candidate in SMU.
Yan Zhang and Jiaqi Wu are former master students majored in Bioinformatics at SMU.
Xiujia Yang and Huikun Zeng is a master student majored in Bioinformatics at SMU.
Jin Xia Ou is a medical technician working at the Division of Laboratory Medicine of Zhujiang Hospital affiliated to SMU.
Chunhong Lan is a lab manager and project coordinator in ZZH lab.
Wei Yang is a professor of the Department of Pathology at the Southern Medical University. His work focuses on the immunometabolism and cancer immunology. Yang has published in Nature, Nature Structural & Molecular Biology, and other journals.
Hong-Wei Zhou is a professor at the Division of Laboratory Medicine of Zhujiang Hospital affiliated to Southern Medical University. His research interest includes microbiome and system biology. Zhou is the chief editor of the journal of Medicine in Microecology and has published on Nature Medicine, Microbiome et al.
References
- 1. Nikolich-Žugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immuno 2004;4:123–32. [DOI] [PubMed] [Google Scholar]
- 2. Hosoi A, Takeda K, Nagaoka K, et al. Increased diversity with reduced “diversity evenness” of tumor infiltrating T-cells for the successful cancer immunotherapy. Sci Rep 2018;8:1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dahal-Koirala S, Risnes LF, Christophersen A, et al. TCR sequencing of single cells reactive to DQ2.5-glia-α2 and DQ2.5-glia-ω2 reveals clonal expansion and epitope-specific V-gene usage. Mucosal Immunol 2016;9:587–96. [DOI] [PubMed] [Google Scholar]
- 4. Delemarre EM, van den Broek T, Mijnheer G, et al. Autologous stem cell transplantation aids autoimmune patients by functional renewal and TCR diversification of regulatory T cells. Blood 2016;127:91–101. [DOI] [PubMed] [Google Scholar]
- 5. Yew PY, Alachkar H, Yamaguchi R, et al. Quantitative characterization of T-cell repertoire in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transplant 2015;50:1227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hou D, Chen C, Seely EJ, et al. High-Throughput Sequencing-Based Immune Repertoire Study during Infectious Disease. Front Immunol 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang L, Betjes M, Klepper M, et al. End-Stage Renal Disease Causes Skewing in the TCR Vbeta-Repertoire Primarily within CD8(+) T Cell Subsets. Front Immunol 2017;8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carey AJ, Hope JL, Mueller YM, et al. Public Clonotypes and Convergent Recombination Characterize the Naïve CD8+ T-Cell Receptor Repertoire of Extremely Preterm Neonates. Front Immunol 2017;8:1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dash P, Fiore-Gartland AJ, Hertz T, et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 2017;547:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Glanville J, Huang H, Nau A, et al. Identifying specificity groups in the T cell receptor repertoire. Nature 2017;547:94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heather JM, Ismail M, Oakes T, et al. High-throughput sequencing of the T-cell receptor repertoire: pitfalls and opportunities. Brief Bioinform 2017. doi: 10.1093/bib/bbx138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alamyar E, Duroux P, Lefranc MP, et al. IMGT((R)) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol 2012;882:569–604. [DOI] [PubMed] [Google Scholar]
- 13. Li S, Lefranc MP, Miles JJ, et al. IMGT/HighV QUEST paradigm for T cell receptor IMGT clonotype diversity and next generation repertoire immunoprofiling. Nat Commun 2013;4:2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alamyar E, Duroux P, Lefranc MP, et al. The IMGT® web portal for immunoglobulin (IG) or antibody and T cell receptor (TR) analysis from NGS high throughput and deep sequencing. Immunome Res 2012;882:569–604. [Google Scholar]
- 15. Thomas N, Heather J, Ndifon W, et al. Decombinator: a tool for fast, efficient gene assignment in T-cell receptor sequences using a finite state machine. Bioinformatics 2013;29:542–50. [DOI] [PubMed] [Google Scholar]
- 16. Ye J, Ma N, Madden TL, et al. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 2013;41:W34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bolotin DA, Shugay M, Mamedov IZ, et al. MiTCR: software for T-cell receptor sequencing data analysis. Nat Methods 2013;10:813–4. [DOI] [PubMed] [Google Scholar]
- 18. Zhang W, Du Y, Su Z, et al. IMonitor: A Robust Pipeline for TCR and BCR Repertoire Analysis. Genetics 2015;201:459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kuchenbecker L, Nienen M, Hecht J, et al. IMSEQ—a fast and error aware approach to immunogenetic sequence analysis. Bioinformatics 2015;31:2963–71. [DOI] [PubMed] [Google Scholar]
- 20. Yu Y, Ceredig R, Seoighe C. LymAnalyzer: a tool for comprehensive analysis of next generation sequencing data of T cell receptors and immunoglobulins. Nucleic Acids Res 2016;44:e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bolotin DA, Poslavsky S, Mitrophanov I, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 2015;12:380–1. [DOI] [PubMed] [Google Scholar]
