

Abstract
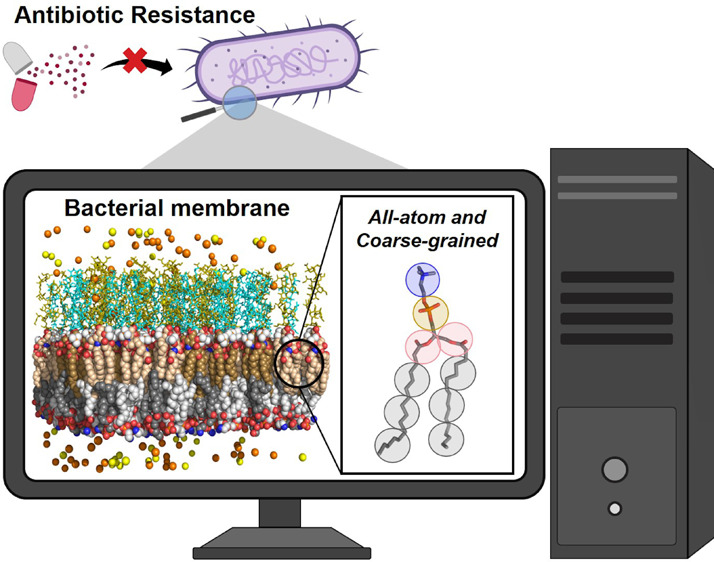
Antimicrobial resistance (AMR) represents a major threat to global public health in the 21st century, dramatically increasing the pandemic expectations in the coming years. The ongoing need to develop new antimicrobial treatments that are effective against multi-drug-resistant pathogens has led the research community to investigate innovative strategies to tackle AMR. The bacterial cell envelope has been identified as one of the key molecular players responsible for antibiotic resistance, attracting considerable interest as a potential target for novel antimicrobials effective against AMR, to be used alone or in combination with other drugs. However, the multicomponent complexity of bacterial membranes provides a heterogeneous morphology, which is typically difficult to study at the molecular level by experimental techniques, in spite of the significant development of fast and efficient experimental protocols. In recent years, computational modeling, in particular, molecular dynamics simulations, has proven to be an effective tool to reveal key aspects in the architecture and membrane organization of bacterial cell walls. Here, after a general overview about bacterial membranes, AMR mechanisms, and experimental approaches to study AMR, we review the state-of-the-art computational approaches to investigate bacterial AMR envelopes, including their limitations and challenges ahead. Representative examples illustrate how these techniques improve our understanding of bacterial membrane resistance mechanisms, hopefully leading to the development of novel antimicrobial drugs escaping from bacterial resistance strategies.
1. AMR in a Global Context
Antimicrobial resistance (AMR) has been identified by the World Health Organization (WHO) as one of the main threats to global health1 and has attracted numerous efforts to tackle it. AMR is the ability of a microorganism (such as bacteria, viruses, and some parasites) to resist the action of an antimicrobial agent (such as antibiotics, antivirals, and antimalarials) that would otherwise successfully kill them or stop growth and proliferation. As a result, standard treatments become ineffective, infections persist, may spread to others, and can seriously compromise surgery and procedures such as chemotherapy and other life-saving achievements of modern medicine, apart from the heavy social and economic burden.1
The WHO has published a list of antibiotic-resistant “priority pathogens”, a catalogue of 12 families of bacteria that pose the greatest threat to human health, divided into three categories according to the urgency for new antibiotics: medium, critical, and high priority. The most critical group includes different patterns of resistance (multi-drug resistant (MDR), extensively drug-resistant (XDR), and pan-drug-resistant (PDR) bacterial strains), often hospital-acquired, associated with serious life-threatening infections worldwide, prolonged illness, higher health care expenditures, and a greater risk of death.2 These strains can cause severe and often deadly bloodstream infections or pneumonia. Although the emergence of resistant microorganisms is driven by bacterial evolution, an accumulation of factors (including misuse of antibiotics in humans and animals, poor hygiene or infection control practices, easiness and frequency of worldwide travel, and transfer of patients between healthcare facilities) has accelerated and transformed AMR into a serious problem of public health worldwide, thus requiring efforts from all nations and sectors to provide an effective solution.1,2
The bacterial cell envelope is currently attracting considerable interest as a potential target of novel antimicrobials. Significant advances in the knowledge and understanding about the architecture and membrane organization are required to efficiently design and discover new antimicrobial agents able to overcome AMR.3−5 In recent years, computational modeling, in particular, molecular dynamics simulations, has shown to be an effective and versatile technique to unveil key aspects of the multicomponent complexity of bacterial membranes.6,7 Here, we start by giving a general overview about bacterial membranes, AMR mechanisms, and experimental approaches to study AMR, to deepen later into the state-of-the-art computational approaches to investigate bacterial envelopes in the context of antimicrobial resistance. Current limitations are discussed, and challenges ahead are shown. Representative examples illustrate how these techniques help to improve our understanding of bacterial membrane resistance mechanisms, with useful insights for the design and development of novel antimicrobial drugs able to overcome AMR.
2. Mechanisms for AMR
Pathogens have rapidly developed various mechanisms of escaping the antimicrobial drugs action. The key mechanisms responsible for resistance to antibiotics in bacteria are classified into four main categories (Figure 1):
Figure 1.

Schematic representation of different mechanism of antimicrobial resistance strategies in bacteria classified into four main categories: (i) limiting drug uptake, (ii) increasing drug extrusion; (iii) modifying drug target structure, and (iv) inactivating the drug structure. LPS, lipopolysaccharide; PBP, penicillin-binding protein. Figure was produced using images from Sevier Medical Art (https://smart.servier.com/), licensed under a Creative Common Attribution 3.0 Generic License (https://creativecommons.org/licenses/by/3.0/).
i. Limiting Drug Uptake
This mechanism is particularly relevant in Gram-negative bacteria, due to the presence of the outer membrane (OM), mainly constituted by lipopolysaccharides (LPS), whose hydrophobicity provides a perfect and impermeable barrier to most small drug molecules. Thus, many hydrophilic antibiotics diffuse across the OM via porin proteins. Outer membrane permeability can be modified, thereby decreasing permeability to antibiotics, either by reducing expression of porins or mutating porin genes, thus altering the porin channel permeability,8 or by forming biofilms which protect the bacteria against the host immune system and also from antibiotic penetration, by increasing the thick and sticky consistency of the biofilm matrix.
ii. Increasing Efflux
Overexpression of efflux pumps, ancient proton-dependent protein complexes, extrudes drugs from inside the cell. As an example, drug extrusion is an effective resistance mechanism in Staphylococcus aureus, where efflux pump NorA confers resistance to fluoroquinolone antibiotics; QacA MFS transporter exports cationic lipophilic drugs, including biocides such as benzalkonium chloride, and LmrS MFS efflux pump exports a variety of agents such as lincomycin, linezolid, chloramphenicol, and trimethoprim.9
iii. Target Site Modification
MDR bacteria often express many antibiotic resistance genes to provide a wide range of protection against common antibiotics. One of the most important examples of a target change is the acquisition of the mecA genes in Staphylococcus aureus, which encode for the penicillin-binding protein PBP-2A, thus conferring resistance to methicillin and to most other β-lactam antibiotics.9 PBPs are responsible for peptidoglycan (PGN) synthesis and assembly, making them excellent targets for selective modification.
