

Key Points
Gene expression–based classification of primary and secondary MF correlates with patients’ clinical and molecular characteristics.
A gene expression signature identifies patient subgroups with different overall survival.
Abstract
Myelofibrosis (MF) belongs to the family of classic Philadelphia-negative myeloproliferative neoplasms (MPNs). It can be primary myelofibrosis (PMF) or secondary myelofibrosis (SMF) evolving from polycythemia vera (PV) or essential thrombocythemia (ET). Despite the differences, PMF and SMF patients are currently managed in the same way, and prediction of survival is based on the same clinical and genetic features. In the last few years, interest has grown concerning the ability of gene expression profiles (GEPs) to provide valuable prognostic information. Here, we studied the GEPs of granulocytes from 114 patients with MF, using a microarray platform to identify correlations with patient characteristics and outcomes. Cox regression analysis led to the identification of 201 survival-related transcripts characterizing patients who are at high risk for death. High-risk patients identified by this gene signature displayed an inferior overall survival and leukemia-free survival, together with clinical and molecular detrimental features included in contemporary prognostic models, such as the presence of high molecular risk mutations. The high-risk group was enriched in post-PV and post-ET MF and JAK2V617F homozygous patients, whereas pre-PMF was more frequent in the low-risk group. These results demonstrate that GEPs in MF patients correlate with their molecular and clinical features, particularly their survival, and represent the proof of concept that GEPs might provide complementary prognostic information to be applied in clinical decision making.
Visual Abstract

Introduction
Classic Philadelphia-negative myeloproliferative neoplasms (MPNs) consist of 3 clinical entities: polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). PMF can be subdivided into a prefibrotic/early stage (pre-PMF) and an overt fibrotic stage (overt PMF), according to the degree of bone marrow (BM) fibrosis.1 Moreover, PV and ET can evolve into secondary myelofibrosis (SMF) giving rise to post-PV myelofibrosis (PPV-MF) and post-ET myelofibrosis (PET-MF).2 These malignancies are grouped together according to their overlapping features. Although the absence of BM fibrosis is considered the chronic phase of these malignancies, myelofibrosis (MF) onset represents the advanced stage that might precede acute myeloid leukemia (AML) transformation, the main cause of death for these patients.3,4
MPNs originate from the mutation of hematopoietic stem cells, which generates a neoplastic clone that gives rise to the myeloproliferative phenotype. Clonal hematopoiesis in MPNs has been demonstrated by the presence of recurrent gene mutations that is shared by the 3 clinical entities.5,6 Mutations in JAK2, calreticulin (CALR), and MPL proto-oncogene thrombopoietin receptor (MPL) genes are considered driver events. Although a JAK2V617F mutation is carried by almost all patients with PV, it occurs at a 55% frequency in PMF and a 65% frequency in ET. CALR indels affect 25% to 35% of PMF patients and 15% to 24% of those with ET, whereas MPL mutations occur in ∼8% and 4% of patients with PMF and ET, respectively. Almost 5% of PMF patients and 10% of those with ET do not harbor any of the 3 driver mutations and are considered triple negative.5,7 Nevertheless, many other somatic mutations and genomic aberrations have been identified that might represent driver events but, in most cases, they accompany other lesions and influence disease outcome.3
PMF and SMF patients are currently managed in the same way because they share common histopathologic features and clinical manifestations. Prediction of survival influences the therapeutic strategy used and is based on prognostic models, such as the International Prognostic Scoring System (IPSS) and Dynamic-IPSS (DIPSS), which have been designed for PMF based on clinical features. Nevertheless, because these tools are not able to accurately distinguish different risk categories in SMF,8,9 the Myelofibrosis Secondary to PV and ET-Prognostic Model (MYSEC-PM)10 was recently created. The need for more specific and more accurate prognostic models in PMF and SMF arises from the observation that only allogeneic stem cell transplantation (ASCT) is potentially curative and is able to prolong survival in patients; however, the incidence of mortality and severe adverse events justify this approach only in high-risk patients.11 New prognostication models have been developed: the Mutation-enhanced International Prognostic Score System (MIPSS70) and the most recent MIPSS70+ version 2.0 integrate clinical information with mutation and cytogenetic data to identify high-risk PMF patients aged ≤ 70 years who are candidates for ASCT.12,13 If cytogenetics is available, a genetically inspired prognostic scoring system (GIPSS) can be applied to predict patients’ survival based on mutations and karyotype information.14
To identify disease subtypes that are characterized by a poor outcome, we studied the correlation between gene expression profiles (GEPs) of PMF and SMF patients and their clinical characteristics, with particular interest in survival. This approach has already been successfully applied in other solid and hematological malignancies, including AML15,16 and myelodysplastic syndromes,17-19 leading to the discovery of gene expression signatures that identify patients with inferior survival. These studies demonstrated that gene expression profiling could be a useful tool for the classification of hematological malignancies, because it might improve the identification of high-risk patients who have a poor prognosis.