- 22. Yang X, Liu D, Lv N, et al. TCRklass: a new K-string-based algorithm for human and mouse TCR repertoire characterization. J Immunol 2015;194:446–54. [DOI] [PubMed] [Google Scholar]
- 23. Gerritsen B, Pandit A, Andeweg AC, et al. RTCR: a pipeline for complete and accurate recovery of T cell repertoires from high throughput sequencing data. Bioinformatics 2016;32:3098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Giraud M, Salson M, Duez M, et al. Fast multiclonal clusterization of V(D) J recombinations from high-throughput sequencing. BMC Genomics 2014;15:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hung S, Chen Y, Chu C, et al. TRIg: a robust alignment pipeline for non-regular T-cell receptor and immunoglobulin sequences. BMC Bioinformatics 2016;17:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shugay M, Britanova OV, Merzlyak EM, et al. Towards error-free profiling of immune repertoires. Nat Methods 2014;11:653–5. [DOI] [PubMed] [Google Scholar]
- 27. Afzal S, Gil-Farina I, Gabriel R, et al. Systematic comparative study of computational methods for T-cell receptor sequencing data analysis. Brief Bioinform 2019;20:222–34. [DOI] [PubMed] [Google Scholar]
- 28. Mamedov IZ, Britanova OV, Zvyagin IV, et al. Preparing unbiased T-cell receptor and antibody cDNA libraries for the deep next generation sequencing profiling. Front Immunol 2013;4:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bolotin DA, Mamedov IZ, Britanova OV, et al. Next generation sequencing for TCR repertoire profiling: platform-specific features and correction algorithms. Eur J Immunol 2012;42:3073–83. [DOI] [PubMed] [Google Scholar]
- 30. Murugan A, Mora T, Walczak AM, et al. Statistical inference of the generation probability of T-cell receptors from sequence repertoires. Proc Natl Acad Sci USA 2012;109:16161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Safonova Y, Lapidus A, Lill J. IgSimulator: a versatile immunosequencing simulator. Bioinformatics 2015;31:3213–5. [DOI] [PubMed] [Google Scholar]
- 32. Marcou Q, Mora T, Walczak AM. High-throughput immune repertoire analysis with IGoR. Nat Commun 2018;9:561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Warren RL, Freeman JD, Zeng T, et al. Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res 2011;21:790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yassai MB, Naumov YN, Naumova EN, et al. A clonotype nomenclature for T cell receptors. Immunogenetics 2009;61:493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mehr R, Sternberg-Simon M, Michaeli M, et al. Models and methods for analysis of lymphocyte repertoire generation, development, selection and evolution. Immunol Lett 2012;148:11–22. [DOI] [PubMed] [Google Scholar]
- 36. Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 2012;7:e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou Q, Su X, Wang A, et al. QC-Chain: fast and holistic quality control method for next-generation sequencing data. PLoS One 2013;8:e60234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen C, Khaleel SS, Huang H, et al. Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code Biol Med 2014;9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Galson JD, Trück J, Fowler A, et al. In-Depth Assessment of Within-Individual and Inter-Individual Variation in the B Cell Receptor Repertoire. Front Immunol 2015;6:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Niu J, Jia Q, Ni Q, et al. Association of CD8+ T lymphocyte repertoire spreading with the severity of DRESS syndrome. Sci Rep 2015;5:9913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heather JM, Best K, Oakes T, et al. Dynamic Perturbations of the T-Cell Receptor Repertoire in Chronic HIV Infection and following Antiretroviral Therapy. Front Immunol 2016;6:644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Turchaninova MA, Davydov A, Britanova OV, et al. High-quality full-length immunoglobulin profiling with unique molecular barcoding. Nat Protoc 2016;11:1599–616. [DOI] [PubMed] [Google Scholar]
- 43. Egorov ES, Merzlyak EM, Shelenkov AA, et al. Quantitative profiling of immune repertoires for minor lymphocyte counts using unique molecular identifiers. J Immunol 2015;194:6155–63. [DOI] [PubMed] [Google Scholar]
- 44. Burgos JD, Moreno-Tovar P. Zipf-scaling behavior in the immune system. Biosystems 1996;39:227–32. [DOI] [PubMed] [Google Scholar]
- 45. Cline J, Braman JC, Hogrefe HH. PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res 1996;24:3546–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang W, Li L, Myers JR, et al. ART: a next-generation sequencing read simulator. Bioinformatics 2012;28:593–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The two simulated datasets were submitted to the NCBI Sequence Read Archive. The accession numbers for Dataset A are from SRR8733522 to SRR8733526. The accession numbers for Dataset B are from SRR8755318 to SRR8755322.