

ABSTRACT
Soil depth represents a strong physiochemical gradient that greatly affects soil-dwelling microorganisms. Fungal communities are typically structured by soil depth, but how other microorganisms are structured is less known. Here, we tested whether depth-dependent variation in soil chemistry affects the distribution and co-occurrence patterns of soil microbial communities. This was investigated by DNA metabarcoding in conjunction with network analyses of bacteria, fungi, as well as other micro-eukaryotes, sampled in four different soil depths in Norwegian birch forests. Strong compositional turnover in microbial assemblages with soil depth was detected for all organismal groups. Significantly greater microbial diversity and fungal biomass appeared in the nutrient-rich organic layer, with sharp decrease towards the less nutrient-rich mineral zones. The proportions of copiotrophic bacteria, Arthropoda and Apicomplexa were markedly higher in the organic layer, while patterns were opposite for oligotrophic bacteria, Cercozoa, Ascomycota and ectomycorrhizal fungi. Network analyses indicated more intensive inter-kingdom co-occurrence patterns in the upper mineral layer (0–5 cm) compared to the above organic and the lower mineral soil, signifying substantial influence of soil depth on biotic interactions. This study supports the view that different microbial groups are adapted to different forest soil strata, with varying level of interactions along the depth gradient.
Keywords: co-occurrences patterns, metabarcoding, microbial communities, microbial interactions, Betula pubescens, boreal birch forest
This study highlights the importance of different soil horizons as well as a combination of markers when assessing microorganisms in forest soil and provide hypotheses about depth-dependent biotic interactions.
INTRODUCTION
Forest soils harbour diverse prokaryotic and eukaryotic microbial assemblages that are crucial for overall ecosystem functioning (Bardgett and van der Putten 2014). Bacteria and fungi are the primary organisms controlling litter decomposition. The filamentous growth and extracellular enzymes secretion ability makes fungi well suited for recalcitrant soil organic matter (SOM) and cellulose decomposition (Lindahl, Taylor and Finlay 2002; Baldrian and Valášková 2008; Bödeker et al. 2014). Other fungi form mutualistic association with plant roots (mycorrhiza), which improves plant access to soil nutrients, and in exchange, they gain carbohydrates from their host plants. Fungi perform a range of functions in the soil and link C and nutrient flow between primary producers and higher trophic levels, making them important players in the microbial food webs (Geisen and Bonkowski 2018). Bacteria preferentially utilise low molecular mass organic compounds produced by biopolymer decomposition via fungi (Štursová et al. 2012). However, bacteria are also able to decompose cellulose and other plant polysaccharides (López-Mondéjar et al. 2016) and study even suggests they are able to decompose lignin (Wilhelm et al. 2018). Micro-eukaryotes other than fungi have long been neglected in surveys of forest ecosystems and barely studied in the topsoil (Geisen et al. 2017; Mahé et al. 2017; Venter, Nitsche and Arndt 2018; Oliverio et al. 2020). This polyphyletic group of organisms is extremely diverse with both multicellular (e.g. invertebrates) and unicellular (e.g. ciliates, cercozoans, apicomplexans and protists) life forms. Protists are among the main consumers of soil bacteria and fungi (Geisen et al. 2017; Seppey et al. 2017). Protists are also predators of soil invertebrates and their diversity correlates with diversity of their hosts (Singer et al. 2020). They form a dynamic hub in the soil microbiome, exerting top-down control over bacterial and fungal populations (Crowther et al. 2013; Xiong et al. 2017). However, we lack comprehensive studies analysing the co-occurrence patterns of all three different organismal groups (bacteria, fungi and micro-eukaryotes) from the same soil niche.
A wide range of edaphic factors can shape the composition of forest soil microbial communities, such as soil pH and nutrients, quantity and quality of litter input, as well as root-derived C (Baldrian 2017). Since nearly all of these factors vary with soil depth, a corresponding shift in the microbial communities is expected. Considering that pH is the major driver for bacterial richness distribution in soil (Fierer and Jackson 2006; Rousk et al. 2010), we also expect sharp change in bacterial richness with depth, where soil pH changes. Indeed, previous studies have shown depth related trends in fungal communities both at species level (Rosling et al. 2003; Baldrian et al. 2012) and functional level (Žifčáková et al. 2017) in coniferous forests. Further, in a reciprocal transplantation experiment, Bödeker et al. (2016) revealed that competition between saprotrophic and ectomycorrhizal fungi was important for regulating their vertical distribution. Depth-dependent structure in fungal communities has previously been observed in both spruce and beech forest from Norway (Asplund et al. 2018). A decrease in diversity, biomass and fungal enzyme activity with increasing depth, as well as higher gene transcription activity in the litter horizon of coniferous forest, have also been reported (Baldrian et al. 2013; Voříšková et al. 2014; Žifčáková et al. 2017). Saprotrophic fungi largely colonize the upper litter and poorly decomposed organic matters due to their ability to utilise recalcitrant plant residues (Lindahl et al. 2007; Voříšková et al. 2014). As the proportion of organic matter decreases with depth, the abundance of saprotrophic fungi decreases. Ectomycorrhizal (ECM) fungi, on the other hand, which are suggested to be key players in forest C dynamics (Clemmensen et al. 2013), increase in the lower humus and mineral soil (Dickie, Xu and Koide 2002). The newly discovered Archaeorhizomycetes, lacking typical mycorrhizal structures but possessing decomposition ability, may dominate in the deeper soils of coniferous forests (Rosling et al. 2011). Most studies both in coniferous and deciduous forest focus on the litter layer and the upper organic soil horizon (Hartmann et al. 2012; Tedersoo et al. 2014; Bahram et al. 2018), but there is evidence for that the deeper mineral soils needs to be included to assess the overall microbial diversity (Rosling et al. 2003; Lindahl et al. 2007; Jumpponen, Jones and Blair 2010; Santalahti et al. 2016; Du et al. 2017). Compared to fungi, there are few studies addressing the vertical distribution of other micro-eukaryotes (Ekelund, Rønn and Christensen 2001; Potapov et al. 2017) and bacteria (Baldrian et al. 2012; Eilers et al. 2012; Hartmann et al. 2012; López-Mondéjar et al. 2015; Du et al. 2017; Pereira et al. 2017) in forest soils, and their distribution have rarely been analyzed through the mineral soil profile (< 30 cm).
Inter-kingdom co-occurrence patterns of soil microbes has been shown to vary depending on soil types, organic C and pH level (Creamer et al. 2016; Xiong et al. 2017; de Araujo et al. 2018), and are tightly linked with above-ground vegetation (Wardle et al. 2004). Recently, Hernandez et al. (2021) demonstrated that in scrub habitats, under stressful environment conditions microbial community network destabilizes by decreasing modularity as well as negative: positive cohesion. Results from co-occurrence network-based analyses does not necessarily reflect biotic interactions. However, such analyses allows us to generate hypotheses about potential biotic associations among community members and enables us to examine cross-kingdom relationships in large microbial community data sets generated in high-throughput sequencing (HTS) studies of environmental DNA (de Vries et al. 2018). By employing network analyses for soil communities we can produce hypotheses about (i) the functional roles of uncultured microorganisms (Fuhrman and Steele 2008; Chaffron et al. 2010); (ii) niche spaces shared by community members (Delmas et al. 2019) and (iii) positive (symbiosis) and negative (e.g. pathogenicity) interactions between community members (Röttjers and Faust 2018).
This study investigates depth dependent associations between microbial taxa bacteria, fungi and non-fungal micro-eukaryotes (referred to in the following as ‘micro-eukaryotes’) in boreal soil and their inter-kingdom co-occurrences patterns. Previous studies have largely focused on a specific organismal group, such as fungi, but here we wanted to assess all the major microbial groups together. This is done by DNA metabarcoding analyses of bacteria, fungi and micro-eukaryotes from various depths in forest soils of five native deciduous birch (Betula pubescens Ehrh.) forests in Western Norway. Our study was aimed 1) to investigate how the various organismal groups differ in composition and diversity through the forest soil column down to 30 cm mineral soil depth and what are their drivers? We here expect that all groups respond strongly to soil depth due to variation in soil nutrients and pH, with highest richness in the top layer; and 2) to assess how inter-kingdom co-occurrence patterns and network architectures, indicative of biotic interactions, vary with soil depth. We expect that the species are more filtered by stressful abiotic conditions and have less biotic interactions in the deep soil profiles, while biotic interactions are more intense in the top layers.
MATERIALS AND METHODS
Site description, experimental setup and sampling
This study was conducted in native deciduous birch (Betula pubescens Ehrh.) forests at five locations in western Norway (61°30' N, 6°12' E; Material and methods S1; Fig. S1, Supporting Information). Distance between the northernmost (Molde) and the southernmost (Jøster II) locations is approx. 320 km. All the locations are positioned in the middle boreal vegetation zone, and occasional grazing as well as selective cutting have occurred through time. At all locations the bedrock is covered by thick moraine deposits (NGU 2020). The soil texture is sandy loam at all locations except for Stranda, where, in addition to the sandy loam, parts of the soil profile are dominated by silt loam. Generally, the soil chemistry did not differ significantly between locations except C: N ratio (Kjønaas et al, submitted). The ground vegetation was dominated by a mix of bilberry (Vaccinium myrtillus L.), grasses, herbs and bryophytes.