Starting from these observations, in this proof-of-concept study, we analyzed the GEP of granulocytes from patients affected by PMF and SMF. To assess the correlation between GEP and survival, we applied a Cox proportional-hazards model to our data set, which led to the identification of a list of genes whose expression can divide patients into 2 subgroups with different outcomes. Overall, our results suggest that gene expression analysis may complement current methods for risk stratification in MF.
Patients and methods
Patients and samples
This study was conducted using granulocyte samples from 114 patients with a diagnosis of PMF or SMF recruited from 5 Italian centers. PMF was diagnosed according to 2016 World Health Organization criteria,1 whereas International Working Group for Myeloproliferative neoplasms Research and Treatment criteria2 were used for the diagnosis of PET-MF and PPV-MF. The study was conducted in accordance with the Declaration of Helsinki and was approved by local ethics committees. All subjects provided informed written consent.
Granulocytes were collected from peripheral blood using a density gradient. Following centrifugation, granulocytes and erythrocytes formed a cell pellet at the bottom of the tube, and granulocytes can be purified using red blood cell lysis reagent. The different units provided frozen granulocyte pellets and lysed cells.
As previously described,12,20 gene mutations were detected in DNA from peripheral blood cells. Real-time quantitative polymerase chain reaction was used to identify JAK2V617F and MPLW515x mutations, whereas CALR mutations were detected by capillary electrophoresis, followed by bidirectional sequencing, and classified as type 1/type 1-like or type 2/type 2-like.21 Next-generation sequencing was used to detect high molecular risk (HMR) mutations (ie, ASXL1, EZH2, IDH1, IDH2 and SRFSF2).
RNA extraction and gene expression profiling
Total cellular RNA was isolated from stored frozen granulocyte pellets using TRIzol Reagent (Invitrogen, ThermoFisher Scientific), following the manufacturer’s instructions. RNA sample concentration was evaluated using a NanoDrop ND-1000 spectrophotometer (ThermoFisher Scientific), and purity was assessed using 260/280 nm and 260/230 ratios. An Agilent 2100 Bioanalyzer (Agilent Technologies) was used to determine the integrity of total RNA.
Gene expression profiling was performed using HG-U219 Array Strips (Affymetrix), as previously described.22 Biotin-labeled complementary RNA was synthesized starting from 100 ng of total RNA using a GeneChip HT 3′ IVT PLUS Reagent Kit (Applied Biosystems), according to the standard protocol supplied, whereas microarray hybridization, staining, and scanning were performed using a GeneAtlas Hybridization, Wash, and Stain Kit (Affymetrix) and a GeneAtlas System (Affymetrix), following the manufacturer’s recommendations.
Partek Genomics Suite (GS) software, version 7.0 (Partek Inc., St. Louis, MO) was used to normalize the probe level data and convert them into expression values by means of robust multiarray average. An exploratory principal component analysis was performed, and the existing batch effect due to the clinical unit of origin was removed using Partek GS. The “Remove batch effect” function in Partek GS is based on a mixed-model analysis of variance and allows adjustment of the gene expression matrix, removing differences between batches. Principal component analysis was then used to verify the batch effect adjustment. Cox regression analysis function was run to identify probesets whose expression correlates with patient survival. Probesets with a P value < .005 were selected for further analysis and were used to perform supervised hierarchical clustering with Partek GS.
