

Keywords: anemia, chronic kidney disease, Na-K-ATPase, reactive oxygen species, red blood cell
Abstract
Chronic kidney disease (CKD) is one of the most prominent diseases affecting our population today. According to the Factsheet published by Centers for Disease Control and Prevention (CDC), it effects approximately 15% of the total population in the United States in some way, shape, or form. Within the myriad of symptomatology associated with CKD, one of the most prevalent factors in terms of affecting quality of life is anemia. Anemia of CKD cannot be completely attributed to one mechanism or cause, but rather has a multifactorial origin in the pathophysiology of CKD. While briefly summarizing well-documented risk factors, this review, as a hypothesis, aims to explore the possible role of Na-K-ATPase and its signaling function [especially recent identified reactive oxygen species (ROS) amplification function] in the interwoven mechanisms of development of the anemia of CKD.
INTRODUCTION
Chronic kidney disease (CKD) may be caused by many risk factors, including diabetes, hypertension, heart disease, hereditary causes, and age, especially over the age of 60 yr. Anemia as a result of CKD was first linked by Dr. Richard Bright (1). Since then, the causes of CKD-related anemia have been found to be multifactorial, especially with moderate or severe kidney function loss (CKD stage 3 or 4) and kidney failure (stage 5) as well as with hypertension, diabetes, and other chronic diseases (2–6). Race and sex may also influence the disease progression (7, 8). Although CKD progression can be a cause and potentially a consequence of anemia, analysis of the Chronic Renal Insufficiency Cohort (CRIC) Study showed that anemia can also be a cause and potentially a consequence of CKD, especially end-stage kidney disease (ESKD) progression (9).
Anemia of CKD involves multiple well-documented pathophysiological factors (Figure 1), including low erythropoietin (EPO) level, iron deficiency, decreased red blood cell (RBC) production, and decreased RBC lifespan (3, 10–14). The regulating factors include oxygen-sensing hypoxia-inducible transcription factor (HIF) (15–20) and liver-derived hormone hepcidin (21–23) under different conditions, such as inflammation, abnormal renal oxygen sensing, blood loss, and decreased RBC lifespan. Circulating EPO levels were normal to slightly increased in anemia of CKD. However, this was still considered inappropriately low as patients with anemia with normal kidney function have ∼10–100 times higher EPO levels (3). It is speculated that it may be due to uremia-induced inhibitors of erythropoiesis, which have yet been identified. Anemia of CKD has also been linked to iron losses. Patients with CKD tend to lose iron (especially in patients who undergo hemodialysis), have impaired dietary absorption of iron from the gastrointestinal tract, and have functional iron deficiency (3, 24). Iron deficiency leads to low serum transferrin saturation and normal-to-high ferritin concentration. Furthermore, hepcidin, which is one of the main hormones responsible for maintaining systemic iron homeostasis, has also been implicated in the pathogenesis of anemia of inflammation. Hepcidin produced by the liver in inflammatory states has been shown to suppress the iron exporter ferroportin that results in decreased iron availability for erythropoiesis to occur. Increased hepcidin production in patients with CKD may be due to increased expression by inflammatory cytokines accompanying reduced renal clearance. There have been studies showing shortened RBC survival in patients with CKD.
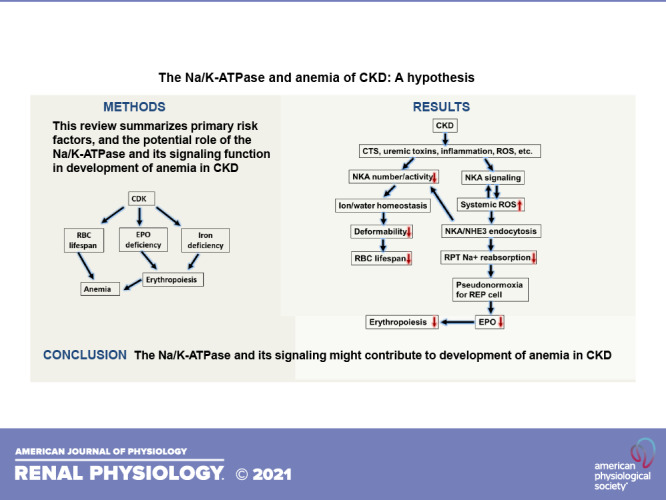
Figure 1.

Illustration of the major causes of anemia of chronic kidney disease (CKD). RBC, red blood cell; GFR, glomerular filtration rate; REP, renal erythropoietin (EPO)-producing cells; HIF, hypoxia-inducible factor.
ANEMIA IN THE CKD POPULATION
The definition of anemia is established based on the concentration of hemoglobin (Hb), which is the oxygen carrying protein housed inside RBCs. This concentration needs to be <13 g/dL in men or <12 g/dL in women for the diagnosis of anemia to apply. CKD is a common affliction among the general population today, affecting ∼15% of the general population based on the Factsheet, “the Chronic Kidney Disease in the United States, 2019” published by CDC (https://tinyurl.com/whbzdhz). Although CKD is age related, it is different in the occurrence of CKD in the following aspects: age >65 yr (38%) versus age 45–64 yr (13%) versus age 18–44 yr (7%), woman (15%) versus men (12%), non-Hispanic Blacks (16%) versus non-Hispanic Whites (13%) versus non-Hispanic Asian (12%). It is also noted that ∼14% of Hispanics have CKD and African Americans are ∼3 times more likely than Whites to develop ESRD. The link between anemia and CKD has been established for nearly 200 years (1). Research has progress on this topic greatly since that time. Today, CKD can be divided into two categories: nondialysis-dependent CKD (NDD-CKD) and dialysis-dependent CKD (DD-CKD). Studies have shown that Health Related Quality of Life (HRQOL) is significantly decreased in patients with severe anemia (Hb <10 g/dL) and has even affected patients with mild-to-moderate anemia (Hb 10–12 g/dL) (25).
While anemia associated with CKD can be considered under the umbrella term of anemia of chronic disease, anemia of CKD is characterized as a unique entity when compared with other chronic forms of anemia. Initially, this distinction was drawn based on inappropriate EPO levels (26). Over time however, the causes of CKD-related anemia have been shown to be multifactorial in nature rather than just based on inappropriate levels of EPO. Many studies have described evidence of iron deficiency in patients suffering from CKD. Decreased RBC lifespan secondary to increased levels of reactive oxygen species (ROS) is also a major contributing factor for this type of anemia. Hyperparathyroidism, bone marrow suppression secondary to uremic toxins, and infection have also been labelled as possible culprits.
ERYTHROPOIETIN DEFICIENCY
One of the key causes behind anemia of CKD is EPO deficiency. This deficiency is due to blunting of the response of the renal EPO-producing cells (REPs, interstitial fibroblast-like cells located in the outer medulla and cortex that produce EPO in an on-off mechanism to simultaneously stimulate renal fibrosis and erythropoiesis in the kidney), as REPs also produce fibrogenic elements, including inflammatory cytokines in CKD. There is a direct link between fibrosis and anemia demonstrated by the loss of EPO-producing ability in myofibroblast-transformed REPs, making it a potential target to cure both fibrosis and anemia simultaneously (27–29). Although the renal fibrosis inhibits EPO production, reduced EPO production might further promote fibrosis and inflammation (30, 31). Premature erythroid cells express the EPO receptor (EPOR) on the cell surface, which when bound by EPO, leads to differentiation and maturation of the erythroid precursor into fully formed RBCs. EPO is released by healthy kidney cells in response to hypoxia, which adds to the oxygen-carrying capacity of the blood and alleviation of the hypoxic state. In this instance, anemia in CKD is related to the fact that differentiation of precursor cells in mature RBCs is blunted by the lack of EPO produced by the diseased kidney. The kidney cannot respond to the hypoxic state of the body due to fibrosed cells, and as RBCs die off due to their natural lifespan, the reduced EPO concentration cannot balance this loss with new mature erythrocytes (32).