At each location, three 144 m2 plots were established within a birch stand in areas with relatively homogeneous soil, topography and vegetation. In July 2016, 20 soil cores per plot were collected down to approx. 30 cm soil depth in a grid sampling design by use of a cylindrical auger (Ø = 2.6 cm). Each soil core was divided into four layers: the forest floor (LFH) and three mineral soil layers based on sampling depth (0-5 cm ‘M1’; 5–15 cm ‘M2’; 15–30 cm ‘M3’). M1 is the organic-mineral interface layer, also termed the ‘Ah’ layer. Samples from each plot and layer were pooled into one composite sample, resulting in altogether 60 samples (5 locations*3 plots*4 depths). After pooling of subsamples and thorough homogenization, the mineral soil samples from each layer were divided into two separate samples (one for DNA/ergosterol and other for chemical analyses). As the LFH sample was difficult to homogenize, the entire sample was allocated to DNA and ergosterol analyses. For chemical analyses of the LFH layer, an additional sample was collected adjacent to the sample collected for DNA analyses using a cylindrical auger (Ø = 6.6 cm). All the samples for DNA and ergosterol analyses were stored at −20°C immediately after collection, whereas the samples for soil chemistry were kept cool during transport and frozen after returning to the lab.
Prior to the DNA and ergosterol analyses, the soil was homogenized by sieving (2 mm sterilised sieve), followed by freeze-drying and pulverizing using FastPrep instrument (MP Biomedicals, Illkirch-Graffenstaden, France). The finely grounded soil fractions were used for analysis of DNA as well as total ergosterol (fungal biomass proxy) using the protocol of Ransedokken et al. (2019).
Soil chemistry analysis
Prior to analyses, the soil samples were thawed, air dried and sieved through a 2 mm sieve. The fine soil fraction was analysed for dry matter (105°C), total C% and N% (Elementar Vario EL with TCD detector), pH (H2O) (PHM 220) and exchangeable elements (Hydrogen (H), Calcium (Ca), Potassium (K), Magnesium (Mg), Manganese (Mn), Sodium (Na), Phosphorous (P) and Sulphur (S)) (in 1M NH4NO3; Thermo Jarell Ash ICP-IRIS HR Duo). The fine soil fraction was finely ground (planet mill) before the C and N analysis. For details see Ogner et al. (1999).
DNA extraction and Illumina sequencing
One gram of homogenized soil was added in 10 ml CTAB buffer, and 600 μl of the CTAB/soil-sludge was transferred to a 2 ml eppendorf tube containing two tungsten-carbide beads. Samples were grinded for one minute (25 Hz) and repeated after flipping the racks, and immediately stored at −20°C. DNA was extracted following the CTAB/chloroform extraction protocol and further purified using the E.Z.N.A soil DNA kit (Omega Biotek, USA) following the manufacturer's protocol. Detailed information about PCR settings and reaction is provided in supplementary material (Material and methods S1). Molecular data was generated from three full Flow-Cell runs and Paired-End (PE: 2 × 300 bp) sequencing with Illumina Miseq. Different primer combinations were used to amplify the 16S rRNA gene for bacteria ITS2 gene for fungi, and 18S rRNA gene for both fungal and micro-eukaryotes. The ITS region, which is the most common DNA barcode for fungi (Schoch et al. 2012), is mostly used to analyze fungal communities (Nilsson et al. 2009). However, various biases, including primer mismatches and length differences, may lead to some groups being excluded (Bellemain et al. 2010; Schadt and Rosling 2015; Tedersoo and Lindahl 2016). Universal 18S primers were used to amplify the overall micro-eukaryotic communities, also including fungi (Material and methods S1). While the highly variable ITS2 marker provide detailed taxonomic information about fungi, mainly at species and genus level, 18S provides taxonomic assignments at higher taxonomic levels. However, the more conserved 18S marker provides a more comprehensive overall picture for micro-eukaryotes, because of less amplification biases (Hugerth et al. 2014). The fastq-formatted sequence data sets for 16S, ITS and 18S markers gene along with barcode mapping files and associated metadata were archived at Zenodo (https://zenedo.org), a scientific data repository developed by CERN, with a single DOI for the project (10.5281/zenodo.4 415 050).
Downstream analyses focused on all the three main microbial groups bacteria, fungi and the highly polyphyletic micro-eukaryotes. The 18S data set contained both fungal and non-fungal reads therefore it was divided into a fungal and a non-fungal data set (the latter hereafter referred to as micro-eukaryotes, for simplicity).
Bioinformatics analyses
To enable comparisons across all data sets, the same bioinformatics pipeline was employed for all data. Raw data was passed through BayesHammer, a bayesian clustering based error correction method (Nikolenko, Korobeynikov and Alekseyev 2013), before merging the PE reads using PEAR v0.9.10 with minimum overlap of 10 bp and Q20 quality score threshold for trimming the low quality part of a read (Zhang et al. 2014). For excluding reads with poor quality we used FASTX-Toolkit v0.0.14 (fastq_quality_filter, http://hannonlab.cshl.edu/fastx_toolkit/index.html) with the parameter settings: minimum Phred quality score = 30, and proportion of bases that must have minimum quality score = 0.9. A second level of quality control was performed using VSEARCH v2.4.3 (Rognes et al. 2016) to remove reads with ambiguous base = 0, length <100 bp and total expected errors (E) >0.5 for all bases. Remaining high quality reads were demultiplexed using the SDM v1.41 program embedded in the LotuS pipeline (Hildebrand et al. 2014). Using FQGREP v0.4.4 (https://github.com/indraniel/fqgrep) and FASTX-Toolkit, reads were oriented in the same direction and primers were trimmed. For fungal ITS data set, the ITS2 region was extracted using ITSx v1.0.11 (Nilsson et al. 2010), followed by removal of reads <100 bp. We then used VSEARCH for dereplication, global singletons removal and clustering (97% similarity threshold for the 16S and ITS2 data sets and 98% for 18S data set). The most abundant sequence of each cluster was designated as the representative sequence. Chimera checking was performed on the representative sequences using uchime_denovo algorithm (Edgar et al. 2011), implemented in VSEARCH, with the minimum divergence parameter = 0.8, abundance skew = 2 and minimum difference in segment = 3. Since we wanted to focus on the more abundant microorganisms, Operational Taxonomic Units (OTUs) with <10 reads were removed, also in order to minimize the impact of sequencing and PCR errors. Taxonomic assignment were made by comparing the representative sequence against the curated reference databases GREENGENE v13.8 (DeSantis et al. 2006; McDonald et al. 2012), UNITE v6 (Kõljalg et al. 2013) and PR2 v4.62 (Guillou et al. 2013) for bacteria, fungi and micro-eukaryotes, respectively. In order to assign a functional guild, the fungal ITS2 OTUs were passed through FUNGUILD (Nguyen et al. 2015).
Bacteria, fungi (18S) and micro-eukaryotes (Fig. S2a, e and g, Supporting Information) showed non-normal distribution of reads, which was not the case for the ITS2 fungal data set (Fig. S2c, Supporting Information). The distribution patterns of reads per OTUs was skewed with few dominating and a long tail of rare OTUs for all groups (Fig. S2b, d, f and h, Supporting Information). Sample-based rarefaction curves of OTU richness showed that the complete richness was not captured in most samples (Fig. S3a–d, Supporting Information). Correspondingly, there was a positive and significant relationship between OTU richness and sequencing depth for bacteria (R2 = 0.58; P < 0.001), fungi (18S) (R2 = 0.52; P < 0.001) and micro-eukaryotes (R2 = 0.74; P < 0.001). Probably due to better sequencing depth, this was not observed in the fungal ITS2 based data set (R2 = 0.16; P = 0.328). To correct for potential sequencing depth biases, all data sets were rarefied prior to diversity analysis (reads per sample for bacteria, fungi (ITS), fungi (18S) and micro-eukaryotes were 6343 (16S), 55 485 (fungi ITS2), 2783 (fungi 18S) and 3430 (micro-eukaryotes), respectively. Two samples with low read numbers were discarded.
Statistical analyses
Unless stated otherwise, statistical analyses were performed in R v3.5.0 (R Core Development Team 2018). Prior to analyses, all soil variables were logarithm-transformed and a Principal Component Analysis (PCA) was used to assess tentative collinearity between environmental variables and soil depth. OTU table containing species data were arcsine-transformed prior to analyses to improve variance homogeneity.
ANOVA followed by Tukey's HSD post-hoc test (package agricolae (Mendiburu and Simon 2015)) was used to examine differences in soil properties, ergosterol content, as well as richness, Shannon diversity and evenness of all the microbial groups, with soil depth. The same test, with Benjamini–Hochberg FDR correction, was used to assess whether the relative proportion of different phyla (from all data sets) and genera varied with soil depth. The results were illustrated using bar plots (phyla) and hierarchical heat plots (genera).
The Bray–Curtis dissimilarity index was used to generate community distance matrices. To address the relative importance of the soil chemistry variability index (i.e. PC1), and the location effect (i.e. PC2) on community composition of all groups of microorganisms, we used multivariate permutational analysis of variance (PERMANOVA), as implemented in the Adonis function of the package vegan (Oksanen et al. 2013). PERMANOVA analyses with 9999 permutations were performed using a forward selection procedure to optimise the final model (Blanchet, Legendre and Borcard 2008). We first tested model for individual above mentioned variables and included significant variables in the final model in order of their R2 values, to assess if remaining variation can also be explained by other variables. Nonmetric Multidimensional Scaling (NMDS) analyses were used to visualize the relative effects of these variables on microbial communities using the metaMDS function of the package vegan. Vectors and centroids of the variables were fitted into NMDS plots using the function envfit, and the ordiellipse function was used to plot the 95% confidence intervals (CI) of the depth.