iv. Drug Inactivation
The most common mechanism of resistance in bacteria is the chemical degradation and inactivation of antibiotics by bacterial enzymes. For example, hydrolysis occurs mainly in the case of β-lactam drugs by the action of β-lactamases, whereas other antibacterial agents suffer from different chemical modifications.8
3. Bacterial Membrane as a Target for Novel Antibacterial Agents
The discovery and development of novel antimicrobial agents to treat resistant bacterial infections generally relies on agents that need to cross the bacterial cell envelope. The main scientific struggle that interferes with the development of new antimicrobials against intrinsically resistant bacteria, especially Gram-negative, is the failure to find compounds that can enter bacterial cells.10 The struggle ultimately stems from an incomplete understanding of compound permeation through the bacterial envelope required for the identification of substances with the ability to overcome bacteria “permeability barriers”.11 The targeted disruption or perturbation of the bacterial envelope, less explored and exploited to develop AMR-targeting antibiotics, presents several advantages and benefits: (i) expected broad spectrum, activity against slow-growing, dormant, and drug-resistant strains; (ii) reduced risk of generating resistance since the bacterial envelope is composed of relatively conserved structures; hence, envelope-targeting antibiotics are likely to maintain prolonged clinical efficacy; and (iii) the antibacterial agent can exert its activity on the surface or in the periplasm without having to completely enter into the pathogen. Despite these advantages, a major challenge is that membrane-disrupting antibiotics need to be selective toward bacterial cell membrane to avoid cytotoxicity in eukaryotic cells.3
3.1. Structure of the Bacterial Cell Walls
The peptidoglycan, also called murein, is constituted by linear glycan chains alternating N-acetylglucosamine and N-acetylmuramic acid cross-linked by peptide stems and surrounding the inner membrane (Figure 2). The PGN general architecture is conserved across bacterial species, despite variations in the composition of both glycan and peptide moieties. Gram-positive bacteria possess a thick PGN layer, whereas in Gram-negative bacteria, a thin PGN layer is surrounded by a further lipid bilayer, the outer membrane (Figure 2).4,5 The OM provides an extra layer of protection, acting as a selective impermeable barrier, conferring extra resistance to stressors, host immune mechanisms, and antimicrobials, whose diffusion is further hindered. Thus, bacterial OM represents one of the main origins of AMR and combines a highly hydrophobic and sophisticated asymmetric lipid bilayer barrier with pore-forming porins of specific size-exclusion properties that provides a path through the OM to small hydrophilic antibiotics, such as β-lactams.8 The main components of the OM, indispensable for the bacterial growth and survival, are LPS, structurally divided into three distinct functional and structural domains, namely, lipid A, core oligosaccharide, and O-antigen (Figure 2).3 The adaptability and flexibility of LPS biosynthesis allow bacteria to react to the selective pressure of the immune system and to adapt environmental threats by significant changes in LPS size and composition. The fine modulation and tuning of the LPS structure can ensure protection, mediate resistance to clinically relevant antimicrobial compounds, and help to evade or reduce immune surveillance by host receptors as Toll-like receptors, NOD, or C-type lectins.4
Figure 2.

Gram-positive and Gram-negative bacterial membranes. All bacteria share the cytoplasmic membrane, in turn, enclosed by a protective and rigid layer of peptidoglycan (PGN). In Gram-positive bacteria, the thick PGN layer forms the basis of the cell envelope.4 In Gram-negative bacteria, the peptidoglycan is surrounded further by an additional asymmetric bilayer, the outer membrane (OM). The inner leaflet of the OM comprises glycerophospholipids, and the external leaflet mainly comprises lipopolysaccharides (LPS), which can cover up to 75% of the cell surface. LPS structures, highly strain-specific, can be roughly divided into three domains: a lipophilic domain (termed lipid A), a core oligosaccharide, and an O-specific polysaccharide (or O-chain). The lipid A anchors the LPS to the OM through hydrophobic and electrostatic interactions. The hopanoids (sterol-like compounds that have been associated with MDR and AMR mechanisms) shown in the figure are present in some, but not all, bacteria.3
Conversely, in Gram-positive bacteria, the thick layer of PGN, able to withstand the turgor pressure exerted against the cell wall, is densely functionalized with teichoic acids (TAs), long anionic glycopolymers largely composed of glycerol-, glucosyl-, or ribitol-phosphate repeats. TAs include lipoteichoic acids (LTAs), which are anchored to the bacterial membrane, and wall teichoic acids (WTAs), which are covalently attached to peptidoglycan. TAs form a highly hydrated, gel-like material affecting bacterial access to ions, nutrients, proteins, and antibiotics.5 Also, Gram-positive bacteria can modulate the structure of their envelope constituents to dampen host immune responses to evade detection by the immune system.4,5
3.2. Compounds Targeting Bacterial Membrane
A viable strategy to enhance protection against bacteria is to block synthesis of bacterial cell envelope components. Among them, the PGN has attracted great attention aimed at developing antibacterial agents to inhibit the intracellular steps of its biosynthesis. For example, MurA-F enzymes, which are involved in the early stages of PGN biosynthesis, represent suitable targets for the design of novel antibiotic compounds, some of them showing promising inhibitory properties and antimicrobial activity.3 Also, the inhibition of lipid II formation, an important membrane-anchored PGN precursor, has been explored, leading to the discovery of novel antibiotic classes such as lantibiotics and defensis.3,5 Recently, the great potential of selective targeting of the Gram-negative bacterial OM has been also shown, focusing on the inhibition of the LPS biosynthesis and transport.4
Antimicrobial peptide (AMP)-based therapies represent suitable alternatives to the currently employed antibiotics. Being produced by the host immune system as a defense mechanism for protection against many pathogens, AMPs are characterized by a significant cytolytic activity and by the ability to form pores in membranes.12 AMPs are rich in lysine and arginine residues, contain more than 30% of hydrophobic residues, and possess amphiphilic secondary structures with variable structural motifs.10 Due to their cationic nature, AMPs can bind anionic bacterial OM, thus provoking its disruption, mainly by two mechanisms: (i) insertion into the lipid bilayer and channel formation typically assuming “barrel stove” or “toroidal” arrangements or (ii) “carpet mechanism”, i.e., aggregation and consequent breaks in the formation on the membrane surface.10,12 Many natural AMPs have entered clinical trials but showed some limitations linked to sensitivity to protein degradation, difficulty to establish their toxicological properties, low in vivo stability, as well as high cost of productions.12 Additionally, smaller and more selective antimicrobials and AMP-inspired compounds have been designed and showed significant inhibitory properties, including cholic and tetramic acid derivatives, carbohydrates, xanthone, quinolone, benzophenone, and porphyrin derivatives.10
4. Studies of AMR Bacterial Membranes: Experimental Approaches
For the reasons above, understanding the structural and functional characteristics of cell envelope components isolated from AMR strains is a fundamental task to (i) comprehend their impact on the antimicrobial permeation, (ii) shed light into molecular mechanisms of drug resistance, and (iii) ultimately to design effective systems able to selectively manipulate, modulate, and remodel the bacterial membrane properties and functionality. Here, an overview of the most common methods to dissect structure and properties of the bacterial cell envelope, alone and in the interaction with potential targets, is reported. The supramolecular arrangements of the cell wall components at different observation scales, from the morphological to the microstructural level (shape, thickness, size of membrane systems containing asymmetric lipids domains, bilayer fluidity, water permeability, LPS/porins interactions), including OM proteins (mainly porins and lipoproteins), may be depicted as follows.
The isolation and structure characterization of cell wall components, like LPS, PGN, lipopeptides, and porins, are performed through a combination of chemical, spectrometric, spectroscopic, and microscopic approaches (Figure 3).12 Membrane model systems, including micelles, supported bilayers, and lipid vesicles such as liposomes, are, in turn, built and used to describe the structural and physicochemical characteristics of the bacterial cell wall. In particular, liposomes constitute the principal model system to analyze the membrane biological properties, as they can be used to closely mimic the specific features of prokaryotic membranes, controlling the lipid content and composition (e.g., inserting LPS in case of Gram-negative bacteria).11
Figure 3.