In response to inappropriate EPO levels in anemia of CKD, new medications were developed and tried to combat this problem. The first example of this treatment came in the form of recombinant human EPO (r-hEPO), which was approved by the Food and Drug Administration in 1989 (33). It demonstrated a dose-dependent response to r-hEPO in a group with established renal insufficiency, specifically to the packed cell volume (PCV). Bone marrow studies on these patients revealed an increase in erythroid, megakaryocyte, and granulocyte progenitor cells. Some adverse effects did occur in these patients, but they were not definitively linked to r-hEPO administration (34).
Erythropoiesis-stimulating agents (ESAs) were developed as a novel treatment for the disease as well. r-hEPO, as well as other ESAs, proved to be beneficial in most patients with anemia of CKD. The majority of patients showed an increase in Hb, decreased need for RBC transfusions, and improved quality of life associated with anemia. However, many of these patients developed complications including higher target Hb levels, increased risk of thrombosis, higher rates of cerebrovascular events, earlier required kidney transplant therapy, and increased mortality than those with lower target hemoglobin levels. It is unsure whether or not the treatment themselves or the higher hemoglobin levels were the culprit for these adverse events, but it was apparent that better treatment was needed. As ESAs can increase cardiovascular events, it was recommended that the lowest dose of EPO should be used to maintain the Hb level between 10 g/dL and 11.5 g/dL to avoid the need of RBC transfusions (when Hb level is too low) (35).
IRON DEFICIENCY AND INFLAMMATION
The next of the major components of the complex interplay resulting in anemia of CKD is iron deficiency. This has similar pathophysiological findings compared with the cause of anemia in chronic disease (12). For humans, ∼80% of daily requirement iron (∼25 mg) is used for erythropoiesis. The major source of iron is the recycling of iron from senescent RBCs (36). The main cause of iron deficiency lies in chronic inflammation caused by the chronic diseases. A battery of inflammatory markers were upregulated in about half of patients with CKD, including but not limited to, C-reactive protein (CRP), tumor necrosis factor-α (TNF-α), interleukin (IL)-1, and IL-6. IL-6 specifically causes an increase of hepcidin in the body (37). The liver-generated hepcidin is a hormone regulating systemic iron balance, which can be upregulated by iron and inflammation, and downregulated by iron deficiency, anemia, and hypoxia (38, 39). In mouse models, silence of hepcidin gene expression leads to toxic levels of high iron in the body, and overexpression of hepcidin gene leads to severe iron deficiency anemia (40, 41). This phenomenon was also observed in patients with hepcidin mutation and hepcidin expression adenomas (42, 43).
The combination of inflammation-promoting hepcidin expression and the decrease in hepcidin clearance by injured kidney leads to a dramatic increase in hepcidin concentration in the later stages of CKD (44, 45). Ferroportin is an iron transporter located on the basolateral membrane of the intestinal epithelium that allows iron absorption by the intestine (46). Mechanistically, ferroportin is the receptor of hepcidin, and binding of hepcidin with ferroportin induces internalization and degradation of ferroportin. This leads to diminished iron absorption by the gastrointestinal tract (47). Experimental treatments have started to arise focusing on the hepcidin-ferroportin interaction. For example, LY2928057 (a humanized IgG4 monoclonal antibody) binds ferroportin and prevents hepcidin-induced ferroportin degradation without affecting iron efflux. Application of the antibody results in decrease of ferritin, with increases of iron and transferrin saturation (48). Another focus is the hepcidin-antibody complexes that could not bind to ferroportin, and, thus, could not affect iron homeostasis in inflammatory states. In the mouse model, the antihepcidin antibody increased serum iron concentrations as well as increased Hb levels 1–2 g/dL over a 7-day period. When treated concomitantly with ESA, the Hb level rose 3–4 g/dL in the same timeframe (49). Increased effectiveness of the antihepcidin antibody with low dose ESA treatment suggested a synergistic relationship secondary to the hepcidin downregulation of EPO. Suppression of hepcidin mRNA showed similar results and could be a treatment to prevent the side effects associated with required intravenous administration of iron (50). One of the important side effects in the setting of CKD, specifically in relation to intravenously administered iron, is the concern that intravenous iron may accelerate kidney damage (51). For treatment of iron deficiency, iron supplement was recommended (iron pill for patients not on hemodialysis and intravenously iron administration for patients who undergo hemodialysis) (35), even though there were questions about dosing and risks (52, 53).
Another causative agent that is elevated in CKD is fibroblast growth factor 23 (FGF23). FGF23 is upregulated in response to hyperphosphatemia and elevated parathyroid hormone (PTH). Specifically, in CKD, as the renal clearance of phosphate is decreased by renal injury, FGF23 is upregulated to correct the increased phosphate load. However, FGF23 normally acts to increase renal clearance of phosphate to induce phosphaturia. This creates a self-perpetuating feedback loop between hyperphosphatemia, FGF23, and CKD (54). In a 5/6 partial nephrectomy (PNx) mouse model to mimic CKD, inhibition of FGF23 increased erythropoiesis, ameliorated iron deficiency by decreasing inflammation, and decreased erythroid apoptosis. This not only highlighted the important role of FGF23 in anemia of CKD, but also lent evidence for FGF23 being an important mediator in erythroid cell life cycles. Furthermore, a lipopolysaccharide (LPS)-inflammation mouse model was used to study the relationship between FGF23 and inflammation-mediated hypoferremia. The study demonstrated that FGF23 was upregulated in the early signaling pathways of inflammation. This led to upregulation of hepcidin, which could be prevented by inhibition of FGF23. It is presumed that the downregulation in hepcidin production following inhibition of FGF23 led to the amelioration of hypoferremia in these mice (55). Despite this study not being directly related to CKD-induced anemia, it suggests that activation of the FGF23 pathway may lead to iron deficiency through upregulation of hepcidin, which is secondary to inflammation. This could lend credence to the idea that treatment with FGF23 inhibition in anemia of CKD might follow a similar pathway in amelioration of iron deficiency in CKD. FGF23 is also directly related to hyperparathyroidism, another player in the multifactorial manifestation of anemia CKD (56). The EPO-FGF23 signaling pathway that directly regulates erythroid progenitor cells in bone marrow was recently reviewed and was found to be linked to CKD, inflammation, iron deficiency, and other factors.
RBC LIFESPAN
Anemia is a disease of imbalance between RBC creation and RBC death. Another major factor in the progression of anemia of CKD is the shortened RBC survivability (57). RBC survivability is the measure of how long an RBC remains active in the body after it is released by the bone marrow following erythrocyte maturation. This survivability time is usually around 120 days in a healthy individual. A new study in 2019 used a new method of measuring RBC lifespan (CO breath measurements) to evaluate at what stage of CKD that diminished survivability is quantifiable (58). This study showed that RBC survivability could be affected as early as stage 1 of CKD (59).