Network analyses
To investigate inter-kingdom co-occurrences patterns, network analyses were performed on core communities (OTUs with >0.5% of total reads, and present in at least three samples) selected from each normalised data set (Material and methods S1). These core OTUs from the three data sets were summarized at the genus level and the samples were normalized separately by subsampling to the lowest number of sequences across all three data sets. The subsampled genus-tables were then merged into one table containing genera of 93 bacteria, 73 fungi and 10 micro-eukaryotes. Co-occurrence networks for the overall data set, as well as for the four soil depths separately, were constructed with SparCC, as implemented in the R package SpiecEasi (Kurtz et al. 2015). SparCC was run with default settings and 500 bootstraps. Only associations with pseudo P-value < 0.05 and correlations > |0.7| were kept. Visualization of networks as well as calculation of network statistics were done with Cytoscape v3.6.1 (Smoot et al. 2011) and the R package igraph (Yu, Chen and Guo 2009). We calculated the flowing network characteristics: density, degree, neighbourhood connectivity, clustering coefficient and average path length (Material and methods S1). Network density is calculated as the proportion of realised possible correlations given the number of genera in the network; degree is a measure of how many correlations each genus form with other genera; neighbourhood connectivity measures how many correlations a ‘neighbour’ genus (i.e. one that is correlated to the focal genus) in turn is correlated to; clustering coefficient describes whether the network can be sectioned into groups of highly correlated organisms; while average path length is the distance (counted as number of edges) between all pairs of associated genera (nodes) divided by the number of genera in the network. A Wilcoxon signed-rank test was used to test whether the network characteristics differed between soil depths.
RESULTS
Data characteristics
Diverse prokaryotic and eukaryotic communities, with altogether 1540 bacterial, 4388 fungal (3461 ITS-based; 927 18S-based) and 2025 micro-eukaryotic OTUs, were detected in the 60 composite soil samples (see Results S2 for details). Ergosterol content, a proxy for fungal biomass, was significantly higher in the upper litter and humus (LFH) layer (0.158 mg g−1) compared to the mineral soil layers (<0.026 mg g−1; Fig. S4, Supporting Information). Likewise, the total C and N content, as well as all exchangeable elements were highest in the LFH layer and decreased with soil depth, while pH had an opposite trend (Fig. S5, Supporting Information). PCA clearly showed that soil depth, as well as all measured edaphic factors, including pH, correlated tightly with the first PC axis (Fig. 1), which can be interpreted as a ‘soil chemistry variability index’. The second PC axis reflected site specific effects, including variation in the C: N ratio across plots and locations. Due to the high collinearity between numerous variables as well as soil depth, PC axes 1 (‘soil chemistry variability index’) and 2 (‘location effects’) were used in the further analyses as proxies for environmental variability.
Figure 1.
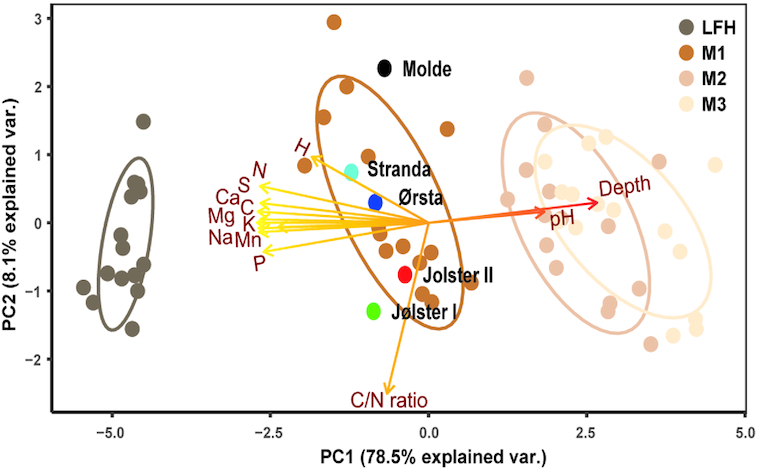
Principal component analysis (PCA) of soil chemistry data along depth gradient. Samples were collected from five different location (Molde, Stranda, Ørsta, Jølster I and Jølster II), along forest floor (LFH) and three mineral soil layers: 0–5 cm (M1), 5–15 cm (M2), 15–30 cm (M3)). The first PC axis (PC1) score of each plot (shown with dark brown to light brown colored circles) was used as a ‘soil chemistry variability index’, as it reflects variation related to the soil nutrients. The second PC axis (PC2) score of each plot was considered as ‘location effect’ (it reflects variation in C: N ratio, related to sampling locations). Ellipses indicate 95% confidence intervals around centroids for each soil depth and black, skyblue, darkblue, red and green colored circles indicate centroid for each sampling location. Following soil chemistry data was used in the analysis: total carbon (C%), total nitrogen (N%), C/N ratio, soil pH and exchangeable elements H, P, Mn, Ca, Mg, Na, S and K.
Microbial diversity and communities’ patterns with soil depth
In line with depth driven changes in soil chemistry, we observed a decrease in richness with soil depth for all microbial groups (Fig. 2A, D, G and J). A similar declining trend was observed for Shannon diversity and the evenness index for all microbial taxa, except for the micro-eukaryotes (18S). However, a positive relationship (R2 = 0.11; P-value < 0.001) between micro-eukaryotic reads and ergosterol content was observed.
Figure 2.
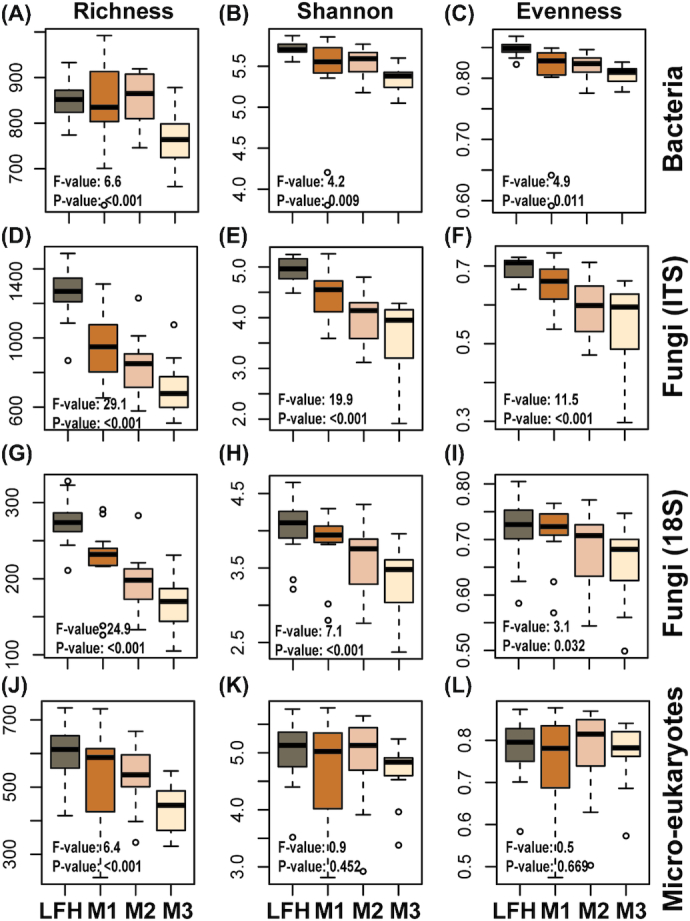
Boxplots showing soil depth related diversity patterns for different microbial groups. Diversity measure such as richness, Shannon index and evenness are shown for bacteria (16S; A–C), fungi (ITS: d-f; 18S: G–I), and micro-eukaryotes (18S: K–L) are shown. Samples were collected along a soil depth gradient (forest floor (LFH) and three mineral soil layers: 0–5 cm (M1), 5–15 cm (M2), 15–30 cm (M3)) from natural birch forest. Statistically significant differences among soil depth were analysed using ANOVA and Tukey's post-hoc test.
All the studied microbial groups showed a strong and consistent pattern of compositional shift with the soil chemistry variability index, as revealed by multivariate PERMANOVA (Table 1) and visualised using NMDS analyses (Fig. 3A–D). The location effect, linked to variability in C: N ratio among sites, had weaker relationships with the community composition. The proportion of OTUs shared among all four soil depths were higher for bacteria (86%; Fig. S6a, Supporting Information) compared to fungi (ITS: 56%; 18S: 48%; Fig. S6b and c, Supporting Information) and micro-eukaryotes (50%; Fig. S6d, Supporting Information).
Table 1.
Results from PERMANOVA analyses, testing to which degree soil chemistry variability index (PC axis 1) and location effect (PC axis 2) can explain compositional variation in the different microbial groups.
| Variables | Df | SS | MS | F-model | R2 | P-value |
|---|---|---|---|---|---|---|
| Bacteria | ||||||
| Soil chemistry variability index | 1 | 1.76 | 1.76 | 33.22 | 0.35 | <0.001 |
| Location effect | 1 | 0.31 | 0.31 | 5.84 | 0.06 | <0.001 |
| Residuals | 57 | 3.03 | 0.05 | 0.59 | ||
| Total | 59 | 5.10 | 1 | |||
| Fungi (ITS) | ||||||
| Soil chemistry variability index | 1 | 2.13 | 2.13 | 11.92 | 0.15 | <0.001 |
| Location effect | 1 | 1.60 | 1.60 | 9.00 | 0.12 | <0.001 |
| Residuals | 57 | 10.17 | 0.18 | 0.73 | ||
| Total | 59 | 13.90 | 1.00 | |||
| Fungi (18S) | ||||||
| Soil chemistry variability index | 1 | 1.42 | 1.42 | 10.23 | 0.14 | <0.001 |
| Location effect | 1 | 0.92 | 0.92 | 6.58 | 0.09 | <0.001 |
| Residuals | 55 | 7.66 | 0.14 | 0.77 | ||
| Total | 57 | 10.00 | 1 | |||
| Micro-eukaryotes | ||||||
| Soil chemistry variability index | 1 | 1.84 | 1.84 | 13.13 | 0.18 | <0.001 |
| Location effect | 1 | 0.56 | 0.56 | 3.98 | 0.06 | <0.001 |
| Residuals | 55 | 7.71 | 0.14 | 0.76 | ||
| Total | 57 | 10.11 | 1.00 |
Figure 3.