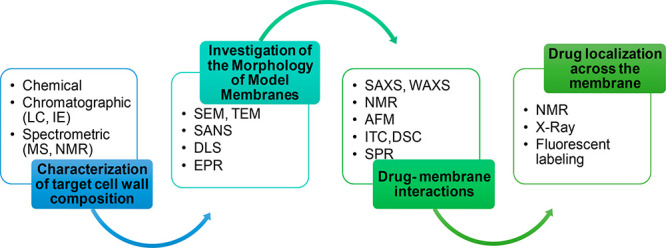
Overview of some of the experimental techniques commonly applied to study bacterial membranes.
Structural and functional investigations of liposomes or membranes is performed by means of dynamic light scattering (DLS) to estimate liposome dimension; electron microscopy (EM) techniques, particularly scanning electron microscopy (SEM) and transmission electron microscopy (TEM), as well as small angle neutron scattering (SANS), are commonly employed to analyze the aggregate morphology and to estimate the thickness of the lipid bilayer.13 The dynamics of the lipid hydrophobic tail in the bilayer can be investigated by electron paramagnetic resonance (EPR).
The study of drug–membrane interactions allows the drug pharmacokinetics and pharmacodynamics, as well as its therapeutics and toxic effects, to be understood when using eukaryotic membrane models.11 The cell envelope behavior upon interaction with identified antimicrobial compounds can be assessed by combining electrophysiological assays, electron microscopy, and calorimetric techniques to gather in-depth information. The modifications arising from the interactions between antibiotics and model membranes can be mapped by synchrotron small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS) and nuclear magnetic resonance (NMR) spectroscopy. Neutron reflectivity (NR) allows one to define at nanometer scale the structure of the reconstituted model membrane once the interaction with the antimicrobial cationic amphiphiles has taken place. Atomic force microscopy (AFM) can be used to analyze the membrane morphological changes induced by the interaction with drugs and to quantify drug-induced membrane disruption.13
Physicochemical techniques such as surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), differential scanning calorimetry (DSC), and fluorescence spectroscopy are essential to define the membrane thermodynamics upon antibiotic interactions, after the evaluations of thermodynamic parameters affecting the membrane fluidity.13 High-resolution NMR techniques, as tr-NOESY and saturation transfer difference (STD), can be exploited to obtain detailed information on the structural requirements necessary for recognition by specific targets, helping the rational design of novel antibacterial compounds.14 The drug localization across the membrane is essential to establish its diffusive properties and can be investigated by direct methods (NMR, X-ray) and, indirectly, for example, using florescent probes.11 Finally, an important parameter to evaluate in the study of potential antibiotics is the lipophilicity, a property that influences the drug pharmacokinetic properties which strictly depends on its interaction with the biological membranes. The lipophilicity in model liposomes can be assessed by spectroscopic methods such as fluorescence spectroscopy.11
Given the AMP ability to form pores across membranes, assays to study membrane fusion and permeability are crucial to design new effective peptides. The depth of internalization inside the membranes can be determined by fluorescence spectroscopy when using peptides containing tryptophan residues.11 For example, in leakage assays, changes of encapsulated ANTS (8-aminonaphthalene-1,3,6-trisulfonic acid disodium salt) fluorescence in liposomes are measured. The observation of the fluorescence signal resulting from the dequenching of ANTS, at various lipid to peptide ratios, allows for the determination of leakage induced by the peptide.15 Furthermore, fluorescence resonance energy transfer (FRET) experiments may aid in understanding the mechanism of interaction and discriminate between fusion and leakage.13
5. Studies of AMR Bacterial Membranes: Computational Approaches
Membrane morphology and its properties, such as phase transition or fluidity, can vary significantly as a function of lipid composition and molecule structure.6 Consequently, a detailed membrane organization is typically difficult to study at a molecular level by experimental techniques, in spite of the significant development of fast and efficient experimental protocols (see section 4). However, these techniques are mainly limited to sample size and time scale, as they provide an averaged structural information about moles of molecules, and within a time window up to micro- and milliseconds. Computational methods based on classical mechanics allow atomic resolution and span the observation time on the size range of pico- and nanoscale by femtosecond steps. In particular, biomolecular simulations have been referred to as computational microscopes, as they reveal molecular aspects (interatomic and intermolecular interactions) not accessible using any experimental microscopy.16 For this reason, molecular dynamics (MD) simulation studies can bring insightful knowledge into AMR mechanisms in bacterial membranes. This review covers the MD simulation studies on AMR bacterial membranes at the following levels of resolution: all-atom (AA), coarse-grained (CG), and hybrid AA/CG models. Figure 4 illustrates the framework of different resolution levels covered by the computational microscopy tools. This means that, depending on the property to be measured, the resolution (time and length scales) must cover a wide range of time and length scales. In the following sections, selected examples illustrating the application of AA and CG MD simulations are shown, especially those devoted to study the influence of membrane composition in the AMR-related physicochemical properties, according to the bacterial strain, as well as to design effective antimicrobial agents. Conformational dynamics of bacterial membrane proteins, such as porins and efflux pumps, are closely determined by the lipid envelope. AA and CG MD simulations have also proven to be indispensable in studying the interactions of membrane proteins with the surrounding lipid environment. However, these studies are out of scope for this review and have been reviewed somewhere else.7,17
Figure 4.

Computational simulations as complementary techniques for different size and time resolutions. (A) Cryo-TEM on outer membranes vesicles of Pseudomonas aeruginosa (PM, plasma membrane; OM, outer membrane; O-IMV, outer–inner membrane vesicles; bar, 200 nm). Reprinted with permission from ref (1000). Copyright 2015 PLOS. (B) Conventional TEM image of negatively stained Shewanella cell at room temperature. Reprinted with permission from ref (1001). Copyright 2008 Cambridge University Press. (C) Coarse-grained lipid vesicle containing 32 OmpF trimers. Reprinted with permission from ref (7). Copyright 2015 Portland Press Ltd. (D) Complex bacterial membrane model including the outer and inner E. coli cell membrane with embedded membrane proteins including the membrane spanning multidrug efflux pump AcrABZ-TolC. Reprinted from ref (1002). Copyright 2017 American Chemical Society. (E) Hybrid model of an atomistic helix embedded in a CG lipid membrane. Reprinted from ref (1003). Copyright 2015 American Chemical Society. (F) Atomistic simulations of electroporation of the E. coli outer membrane. Reprinted from ref (18). Copyright 2011 American Chemical Society.
5.1. All-Atom MD Simulation of AMR Bacterial Membranes
In the past few years, the development of new and powerful computational algorithms has allowed to include atomistic LPS models and complex phospholipids into heterogeneous membranes simulations, such as lysylphosphatidylglycerol and diphosphatidylglycerol lipids (Figure 5). Atomistic models of LPSs and phospholipids have been reported for each of the three most widely used families of all-atom force fields (FF), CHARMM and AMBER, and the united-atom force field, GROMOS.19 All-atom MD simulations performed to address the study of AMR bacterial membranes could be classified into two main general categories: (1) those aiming at studying the membrane biophysics and properties related to the membrane components, such as LPS and phospholipids, and (2) those aiming the characterization of the mechanisms of action of antimicrobial drugs and the design of effective nonresistant novel antimicrobial agents.
Figure 5.

Atomistic representation of some representative membrane components and their corresponding CG mapping scheme. Phospholipids: (A) 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC); (B) dipalmitoylphosphatidylcholine (DPPC); (C) 1-stearoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (SOPE); and (D) 1-stearoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (SOPG); (E) 1,3-bis(sn-3′-phosphatidyl)-sn-glycerol (cardiolipin); (F) lipid A of E. coli; (G) lipid A of S. minnesota. Ellipsoids and circles represent the corresponding CG beads (represented in different colors and assigned to different codes for clarification).