Eryptosis, which is the term for suicide of erythrocytes coined by Lang et al. (60), is triggered by multiple mechanisms. One such mechanism is triggered when calcium is inducted into the erythrocyte via unspecific calcium cation channels [probably through the RBC plasma membrane calcium pump PMCA (60)]. Conversely, these calcium cation channels are inhibited by erythropoietin. However, in the setting of CKD or renal failure, there is a decreased concentration of EPO in the body (32, 61). The induction of calcium into the cell triggers specific enzymes within the erythrocyte, such as flippase, floppase, and scramblease. These enzymes regulate the RBC’s ability to translocate phosphatidylserine onto membrane surface (62). Phosphatidylserine itself usually resides on the inner leaflet of the cellular membrane. When it is translocated on the outside of the cell membrane, it becomes a marker for RBC apoptosis, that is, eryptosis (63). During this eryptotic process, as the erythrocytes undergo apoptosis, the cellular membranes become distorted, which creates blebs and microvesicles. The phosphatidylserine now translocated on the surface of these microvesicles also make it easier to bind to endothelial cells of the veins and arteries (61, 64). This could also contribute to the multifactorial development of cardiovascular compromise in patients with CKD.
OXIDATIVE STRESS
Oxidative stress [ROS and reactive nitrogen species (RNS)] is also a major contributing factor leading to eryptosis in CKD (61, 65). Following suit with the already complicated nature of anemia in CKD, the formation of ROS is also multifactorial in nature. Kidneys are very sensitive to oxygen change since they have high oxygen demand, mainly driven by sodium reabsorption in renal tubules (especially proximal tubules) (66–69). When kidneys sense hypoxia, renal EPO-producing cells (REP cells; located in the outer medulla and cortex) produce EPO to stimulate erythropoiesis (11, 27, 31). In the setting of CKD, the abnormal oxygen-sensing mechanism keeps the kidneys in a hypoxia/pseudonormoxia state to suppress HIF signaling, leading to inhibition of EPO expression and iron reabsorption/availability (11, 19, 23, 70). On the other hand, uremic toxins, such as acrolein and indoxyl sulfate, are upregulated by limited kidney clearance and lead to an increase in oxidative stress (71–74). Previous studies have shown that retention of uremic toxins in CKD or the inability of the kidneys to remove putative uremic mediators leads to the increased ROS and subsequent oxidative stress and inflammation (75). Acrolein is a breakdown product of LPS that is usually removed by the kidney. However, in the presence of kidney damage, it begins to accumulate in the body leading to increased ROS and oxidative stress in cellular and clinical models (73, 76, 77). Acrolein also upregulates the presence of ceramide on the cell surface of erythrocytes, making them more susceptible to eryptosis (76).
Indoxyl sulfate, which can cause renal inflammation and dysregulation of oxygen sensing in REP cells, is also a contributor in ROS generation in anemia of CKD. Indoxyl sulfate is a uremic toxin that accumulates in the body with the progression of CKD and further worsens CKD progression (72, 78). The importance of indoxyl sulfate in anemia of CKD is further enhanced by its inhibition of EPO expression through inhibition of EPO mRNA expression and transcription factor HIF-α, which directly controls the transcription of EPO (79, 80). Indoxyl sulfate is transported into the erythrocytes by organic anion transporter 2 (OAT2). When OAT2 is inhibited by ketoprofen, indoxyl sulfate-induced oxidative stress on human RBCs (thus eryptosis) is reduced, which is related to the regulation of NADPH oxidase (72). Accumulated evidence suggested a multifactorial effect of indoxyl sulfate on oxidative stress in CKD. It was shown that not only is indoxyl sulfate involved in the activation of NADPH oxidase, but it also inhibited antioxidative processes by downregulating total glutathione. Glutathione is one of the most potent nonenzymatic antioxidants levels in endothelial cells (81). Although the underlying molecular mechanisms are not yet fully known, the retention of uremic toxins in patients with this clinical disorder shifts the redox balance, favoring free radicals and pro-oxidant state (71). This redox imbalance further results in proinflammatory cytokine production and this inflammatory milieu positively correlates with increasing stages of CKD (82). Several studies have provided evidence of uremia-associated oxidant stress and inflammation, including increases in CRP and nitric oxide (NO), and resulting in activation of the inflammatory NF-κB pathway (83). This results in notable increases of inflammatory cytokines, such as IL-1, IL-6, TNF-α, and monocyte chemoattractant protein-1. Although excessive accumulation of uremic toxin certainly contributes to the aggravation of diseased phenotype by increasing oxidative stress, several other sources of ROS generation have been implicated in CKD and renal failure-associated cardiomyopathy. The pathophysiological mechanism underlying the increased ROS production in CKD is also mediated by the activation of NADPH oxidase, xanthine oxidase, and mitochondrial dysfunction as well as decreased activity of serum antioxidant, paraoxonase-1 (PON1) activity (75, 84). Even though RBCs have a potent antioxidant system, circulating RBCs are continuously exposed to ROS that can damage the RBC membrane. This may lead to aging and decreased deformability, thereby inhibiting its ability to pass through narrow capillaries in the microcirculation. RBC deformability was found to be decreased in several other disease states associated with oxidative stress, such as hypertension, diabetes mellitus, and aging (60, 85–89).
Na-K-ATPASE AND ITS SIGNALING-ROS AXIS IN THE DEVELOPMENT OF ANEMIA OF CKD: A HYPOTHESIS
Na-K-ATPase is a plasmalemmal ion transporter that is expressed in all known mammalian cells. As the primary transporter, the Na+ gradient across the plasma membrane is the driving force for other Na+-coupled transporters. Therefore, Na-K-ATPase could contribute to anemia of CKD in several aspects.
As the primary ion transporter, Na-K-ATPase maintains cellular ion homeostasis. The number/activity of RBC Na-K-ATPase controls the surface area-to-volume ratio and cytoplasmic rheology, two of the major factors contributing to RBC deformability and clearance (60, 89–91). Decrease in number/activity of Na-K-ATPase and increase in ROS are two major contributors in RBC aging and shortened RBC lifespan (60, 85). The number/activity of RBC Na-K-ATPase was significantly decreased not only by aging, but also in patients with CKD with end-stage renal failure on dialysis and type 2 diabetes mellitus as well (92, 93). Moreover, CKD patients and hypertensive patients tend to have increased circulating cardiotonic steroids (CTS, a group of compounds with similar structure functioning as specific inhibitors and ligands of Na-K-ATPase) (94, 95). CTS had been classified as a new class of hormone, making Na-K-ATPase a potential therapeutic target (96–99).