Classification model construction
The classification model was built starting from the list of probesets resulting from Cox regression analysis and risk classes defined with hierarchical clustering. The “nearest shrunken centroids” supervised learning technique, implemented in the pamr.train function of the R package pamr (v.1.56.1), was applied for the classification model generation. A standardized centroid was calculated for each risk class and then it was shrunk toward the overall centroid by 30 different thresholds. The threshold giving the lowest error rate was selected. The “nearest shrunken centroids” technique made it possible to optimize the number of probesets by excluding from the prediction rule those whose expression was not sufficiently different between the 2 groups (and thus not contributing to the classification). Briefly, the centroid is defined as the median expression of each probeset in each class divided by the standard deviation in each class. Each of the class centroids was shrunk toward the overall centroid by an amount called threshold. This shrinkage consists of moving the centroid toward 0 by threshold, setting it equal to 0 if it hits 0. If a probeset was shrunk to 0 for all classes, then it did not survive the threshold.
The resulting model was cross-validated with a k-fold cross-validation method, with k = 20, using the pamr.cv function in the R package pamr. The whole data set was split into k smaller sets and then several models were defined using k − 1 of the folds. The remaining data were used for testing the model, repeating the procedure until each fold was used as the test set. The class of the test samples was predicted based on the nearest centroid.
To further optimize the number of probesets, model generation and cross-validation were repeated m − 1 times (where m is the total number of probesets). As described previously,23 starting from 2 probesets, each time 1 probeset was sequentially added from the top of the rank-ordered probeset list based on the hazard ratio (HR), until all of the probesets were used. The model’s performance was assessed with the use of the cross-validated misclassification error calculated with the pamr.cv function from the pamr R package; the model with the optimized set of probesets that provided the lowest error was selected.
Statistical analysis
Differences in the distribution of numerical variables were evaluated using the Mann-Whitney U test or the Kruskal-Wallis test for the comparison of 2 groups or >2 groups, respectively. Categorical variables were compared using the χ2 test or Fisher’s exact test. Overall survival (OS) was calculated from the date of sample collection to the date of death or last follow-up. When calculating leukemia-free survival (LFS), the date of leukemic transformation was used in place of the date of death. OS and LFS analyses were performed with the Kaplan-Meier method, and the log-rank test was used to compare curves. Univariate and multivariate analyses were carried out using Cox proportional-hazards regression for OS. Analyses were performed using R version 3.4.1. A P value < .05 was considered statistically significant.
Results
Patients
A total of 114 patients (35 pre-PMF, 37 overt PMF, 26 PET-MF, and 16 PPV-MF) was studied. Table 1 shows the distribution of patients with regard to MF clinical subtype. No difference in terms of OS was detected among these groups (Table 1; supplemental Figure 1), whereas some dissimilarities were evident in terms of the clinical characteristics. Pre-PMF patients displayed increased hemoglobin (Hb) levels and were less frequently anemic; platelet count was increased in this group, and splenomegaly was more frequent.24,25 Hb was also increased in PPV-MF, as were white blood cell counts. Anemia was less frequent in these patients, whereas leukocytosis was more common.20,26 PET-MF patients showed increased platelet counts.20,26 All PPV-MF patients harbored a JAK2V617F mutation, with the exception of 1 patient who displayed a mutation on exon 12. Patients with pre-PMF displayed an increased frequency of SRSF2 mutations and >2 HMR mutations. Leukemic transformation occurred in 13 patients, whereas 49 died of causes related to the disease.
Table 1.