Nonmetric multidimensional scaling (NMDS) plots displaying the community structure of the different microbial groups. Bacterial (A), fungal (ITS: B; 18S: C) and micro-eukaryotic (D) community compositional patterns among samples from different soil depth (forest floor (LFH) and three mineral soil layers: 0–5 cm (M1), 5–15 cm (M2), and 15–30 cm (M3)), as revealed by NMDS ordination analysis. The ordination plots are based on all Operational Taxonomic Units (OTUs) present in the respective microbial groups. The stress value for NMDS ordination was 0.104 for bacteria, 0.166 for fungi (ITS), 0.193 for fungi (18S) and 0.135 for micro-eukaryotes. Ellipses indicate 95% confidence intervals around centroids for each soil depth. Arrows point in the direction of maximum increase of the variables and size of circle indicates number of OTUs richness. All variables and factor shown in the panels had significant effects (P < 0.05) on the ordination configuration.
Overall, the prokaryotic communities were dominated by Proteobacteria (32% reads; 31% OTUs) and Acidobacteria (25% reads; 21% OTUs). The shift in bacterial composition from organic to mineral soil layers was mainly driven by changes in Proteobacteria, Actinobacteria, Verrucomicrobia and Bacteriodetes, being significantly more proportionally abundant in the upper LFH layer, while Acidobacteria, Firmicutes and Chlorofexi were proportionally more abundant in the deeper mineral soil layers (Fig. 4A). Similarly, proportions of certain genera also showed consistent patterns with soil depth (Fig. 5A). The proportions of the genera Povalibacter, Roseiarcus, Rhizomicrobium, Reyranella, Burkholderia and Phenylobacterium (Proteobacteria); Granulicelia (Acidobacter) were significantly higher in upper LFH layer (Fig. 5A) while the proportions of Steroidobacter, Nitrospirillum, Blastochloris and Afipia (Proteobacteria); Gp2, Gp6 and Acanthopleuribacter (Acidobacteria) and Tepidibacillus (Planctomycetes) were higher in the lower mineral layers.
Figure 4.

Barplots displaying abundances distribution of the different microbial taxa (phyla). Plots for relative abundances of the bacterial (A), fungal (ITS: B; 18S: C), and micro-eukaryotic (D) taxa with soil depth (forest floor (LFH) and three mineral soil layers: 0–5 cm (M1), 5–15 cm (M2), and 15–30 cm (M3)) are shown here. Statistically significant differences among soil depth was analysed using ANOVA and Tukey´s post-hoc test. Note the significant difference in distribution of fungal groups (Ascomycota) as revealed by the 18S rRNA gene markers (C) but pattern absent while using ITS (B).
Figure 5.

Hierarchical heatplots illustrating abundances distribution of the different microbial taxa (genus or class level). Plots for bacterial (A), fungal (ITS: B; 18S: C) and micro-eukaryotic (D) taxa with different soil depth (forest floor (LFH) and three mineral soil layers: 0–5 cm (M1), 5–15 cm (M2), and 15–30 cm (M3)) are shown here. All taxa shown in the plots are analysed using ANOVA and Tukey´s post-hoc test and differ significantly (P <<0.05i>) in abundances among soil depth. Colour gradients in the plots from white to grey to black indicates increasing dominance of the taxonomic groups in particular soil depth. Different color in bacterial and fungal genus name indicates respective phyla and in case of micro-eukaryotes color represent different taxa at higher taxonomic level. Different colors are used to indicate phyla for bacteria, fungi and micro-eukaryotes
As revealed by the 18S primers, fungi dominated among the eukaryotes, making up 50% of the reads and 30% of the OTUs. In this data set, the proportion of Ascomycota reads was significantly higher in the lower mineral soil layer compared to the upper LFH layer (Fig. 4C), a shift largely driven by significantly higher proportions of Archaeorhizomycetes reads in the deeper mineral soils (Fig. 5C). In contrast, Chytridiomycota (Fig. 4C) made up a significantly higher proportion of reads in the upper organic layer, while the other fungal phyla, Basidiomycota, Mucoromycota and Cryptomycota, showed rather uniform distribution with soil depth in the 18S data set. When the ITS2 region was used to survey the fungal communities, a contrasting pattern was detected for some groups (Figs 4B and 5B), especially so for the Archaeorhizomycetes, which was only recovered as dominating taxa while using the 18S primers (Fig. 5C). In contrast to the 18S data, basidiomycetes (50% reads; 29% OTUs) were the most abundant in the ITS2 data, but ascomycetes still included a higher OTU richness (45% reads; 60% OTUs). In the ITS2 data set, relatively higher proportion of symbiotrophic fungi such as Inocybe, Gyroporus (both ECM) appeared in the mineral soil layers (Figs 4B and 5B). Several fungal genera had significant shifts in proportions with soil depth (Fig. 5B). Clavaria were proportionally more abundant in the lower mineral layer, whereas Cortinarius, Lactarius, Tomentella (all ECM) and Mycena were proportionally more abundant in upper LFH layer. The Ascomycota genera Oidiodendron, Capronia, Leohumicola and Hydnotrya (ECM) were proportionally more common in the lower mineral layer.
Among the other micro-eukaryotes, Metazoa was the most dominating group (28% reads; 17% OTUs) followed by Cercozoa (11% reads; 27% OTUs) and Ciliophora (4% reads; 8% OTUs). As for bacteria and fungi, they all displayed significant depth related changes in their proportions (Fig. 4D). In the upper layer, the proportion of metazoan reads, and specifically so Arachnida, was significantly higher along with Conosea (Variosea). Additionally, apixomplexan parasites belonging to Gregarines were also proportionally more abundant in the upper organic layer. The cercozoans Filosa-sarcomonadea (Sandonidae, Cercomonadidae and Allapsidae) and Filosa-Inbricatia, and the ciliates Spirotrichea (Oxytrichidae) and Colpodea made up a higher proportion of the reads in the lower mineral layers (Fig. 5D).
Inter-kingdom co-occurrence patterns with soil depth
The network analyses revealed a higher number of positively or negatively correlated (co-occurring) genera in the top layers, compared to the deepest layer M3, with M1 having the highest number of correlations (Fig. 6A–D, Figs S7–S10, Supporting Information). In line with this, the network statistics network density, degree and neighbourhood connectivity were on average highest in M1 (Table S1, Supporting Information; Fig 6E and F). Likewise, the average path length was lowest in the M1 layer and increased with increasing depth (Fig. 6h). A low average path length indicates that most taxa in the network are connected through few intermediate taxa. The network statistics suggest that organisms in the M1 layer form more association with each other compared to in the other soil layers. Further, the clustering coefficient, which describes whether the network can be sectioned into clusters of highly correlated genera, was also highest in M1 (Fig. 6G). A Wilcoxon signed-rank test showed that the M1 layer was significantly different for most of the network statistics (Table S2, Supporting Information) and 14 genera were exclusively detected in this layer (Fig. S11, Supporting Information). There was no significant difference between the LFH layer and the M3 layer in the network topology (Table S2, Supporting Information). These two layers have 30 genera in common (Fig. S11, Supporting Information), and only 10 and 6 genera were exclusively detected in the LFH and M3 layers, respectively. These two layers are thus predominantly dominated by similar taxonomic composition. The proportion of positive correlations was highest for the above LFH (73.8%) and M1 layers (73.5%), and decreased towards the M3 layer (57.0%), where the relative proportion of negative correlations was higher (43%) (Fig. 6A–D). Taxa belonging to Firmicutes (Vagococcus, Bacillus, Enterococcus, Paenibacillus and Macrococcus), Acidobacteria (Gp1 and Gp3) and a yeast group (Candida), showed higher numbers of co-occurrences in the upper M1 compared to other layers.
Figure 6.
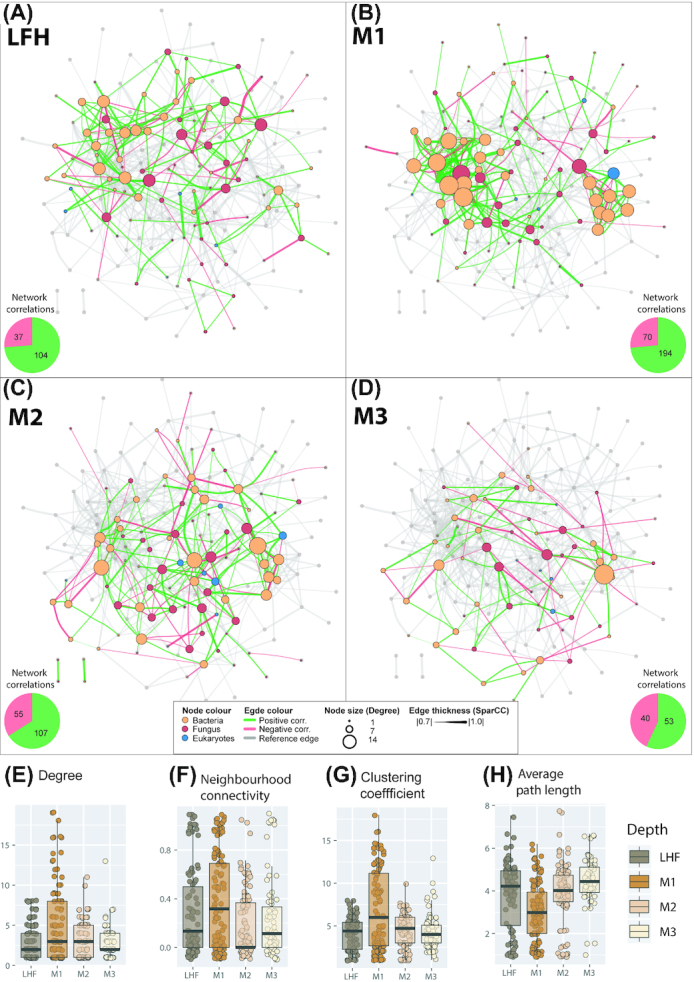
Inter-kingdom correlation patterns of bacterial, fungal and eukaryotic genera. The network is based on a SparCC correlation analysis for the forest floor (LFH) (A) and mineral soil layers 0–5 cm (M1) (B), 5–15 cm (M2) (C) and 15–30 cm (M3) (D). Pie charts from each panel represents number of total correlations (positive as green and negative as red) in Network from each depth. Positive correlations (SparCC > 0.7, P < 0.05) are drawn as green edges and negative correlations (SparCC < 0.7, P < 0.05) are drawn as red edges. Nodes represents genera and are coloured according to taxonomic group: bacteria in orange, fungi in red and micro-eukaryotes in blue. The size of a node is proportional to connection it forms with other nodes. The thickness of the connection between two nodes is proportional to the value of correlation coefficients. The network in transparent grey is a reference network combining the correlations for all four depth layers. Boxplots (F–H) show the upper and lower quartile, and the average value for important network statistics for all four depths: degree (E), neighbourhood connectivity (F), clustering coefficient (G) and average path length (H). The degree of a genus is the number of co-occurrences it has with other genera (i.e. the number of connections (edges) formed by a node to other nodes). The neighbourhood connectivity is the average connectivity (correlations) of neighbours of a given node (i.e. nodes correlated to a given node can themselves be correlated to other nodes). The clustering coefficient describes whether the network can be sectioned into clusters of highly interconnected organisms. Highly clustered networks are those that contain groups of statistically associated organisms. A high clustering coefficient is an indication of a high degree of interactions and associations. The Average path length is the distance (counted as number of edges) between all pairs of associated genera (divided by the number of genera in the network. A low average path length indicates that most species in the network are connected through a few intermediates’ species.