Other than an ion transporter, Na-K-ATPase had a secondary function of signaling when its α1 subunit interacts with tyrosine kinase c-Src to form a Na-K-ATPase/c-Src complex (100, 101). In pig renal proximal tubule LLC-PK1 cells, Na-K-ATPase signaling is redox sensitive, and activation of Na-K-ATPase signaling stimulates ROS generation and endocytosis of Na-K-ATPase. This leads to decreased number/activity of functional membrane Na-K-ATPase (102, 103). Interestingly, this redox-sensitive Na-K-ATPase signaling can be activated by both CTS and/or ROS, which triggers a signaling cascade that results in the amplification of ROS generation in a feed forward mechanism (104–106). Moreover, inhibition of Na-K-ATPase signaling function is able to ameliorate the increases in systemic oxidative stress (104, 107). This signaling mechanism has been demonstrated in the development of uremic cardiomyopathy and anemia in the 5/6th PNx mouse model. The model showed that in addition to increased plasma levels of indoxyl sulfate and inflammatory markers, the blockage of the signaling cascade ameliorated the anemia caused by CKD (104, 107, 108). Furthermore, this signaling mechanism also induces other pathophysiological alterations with upregulation of inflammatory cytokines (109–112). In preadipocyte 3T3-L1 cells and MSCs-derived adipocytes, indoxyl sulfate and p-cresol sulfate can also stimulate the Na-K-ATPase signaling-mediated oxidant amplification loop, suggesting that this mechanism is not limited to renal proximal tubular cells (71). In RBCs, increase in oxidative stress can also active Src kinases (113), but it is not clear whether they are Na-K-ATPase associated or not. The link between CTS/ROS and the Na-K-ATPase/c-Src signaling leading to upregulation of ROS went one step further and identified a peptide, NaKtide (or cell permeable variant pNaKtide with a HIV TAT lead sequence). This peptide binds to the c-Src kinase domain and functions as a c-Src antagonist, effectively inhibiting c-Src activation as well as excessive generation of ROS in CKD (107). NaKtide was developed from the 20-amino acid sequence of the ND1 segment of the Na-K-ATPase α1-subunit that targets c-Src kinase domain to prevent c-Src phosphorylation. pNaKtide was further developed by adding a 13-amino acid HIV-TAT leading sequence to facilitate its cellular permeation (114, 115). Recent studies have further shown that systemic administration of pNaKtide attenuates obesity and associated metabolic syndrome, nonalcoholic steatohepatitis, and atherosclerosis (110, 112). These studies extensively elucidated that the inhibition of Na-K-ATPase signaling by pNaKtide restores systemic redox imbalance and attenuates the release of inflammatory cytokines. This leads to improvement of the diseased phenotype.
Furthermore, Na-K-ATPase itself is able to sense changes of ROS/RNS (34, 104). These observations make Na-K-ATPase and its signaling function a potent regulating candidate of systemic oxidative stress and inflammation. Thus, this also affects the RBC redox status and lifespan as well as erythropoiesis in the settings of CKD and ESRD (Fig. 2). It stands to reason then that, with the increased generation of ROS by this mechanism as well in the setting of CKD, it would be by another mechanism in which eryptosis would be increased. It would also be reasonable to suggest that treatment with pNaKtide could possibly lead to a reduction in RBC loss, and thus, be beneficial in the setting of anemia of CKD.
Figure 2.

Illustration of the possible roles of Na-K-ATPase (NKA) and its signaling on red blood cell (RBC) lifespan. Both cardiotonic steroids (CTS) and ROS induce a E2-prone conformational change that leads to inhibition of NKA activity. On the other hand, both CTS and ROS activate NKA signaling that leads to stimulation of ROS generation and NKA endocytosis. Taking together, NKA and its signaling weaken the sodium gradient status across the membrane and impair iron and nutrient transport by other secondary transporters. Eventually, RBC clearance is increased through decreased in deformability. NHE3, Na/H exchanger isoform 4; GFR, glomerular filtration rate; HIF, hypoxia-inducible factor.
CONCLUSIONS
Mechanisms of anemia in CKD have been shown to be interwoven and multifactorial. EPO deficiency caused by CKD leads to decreased differentiation and maturation of erythroid precursors, which results in anemia. Current medications include ESAs treatment to increase erythropoiesis and decrease hepcidin level, which may cause side effects at high doses. Iron deficiency in CKD involved increased inflammatory cytokines and hepcidin. This causes limitations of iron absorption and availability as well as EPO resistance. Current medications include iron supplement (iron pill and iron IV administration) and HIF-PHD (prolyl-hydroxylase domain-containing proteins) inhibitors to elevate iron availability, increase iron uptake, and increase endogenous EPO release. RBC survivability in CKD involves inflammatory cytokines, oxidative stress, and uremic toxins, which affect RBC deformability/clearance and lifespan. Other than ESAs and HIF-PHD inhibitors, FGF23 might be a potential therapeutic target. FGF23 is upregulated in inflammatory states and inhibiting its upregulation resulted in increased erythropoiesis, amelioration of iron deficiency, and decreased eryptosis.
Recent studies regarding Na-K-ATPase signaling have shed insight on another possible explanation for development of anemia of CKD. Na-K-ATPase not only functions as a physiological ion transporter, but it also functions as a signaling transducer leading to generation of ROS and oxidative modification of protein. The (p)NaKtide, which was created from the N domain of the Na-K-ATPase α1-subunit, is able to inhibit the Na-K-ATPase signaling-mediated feedforward amplification loop in the generation of ROS, resulting in prevention of PNx-mediated anemia and attenuation of inflammation and fibrosis (107, 111, 112). Further research on ROS and its effects on anemia should investigate if there is indeed a relationship with Na-K-ATPase signaling, ROS generation, and its effects on anemia of CKD. If there is a relationship in this area, treatments targeting this pathway may provide a breakthrough in future treatment of anemia of CKD (Fig. 2).
GRANTS
This work was funded by National Institutes of Health Grants 1R15DK106666 (to J.L.), 1R15HL150721 (to K.S.), and RO1HL071556 (to J.I.S.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.D.M., J.C., M.C., Y.N., F.B., and J.L. drafted manuscript; J.L. and J.I.S. edited, revised and approved final version of manuscript.
REFERENCES
- 1.Bright R. Cases and observations illustrative of renal disease, accompanied with the secretion of albuminous urine. Med Chir Rev 25: 23–35, 1836. [PMC free article] [PubMed] [Google Scholar]
- 2.Astor BC, Muntner P, Levin A, Eustace JA, Coresh J. Association of kidney function with anemia: the Third National Health and Nutrition Examination Survey (1988-1994). Arch Intern Med 162: 1401–1408, 2002. doi: 10.1001/archinte.162.12.1401. [DOI] [PubMed] [Google Scholar]