Clinical and laboratory characteristics of patients included in our dataset, divided according to diagnosis
| Variable | Pre-PMF (n = 35) | Overt PMF (n = 37) | PET-MF (n = 26) | PPV-MF (n = 16) | P |
|---|---|---|---|---|---|
| Follow-up, median (95% CI), y | 6.88 (3.39-NA) | 5.54 (3.26-6.36) | 4.55 (2.91-NA) | 4.18 (1.72-6.79) | 6.15E-01 |
| Males | 19 (54.3) | 21 (56.8) | 13 (50.0) | 7 (43.8) | 8.33E-01 |
| Age | |||||
| Median (range), y | 62.90 (34.6-80.9) | 63.80 (31.1-84.3) | 65.80 (32.1-81.0) | 71.10 (42.6-85.6) | 9.62E-02 |
| >65 y | 13 (37.1) | 15 (40.5) | 14 (53.8) | 11 (68.8) | 1.38E-01 |
| Hb | |||||
| Median (range), g/dL | 12.40 (8.0-16.6) | 11.20 (5.2-15.3) | 10.75 (6.5-14.6) | 12.55 (9.2-15.9) | 2.52E-04 |
| <10 g/dL | 2 (5.7) | 9 (24.3) | 9 (34.6) | 2 (12.5) | 2.75E-02 |
| Leukocytes | |||||
| Median (range), ×109/L | 8.70 (3.6-41.0) | 10.00 (2.8-89.0) | 9.58 (2.3-104.0) | 14.90 (5.9-88.7) | 1.35E-02 |
| >25 × 109/L | 3 (8.8) | 6 (16.2) | 3 (11.5) | 7 (46.7) | 9.09E-03 |
| Platelets | |||||
| Median (range), ×109/L | 410.0 (72-1299) | 179.0 (22-1252) | 377.5 (61-1568) | 224.5 (20-1271) | 6.54E-03 |
| <100 × 109/L | 2 (5.9) | 9 (25.0) | 2 (8.3) | 1 (6.2) | 6.19E-02 |
| Circulating blasts ≥1% | 5 (15.2) | 8 (25.8) | 8 (34.8) | 3 (20.0) | 3.77E-01 |
| Circulating blasts ≥2% | 5 (15.2) | 4 (12.9) | 6 (26.1) | 2 (13.3) | 5.81E-01 |
| BM fibrosis grade ≥2 | — | 33 (97.1) | 23 (100) | 13 (92.9) | 4.44E-01 |
| Constitutional symptoms | 5 (14.7) | 11 (30.6) | 7 (26.9) | 5 (31.2) | 4.08E-01 |
| Splenomegaly | 16 (45.7) | 31 (86.1) | 22 (84.6) | 10 (71.4) | 6.24E-04 |
| Driver mutation | |||||
| JAK2 V617F | 12 (34.3) | 16 (43.2) | 11 (42.3) | 15 (93.8) | 8.04E-04 |
| JAK2 ex12 | 0 | 0 | 0 | 1 (6.2) | — |
| CALR unspecified | 0 | 0 | 1 (3.8) | 0 | — |
| CALR type 1 | 4 (11.4) | 10 (27.0) | 7 (26.9) | 0 | 1.99E-01 |
| CALR type 2 | 5 (14.3) | 3 (8.1) | 0 | 0 | 1.31E-01 |
| MPL | 5 (14.3) | 3 (8.1) | 3 (11.5) | 0 | 7.07E-01 |
| Triple negative | 9 (25.7) | 5 (13.5) | 4 (15.4) | 0 | 3.69E-01 |
| ASXL1 mutation (n evaluable, total = 85) | 25 | 32 | 15 | 13 | |
| n (%) | 11 (44.0) | 10 (31.2) | 5 (33.3) | 6 (46.2) | 6.81E-01 |
| EZH2 mutation (n evaluable, total = 82) | 24 | 27 | 16 | 15 | |
| n (%) | 3 (12.5) | 2 (7.4) | 0 | 1 (6.7) | 5.27E-01 |
| SRSF2 mutation (n evaluable, total = 81) | 23 | 27 | 16 | 15 | |
| n (%) | 7 (30.4) | 1 (3.7) | 1 (6.2) | 0 | 5.98E-03 |
| IDH1/2 mutation (n evaluable, total = 81) | 23 | 27 | 16 | 15 | |
| n (%) | 2 (8.7) | 1 (6.7) | 0 | 1 (6.7) | 6.36E-01 |
| HMR (n evaluable, total = 86) | 25 | 29 | 17 | 15 | |
| n (%) | 14 (56.0) | 12 (41.4) | 6 (35.3) | 7 (46.7) | 5.66E-01 |
| ≥2 | 8 (32.0) | 2 (6.9) | 0 | 1 (6.7) | 6.82E-03 |
| DIPSS (n evaluable, total = 107) | 31 | 36 | 25 | 15 | |