DISCUSSION
As hypothesised, soil depth represents a strong and complex environmental gradient with multiple edaphic factors changing concurrently with depth in the investigated birch stands. A strong niche partitioning with depth is reflected in both diversity and structure of the bacterial, fungal as well as micro-eukaryotic communities. Further, the soil gradient also affected inter-kingdom co-occurrence patterns among soil horizons, and most dense as well as complex network, with higher degree and neighborhood connectivity, was observed in the upper mineral soil layer (M1).
In addition to soil depth, some of the variation in community composition could be attributed to environmental differences among locations (location effects), including differences in C: N-ratio, aspect and latitude. Two locations (Molde and Stranda) were south-facing, two (Jølster I and II) north-facing and the last one (Ørsta) east-facing, which lead to temperature differences. The systematic variation in C: N-ratio may be induced by a gradient in N deposition (Fischer et al. 2007).
The two different markers (ITS2 vs 18S) used for fungi provided a slightly different view of the fungal communities, especially when it comes to Archaeorhizomycetes (Ascomycota) and chytrids. These two groups were not captured fully by the ITS2 primers, indicating primer biases discriminating against e.g. Archaeorhizomycetes and chytrids (Rosling et al. 2011; Ihrmark et al. 2012; Tedersoo et al. 2014; Schadt and Rosling 2015; Nilsson et al. 2019). The 18S universal primers employed in this study seems to amplify most eukaryotes (Hadziavdic et al. 2014), providing a more comprehensive picture of the fungal community composition compared to the ITS2 data set. However, there might also be primer biases associated with the 18S primers (Anderson, Campbell and Prosser 2003). Nevertheless, our results highlights the importance of using different markers to better capture the diverse microbial communities in soil.
Depth matters: biomass, diversity patterns and soil characteristics
One of the most evident changes through the soil profile was the distinct decrease in fungal biomass with depth in the deciduous forest, corroborating findings of previous studies from coniferous forest (Hartmann et al. 2012; Voříšková et al. 2014). A similar declining trend in bacterial biomass with depth in deciduous forest soils has also been reported (Hartmann et al. 2012). Using traditional culturing and quantitative methods, a sharp decline in protozoan biomass with depth has been observed in Danish deciduous and coniferous forest sites (Ekelund, Rønn and Christensen 2001). Thus, there is mounting evidence of a general pattern of decreasing biomass with increasing forest soil depth, regardless of microbial groups.
We observed significant declines in diversity patterns with depth across different microbial groups, with a stronger pattern for fungi compared to bacteria and other micro-eukaryotes. Our findings are in line with studies from boreal coniferous forest (Rosling et al. 2003; Lindahl et al. 2007; Baldrian et al. 2012; Hartmann et al. 2012; Santalahti et al. 2016; Kyaschenko et al. 2017). Similarly, studies from deciduous mountainous hardwood (Du et al. 2017), temperate oak forest (Voříšková et al. 2014) and tallgrass prairie (Jumpponen, Jones and Blair 2010) ecosystem also consistently revealed declining patterns for fungal richness. These patterns are not surprising considering the observed lower biomass of fungi (this study) and bacteria (see above) in deeper mineral soil. Although micro-eukaryotes are important in soil food webs, the effects of soil depth on their diversity has rarely been investigated (Potapov et al. 2017). A synchronous decrease in diversity of bacteria and fungi together with micro-eukaryotes suggest key role of micro-eukaryotic organisms in the food webs since they are highly dependent on bacteria and fungi as C sources (Crowther et al. 2013; Geisen et al. 2016; Seppey et al. 2017; Xiong et al. 2017).
The variation in soil chemistry affects micro-eukaryotic diversity (Tedersoo et al. 2015), and the observed diversity patterns showed a high correlation with the soil gradient patterns. The C concentration and soil nutrients availability are considerably higher in upper organic layers. Further, the easily decomposable labile fraction of C has been found to decrease with soil depth at the same sampling locations (Hansen et al. submitted). In agreement with previous studies targeting bacterial richness from coniferous forest (Eilers et al. 2012; Hartmann et al. 2012), we found a decline in bacterial richness with soil depth. There is a large variation in microclimatic conditions and diverse resources of organic matter (Schurig et al. 2013) at the soil surface, that may support a high diversity of ecological niches as compared to the deeper soil. Further, only few bacterial groups are well-adapted to grow under the oligotrophic conditions that characterize the deeper mineral soil. The decline in fungal biomass and diversity in deeper mineral soil may also affect the bacterial richness, as some bacterial groups feed on fungi through their ability to secrete protein and cause hyphal decay (Swain et al. 2017). Together, our results indicate that variation in environmental conditions with soil depth represent an ecological filter, and that many surface-dwelling organisms do not thrive in the nutrient poor environments of the deeper soil horizons. Our results also suggest that, independent of organismal groups, a considerable part of the diversity cannot be recovered when examining only the topsoil layers, since different soil horizon host diverse microbial assemblages.
Depth matters: community compositional patterns
Our observation of a clear shift in fungal communities with soil depth, agrees with previous studies from coniferous forests (Rosling et al. 2003; Hartmann et al. 2012; Hobbie et al. 2014b; Santalahti et al. 2016). Saprotrophic fungi, including Mycena spp. (common litter basidiomycetes) with demonstrated peroxidase activity (Kyaschenko et al. 2017) and the common leaf endophytes Phialocephala spp., dominated in the top layer. Here, fresh litter is supplied by aboveground vegetation and the saprotrophic fungi may out-compete the ectomycorrhizal fungi (Fernandez and Kennedy 2016). In accordance with previous studies, we detected an overall higher dominance of symbiotrophic fungi in the deeper mineral soils. However, the ectomycorrhizal genera Cortinarius, Tomentella and Lactarius, were significantly more abundant in the organic layer. This can be linked with the ability of Cortinarius and Lactarius to infect root tips in the organic layer and also their high extrametrical mycelium production rate generating more biomass (Genney, Anderson and Alexander 2006; Anderson, Genney and Alexander 2014). Further, some species of Cortinarius may secrete peroxidase in order to access and decompose organically bound N present in the organic layers (Bödeker et al. 2014). In an experiment using litter bags with 15N-labelled beech leaf, Pena et al. (2013) showed high abundance of Tomentella in leaf litter and provided experimental evidence that they are key players in decomposition and N capture from decaying leaves. On the other hand, the dominance of the ectomycorrhizal genera Inocybe, Gyroporus and Russula increased in the deeper mineral soil, where the substrate becomes increasingly depleted and the symbiotrophic fungi likely are highly dependent on host-derived C. This has also been validated using a isotope tracer technique, conducted at the Duke free CO2 enrichment (FACE) experiment (Hobbie et al. 2014a; Hobbie et al. 2014b). Here they showed that ECM fungi with hydrophobic exploration types (e.g. Cortinarius and Tricholoma) preferentially utilise N from the deeper humus layer whereas fungi with hydrophilic exploration types (e.g. Russula and Lactarius) acquired N from the forest litter layer. Further, they also showed that Lactarius and Russula only incorporated fresh photosynthate as C source (litter-derived), whereas Inocybe and Cortinarius also forage on soil-derived C. This suggests that different fungal species and genera have different niche preferences due to their nutrient acquisition strategies (Lindahl et al. 2007). The 18S marker demonstrated a significantly higher abundance of Archaeorhizomycetes in deeper soil. Although we lack a complete understanding of the Archaeorhizomycetes ecology (Rosling et al. 2011), they may act as root-associated mutualists (Menkis et al. 2014). The high abundance of Archaeorhizomycetes in the deeper mineral soil may also indicate an adaption to nutrient limited and stressful environments conditions, as suggested in previous studies (Sterkenburg et al. 2015; Pinto-Figueroa et al. 2019).