- 3.Babitt JL, Lin HY. Mechanisms of anemia in CKD. J Am Soc Nephrol 23: 1631–1634, 2012. doi: 10.1681/ASN.2011111078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen TK, Estrella MM, Astor BC, Greene T, Wang X, Grams ME, Appel LJ. Longitudinal changes in hematocrit in hypertensive chronic kidney disease: results from the African-American Study of Kidney Disease and Hypertension (AASK). Nephrol Dial Transplant 30: 1329–1335, 2015. doi: 10.1093/ndt/gfv037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McClellan W, Aronoff SL, Bolton WK, Hood S, Lorber DL, Tang KL, Tse TF, Wasserman B, Leiserowitz M. The prevalence of anemia in patients with chronic kidney disease. Curr Med Res Opin 20: 1501–1510, 2004. doi: 10.1185/030079904X2763. [DOI] [PubMed] [Google Scholar]
- 6.Stauffer ME, Fan T. Prevalence of anemia in chronic kidney disease in the United States. PLoS One 9: e84943, 2014. doi: 10.1371/journal.pone.0084943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang P-Y, Chien L-N, Lin Y-F, Wu M-S, Chiu W-T, Chiou H-Y. Risk factors of gender for renal progression in patients with early chronic kidney disease. Medicine (Baltimore) 95: e4203, 2016. doi: 10.1097/MD.0000000000004203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel KV, Harris TB, Faulhaber M, Angleman SB, Connelly S, Bauer DC, Kuller LH, Newman AB, Guralnik JM. Racial variation in the relationship of anemia with mortality and mobility disability among older adults. Blood 109: 4663–4670, 2007. doi: 10.1182/blood-2006-10-055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saraf SL, Hsu JY, Ricardo AC, Mehta R, Chen J, Chen TK, Fischer MJ, Hamm L, Sondheimer J, Weir MR, Zhang X, Wolf M, Lash JP. Anemia and incident end-stage kidney disease. Kidney360 1: 623–630, 2020. doi: 10.34067/KID.0000852020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batchelor EK, Kapitsinou P, Pergola PE, Kovesdy CP, Jalal DI. Iron deficiency in chronic kidney disease: updates on pathophysiology, diagnosis, and treatment. J Am Soc Nephrol 31: 456–468, 2020. doi: 10.1681/ASN.2019020213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koury MJ, Haase VH. Anaemia in kidney disease: harnessing hypoxia responses for therapy. Nat Rev Nephrol 11: 394–410, 2015. doi: 10.1038/nrneph.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopez A, Cacoub P, Macdougall IC, Peyrin-Biroulet L. Iron deficiency anaemia. Lancet 387: 907–916, 2016. doi: 10.1016/S0140-6736(15)60865-0. [DOI] [PubMed] [Google Scholar]
- 13.Percy MJ, Furlow PW, Beer PA, Lappin TR, McMullin MF, Lee FS. A novel erythrocytosis-associated PHD2 mutation suggests the location of a HIF binding groove. Blood 110: 2193–2196, 2007. doi: 10.1182/blood-2007-04-084434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Semenza GL. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood 114: 2015–2019, 2009. doi: 10.1182/blood-2009-05-189985. [DOI] [PubMed] [Google Scholar]
- 15.Haase VH. Hypoxic regulation of erythropoiesis and iron metabolism. Am J Physiol Renal Physiol 299: F1–F13, 2010. doi: 10.1152/ajprenal.00174.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplan JM, Sharma N, Dikdan S. Hypoxia-inducible factor and its role in the management of anemia in chronic kidney disease. Int J Mol Sci 19: 389, 2018. doi: 10.3390/ijms19020389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lappin TR, Lee FS. Update on mutations in the HIF: EPO pathway and their role in erythrocytosis. Blood Rev 37: 100590, 2019. doi: 10.1016/j.blre.2019.100590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee FS, Percy MJ. The HIF pathway and erythrocytosis. Annu Rev Pathol 6: 165–192, 2011. doi: 10.1146/annurev-pathol-011110-130321. [DOI] [PubMed] [Google Scholar]
- 19.Liu Q, Davidoff O, Niss K, Haase VH. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest 122: 4635–4644, 2012. doi: 10.1172/JCI63924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wenger RH, Hoogewijs D. Regulated oxygen sensing by protein hydroxylation in renal erythropoietin-producing cells. Am J Physiol Renal Physiol 298: F1287–F1296, 2010. doi: 10.1152/ajprenal.00736.2009. [DOI] [PubMed] [Google Scholar]
- 21.Babitt JL, Lin HY. Molecular mechanisms of hepcidin regulation: implications for the anemia of CKD. Am J Kidney Dis 55: 726–741, 2010. doi: 10.1053/j.ajkd.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ganz T, Nemeth E. Iron balance and the role of hepcidin in chronic kidney disease. Semin Nephrol 36: 87–93, 2016. doi: 10.1016/j.semnephrol.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vokurka M, Krijt J, Sulc K, Necas E. Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis. Physiol Res 55: 667–674, 2006. [DOI] [PubMed] [Google Scholar]
- 24.Fudin R, Jaichenko J, Shostak A, Bennett M, Gotloib L. Correction of uremic iron deficiency anemia in hemodialyzed patients: a prospective study. Nephron 79: 299–305, 1998. doi: 10.1159/000045053. [DOI] [PubMed] [Google Scholar]
- 25.Hoshino J, Muenz D, Zee J, Sukul N, Speyer E, Guedes M, Lopes AA, Asahi K, van Haalen H, James G, Dhalwani N, Pecoits-Filho R, Bieber B, Robinson BM, Pisoni RL; CKDopps Investigators. Associations of hemoglobin levels with health-related quality of life, physical activity, and clinical outcomes in persons with stage 3-5 nondialysis CKD. J Ren Nutr 30: 404–414, 2020. doi: 10.1053/j.jrn.2019.11.003. [DOI] [PubMed] [Google Scholar]
- 26.Yilmaz MI, Solak Y, Covic A, Goldsmith D, Kanbay M. Renal anemia of inflammation: the name is self-explanatory. Blood Purif 32: 220–225, 2011. doi: 10.1159/000328037. [DOI] [PubMed] [Google Scholar]
- 27.Pan X, Suzuki N, Hirano I, Yamazaki S, Minegishi N, Yamamoto M. Isolation and characterization of renal erythropoietin-producing cells from genetically produced anemia mice. PLoS One 6: e25839, 2011. doi: 10.1371/journal.pone.0025839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Souma T, Suzuki N, Yamamoto M. Renal erythropoietin-producing cells in health and disease. Front Physiol 6: 167, 2015. doi: 10.3389/fphys.2015.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamazaki S, Souma T, Hirano I, Pan X, Minegishi N, Suzuki N, Yamamoto M. A mouse model of adult-onset anaemia due to erythropoietin deficiency. Nat Commun 4: 1950, 2013. doi: 10.1038/ncomms2950. [DOI] [PubMed] [Google Scholar]
- 30.Noguchi CT, Wang L, Rogers HM, Teng R, Jia Y. Survival and proliferative roles of erythropoietin beyond the erythroid lineage. Expert Rev Mol Med 10: e36, 2008. doi: 10.1017/S1462399408000860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Souma T, Yamazaki S, Moriguchi T, Suzuki N, Hirano I, Pan X, Minegishi N, Abe M, Kiyomoto H, Ito S, Yamamoto M. Plasticity of renal erythropoietin-producing cells governs fibrosis. J Am Soc Nephrol 24: 1599–1616, 2013. doi: 10.1681/ASN.2013010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vázquez-Méndez E, Gutiérrez-Mercado Y, Mendieta Condado E, Gálvez-Gastélum F, Esquivel H, Sánchez-Toscano Y, Morales-Martínez C, Canales-Aguirre A, Márquez-Aguirre A. Recombinant erythropoietin provides protection against renal fibrosis in adenine-induced chronic kidney disease. Mediators Inflamm 2020: 8937657–8937611, 2020. doi: 10.1155/2020/8937657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 12: 5447–5454, 1992. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bogdanova A, Petrushanko IY, Hernansanz-Agustín P, Martínez-Ruiz A. Oxygen sensing” by Na,K-ATPase: these miraculous thiols. Front Physiol 7: 314, 2016. doi: 10.3389/fphys.2016.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McMurray JJV, Parfrey PS, Anderson JW, Aljama P, Berns JS, Bohlius J, Drüeke TB, Finkelstein FO, Fishbane S, Ganz T, MacDougall IC, McDonald RA, McMahon LP, Obrador GT, Strippoli GFM, Weiss G, Wiȩcek A. Kidney disease: Improving global outcomes (KDIGO) anemia work group. KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney Int Suppl 2: 279–335, 2012. doi: 10.1038/kisup.2012.37. [DOI] [Google Scholar]