| Low | 14 (45.2) | 9 (25.0) | 4 (16.0) | 3 (20.0) | |
| Intermediate-1 | 10 (32.3) | 13 (36.1) | 13 (52.0) | 5 (33.3) | |
| Intermediate-2 | 3 (9.7) | 12 (33.3) | 4 (16.0) | 5 (33.3) | |
| High | 4 (12.9) | 2 (5.6) | 4 (16.0) | 2 (13.3) | 3.89E-01 |
| MIPSS70 (n evaluable, total = 73) | 21 | 25 | 15 | 12 | |
| Low | 10 (47.6) | 2 (8.0) | 0 | 1 (8.3) | |
| Intermediate | 4 (19.0) | 14 (56.0) | 10 (66.7) | 5 (41.7) | |
| High | 7 (33.3) | 9 (36.0) | 5 (33.3) | 6 (50.0) | 1.71E-03 |
| MYSEC-PM (n evaluable, total = 41) | 23 | 15 | |||
| Low | — | — | 7 (30.4) | 2 (13.3) | |
| Intermediate-1 | — | — | 9 (39.1) | 7 (46.7) | |
| Intermediate-2 | — | — | 4 (17.4) | 3 (20.0) | |
| High | — | — | 3 (13.0) | 3 (20.0) | 6.70E-01 |
| Progression to leukemia | 8 (22.9) | 3 (8.1) | 2 (7.7) | 0 | 6.14E-02 |
| Death | 13 (37.1) | 17 (45.9) | 9 (34.6) | 10 (62.5) | 2.78E-01 |
Unless otherwise noted, data are n (%). Significant P values (<.05) are highlighted in bold.
NA, not available. —, missing value.
Identification of a gene signature that correlates with OS
Cox-regression analysis identified 832 probesets (corresponding to 596 genes) that correlated with patient survival; among them, 433 genes were associated with inferior survival (supplemental Table 1). According to hierarchical clustering (Figure 1A), this list splits our data set into 2 main branches composed of 62 (left) and 52 (right) samples. As shown in Figure 1B and supplemental Table 2, the cluster on the right was characterized by significantly inferior OS (P = 4.38E-6, log-rank test). Moreover, the frequency of dead patients was significantly higher in the right branch (P = 3.08E-4, Fisher’s exact test) (supplemental Table 2); therefore, we named this cluster the high-risk group, whereas the other one was termed the low-risk group.
Figure 1.
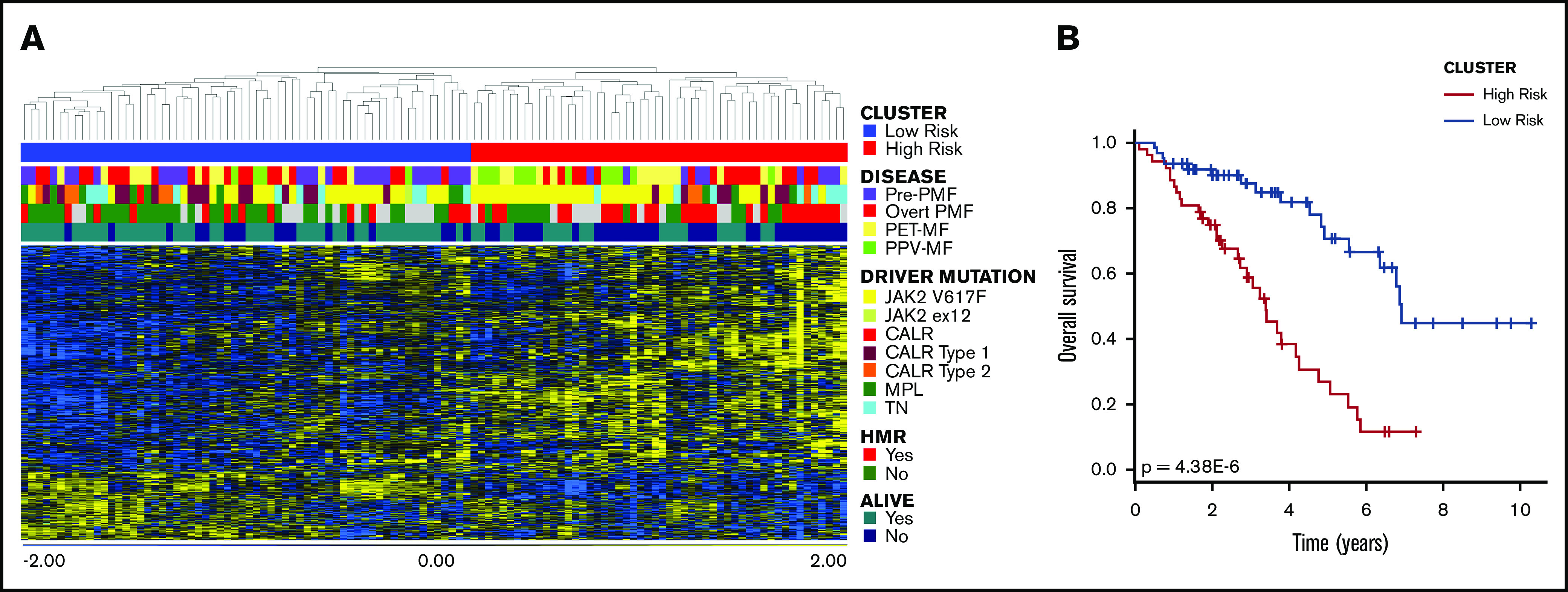
Cox-regression analysis. (A) Hierarchical clustering of samples according to the expression of 832 probesets that correlated with OS. The dataset was divided into 2 clusters. (B) The red cluster (on the right in the heatmap) displayed significantly inferior survival compared with the other one, as demonstrated by the result of log-rank test. Therefore, we defined the blue group as low-risk, whereas the red one was named high-risk.
The majority of pre-PMF samples were included in the low-risk cluster, whereas SMF was more frequent in the high-risk group (Figure 1A; supplemental Table 1). The high-risk group was also enriched in patients harboring JAK2V617F mutations; the frequency of homozygous mutations was increased in this subgroup, whereas JAK2 heterozygosity was more frequent in the low-risk group. Moreover, 26 of 52 samples (65%) in the high-risk group harbored ≥1 HMR mutation (Figure 1A; supplemental Table 2). Despite