There has been numerous studies focusing on the vertical distribution of fungi in deciduous forest soils, but the literature is more limited for bacteria (Baldrian et al. 2012; Lladó, López-Mondéjar and Baldrian 2017) and specifically for micro-eukaryotes (Oliverio et al. 2020). As expected, these organismal groups were also structured by soil depth. Vertical differences in bacterial community composition have previously been reported from sub-boreal spruce (Hartmann et al. 2012) and pine dominated montane forest (Eilers et al. 2012), where different bacterial taxa tended to show variable abundances with soil depth. We found that the most abundant group, Proteobacteria (32% of the reads), was significantly more abundant in the organic layer of these deciduous forests, which is in agreement with previous studies in coniferous forests (Baldrian et al. 2012; López-Mondéjar et al. 2015). In these studies, however, the sampling was limited to the litter and humus layers, whereas we investigated communities down to 30 cm mineral soil depth. Changes in the relative abundance of Verrucomicrobia and Bacteriodetes with depth were particularly striking, both being more abundant in the organic layer. A similar pattern has been reported previously from coniferous forests for Bacteriodetes (Eilers et al. 2012). Both Bacteriodetes and Proteobacteria are typically copiotrophic bacteria, found commonly in combined C- and nutrient-rich environments (Goldfarb et al. 2011). Observed higher dominance of Acidobacteria taxa Gp2 and Gp6 in the C- and nutrient-poor deeper mineral soil is in agreement with Fierer, Bradford and Jackson (2007), where higher abundances was observed in C-poor bulk soils compared to rhizosphere. As the soil substrate quality become poor and resistant to degradation in deeper soil, copiotrophic bacteria are replaced by oligotrophic bacteria, such as Acidobacteria, that are able to cope with more recalcitrant substrates, or Actinobacteria with a high metabolic plasticity (VanInsberghe et al. 2013).
Although the diversity patterns showed limited changes with soil depth for the micro-eukaryotes, there was still decline in abundance of nematodes, arthropods and Apicomplexa with soil depth. Root-derived carbon enters the soil animal food web via different pathways: animals can either feed (i) directly on living or dead roots, (ii) on bacteria living on root exudates or (iii) on fungi that acquire carbon from roots, i.e. mycorrhizae. The positive correlation between fungal biomass and micro-eukaryotic abundance may reflect the fungi's importance in driving the community patterns. Since several eukaryotic organisms, including protists (e.g. Cercomonas and Lecythium), testate (e.g. Cryptodifflugia) and naked (e.g. Acanthamoeba and Leptomyxa) amoebae (Dumack, Müller and Bonkowski 2016; Geisen et al. 2016), exclusively feed on fungi, changes in micro-eukaryotic communities with declining fungal richness and biomass is expected. High abundance of parasitic Apicomplexa (Gregarines) in the organic layer possibly reflects host dependencies (ex. Arthropoda) in the same depth (Bates et al. 2013; Mahé et al. 2017). In contrast, the higher abundances of Catenulida (Metazoa; flat-worms) and Marionina (Annelid; ringed-worms) in mineral soil may be due to their burrowing ability.
Depth matters: inter-kingdom co-occurrence patterns
Co-occurrence and network analyses can be used to explore putative interactions among microbial communities. Positive correlations mean that genera co-occur more than by chance, which may potentially be due to mutualism, parasitism, predation or alternatively, shared niche preferences. On the other hand, negative correlations may suggest competitive exclusion, or alternatively, preference for different niches (Röttjers and Faust 2018). Despite an overall higher microbial diversity in the forest floor (LHF), the relative amounts of positive correlations between genera was similar in the organic and the upper mineral soil, whereas the total amount of correlations (positive or negative) was higher in the upper mineral layer. In addition to the highest number of co-occurrences in the upper mineral layer, the network here was most dense, with higher degree and neighbourhood connectivity. The structural properties of network statistics suggest that organisms in the upper mineral layer form more association with each other compared to upper organic and deeper mineral soil layers. The LFH horizon is likely the most unstable and disturbed environment due to fresh litter supply from aboveground vegetation, and is highly influenced by fluctuating climatic conditions such as precipitation and temperature. Compared to forest floor, in the upper mineral soil the variability in the environmental conditions is lower whereas the amount of available plant-derived C is higher compared to deeper mineral layers. Therefore, organisms with competitive trait (C strategy) is expected to be more common in this soil layer. This variability in microbial trait strategy may result in a shift in the microbial communities from organic to the upper mineral layer. While comparing network complexity among arable, grass and forest ecosystem, Creamer et al. (2016), observed a denser microbial network in relatively stable forest soils, where a developed food web is expected. Since communities in the C-poor deeper mineral soil are relatively stable due to less environmental fluctuations and microbial organisms themselves potentially represent a relatively more important carbon source for each other (Crowther et al. 2013; Xiong et al. 2017). This may provide higher connectivity among microbial communities with more biotic interactions opportunities for C exchange and their survival (Milici et al. 2016). We also would like to highlight that seasonal variation may affect network architecture along depth gradients (Ings et al. 2009), but as our data were collected at one time point, they can to be used to validate the impact of time on the network architecture.
As expected, in the mineral soil we found that the network density as well as the proportion of positive correlations decreased downwards in the soil profile and, correspondingly, negative correlations increased. These findings are corresponding with Creamer et al. (2016), who showed that microbial networks are relatively denser in nutrient-rich soils, compared to nutrient-poor arable soil and positive interactions are stronger in soils with lower organic matter. The relative increase in negative correlations with soil depth may, on one hand, indicate that the relevant taxa have different niches preference, and/or that competition for the same resources is increasing with depth, as suggested by Weiss et al. (2016). Only a few organisms with highly specialized enzymatic capabilities can decompose organic matter in the lower mineral soil where nutrient availability is low. This, in turn, means less freely accessible C to other organisms as well as potentially lower abundance of organisms as a C source through predation and parasitism. In the lower mineral soil there is less opportunity for organisms to interact due to the lower species richness of all organismal groups, which may explain the lower network complexity in the lower mineral (M2 and M3) horizons.
Based on the network analyses alone, we cannot separate the dominant process(es) behind the positive correlations, i.e. whether this is an indication of shared niche preferences in the soil or biotic interactions in the form of synergetic relationships, commensalism, parasitism or proto-cooperation. Which process(es) are structuring the networks are also expected to vary for different organisms. For instance, members of the ubiquitous soil bacterial group Firmicutes (Barberán et al. 2011) such as Vagococcus, Bacillus, Enterococcus, Paenibacillus and Macrococcus, which are copiotrophic in nature, tended to co-occur more in the upper mineral layer than expected by chance. This may be because they share a specific (and yet undefined) niche. The soil yeast fungus Candida, also showed a high co-occurrence with members of Firmicutes in the upper mineral layer. This could suggest a putative role of the Candida yeasts as a nutrient and energy source for the predatory bacteria (Botha 2011) or vice versa, and further they can be opportunists feeding on the product decomposed by other bacterial groups (Mašínová, Yurkov and Baldrian 2018).
CONCLUSIONS
In this study, we demonstrate vertical niche partitioning for bacterial, fungal and other micro-eukaryotic communities in forest soils, mirroring concurrent changes in soil properties (H1). Dominant taxa varied across the soil profile, and diversity declined significantly with depth independent of microbial groups. These results suggest that different soil horizons should be taken into consideration when assessing microorganisms in soil. We demonstrate that inter-kingdom co-occurrence patterns vary dependent on soil layer (H2) and the most complex network occurs in upper mineral soil layer. Although the results are correlative in nature, they provide hypotheses about depth-dependent biotic interactions in forest soils, calling for more detailed experimental studies. Based on our results we also suggest to use a broader set of primers and DNA-markers in future community studies to capture a more comprehensive picture of the soil microbiota.
Supplementary Material
ACKNOWLEDGEMENTS
The work was funded by the Research Council of Norway [Grant numbers 255307 and 240859]. Thanks are due to Helge Meissner and Jan Světlik for assistance in the field, Monica Fongen, Jan-Erik Jacobsen, and Helge Meissner for soil sample pre-treatment and chemical analyses, and Stephanie Eisner for calculating metrological data. We acknowledge Tobias Guldberg Frøslev for providing us varying size (7-9 bp) Multiplex Identification DNA-tags sequences and Line Nybakken for ergosterol analysis.
Contributor Information
Sunil Mundra, Section for Genetics and Evolutionary Biology (EvoGene), Department of Biosciences, University of Oslo, NO-0316 Oslo, Norway; Department of Biology, College of Science, United Arab Emirates University, Al-Ain, Abu-Dhabi, UAE.
O Janne Kjønaas, NIBIO, Department of Terrestrial Ecology, NO-1431 Ås, Norway.
Luis N Morgado, Section for Genetics and Evolutionary Biology (EvoGene), Department of Biosciences, University of Oslo, NO-0316 Oslo, Norway; Naturalis Biodiversity Center, 2300 RA Leiden, the Netherlands.
Anders Kristian Krabberød, Section for Genetics and Evolutionary Biology (EvoGene), Department of Biosciences, University of Oslo, NO-0316 Oslo, Norway.
Yngvild Ransedokken, Faculty of Environmental and Natural Resource Management, Norwegian University of Life Sciences, NO-1432 Ås, Norway.
Håvard Kauserud, Section for Genetics and Evolutionary Biology (EvoGene), Department of Biosciences, University of Oslo, NO-0316 Oslo, Norway.
Conflicts of interest
None declared.