Examples from the first category include the work by Wu et al. pioneering the simulation of several all-atom bilayers of Escherichia coli composed of E. coli LPSs with various O-antigen polysaccharide chain lengths.20 Water molecules were able to penetrate inside the inner core region of all bilayer systems, indicating that sugar residues in the core and O-antigen region were fully hydrated. Thus, the core and O-antigen sugar residues provide a hydrophilic barrier that protects against hydrophobic molecules, but it does not prevent the rapid diffusion of small polar molecules, like water molecules. This explained why Gram-negative bacteria are generally more resistant to hydrophobic antibiotics than their Gram-positive counterparts; the hydrophilic polysaccharide component is responsible for the exclusion of hydrophobic molecules and the hydrophobic lipid A of LPS limits the entry of hydrophilic compounds.
Kim et al.21 simulated membrane bilayers with 21 different lipid A types from 12 bacterial species to investigate membrane properties, considering the influence of different neutralizing ion types (Ca2+, K+, and Na+). The authors concluded that lipid A acyl chains were better packed with higher chain number. Ca2+ ions resided longer on the lipid A headgroups than K+ and Na+ ion types, as they prevent repulsion between the negatively charged groups of adjacent LPS molecules, leading to lower lipid A lateral diffusion and a higher lipid A compressibility. These observations could explain why chelating agents that bind divalent cations (e.g., EDTA) can cause destabilization of the OM and increase bacteria susceptibility to antibiotics.
Rice and Wereszczynski studied the effect of Salmonella enterica LPS modifications on the structure and properties of the bacterial OM S. enterica through the PhoPQ pathway. They observed a 2-fold decrease in susceptibility to the antibiotic drug novobiocin compared to that with the null mutant, showing that LPS modification through the PhoPQ system results in a stronger permeability barrier.22 Among the lipid A modifications, 2-hydroxylation, palmitoylation, and incorporation of aminoarabinose onto the 4′-phosphate group, the latter radically altered the localization of Ca2+ counterions and water near the lipid A headgroup and decreased the net charge of the lipid A, strengthening adjacent LPS–LPS hydrogen-bonding networks. All this leads to a reduced permeability to large lipophilic agents. In addition, the decreased net charge conferred by aminoarabinose may help to protect bacteria from cationic AMPs.
On to the second category, i.e., all-atom MD simulations of AMR bacterial membranes applied to the study, rationalization and design of novel antimicrobial drugs, interesting examples are described as follows. Piggot et al. studied the molecular rearrangements and pore formation process that occur during electroporation in a model of Gram-positive (Staphylococcus aureus) and Gram-negative (E. coli) bacteria (Figure 6).18 A series of MD simulations varying external electric fields were performed in order to provide a route for large drugs to enter through the bacteria membrane and were validated by in vitro experiments. Authors observed that S. aureus cell membrane was less resistant to poration than the E. coli outer membrane. The higher resistance arose from the reduced mobility of E. coli LPS molecules, due to tight cross-linking by cations and extended LPS–LPS hydrogen-bonding networks.
Figure 6.

Pore formation by electroporation (left panel) and unbiased AA MD (right panel). (A–D) Sequence of events in the electroporation of the E. coli outer membrane with an electric field of 0.4 V/nm applied. Phosphatidylethanolamine lipids are shown in orange, PGN layer in green, 2,3-diphosphoglycerate in blue, and LPS in yellow. Lipid tails have been omitted for clarity. Reprinted from ref (18). Copyright 2011 American Chemical Society. (E,F) Assemblies of two and four complexes of MU1140–lipid II in a bacterial membrane at the beginning and end of the 500 ns simulations, respectively. Reprinted with permission from ref (23). Copyright 2019 Royal Society of Chemistry. MU1140 chains are colored by residue types; lipid II chains are highlighted in yellow, and the water molecules are highlighted as red spheres. Phosphorus atoms of POPE are shown as green spheres and POPG phosphorus atoms as gray spheres. (G) Same as (F) but showing the water molecules above and below the lipid layers.
MD simulations have also been used to investigate the dynamic properties of several antimicrobial drugs, with special focus on AMPs, but not only, and the relationship between the molecular mechanism and their properties such as partition coefficient and diffusion constants across membranes. For example, Pasenkiewicz-Gierula et al.6 have reviewed the computational studies on the interactions of antibacterial membrane active compounds with OM models, including magainins, polymyxins, melittin, CM15 (a chimeric peptide containing fragments of melittin and cecropin A), and the peptide dendrimer BALY (Figure 7).10
Figure 7.

Chemical structure of selected drugs related to AMR.
As described above, many AMPs selectively target and form channels (pores) in bacterial membranes. Pokhrel et al. explored the mechanisms of membrane pore formation by all-atom molecular simulations of the MU1140–lipid II complex in a model of Gram-positive bacterial membrane, composed of POPE and POPG.23 MU1140 (Figure 7) is a promising antimicrobial lanthipeptide effective against Gram-positive bacteria that targets and sequesters lipid II by inserting itself inside the membrane, thus leading to bacteria cell lysis. MU1140, in complex with lipid II, was able to form water-permeating membrane pores and promote membrane distortion and lipid relocation toward the central region of the lipid bilayer (Figure 6). These investigations provided an atomistic level insight into a novel action mechanism of MU1140.
All-atom MD simulations in combination with umbrella sampling can be used to sample the conformational space and to study the preferred location of AMPs in the bacteria membrane.6 Unbiased atomistic simulations of AMP maculatin (Figure 7) have been performed at high temperatures (90–150°) in different bilayer models consisting of DMPC, DPPC, and DSPC lipids, yielding the mechanism of spontaneous pore assembly.24 Maculatin formed an ensemble of temporal low oligomeric channel-like pores, which mimic integral membrane protein channels in the structure. These pores continuously assemble and disband in the membrane. When maculatin inserted, the membrane translocation barrier (∼15–20 kcal mol–1) was overcome by peptides’ cooperative insertion, through membrane defects induced by maculatin charged and polar side chains. The diversity of the observed pore architectures formed by maculatin peptide and their variation upon minor perturbation of the peptide sequence reveal a key feature in preventing bacterial resistance and could explain why sequence–function relationship in AMPs still remains unknown.
For a large number of peptides, translocation, leakage of vesicles, and antimicrobial activity have been observed, but with no pore formation. Simulated annealing MD have been used to understand the AMP behavior concerning pore formation.24 In this respect, simulated annealing MD of AMP PGLa (Figure 7) in two bilayer models consisting of pure DMPC and a mixture of DMPC/DMPG have been performed.24 PGLa possesses an amidated C-terminus, which increases its resistance to proteases. Remarkably, PGLa-H, a naturally produced C-terminus fragment of PGLa, shows moderate antibacterial activity against multi-drug-resistant clinical isolates of Gram-negative and -positive bacteria. Ulmschneider et al. observed PGLa spontaneously translocated across the membrane individually on a time scale of tens of microseconds, without forming pores.24 Instead of stable pores, short-lived water bridges occurred when two or three peptides connected at their termini, allowing both ion translocation and lipid flip-flop via a brushlike mechanism usually involving the C-terminus of one peptide. The results can explain why for many AMPs no channel formation has been observed experimentally, despite clear experimental evidence of membrane leakage and antimicrobial activity.
In a further step, detailed structural and dynamic information gathered from MD simulations can be utilized for the de novo design of AMPs. Chen et al. performed a rational design approach based on folding–partitioning–assembly atomistic simulations and applied it to develop a potent new pore-forming AMP sequence, consisting of only four types of amino acids: LDKA.25 The final designed peptide forms large pores in membranes and exhibits low micromolar activity against common Gram-positive and Gram-negative pathogenic bacteria.
Besides investigations in AMPs, AA MD simulations of nonpeptidic compounds targeting bacterial membranes are also opening new avenues of research to find novel drugs able to fight AMR. For instance, Dias et al. recently discovered deoxyglycosides acting as potent bactericides by targeting phosphatidylethanolamine (PE)-rich membranes, thereby promoting bacterial membrane disruption through PE lamellar-to-inverted hexagonal phase transition.26 This mechanism circumvents the cytotoxicity of other membrane-disrupting antibiotics, as eukaryotic cells do not have exposed PE. The impact of deoxyglycosides in the membrane structural properties was investigated by AA MD simulations. Stability of preformed membrane pores was devised, and pore closure was observed over the simulation trajectory.