- 36.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell 117: 285–297, 2004. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 37.Plastina JC, Obara VY, Barbosa DS, Morimoto HK, Reiche EM, Graciano A, Delfino VD. Functional iron deficiency in patients on hemodialysis: prevalence, nutritional assessment, and biomarkers of oxidative stress and inflammation. J Braz Nephrol 41: 472–480, 2019. doi: 10.1590/2175-8239-jbn-2018-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 110: 1037–1044, 2002. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zumbrennen-Bullough K, Babitt JL. The iron cycle in chronic kidney disease (CKD): from genetics and experimental models to CKD patients. Nephrol Dial Transplant 29: 263–273, 2014. doi: 10.1093/ndt/gft443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA 98: 8780–8785, 2001. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, Grandchamp B, Sirito M, Sawadogo M, Kahn A, Vaulont S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci USA 99: 4596–4601, 2002. doi: 10.1073/pnas.072632499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, Loukopoulos D, Camaschella C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet 33: 21–22, 2003. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 43.Weinstein DA, Roy CN, Fleming MD, Loda MF, Wolfsdorf JI, Andrews NC. Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood 100: 3776–3781, 2002. doi: 10.1182/blood-2002-04-1260. [DOI] [PubMed] [Google Scholar]
- 44.Mercadel L, Metzger M, Haymann JP, Thervet E, Boffa J-J, Flamant M, Vrtovsnik F, Houillier P, Froissart M, Stengel B; NephroTest Study Group. The relation of hepcidin to iron disorders, inflammation and hemoglobin in chronic kidney disease. PLoS One 9: e99781, 2014. doi: 10.1371/journal.pone.0099781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaritsky J, Young B, Wang H-J, Westerman M, Olbina G, Nemeth E, Ganz T, Rivera S, Nissenson AR, Salusky IB. Hepcidin—a potential novel biomarker for iron status in chronic kidney disease. Clin J Am Soc Nephrol 4: 1051–1056, 2009. doi: 10.2215/CJN.05931108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Domenico ID, Ward DM, Langelier C, Vaughn MB, Nemeth E, Sundquist WI, Ganz T, Musci G, Kaplan J. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell 18: 2569–2578, 2007. doi: 10.1091/mbc.e07-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chawla LS, Krishnan M. Causes and consequences of inflammation on anemia management in hemodialysis patients. Hemodial Int 13: 222–234, 2009. doi: 10.1111/j.1542-4758.2009.00352.x. [DOI] [PubMed] [Google Scholar]
- 48.Barrington P, Sheetz MJ, Callies S, Waters DG, Berg PH, Pappas D, Marbury TC, Decker BS, Berg JK. Safety, tolerability, pharmacokinetics and pharmacodynamics of an anti-ferroportin antibody in patients with anemia due to chronic renal failure. Blood 128: 1280–1280, 2016. doi: 10.1182/blood.V128.22.1280.1280. [DOI] [Google Scholar]
- 49.Cooke KS, Hinkle B, Salimi-Moosavi H, Foltz I, King C, Rathanaswami P, Winters A, Steavenson S, Begley CG, Molineux G, Sasu BJ. A fully human anti-hepcidin antibody modulates iron metabolism in both mice and nonhuman primates. Blood 122: 3054–3061, 2013. doi: 10.1182/blood-2013-06-505792. [DOI] [PubMed] [Google Scholar]
- 50.Sasu BJ, Cooke KS, Arvedson TL, Plewa C, Ellison AR, Sheng J, Winters A, Juan T, Li H, Begley CG, Molineux G. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood 115: 3616–3624, 2010. doi: 10.1182/blood-2009-09-245977. [DOI] [PubMed] [Google Scholar]
- 51.Pisani A, Riccio E, Sabbatini M, Andreucci MD, Rio AD.Visciano B. . Effect of oral liposomal iron versus intravenous iron for treatment of iron deficiency anaemia in CKD patients: a randomized trial. Nephrol Dial Transplant 30: 645–652, 2015. doi: 10.1093/ndt/gfu357. [DOI] [PubMed] [Google Scholar]
- 52.Gaweda AE, Ginzburg YZ, Chait Y, Germain MJ, Aronoff GR, Rachmilewitz E. Iron dosing in kidney disease: inconsistency of evidence and clinical practice. Nephrol Dial Transplant 30: 187–196, 2015. doi: 10.1093/ndt/gfu104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rostoker G. When should iron supplementation in dialysis patients be avoided, minimized or withdrawn? Semin Dial 32: 22–29, 2019. doi: 10.1111/sdi.12732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fliser D, Kollerits B, Neyer U, Ankerst DP, Lhotta K, Lingenhel A, Ritz E, Fibroblast KF. Growth factor 23 (FGF23) predicts progression of chronic kidney disease: the mild to moderate kidney disease (MMKD) study. J Am Soc Nephrol 18: 2600–2608, 2007. doi: 10.1681/ASN.2006080936. [DOI] [PubMed] [Google Scholar]
- 55.Agoro R, Park MY, Le Henaff C, Jankauskas S, Gaias A, Chen G, Mohammadi M, Sitara D. C-FGF23 peptide alleviates hypoferremia during acute inflammation. Haematologica, 106(2):391–403, 2021. doi: 10.3324/haematol.2019.237040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krajisnik T, Olauson H, Mirza MA, Hellman P, Akerstrom G, Westin G, Larsson TE, Bjorklund P. Parathyroid Klotho and FGF-receptor 1 expression decline with renal function in hyperparathyroid patients with chronic kidney disease and kidney transplant recipients. Kidney Int 78: 1024–1032, 2010. doi: 10.1038/ki.2010.260. [DOI] [PubMed] [Google Scholar]
- 57.Joske RA, McAlister JM, Prankerd TA. Isotope investigations of red cell production and destruction in chronic renal disease. Clin Sci 15: 511–522, 1956. [PubMed] [Google Scholar]
- 58.Zhang HD, Ma YJ, Liu QF, Ye TZ, Meng FY, Zhou YW, Yu GP, Yang JP, Jiang H, Wang QS, Li GP, Ji YQ, Zhu GL, Du LT, Ji KM. Human erythrocyte lifespan measured by Levitt's CO breath test with newly developed automatic instrument. J Breath Res 12: 36003, 2018. doi: 10.1088/1752-7163/aaacf1. [DOI] [PubMed] [Google Scholar]
- 59.Li JH, Luo JF, Jiang Y, Ma YJ, Ji YQ, Zhu GL, Zhou C, Chu HW, Zhang HD. Red blood cell lifespan shortening in patients with early-stage chronic kidney disease. Kidney Blood Press Res 44: 1158–1165, 2019. doi: 10.1159/000502525. [DOI] [PubMed] [Google Scholar]
- 60.Lew VL, Tiffert T. On the mechanism of human red blood cell longevity: roles of calcium, the sodium pump, PIEZO1, and Gardos Channels. Front Physiol 8: 977, 2017. doi: 10.3389/fphys.2017.00977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lang E, Qadri SM, Lang F. Killing me softly – suicidal erythrocyte death. Int J Biochem Cell Biol 44: 1236–1243, 2012. doi: 10.1016/j.biocel.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 62.Woon LA, Holland JW, Kable EP, Roufogalis BD. Ca2+sensitivity of phospholipid scrambling in human red cell ghosts. Cell Calcium 25: 313–320, 1999. doi: 10.1054/ceca.1999.0029. [DOI] [PubMed] [Google Scholar]