REFERENCES
- Anderson IC, Campbell CD, Prosser JI.. Potential bias of fungal 18S rDNA and internal transcribed spacer polymerase chain reaction primers for estimating fungal biodiversity in soil. Environ Microbiol. 2003;5:36–47. [DOI] [PubMed] [Google Scholar]
- Anderson IC, Genney DR, Alexander IJ.. Fine-scale diversity and distribution of ectomycorrhizal fungal mycelium in a Scots pine forest. New Phytol. 2014;201:1423–30. [DOI] [PubMed] [Google Scholar]
- Asplund J, Kauserud H, Ohlson Met al. Spruce and beech as local determinants of forest fungal community structure in litter, humus and mineral soil. FEMS Microbiol Ecol. 2018;95:fiy232. [DOI] [PubMed] [Google Scholar]
- Bahram M, Hildebrand F, Forslund SKet al. Structure and function of the global topsoil microbiome. Nature. 2018;560:233–7. [DOI] [PubMed] [Google Scholar]
- Baldrian P, Kolařík M, Štursová Met al. Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J. 2012;6:248–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldrian P, Valášková V.. Degradation of cellulose by basidiomycetous fungi. FEMS Microbiol Rev. 2008;32:501–21. [DOI] [PubMed] [Google Scholar]
- Baldrian P, Větrovský T, Cajthaml Tet al. Estimation of fungal biomass in forest litter and soil. Fungal Ecology. 2013;6:1–11. [Google Scholar]
- Baldrian P. Forest microbiome: diversity, complexity and dynamics. FEMS Microbiol Rev. 2017;41:109–30. [DOI] [PubMed] [Google Scholar]
- Barberán A, Bates ST, Casamayor EOet al. Using network analysis to explore co-occurrence patterns in soil microbial communities. Isme J. 2011;6:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardgett RD, van der Putten WH.. Belowground biodiversity and ecosystem functioning. Nature. 2014;515:505. [DOI] [PubMed] [Google Scholar]
- Bates ST, Clemente JC, Flores GEet al. Global biogeography of highly diverse protistan communities in soil. ISME J. 2013;7:652–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellemain E, Carlsen T, Brochmann Cet al. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol. 2010;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet FG, Legendre P, Borcard D.. Forward selection of explanatory variables. Ecology. 2008;89:2623–32. [DOI] [PubMed] [Google Scholar]
- Botha A. The importance and ecology of yeasts in soil. Soil Biol Biochem. 2011;43:1–8. [Google Scholar]
- Bödeker ITM, Clemmensen KE, de Boer Wet al. Ectomycorrhizal Cortinarius species participate in enzymatic oxidation of humus in northern forest ecosystems. New Phytol. 2014;203:245–56. [DOI] [PubMed] [Google Scholar]
- Bödeker ITM, Lindahl BD, Olson Ået al. Mycorrhizal and saprotrophic fungal guilds compete for the same organic substrates but affect decomposition differently. Funct Ecol. 2016;30:1967–78. [Google Scholar]
- Chaffron S, Rehrauer H, Pernthaler Jet al. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 2010;20:947–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemmensen KE, Bahr A, Ovaskainen Oet al. Roots and Associated Fungi Drive Long-Term Carbon Sequestration in Boreal Forest. Science. 2013;339:1615–8. [DOI] [PubMed] [Google Scholar]
- Creamer RE, Hannula SE, Leeuwen JPVet al. Ecological network analysis reveals the inter-connection between soil biodiversity and ecosystem function as affected by land use across Europe. Applied Soil Ecology. 2016;97:112–24. [Google Scholar]
- Crowther TW, Stanton DWG, Thomas SMet al. Top-down control of soil fungal community composition by a globally distributed keystone consumer. Ecology. 2013;94:2518–28. [DOI] [PubMed] [Google Scholar]
- de Araujo ASF, Mendes LW, Lemos LNet al. Protist species richness and soil microbiome complexity increase towards climax vegetation in the Brazilian Cerrado. Commun Biol. 2018;1:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas E, Besson M, Brice M-Het al. Analysing ecological networks of species interactions. Biological Reviews. 2019;94:16–36. [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen Net al. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl Environ Microbiol. 2006;72:5069–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries FT, Griffiths RI, Bailey Met al. Soil bacterial networks are less stable under drought than fungal networks. Nat Commun. 2018;9:3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickie IA, Xu B, Koide RT.. Vertical niche differentiation of ectomycorrhizal hyphae in soil as shown by T-RFLP analysis. New Phytol. 2002;156:527–35. [DOI] [PubMed] [Google Scholar]
- Du C, Geng Z, Wang Qet al. Variations in bacterial and fungal communities through soil depth profiles in a Betula albosinensis forest. J Microbiol. 2017;55:684–93. [DOI] [PubMed] [Google Scholar]
- Dumack K, Müller MEH, Bonkowski M.. Description of Lecythium terrestris sp. nov. (Chlamydophryidae, Cercozoa), a soil dwelling protist feeding on fungi and algae. Protist. 2016;167:93–105. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, CJ C.et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers KG, Debenport S, Anderson Set al. Digging deeper to find unique microbial communities: The strong effect of depth on the structure of bacterial and archaeal communities in soil. Soil Biol Biochem. 2012;50:58–65. [Google Scholar]
- Ekelund F, Rønn R, Christensen S.. Distribution with depth of protozoa, bacteria and fungi in soil profiles from three Danish forest sites. Soil Biol Biochem. 2001;33:475–81. [Google Scholar]
- Fernandez CW, Kennedy PG.. Revisiting the ‘Gadgil effect’: do interguild fungal interactions control carbon cycling in forest soils?. New Phytol. 2016;209:1382–94. [DOI] [PubMed] [Google Scholar]
- Fierer N, Bradford MA, Jackson RB.. Toward an ecological classification of soil bacteria. Ecology. 2007;88:1354–64. [DOI] [PubMed] [Google Scholar]
- Fierer N, Jackson RB. The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA. 2006;103:626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer R, Mues V, Ulrich Eet al. Monitoring of atmospheric deposition in European forests and an overview on its implication on forest condition. Appl Geochem. 2007;22:1129–39. [Google Scholar]
- Fuhrman JA, Steele JA.. Community structure of marine bacterioplankton: patterns, networks, and relationships to function. Aquat Microb Ecol. 2008;53:69–81. [Google Scholar]
- Geisen S, Bonkowski M.. Methodological advances to study the diversity of soil protists and their functioning in soil food webs. Applied Soil Ecology. 2018;123:328–33. [Google Scholar]
- Geisen S, Koller R, Hünninghaus Met al. The soil food web revisited: Diverse and widespread mycophagous soil protists. Soil Biol Biochem. 2016;94:10–8. [Google Scholar]
- Geisen S, Mitchell EAD, Wilkinson DMet al. Soil protistology rebooted: 30 fundamental questions to start with. Soil Biol Biochem. 2017;111:94–103. [Google Scholar]
- Genney DR, Anderson IC, Alexander IJ.. Fine-scale distribution of pine ectomycorrhizas and their extramatrical mycelium. New Phytol. 2006;170:381–90. [DOI] [PubMed] [Google Scholar]
- Goldfarb KC, Karaoz U, Hanson CAet al. Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front Microbiol. 2011;2:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillou L, Bachar D, Audic Set al. The Protist Ribosomal Reference database (PR(2)): a catalog of unicellular eukaryote Small Sub-Unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 2013;41:D597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadziavdic K, Lekang K, Lanzen Aet al. Characterization of the 18S rRNA gene for designing universal eukaryote specific primers. PLoS One. 2014;9:e87624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M, Howes CG, VanInsberghe Det al. Significant and persistent impact of timber harvesting on soil microbial communities in Northern coniferous forests. The Isme Journal. 2012;6:2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez DJ, David AS, Menges ESet al. Environmental stress destabilizes microbial networks. ISME J. 2021, DOI 10.1038/s41396-020-00882-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand F, Tadeo R, Voigt AYet al. LotuS: an efficient and user-friendly OTU processing pipeline. Microbiome. 2014;2:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbie EA, Hofmockel KS, van Diepen LTAet al. Fungal carbon sources in a pine forest: evidence from a 13C-labeled global change experiment. Fungal Ecology. 2014a;10:91–100. [DOI] [PubMed] [Google Scholar]
- Hobbie EA, van Diepen LTA, Lilleskov EAet al. Fungal functioning in a pine forest: evidence from a 15N-labeled global change experiment. New Phytol. 2014b;201:1431–9. [DOI] [PubMed] [Google Scholar]
- Hugerth LW, Muller EEL, Hu YOOet al. Systematic Design of 18S rRNA Gene Primers for Determining Eukaryotic Diversity in Microbial Consortia. PLoS One. 2014;9:e95567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrmark K, Bödeker ITM, Cruz-Martinez Ket al. New primers to amplify the fungal ITS2 region – evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol Ecol. 2012;82:666–77. [DOI] [PubMed] [Google Scholar]
- Ings TC, Montoya JM, Bascompte Jet al. Review: Ecological networks – beyond food webs. J Anim Ecol. 2009;78:253–69. [DOI] [PubMed] [Google Scholar]
- Jumpponen A, Jones KL, Blair J.. Vertical distribution of fungal communities in tallgrass prairie soil. Mycologia. 2010;102:1027–41. [DOI] [PubMed] [Google Scholar]
- Kurtz ZD, Müller CL, Miraldi ERet al. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLoS Comp Biol. 2015;11:e1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyaschenko J, Clemmensen KE, Karltun Eet al. Below-ground organic matter accumulation along a boreal forest fertility gradient relates to guild interaction within fungal communities. Ecol Lett. 2017;20:1546–55. [DOI] [PubMed] [Google Scholar]
- Kõljalg U, Nilsson RH, Abarenkov Ket al. Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol. 2013;22:5271–7. [DOI] [PubMed] [Google Scholar]
- Lindahl BD, Ihrmark K, Boberg Jet al. Spatial separation of litter decomposition and mycorrhizal nitrogen uptake in a boreal forest. New Phytol. 2007;173:611–20. [DOI] [PubMed] [Google Scholar]