5.2. Coarse-Grained as a Large-Scale Method for Simulation of AMR Bacterial Membranes
By now, realistic membranes, where size and variety of lipid membrane have a direct effect on observable properties, such as membrane permeability, membrane curvature, or time-scale-related phenomena, such as unbiased pore formation, are not accessible from atomistic MD simulations. This is because the time scales achievable in simulations must reach up to the millisecond range and even longer and, also, because the computational model must accurately reproduce what is measured experimentally. Therefore, since all-atom MD simulations have still some limitations because the computational power to gain a significant sampling, coarse-grained MD models with a compromised atoms-to-bead mapping are required to observe relevant structure–property relationships from computer simulations (Figure 5). We would limit the concept of coarse graining by making reference to the omission of irrelevant degrees of freedom of a system, in order to simplify the model, i.e., coarse-grained force fields (CG FF) that groups atoms into beads. Detailed concepts and further read can be found in the literature.19,27
The popular MARTINI FF is one of the most common CG FF for a wide range of simulations of proteins and membranes (as well as polymers). It can reproduce the accurate dynamic behavior of lipid bilayers and can be used to explore interactions between peptides and membranes at time and length scales hardly accessed by all-atom MD simulations. The loss of resolution in the MARTINI model compared to atomistic force fields brings along different limitations and challenges. Except for the obvious loss in structural detail, there are a few problems that especially are worth mentioning. First, the grouping together of four heavy atoms (non-hydrogen atoms) reduces the entropy in a molecule. In order to obtain correct free energies, this is corrected by adapting the enthalpic interactions.28 As a result, the balance between entropy and enthalpy will be disturbed and separating these two contributions has to be done with the greatest care. In spite of that, a reliable collection of data supported by experimental methods and vice versa can be found in the literature. On this respect, CG approaches aiming to uncover the molecular interactions and forces governing the AMR mechanism are in a continuous progress, becoming an active research field. The research around CG on AMR could be grouped in two categories: (1) membrane biophysics related to membrane components, such as LPS and phospholipids, and (2) discovery and design of effective antimicrobial agents.
In the first group, we can find those studies dedicated to membrane component-dependent biophysical properties of bacterial membrane. Khalid et al. studied the role of LPS O-antigen on the mechanical properties in the E. coli OM.29 They found by CG MD simulation that LPS structures are sensitive to the lipids surrounding them. In addition, the presence of O-antigen decreases the molecular mobility in the OM with a considerable reduction of surface tension. Ma et al. studied the LPS composition effect on the OM of Pseudomona aeruginosa.30 They demonstrated the phase transition temperature dependency on lipid composition of OM, for instance, membranes rich in LPS showed lower melting points, higher area per lipid, and higher disorder compared to membranes with simple phospholipids. Also, Ma et al., modeled the lipid A structures of 8 bacterial species (Helicobacter pylori, Porphyromonas gingivalis, Bacterioides fragilis, Bordetella pertussis, Chlamydia trachomatis, Campylobacter jejuni, Neisseria meningitidis, and Salmonella minnesota) and characterized and compared their membrane properties using CG MD simulations. They concluded that longer acyl chain lipids A have smaller area per lipid and a higher phase transition temperature. The membrane composition and charge of the inner membrane can influence the phase transition temperature. Divalent ions stay on the membrane surface and act as chelating agents.31 These works illustrate how these computational approaches contribute to establish a correlation between membrane composition, structure and biophysical properties involved in AMR.
A second group of research articles, related to the discovery and design of effective antimicrobial agents, are mainly devoted to AMPs. Zhao et al. combined computational and experimental techniques to study a complete translocation process of the AMP Bac2A using a computationally designed Bac2A-based peptide library.33 The library, based on a complete single-point substitution of the Bac2A amino acid sequence, was synthesized, tested, and provided an excellent model system for computationally probing the sequence–structure activity of AMPs, and for the rational design of new AMPs with improved antibiotic activity. The authors used CG MD simulations with adaptive biasing force method and the umbrella sampling technique to investigate the translocation of a total of 91 peptides with different amino acid substitutions through a mixed anionic POPE/POPG (3:1) bilayer and a neutral POPC bilayer. The potential of mean force associated with peptide translocation process was calculated to directly determine the free energy barrier required to transfer the peptides from the water phase to the water–membrane interface, and to the membrane interior (Figure 8). Peptides identified with enhanced membrane association were synthesized and evaluated by both bacterial inhibition and hemolysis assays. They confirmed that the balanced substitution of charged residue (Arg) and hydrophobic residue (Trp) in Bac2A achieves the best antimicrobial activity while minimizing red blood cell lysis.33 Horn et al. revealed the molecular mechanisms of action of the potent synthetic antimicrobial lipopeptide C16-KGGK, via CG MD simulations with the MARTINI force field, and a total simulation time of nearly 46 μs. The C16-KGGK lipopeptide exhibits micromolar minimum inhibitory concentrations and excellent selectivity for bacterial membranes, including the gentamicin-resistant Acinetobacter baumannii strain.34 They found that lipopeptides aggregated to form micelles in solution, prior to membrane binding. Furthermore, upon binding to the surface of the bilayer, C16-KGGK altered the local lipid organization by recruiting negatively charged POPG lipids to the site of binding.35
Figure 8.

Antimicrobial agent insertion by an unbiased AA MD (top panel) and a PMF method (bottom panel). (A) PMB1 benzyl group penetrates the lipid core. Positions of Re LPS phosphate groups and a representative PMB1 benzyl group are shown as black and blue lines, respectively. The coordinates are with respect to the bilayer normal; distances are relative to the bilayer center. The temperature was 310 K; pressure was 1 bar; ambient ions were Ca2+ ions. (B) Side-view snapshot for the insertion event; perspective is reversed relative to (A) for clarity. The inset shows the two-dimensional Voronoi tessellation for Re LPS headgroups as PMB1 enters the lipid core; projected polygons are colored cyan if they represent lipids adjacent to the embedded PMB1 benzyl group. (C) Schematic of membrane penetration of a given peptide with different C-/N-terminus insertion pathways. The partition of AMPs into different lipid bilayers produced different PMF profiles with three distinct MID, MAX, and MIN values, which help to determine insertion free energy barriers and to predict both antibacterial and hemolytic activities. (D) Map of the predicted cell selectivity (i.e., therapeutic index) of some studied AMPs onto the two reaction coordinates of hydrophobicity and net charge of the peptides. Cell selectivity from high to low values is presented by a red-green-blue color scale. Simultaneous increase of hydrophobicity and charge characters together indicates improved cell selectivity. Panels (A,B) reprinted from ref (32). Copyright 2017 American Chemical Society. Panels (C,D) reprinted from ref (33). Copyright 2013 American Chemical Society.
Finally, this active research field is also focusing on describing the biophysical implications of AMPs-LPS interactions in a bacterial membrane. For example, Jefferies et al. studied the cyclic peptide polimixin B1 (PMB1, Figure 7) by CG MD simulations on a E. coli bilayer containing LPS. Polymyxins are nonribosomal peptide antibiotics used as the last-resort drug for treatment of multi-drug-resistant Gram-negative bacteria. In particular, PMB1 is one of the most potent antimicrobial peptides that targets Gram-negative bacteria.32 During the CG MD simulation studies, the PMB1 peptide, in contact with the LPS membrane surface, showed to increase order within the lipopolysaccharide bilayers by inducing the formation of crystalline patches at different temperatures (Figure 8). This observation has several consequences on membrane process that affect cell viability including protein sorting, signal transduction, molecular transport, enzymatic activities and immune responses.