- 63.Nguyen DB, Wagner-Britz L, Maia S, Steffen P, Wagner C, Kaestner L, Bernhardt I. Regulation of phosphatidylserine exposure in red blood cells. Cell Physiol Biochem 28: 847–856, 2011. doi: 10.1159/000335798. [DOI] [PubMed] [Google Scholar]
- 64.Chung S-M, Bae O-N, Lim K-M, Noh J-Y, Lee M-Y, Jung Y-S, Chung J-H. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler Thromb Vasc Biol 27: 414–421, 2007. doi: 10.1161/01.ATV.0000252898.48084.6a. [DOI] [PubMed] [Google Scholar]
- 65.Kuhn V, Diederich L, Keller TC, Kramer CM, Lückstädt W, Panknin C, Suvorava T, Isakson BE, Kelm M, Cortese-Krott MM. Red blood cell function and dysfunction: redox regulation, nitric oxide metabolism. Antioxid Redox Signal 26: 718–742, 2017. doi: 10.1089/ars.2016.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Halperin ML, Cheema-Dhadli S, Lin SH, Kamel KS. Properties permitting the renal cortex to be the oxygen sensor for the release of erythropoietin: clinical implications. Clin J Am Soc Nephrol 1: 1049–1053, 2006. doi: 10.2215/CJN.00100106. [DOI] [PubMed] [Google Scholar]
- 67.Kiil F, Aukland K, Refsum HE. Renal sodium transport and oxygen consumption. Am J Physiol 201: 511–516, 1961. doi: 10.1152/ajplegacy.1961.201.3.511. [DOI] [PubMed] [Google Scholar]
- 68.Lassen NA, Munck O, Thaysen JH. Oxygen consumption and sodium reabsorption in the kidney. Acta Physiol Scand 51: 371–384, 1961. doi: 10.1111/j.1748-1716.1961.tb02147.x. [DOI] [PubMed] [Google Scholar]
- 69.Thaysen JH, Lassen NA, Munck O. Sodium transport and oxygen consumption in the mammalian kidney. Nature 190: 919–921, 1961. doi: 10.1038/190919a0. [DOI] [PubMed] [Google Scholar]
- 70.Mastrogiannaki M, Matak P, Keith B, Simon MC, Vaulont S, Peyssonnaux C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest 119: 1159–1166, 2009. doi: 10.1172/JCI38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bartlett DE, Miller RB, Thiesfeldt S, Lakhani HV, Khanal T, Pratt RD, Cottrill CL, Klug RL, Adkins NS, Bown PC, Nease DB, Shapiro JI, Sodhi K. Uremic toxins activates Na/K-ATPase oxidant amplification loop causing phenotypic changes in adipocytes in in vitro models. Int J Mol Sci 19: 2685, 2018. doi: 10.3390/ijms19092685. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Dias GF, Bonan NB, Steiner TM, Tozoni SS, Rodrigues S, Nakao LS, Kuntsevich V, Pecoits Filho R, Kotanko P, Moreno-Amaral AN. Indoxyl sulfate, a uremic toxin, stimulates reactive oxygen species production and erythrocyte cell death supposedly by an organic anion transporter 2 (OAT2) and NADPH oxidase activity-dependent pathways. Toxins (Basel) 10: 280, 2018. doi: 10.3390/toxins10070280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Igarashi K, Ueda S, Yoshida K, Kashiwagi K. Polyamines in renal failure. Amino Acids 31: 477–483, 2006. doi: 10.1007/s00726-006-0264-7. [DOI] [PubMed] [Google Scholar]
- 74.Li L, Jiang L, Geng C, Cao J, Zhong L. The role of oxidative stress in acrolein-induced DNA damage in HepG2 cells. Free Radic Res 42: 354–361, 2008. doi: 10.1080/10715760802008114. [DOI] [PubMed] [Google Scholar]
- 75.Wang X, Shapiro JI. Evolving concepts in the pathogenesis of uraemic cardiomyopathy. Nat Rev Nephrol 15: 159–175, 2019. doi: 10.1038/s41581-018-0101-8. [DOI] [PubMed] [Google Scholar]
- 76.Ahmed MS, Langer H, Abed M, Voelkl J, Lang F. The uremic toxin acrolein promotes suicidal erythrocyte death. Kidney Blood Press Res 37: 158–167, 2013. doi: 10.1159/000350141. [DOI] [PubMed] [Google Scholar]
- 77.Kwolek-Mirek M, Zadrąg-Tęcza R, Bednarska S, Bartosz G. Acrolein-induced oxidative stress and cell death exhibiting features of apoptosis in the yeast Saccharomyces cerevisiae deficient in SOD1. Cell Biochem Biophys 71: 1525–1536, 2015. doi: 10.1007/s12013-014-0376-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vanholder R, Schepers E, Pletinck A, Nagler EV, Glorieux G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: a systematic review. J Am Soc Nephrol 25: 1897–1907, 2014. doi: 10.1681/ASN.2013101062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chiang C-K, Tanaka T, Inagi R, Fujita T, Nangaku M. Indoxyl sulfate, a representative uremic toxin, suppresses erythropoietin production in a HIF-dependent manner. Lab Invest 91: 1564–1571, 2011. doi: 10.1038/labinvest.2011.114. [DOI] [PubMed] [Google Scholar]
- 80.Wu C-J, Chen C-Y, Lai T-S, Wu P-C, Chuang C-K, Sun F-J, Liu H-L, Chen H-H, Yeh H-I, Lin C-S, Lin C-J. The role of indoxyl sulfate in renal anemia in patients with chronic kidney disease. Oncotarget 8: 83030–83037, 2017. doi: 10.18632/oncotarget.18789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dou L, Jourde-Chiche N, Faure V, Cerini C, Berland Y, Dignat-George F, Brunet P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J Thromb Haemost 5: 1302–1308, 2007. doi: 10.1111/j.1538-7836.2007.02540.x. [DOI] [PubMed] [Google Scholar]
- 82.Podkowińska A, Formanowicz D. Chronic kidney disease as oxidative stress- and inflammatory-mediated cardiovascular disease. Antioxidants (Basel) 9: 752, 2020. doi: 10.3390/antiox9080752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rapa SF, Di Iorio BR, Campiglia P, Heidland A, Marzocco S. Inflammation and oxidative stress in chronic kidney disease-potential therapeutic role of minerals, vitamins and plant-derived metabolites. Int J Mol Sci 21: 263, 2019. doi: 10.3390/ijms21010263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Duni A, Liakopoulos V, Rapsomanikis KP, Dounousi E. Chronic kidney disease and disproportionally increased cardiovascular damage: does oxidative stress explain the burden? Oxid Med Cell Longev 2017: 9036450, 2017. doi: 10.1155/2017/9036450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bogdanova A, Lutz H. Mechanisms tagging senescent red blood cells for clearance in healthy humans. Front Physiol 4: 387, 2013. doi: 10.3389/fphys.2013.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Diederich L, Suvorava T, Sansone R, Keller TC, Barbarino F, Sutton TR, Kramer CM, Lückstädt W, Isakson BE, Gohlke H, Feelisch M, Kelm M, Cortese-Krott MM. On the effects of reactive oxygen species and nitric oxide on red blood cell deformability. Front Physiol 9: 332, 2018. doi: 10.3389/fphys.2018.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mohanty J, Nagababu E, Rifkind J. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front Physiol 5, 2014. doi: 10.3389/fphys.2014.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pries AR, Secomb TW, Gaehtgens P. Biophysical aspects of blood flow in the microvasculature. Cardiovasc Res 32: 654–667, 1996. doi: 10.1016/0008-6363(96)00065-X. [DOI] [PubMed] [Google Scholar]
- 89.Radosinska J, Vrbjar N. The role of red blood cell deformability and Na,K-ATPase function in selected risk factors of cardiovascular diseases in humans: focus on hypertension, diabetes mellitus and hypercholesterolemia. Physiol Res 65, Suppl 1: S43–S54, 2016. [DOI] [PubMed] [Google Scholar]