- Lindahl BO, Taylor AFS, Finlay RD.. Defining nutritional constraints on carbon cycling in boreal forests – towards a less `phytocentric' perspective. Plant Soil. 2002;242:123–35. [Google Scholar]
- Lladó S, López-Mondéjar R, Baldrian P.. Forest Soil Bacteria: Diversity, Involvement in Ecosystem Processes, and Response to Global Change. Microbiol Mol Biol Rev. 2017;81:e00063–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Mondéjar R, Voříšková J, Větrovský Tet al. The bacterial community inhabiting temperate deciduous forests is vertically stratified and undergoes seasonal dynamics. Soil Biol Biochem. 2015;87:43–50. [Google Scholar]
- López-Mondéjar R, Zühlke D, Becher Det al. Cellulose and hemicellulose decomposition by forest soil bacteria proceeds by the action of structurally variable enzymatic systems. Sci Rep. 2016;6:25279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahé F, de Vargas C, Bass Det al. Parasites dominate hyperdiverse soil protist communities in Neotropical rainforests. Nat Ecol Amp; Evol. 2017;1:0091. [DOI] [PubMed] [Google Scholar]
- Mašínová T, Yurkov A, Baldrian P.. Forest soil yeasts: Decomposition potential and the utilization of carbon sources. Fungal Ecol. 2018;34:10–9. [Google Scholar]
- McDonald D, Price MN, Goodrich Jet al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendiburu Fd, Simon R.. Agricolae - Ten years of an open source statistical tool for experiments in breeding, agriculture and biology. PeerJ PrePrints. 2015;3:e1748v1:17. [Google Scholar]
- Menkis A, Urbina H, James TYet al. Archaeorhizomyces borealis sp. nov. and a sequence-based classification of related soil fungal species. Fungal Biol. 2014;118:943–55. [DOI] [PubMed] [Google Scholar]
- Milici M, Deng Z-L, Tomasch Jet al. Co-occurrence Analysis of Microbial Taxa in the Atlantic Ocean Reveals High Connectivity in the Free-Living Bacterioplankton. Front Microbiol. 2016;7:649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NGU Norge og Svalbard med havområder (Areal information on Norway and Svalbard including ocean), accessed 19 January2020, 2020. [Google Scholar]
- Nguyen NH, Song Z, Bates STet al. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2015;20:241–8. [Google Scholar]
- Nikolenko SI, Korobeynikov AI, Alekseyev MA.. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genomics. 2013;14:S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson RH, Anslan S, Bahram Met al. Mycobiome diversity: high-throughput sequencing and identification of fungi. Nat Rev Microbiol. 2019;17:95–109. [DOI] [PubMed] [Google Scholar]
- Nilsson RH, Ryberg M, Abarenkov Ket al. The ITS region as a target for characterization of fungal communities using emerging sequencing technologies. FEMS Microbiol Lett. 2009;296:97–101. [DOI] [PubMed] [Google Scholar]
- Nilsson RH, Veldre V, Hartmann Met al. An open source software package for automated extraction of ITS1 and ITS2 from fungal ITS sequences for use in high-throughput community assays and molecular ecology. Fungal Ecology. 2010;3:284–7. [Google Scholar]
- Ogner G, Wickstrøm T, Remedios Get al. The chemical analysis program of the Norwegian Forest Research Institute 2000. Norwegian Forest Research Institute: Ås, Norway, 1999. [Google Scholar]
- Oksanen J, Blanchet FG, Kindt Ret al. Vegan: community ecology package. R package version 2.0-7, DOI citeulike-article-id:10213170. http://cran.r-project.org/, 2013. [Google Scholar]
- Oliverio AM, Geisen S, Delgado-Baquerizo Met al. The global-scale distributions of soil protists and their contributions to belowground systems. Sci Adv. 2020;6:eaax8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena R, Tejedor J, Zeller Bet al. Interspecific temporal and spatial differences in the acquisition of litter-derived nitrogen by ectomycorrhizal fungal assemblages. New Phytol. 2013;199:520–8. [DOI] [PubMed] [Google Scholar]
- Pereira APdA, Andrade PAMd, Bini Det al. Shifts in the bacterial community composition along deep soil profiles in monospecific and mixed stands of Eucalyptus grandis and Acacia mangium. PLoS One. 2017;12:e0180371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto-Figueroa EA, Seddon E, Yashiro Eet al. Archaeorhizomycetes spatial distribution in soils along wide elevational and environmental gradients reveal co-abundance patterns with other fungal saprobes and potential weathering capacities. Front Microbiol. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapov AM, Goncharov AA, Semenina EEet al. Arthropods in the subsoil: abundance and vertical distribution as related to soil organic matter, microbial biomass and plant roots. Eur J Soil Biol. 2017;82:88–97. [Google Scholar]
- Ransedokken Y, Asplund J, Ohlson Met al. Vertical distribution of soil carbon in boreal forest under European beech and Norway spruce. Eur J Forest Res. 2019;138:353–61. [Google Scholar]
- R Core Development Team . R: A language and environment for statistical computing. http://www.R-project.org: R Foundation for Statistical Computing, Vienna, 2018. [Google Scholar]
- Rognes T, Flouri T, Nichols Bet al. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosling A, Cox F, Cruz-Martinez Ket al. Archaeorhizomycetes: Unearthing an Ancient Class of Ubiquitous Soil Fungi. Science. 2011;333:876–9. [DOI] [PubMed] [Google Scholar]
- Rosling A, Landeweert R, Lindahl BDet al. Vertical distribution of ectomycorrhizal fungal taxa in a podzol soil profile. New Phytol. 2003;159:775–83. [DOI] [PubMed] [Google Scholar]
- Rousk J, Baath E, Brookes PCet al. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010;4:1340–51. [DOI] [PubMed] [Google Scholar]
- Röttjers L, Faust K.. From hairballs to hypotheses–biological insights from microbial networks. FEMS Microbiol Rev. 2018;42:761–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santalahti M, Sun H, Jumpponen Aet al. Vertical and seasonal dynamics of fungal communities in boreal Scots pine forest soil. FEMS Microbiol Ecol. 2016;92:fiw170. [DOI] [PubMed] [Google Scholar]
- Schadt CW, Rosling A.. Comment on “Global diversity and geography of soil fungi”. Science. 2015;348:1438. [DOI] [PubMed] [Google Scholar]
- Schoch CL, Seifert KA, Huhndorf Set al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci. 2012;109:6241–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurig C, Smittenberg RH, Berger Jet al. Microbial cell-envelope fragments and the formation of soil organic matter: a case study from a glacier forefield. Biogeochemistry. 2013;113:595–612. [Google Scholar]
- Seppey CVW, Singer D, Dumack Ket al. Distribution patterns of soil microbial eukaryotes suggests widespread algivory by phagotrophic protists as an alternative pathway for nutrient cycling. Soil Biol Biochem. 2017;112:68–76. [Google Scholar]
- Singer D, Duckert C, Heděnec Pet al. High-throughput sequencing of litter and moss eDNA reveals a positive correlation between the diversity of Apicomplexa and their invertebrate hosts across alpine habitats. Soil Biol Biochem. 2020;147:107837. [Google Scholar]
- Smoot ME, Ono K, Ruscheinski Jet al. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterkenburg E, Bahr A, Brandström Durling Met al. Changes in fungal communities along a boreal forest soil fertility gradient. New Phytol. 2015;207:1145–58. [DOI] [PubMed] [Google Scholar]
- Swain DM, Yadav SK, Tyagi Iet al. A prophage tail-like protein is deployed by Burkholderia bacteria to feed on fungi. Nat Commun. 2017;8:404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Štursová M, Zifcakova L, Leigh MBet al. Cellulose utilization in forest litter and soil: identification of bacterial and fungal decomposers. FEMS Microbiol Ecol. 2012;80:735–46. [DOI] [PubMed] [Google Scholar]
- Tedersoo L, Bahram M, Cajthaml Tet al. Tree diversity and species identity effects on soil fungi, protists and animals are context dependent. Isme J. 2015;10:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedersoo L, Bahram M, Põlme Set al. Global diversity and geography of soil fungi. Science. 2014;346. [DOI] [PubMed] [Google Scholar]
- Tedersoo L, Lindahl B.. Fungal identification biases in microbiome projects. Environ Microbiol Rep. 2016;8:774–9. [DOI] [PubMed] [Google Scholar]
- VanInsberghe D, Hartmann M, Stewart GRet al. Isolation of a substantial proportion of forest soil bacterial communities detected via pyrotag sequencing. Appl Environ Microbiol. 2013;79:2096–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter PC, Nitsche F, Arndt H.. The hidden diversity of flagellated protists in soil. Protist. 2018;169:432–49. [DOI] [PubMed] [Google Scholar]
- Voříšková J, Brabcová V, Cajthaml Tet al. Seasonal dynamics of fungal communities in a temperate oak forest soil. New Phytol. 2014;201:269–78. [DOI] [PubMed] [Google Scholar]
- Wardle DA, Bardgett RD, Klironomos JNet al. Ecological linkages between aboveground and belowground biota. Science. 2004;304:1629. [DOI] [PubMed] [Google Scholar]
- Weiss S, Van Treuren W, Lozupone Cet al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016;10:1669–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm RC, Singh R, Eltis LDet al. Bacterial contributions to delignification and lignocellulose degradation in forest soils with metagenomic and quantitative stable isotope probing. ISME J. 2018, DOI 10.1038/s41396-018-0279-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Jousset A, Guo Set al. Soil protist communities form a dynamic hub in the soil microbiome. ISME J. 2017;12:634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Chen Y-s, Guo Y-c.. Design of integrated system for heterogeneous network query terminal. J Comp App. 2009;29:2191–21. [Google Scholar]
- Zhang J, Kobert K, Flouri Tet al. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30:614–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Žifčáková L, Větrovský T, Lombard Vet al. Feed in summer, rest in winter: microbial carbon utilization in forest topsoil. Microbiome. 2017;5:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.