It can be observed that, in the past few years, the sophistication and complexity of membrane models have improved considerably, such that the heterogeneity of the lipid and protein composition of the membranes can now be considered at the CG level. This means that the relevant biology around the AMR mechanisms on bacteria cell is now being accounted for the models, and therefore, linking the in silico and in vitro experiments. Nevertheless, it is worth to mention that progress on CG approaches seems to be strongly dependent on a unique CG force field, MARTINI. Very potent and versatile, it still leaves room for improvement and development of new FFs able to tackle relevant membrane features, as the inclusion of the pH variation, of vital relevance in bacteria environment, or the observation of conformational changes of AMPs secondary structures in situ, either while the AMP is located on the bacterial membrane surface or embedded into the membrane. This review encourages to the development of implemented CG force fields that allows the address these challenging problems of paramount importance in AMR understanding.
5.3. Hybrid All-Atom and Coarse-Grained Simulations for AMR Bacterial Membranes
Hybrid simulations, in which part of the system is represented at the atomic level and the remaining part at CG level, offer a powerful way to combine the accuracy associated with the atomistic force fields to the sampling speed obtained with CG potentials. Orsi et al. studied the passive transport phenomena across biomembranes of a set of small molecules representing common chemical functional groups through a DMPC bilayer. They used coarse-grained simulations to develop the bilayer and all-atom simulations to represent the set of small molecules. Free energy profiles, diffusion and resistance parameters, and permeability coefficients were analyzed and compared, leading to accurately reproduction of the experimental measures. They demonstrated this multiresolution approach could be transferred to other systems, so that it can be applied to determine membrane permeability of drug molecules and AMPs.36 Shi et al. built a mixed all-atom and coarse-grained model of gramicidin A (gA) embedded in a lipid bilayer. Gramicidin A is a well-characterized ion channel, so it serves as a prototypical model for the study of more complex ion channels. The interspersing l- and d-amino acid structure, together with the modified termini, make gA less susceptible to degradation by proteolysis. Also, by targeting the plasma membrane, decreases the probability for bacteria to develop resistance toward gA. Moreover, synthetic gA mutants that display cationic side chains, exhibit varied activities toward methicillin-resistant S. aureus. This multiscale CG method was sufficiently flexible and accurate to reproduce the interactions between the peptide and the hydrophobic lipid tails.37
6. Conclusions, Challenges, and Future Perspectives
The urgent need to develop new antimicrobial treatments that are effective to overcome AMR bacterial infections has led the research community to focus on the bacterial cell envelope as one of the key molecular actors responsible for antibiotic resistance, attracting considerable interest as a potential target of novel antimicrobials. Valuable morphology-dependent AMR mechanisms information can be obtained from the computational modeling techniques, by tuning the resolution scale according to a particular phenomenon and property to be measured. More important, a large number of properties measured by experimental techniques can be complementary validated and predicted by the study of the classical forces that govern the journey of the molecules once inside the bacterial membrane, from the dynamics and molecular organization of the bacterial membrane components to membrane transport phenomena affected by antimicrobial agents. Atomistic simulations represent a continuous-growing area of application in the scope of bacterial membranes, particularly when combined with experimental data. In the past few years, several computational studies have demonstrated the importance in addressing the issue of biochemical complexity of bacterial membranes by including complex models of bacterial cell envelopes. Development of new and powerful computational algorithms has allowed the inclusion of atomistic LPS models and complex phospholipids, which provide a detailed knowledge of the bacterial membrane structure and dynamics previously unknown. Atomistic MD simulations, in combination with other computational techniques (e.g., umbrella sampling, adaptive biasing force), have also been used to investigate the dynamic properties of several antimicrobial drugs, including AMPs, for which MD simulations can accurately reproduce, at the atomic level, experimental ensemble averages, partition coefficient, and diffusion constants across membranes, revealing the molecular mechanisms of drug transport through the bacterial membrane. On the other hand, the study of bacterial membranes by CG MD simulations have set a platform to investigate the factors involved in the molecular mechanisms of AMR and to provide a molecular view of such of heterogeneous architecture. CG MD simulations, in combination with in vitro assays, can be used to design antimicrobial drugs considering a realistic bacterial membrane. Also, illustrative examples show how spontaneous processes that happen at large time scale such as pore formation and peptide translocation are captured by CG MD. Hybrid AA/CG MD simulations exhibit a powerful way to combine the accuracy associated with the atomistic force fields with the sampling speed obtained with CG potentials. This review highlights how a precise structure elucidation is a limited, but not trivial, factor, yet relying on the classical computational methods. Anyway, a new era for lipidomics is providing trusting data about bacterial membrane composition. The vision of MD simulations to provide reliable, quantitative, and mechanistic predictions of the action mechanism of antimicrobial drugs is rapidly becoming a reality and will transform the way these agents are designed, selected, and optimized for tackling the AMR.
Acknowledgments
This work was financially supported by the Spanish Ministry for Science and Innovation (Grant Nos. CTQ2017-88353-R and PRE2018-086249 to A.M.R) and RES-BSC QSB-2020-2-0017. FSE, PON Ricerca e Innovazione 2014-2020, Azione I.1 “Dottorati Innovativi con caratterizzazione Industriale” is acknowledged for funding the Ph.D. grant to R.E.F. S.H.J. is gratefully acknowledged for his relentless support.
Glossary
Abbreviations
- AA
all-atom
- AFM
atomic force microscopy
- AMPs
antimicrobial peptides
- AMR
antimicrobial resistance
- ANTS
8-aminonaphthalene-1,3,6-trisulfonic acid disodium salt
- CG
coarse-grained
- DLS
light scattering
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- DMPG
1,2-dimyristoyl-sn-glycero-3-phosphoglycerol
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DPPC
dipalmitoylphosphatidylcholine
- DSC
differential scanning calorimetry
- EDTA
ethylenedinitrilotetraacetic acid
- EM
electron microscopy
- EPR
electron paramagnetic resonance
- FF
force fields
- FRET
fluorescence resonance energy transfer
- gA
gramicidin A
- IM
inner membrane
- ITC
isothermal titration calorimetry
- LPS
lipopolysaccharides
- LTAs
lipoteichoic acids
- MATE
toxic-compound extrusion
- MD
molecular dynamics
- MDR
multi-drug-resistant
- NMR
nuclear magnetic resonance
- NR
neutron reflectivity
- O-IMV
outer–inner membrane vesicles
- OM
outer membrane
- PBP
penicillin-binding protein
- PDR
pan-drug-resistant
- PE
phosphatidylethanolamine
- PGN
peptidoglycan
- PM
plasma membrane
- PMB1
polimixin B1
- POPE
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylglycerol
- SANS
small-angle neutron scattering
- SAXS
synchrotron small-angle X-ray scattering
- SEM
scanning electron microscopy
- SOPE
1-stearoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- SOPG
1-stearoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- SPR
surface plasmon resonance
- STD
saturation transfer difference
- TA
teichoic acids
- TEM
transmission electron microscopy
- WAXS
wide-angle X-ray scattering
- WHO
World Health Organization
- WTAs
wall teichoic acids
- XDR
extensively drug-resistant
Biographies

Alejandra Matamoros-Recio received a postgraduate from the University of Alcalá, Madrid (M.Sc. in Drug Discovery 2017). She is a predoctoral research fellow in the “Computational Biological Chemistry” group led by Dr. Sonsoles Martín-Santamaría, at CIB Margarita Salas, CSIC (Spain), pursuing a Ph.D. at the University Complutense of Madrid. Her research project lies at the interface between Chemistry and Biology, by means of molecular modeling and computational chemistry applied to the understanding of ligand–receptor interactions and molecular recognition processes relevant for drug design, with a particular focus on Toll-like receptors and AMR-related receptors.