- 90.Huisjes R, Bogdanova A, van Solinge WW, Schiffelers RM, Kaestner L, van Wijk R. Squeezing for life – properties of red blood cell deformability. Front Physiol 9: 656, 2018. doi: 10.3389/fphys.2018.00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tomaiuolo G. Biomechanical properties of red blood cells in health and disease towards microfluidics. Biomicrofluidics 8: 51501, 2014. doi: 10.1063/1.4895755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cheng JT, Kahn T, Kaji DM. Mechanism of alteration of sodium potassium pump of erythrocytes from patients with chronic renal failure. J Clin Invest 74: 1811–1820, 1984. doi: 10.1172/JCI111600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Koc B, Erten V, Yilmaz MI, Sonmez A, Kocar IH. The relationship between red blood cell Na/K-ATPase activities and diabetic complications in patients with type 2 diabetes mellitus. Endocrine 21: 273–278, 2003. doi: 10.1385/ENDO:21:3:273. [DOI] [PubMed] [Google Scholar]
- 94.Kolmakova EV, Haller ST, Kennedy DJ, Isachkina AN, Budny GV, Frolova EV, Piecha G, Nikitina ER, Malhotra D, Fedorova OV, Shapiro JI, Bagrov AY. Endogenous cardiotonic steroids in chronic renal failure. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association. Eur Renal Assoc 26: 2912–2919, 2011. doi: 10.1093/ndt/gfq772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Komiyama Y, Dong XH, Nishimura N, Masaki H, Yoshika M, Masuda M, Takahashi H. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin Biochem 38: 36–45, 2005. doi: 10.1016/j.clinbiochem.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 96.Aperia A. New roles for an old enzyme: Na,K-ATPase emerges as an interesting drug target. J Intern Med 261: 44–52, 2007. doi: 10.1111/j.1365-2796.2006.01745.x. [DOI] [PubMed] [Google Scholar]
- 97.Bagrov AY, Shapiro JI. Endogenous digitalis: pathophysiologic roles and therapeutic applications. Nat Clin Pract Nephrol 4: 378–392, 2008. doi: 10.1038/ncpneph0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev 61: 9–38, 2009. doi: 10.1124/pr.108.000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schoner W. Endogenous cardiac glycosides, a new class of steroid hormones. Eur J Biochem 269: 2440–2448, 2002. doi: 10.1046/j.1432-1033.2002.02911.x. [DOI] [PubMed] [Google Scholar]
- 100.Nie Y, Bai F, Chaudhry MA, Pratt R, Shapiro JI, Liu J. The Na/K-ATPase alpha1 and c-Src form signaling complex under native condition: a crosslinking approach. Sci Rep 10: 6006, 2020. doi: 10.1038/s41598-020-61920-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M, Maksimova E, Huang X-Y, Xie Z-J. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol Biol Cell 17: 317–326, 2006. doi: 10.1091/mbc.e05-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu J, Kesiry R, Periyasamy SM, Malhotra D, Xie Z, Shapiro JI. Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent mechanism. Kidney Int 66: 227–241, 2004. doi: 10.1111/j.1523-1755.2004.00723.x. [DOI] [PubMed] [Google Scholar]
- 103.Liu J, Liang M, Liu L, Malhotra D, Xie Z, Shapiro JI. Ouabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int 67: 1844–1854, 2005. doi: 10.1111/j.1523-1755.2005.00283.x. [DOI] [PubMed] [Google Scholar]
- 104.Liu J, Nie Y, Chaudhry M, Bai F, Chuang J, Sodhi K, Shapiro JI. The redox-sensitive Na/K-ATPase signaling in uremic cardiomyopathy. Int J Mol Sci 21: 1256, 2020. doi: 10.3390/ijms21041256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pratt RD, Brickman CR, Cottrill CL, Shapiro JI, Liu J. The Na/K-ATPase signaling: from specific ligands to general reactive oxygen species. Int J Mol Sci 19: 2600, 2018. doi: 10.3390/ijms19092600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yan Y, Shapiro AP, Haller S, Katragadda V, Liu L, Tian J, Basrur V, Malhotra D, Xie ZJ, Abraham NG, Shapiro JI, Liu J. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J Biol Chem 288: 34249–34258, 2013. doi: 10.1074/jbc.M113.461020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu J, Tian J, Chaudhry M, Maxwell K, Yan Y, Wang X, Shah PT, Khawaja AA, Martin R, Robinette TJ, El-Hamdani A, Dodrill MW, Sodhi K, Drummond CA, Haller ST, Kennedy DJ, Abraham NG, Xie Z, Shapiro JI. Attenuation of Na/K-ATPase mediated oxidant amplification with pNaKtide ameliorates experimental uremic cardiomyopathy. Sci Rep 6: 34592, 2016. [Erratum in Sci Rep 7: 6893, 2017]. doi: 10.1038/srep34592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sodhi K, Wang X, Chaudhry MA, Lakhani HV, Zehra M, Pratt R, Nawab A, Cottrill CL, Snoad B, Bai F, Denvir J, Liu J, Sanabria JR, Xie Z, Abraham NG, Shapiro JI. Central role for adipocyte Na,K-ATPase oxidant amplification loop in the pathogenesis of experimental uremic cardiomyopathy. J Am Soc Nephrol 31: 1746–1760, 2020. doi: 10.1681/ASN.2019101070. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 109.Pratt RD, Brickman C, Nawab A, Cottrill C, Snoad B, Lakhani HV, Jelcick A, Henderson B, Bhardwaj NN, Sanabria JR, Liu J, Xie Z, Abraham NG, Shapiro JI, Sodhi K. The adipocyte Na/K-ATPase oxidant amplification loop is the central regulator of western diet-induced obesity and associated comorbidities. Sci Rep 9: 7927–7927, 2019. [Erratum in Sci Rep 10: 19561, 2020]. doi: 10.1038/s41598-019-44350-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 110.Sodhi K, Maxwell K, Yan Y, Liu J, Chaudhry MA, Getty M, Xie Z, Abraham NG, Shapiro JI. pNaKtide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci Adv 1: e1500781, 2015. doi: 10.1126/sciadv.1500781. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 111.Sodhi K, Nichols A, Mallick A, Klug RL, Liu J, Wang X, Srikanthan K, Goguet-Rubio P, Nawab A, Pratt R, Lilly MN, Sanabria JR, Xie Z, Abraham NG, Shapiro JI. The Na/K-ATPase oxidant amplification loop regulates aging. Sci Rep 8: 9721–9721, 2018. doi: 10.1038/s41598-018-26768-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 112.Sodhi K, Srikanthan K, Goguet-Rubio P, Nichols A, Mallick A, Nawab A, Martin R, Shah PT, Chaudhry M, Sigdel S, El-Hamdani M, Liu J, Xie Z, Abraham NG, Shapiro JI. pNaKtide attenuates steatohepatitis and atherosclerosis by blocking Na/K-ATPase/ROS amplification in C57Bl6 and ApoE knockout mice fed a western diet. Sci Rep 7: 193–193, 2017. doi: 10.1038/s41598-017-00306-5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 113.Serafini M, Mallozzi C, Di Stasi AM, Minetti M. Peroxynitrite-dependent upregulation of Src kinases in red blood cells: strategies to study the activation mechanisms. In: Methods in Enzymology Academic Press, 2005, p. 215–229. [DOI] [PubMed] [Google Scholar]
- 114.Li Z, Cai T, Tian J, Xie JX, Zhao X, Liu L, Shapiro JI, Xie Z. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes Ouabain-activated signal transduction in cultured cells. J Biol Chem 284: 21066–21076, 2009. doi: 10.1074/jbc.M109.013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li Z, Zhang Z, Xie JX, Li X, Tian J, Cai T, Cui H, Ding H, Shapiro JI, Xie Z. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. J Biol Chem 286: 32394–32403, 2011. doi: 10.1074/jbc.M110.207597. [DOI] [PMC free article] [PubMed] [Google Scholar]