Juan Felipe Franco-Gonzalez received his B.Sc. in Chemistry in 2007 from University of Antioquia (Colombia) and worked as a teaching assistant at the same university during 2007–2010. He earned his Ph.D. in Biophysics in 2015 from Autónoma University of Madrid, UAM/IEM-CSIC (Spain). During 2015–2018, he joined the group of Prof. Igor Zozoulenko at LOE from Linköping University (Sweden), where he specialized on simulating and modeling of structure and morphology of conducting polymers. Since 2018, he has focused on computer-aided drug design and membrane biophysics. He joined the group of Dr. Manuela G. López at Department of Pharmacology in UAM during 2018–2019, and currently, he is a postdoctoral fellow working with Dr. Martín-Santamaría at CIB Margarita Salas, CSIC (Spain). His main research interests focus on the implementation of atomistic and coarse-grained computer simulations to investigate structure–property relationships in biointerfaces in order to unravel breakthrough discoveries in a broad range of applications, from organic electronics to drug design.

Rosa Ester Forgione is a Ph.D. student at the Department of Chemical Sciences, University of Naples “Federico II”. Her research project mainly concerns the interplay between eukaryotic receptors and glycans with a particular focus on the biological roles of sialic acid. In this context, she has gained experience in various techniques including NMR spectroscopy, biophysical methods, and computational methodologies.

Angel Torres-Mozas received a graduate degree in Biochemistry from Seville University (M.Sc in Drug Discovery 2020). He partook in the Computational Biological Chemistry lab of CIB-CSIC as a M.Sc student. His research interest focuses on Computational Chemistry and Biology.

Alba Silipo is Full Professor of Organic Chemistry at the Department of Chemical Sciences, University of Napoli “Federico II”. She has a M.D. and a Ph.D. in Chemistry. Her research has produced more than 140 papers in international peer reviewed journals, and it is focused on structural glycobiology. In particular, she is interested in (1) structural characterization of microbial glycoconjugates, (2) conformational analysis of glycoconjugates by NMR and computational techniques, and (3) study of the protein–glycan interaction by NMR spectroscopy and computational approaches. She has been recently awarded for Carbohydrate Research Award for Creativity in Carbohydrate Chemistry; III European prize of the RSEQ Chemical Biology Division (GEQB) to young group leaders, 2017; Recipient of the IEIIS Nowotny Award, 2016; Medaglia Giacomo Ciamician from SCI (Italian Chemical Society) in 2012.

Sonsoles Martín-Santamaría (Ph.D. in Pharmaceutical Chemistry) is Staff Researcher at the Spanish Research Council (CSIC). Since 2012, she leads the “Computational Chemical Biology” group devoted to the study of the molecular recognition processes involving Pattern Recognition Receptors (PRRs), as main players in immune response. By means of computational chemistry and biomolecular simulations, the work is mainly focused on Toll-like receptors, lectins, and AMR-related receptors, aiming to understand ligand(carbohydrate)–protein, protein–protein, and protein–membrane interactions as a source of novel immune modulators. She is the Secretary General of the Royal Society of Chemistry of Spain and Chair of the EuChemS Division for Chemistry in Life Sciences.
Author Contributions
# A.M.-R. and J.F.F.-G. contributed equally to this review.
The authors declare no competing financial interest.
Dedication
This paper is dedicated to Prof. Jesús Jiménez-Barbero for his 60th birthday.
References
- World Health Organization . Antimicrobial Resistance. https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed December 29, 2020).
- Asokan G. V.; Ramadhan T.; Ahmed E.; Sanad H. WHO Global Priority Pathogens List: A Bibliometric Analysis of Medline-Pubmed for Knowledge Mobilization to Infection Prevention and Control Practices in Bahrain. Oman Med. J. 2019, 34, 184–193. 10.5001/omj.2019.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epand R. M.; Walker C.; Epand R. F.; Magarvey N. A. Molecular Mechanisms of Membrane Targeting Antibiotics. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 980–987. 10.1016/j.bbamem.2015.10.018. [DOI] [PubMed] [Google Scholar]
- Mingeot-Leclercq M. P.; Décout J. L. Bacterial Lipid Membranes as Promising Targets to Fight Antimicrobial Resistance, Molecular Foundations and Illustration through the Renewal of Aminoglycoside Antibiotics and Emergence of Amphiphilic Aminoglycosides. MedChemComm 2016, 7, 586–611. 10.1039/C5MD00503E. [DOI] [Google Scholar]
- Dörr T.; Moynihan P. J.; Mayer C. Editorial: Bacterial Cell Wall Structure and Dynamics. Front. Microbiol. 2019, 10, 2051–2055. 10.3389/fmicb.2019.02051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasenkiewicz-Gierula M.; Baczynski K.; Markiewicz M.; Murzyn K. Computer Modelling Studies of the Bilayer/Water Interface. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2305–2321. 10.1016/j.bbamem.2016.01.024. [DOI] [PubMed] [Google Scholar]
- Khalid S.; Berglund N. A.; Holdbrook D. A.; Leung Y. M.; Parkin J. The Membranes of Gram-Negative Bacteria: Progress in Molecular Modelling and Simulation. Biochem. Soc. Trans. 2015, 43, 162–167. 10.1042/BST20140262. [DOI] [PubMed] [Google Scholar]
- C Reygaert W. An Overview of the Antimicrobial Resistance Mechanisms of Bacteria. AIMS Microbiol. 2018, 4, 482–501. 10.3934/microbiol.2018.3.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato G. Strategies to Overcome Antimicrobial Resistance (AMR) Making Use of Non-Essential Target Inhibitors: A Review. Int. J. Mol. Sci. 2019, 20, 5844–5869. 10.3390/ijms20235844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias C.; Rauter A. P. Membrane-Targeting Antibiotics: Recent Developments Outside the Peptide Space. Future Med. Chem. 2019, 11, 211–228. 10.4155/fmc-2018-0254. [DOI] [PubMed] [Google Scholar]
- Pinheiro M.; Magalhães J.; Reis S. Antibiotic Interactions Using Liposomes as Model Lipid Membranes. Chem. Phys. Lipids 2019, 222, 36–46. 10.1016/j.chemphyslip.2019.05.002. [DOI] [PubMed] [Google Scholar]
- Lee T. H.; Hofferek V.; Separovic F.; Reid G. E.; Aguilar M. I. The Role of Bacterial Lipid Diversity and Membrane Properties in Modulating Antimicrobial Peptide Activity and Drug Resistance. Curr. Opin. Chem. Biol. 2019, 52, 85–92. 10.1016/j.cbpa.2019.05.025. [DOI] [PubMed] [Google Scholar]
- Bunea A. I.; Harloff-Helleberg S.; Taboryski R.; Nielsen H. M. Membrane Interactions in Drug Delivery: Model Cell Membranes and Orthogonal Techniques. Adv. Colloid Interface Sci. 2020, 281, 102177. 10.1016/j.cis.2020.102177. [DOI] [PubMed] [Google Scholar]
- Tamigney Kenfack M.; Mazur M.; Nualnoi T.; Shaffer T. L.; Ngassimou A.; Blériot Y.; Marrot J.; Marchetti R.; Sintiprungrat K.; Chantratita N.; Silipo A.; Molinaro A.; AuCoin D. P.; Burtnick M. N.; Brett P. J.; Gauthier C. Deciphering Minimal Antigenic Epitopes Associated with Burkholderia Pseudomallei and Burkholderia Mallei Lipopolysaccharide O-Antigens. Nat. Commun. 2017, 8, 115. 10.1038/s41467-017-00173-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley W. C. Determining the Effects of Membrane-Interacting Peptides on Membrane Integrity. Methods Mol. Biol. 2015, 1324, 89–106. 10.1007/978-1-4939-2806-4_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror R. O.; Dirks R. M.; Grossman J. P.; Xu H.; Shaw D. E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- Pavlova A.; Hwang H.; Lundquist K.; Balusek C.; Gumbart J. C. Living on the Edge: Simulations of Bacterial Outer-Membrane Proteins. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1753–1759. 10.1016/j.bbamem.2016.01.020. [DOI] [PubMed] [Google Scholar]
- Perez-Cruz C.; Delgado L.; Lopez-Iglesias C.; Mercade E. Outer-Inner Membrane Vesicles Naturally Secreted by Gram-Negative Pathogenic Bacteria. PLoS One 2015, 10, e0116896. 10.1371/journal.pone.0116896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohnalkova A; Marshall M.; Fredrickson J. Cryo TEM: A New Perspective to the Biogeochemical Associations of Bacteria and Minerals. Microsc. Microanal. 2008, 14, 1298–1299. 10.1017/S1431927608082536. [DOI] [Google Scholar]
- Hsu P.-C.; Samsudin F.; Shearer J.; Khalid S. It Is Complicated: Curvature, Diffusion, and Lipid Sorting within the Two Membranes of Escherichia coli. J. Phys. Chem. Lett. 2017, 8, 5513–5518. 10.1021/acs.jpclett.7b02432. [DOI] [PubMed] [Google Scholar]
- Genheden S.; Essex J. W. A Simple and Transferable All-Atom/Coarse-Grained Hybrid Model to Study Membrane Processes. J. Chem. Theory Comput. 2015, 11, 4749–4759. 10.1021/acs.jctc.5b00469. [DOI] [PubMed] [Google Scholar]
- Piggot T. J.; Holdbrook D. A.; Khalid S. Electroporation of the E. Coli and S. Aureus Membranes: Molecular Dynamics Simulations of Complex Bacterial Membranes. J. Phys. Chem. B 2011, 115, 13381–13388. 10.1021/jp207013v. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Corradi V.; Souza P. C. T.; Ingólfsson H. I.; Tieleman D. P.; Sansom M. S. P. Computational Modeling of Realistic Cell Membranes. Chem. Rev. 2019, 119, 6184–6226. 10.1021/acs.chemrev.8b00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E. L.; Engström O.; Jo S.; Stuhlsatz D.; Yeom M. S.; Klauda J. B.; Widmalm G.; Im W. Molecular Dynamics and NMR Spectroscopy Studies of E. Coli Lipopolysaccharide Structure and Dynamics. Biophys. J. 2013, 105, 1444–1455. 10.1016/j.bpj.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Patel D. S.; Park S.; Slusky J.; Klauda J. B.; Widmalm G.; Im W. Bilayer Properties of Lipid A from Various Gram-Negative Bacteria. Biophys. J. 2016, 111, 1750–1760. 10.1016/j.bpj.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice A.; Wereszczynski J. Atomistic Scale Effects of Lipopolysaccharide Modifications on Bacterial Outer Membrane Defenses. Biophys. J. 2018, 114, 1389–1399. 10.1016/j.bpj.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokhrel R.; Bhattarai N.; Baral P.; Gerstman B. S.; Park J. H.; Handfield M.; Chapagain P. P. Molecular Mechanisms of Pore Formation and Membrane Disruption by the Antimicrobial Lantibiotic Peptide Mutacin 1140. Phys. Chem. Chem. Phys. 2019, 21, 12530–12539. 10.1039/C9CP01558B. [DOI] [PubMed] [Google Scholar]
- Ulmschneider J. P.; Ulmschneider M. B. Molecular Dynamics Simulations Are Redefining Our View of Peptides Interacting with Biological Membranes. Acc. Chem. Res. 2018, 51, 1106–1116. 10.1021/acs.accounts.7b00613. [DOI] [PubMed] [Google Scholar]
- Chen C. H.; Starr C. G.; Troendle E.; Wiedman G.; Wimley W. C.; Ulmschneider J. P.; Ulmschneider M. B. Simulation-Guided Rational de Novo Design of a Small Pore-Forming Antimicrobial Peptide. J. Am. Chem. Soc. 2019, 141, 4839–4848. 10.1021/jacs.8b11939. [DOI] [PubMed] [Google Scholar]
- Dias C.; Pais J. P.; Nunes R.; Blázquez-Sánchez M. T.; Marquês J. T.; Almeida A. F.; Serra P.; Xavier N. M.; Vila-Viçosa D.; Machuqueiro M.; Viana A. S.; Martins A.; Santos M. S.; Pelerito A.; Dias R.; Tenreiro R.; Oliveira M. C.; Contino M.; Colabufo N. A.; de Almeida R. F. M.; Rauter A. P. Sugar-Based Bactericides Targeting Phosphatidylethanolamine-Enriched Membranes. Nat. Commun. 2018, 9, 4857. 10.1038/s41467-018-06488-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coarse-Graining of Condensed Phase and Biomolecular Systems, 1st ed.; Voth G. A., Ed.; CRC Press: Boca Raton, FL, 2008. [Google Scholar]
- Baron R.; De Vries A. H.; Hünenberger P. H.; Van Gunsteren W. F. Comparison of Atomic-Level and Coarse-Grained Models for Liquid Hydrocarbons from Molecular Dynamics Configurational Entropy Estimates. J. Phys. Chem. B 2006, 110, 8464–8473. 10.1021/jp055888y. [DOI] [PubMed] [Google Scholar]
- Jefferies D.; Shearer J.; Khalid S. Role of O-Antigen in Response to Mechanical Stress of the E. Coli Outer Membrane: Insights from Coarse-Grained MD Simulations. J. Phys. Chem. B 2019, 123, 3567–3575. 10.1021/acs.jpcb.8b12168. [DOI] [PubMed] [Google Scholar]
- Ma H.; Irudayanathan F. J.; Jiang W.; Nangia S. Simulating Gram-Negative Bacterial Outer Membrane: A Coarse Grain Model. J. Phys. Chem. B 2015, 119, 14668–14682. 10.1021/acs.jpcb.5b07122. [DOI] [PubMed] [Google Scholar]
- Ma H.; Cummins D. D.; Edelstein N. B.; Gomez J.; Khan A.; Llewellyn M. D.; Picudella T.; Willsey S. R.; Nangia S. Modeling Diversity in Structures of Bacterial Outer Membrane Lipids. J. Chem. Theory Comput. 2017, 13, 811–824. 10.1021/acs.jctc.6b00856. [DOI] [PubMed] [Google Scholar]
- Jefferies D.; Hsu P. C.; Khalid S. Through the Lipopolysaccharide Glass: A Potent Antimicrobial Peptide Induces Phase Changes in Membranes. Biochemistry 2017, 56, 1672–1679. 10.1021/acs.biochem.6b01063. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Zhao C.; Liang G.; Zhang M.; Zheng J. Engineering Antimicrobial Peptides with Improved Antimicrobial and Hemolytic Activities. J. Chem. Inf. Model. 2013, 53, 3280–3296. 10.1021/ci400477e. [DOI] [PubMed] [Google Scholar]
- Horn J. N.; Sengillo J. D.; Lin D.; Romo T. D.; Grossfield A. Characterization of a Potent Antimicrobial Lipopeptide via Coarse-Grained Molecular Dynamics. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 212–218. 10.1016/j.bbamem.2011.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn J. N.; Romo T. D.; Grossfield A. Simulating the Mechanism of Antimicrobial Lipopeptides with All-Atom Molecular Dynamics. Biochemistry 2013, 52, 5604–5610. 10.1021/bi400773q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi M.; Sanderson W. E.; Essex J. W. Permeability of Small Molecules through a Lipid Bilayer: A Multiscale Simulation Study. J. Phys. Chem. B 2009, 113, 12019–12029. 10.1021/jp903248s. [DOI] [PubMed] [Google Scholar]
- Shi Q.; Izvekov S.; Voth G. A. Mixed Atomistic and Coarse-Grained Molecular Dynamics: Simulation of a Membrane-Bound Ion Channel. J. Phys. Chem. B 2006, 110, 15045–15048. 10.1021/jp062700h. [DOI] [PubMed] [Google Scholar]