this, we identified a significant difference only in the distribution of patients with ASXL1 mutations (P value = 2.79E-4, Fisher’s exact test). Considering clinical characteristics, the high-risk group displayed features of patients with predictable inferior survival. Indeed, the median age at the time of sample collection was higher in this group compared with the low-risk group. White blood cell count was increased in high-risk patients, whereas Hb levels and platelet count were decreased compared with the low-risk group. The high-risk group had greater frequencies of patients with >1% or 2% circulating blasts, splenomegaly, and BM fibrosis grade ≥ 2 (supplemental Table 2). Taken together, these results demonstrated that Cox-regression analysis led to the identification of genes whose expression correlates with OS in MF patients. These genes identified 2 patient subgroups characterized by high-level differences in terms of clinical and molecular features and OS.
Gene expression–based classification correlates with patients’ clinical and molecular features
Starting with the list of 832 probesets derived from Cox-regression analysis, we constructed a gene expression–based classifier for the 2 risk categories defined according to hierarchical clustering. As detailed in the Patients and methods section, we applied a supervised learning technique, the “nearest shrunken centroids” method, to construct the model and cross-validated it using a 20-fold cross-validation strategy exploiting the pamr.cv R function to estimate the misclassification error and build a robust classifier. We obtained an optimized model using the first 351 probesets of the list, of which 273 (corresponding to 201 genes) survived the cross-validation threshold (supplemental Table 3).
According to this classification, the low-risk and high-risk groups were identified, which contained 60 and 54 patients, respectively (Table 2). Patients in the high-risk group displayed several detrimental characteristics. The high-risk class was enriched in samples derived from dead patients (Table 2) and was characterized by a significantly inferior survival compared with the low-risk group (P value = 1.78E-7, log-rank test) (Figure 2A; Table 2). Moreover, the low-risk group was enriched in pre-PMF samples, whereas overt PMF samples were distributed equally between the 2 classes. On the contrary, higher percentages of PET-MF and PPV-MF were present in the high-risk group. Taking into account molecular characteristics, the high-risk group was enriched in patients harboring the JAK2V617F mutation (Table 2); in particular, homozygosity was more frequent in this group compared with the low-risk one. The frequency of patients with ≥1 HMR mutation was increased in the high-risk group; again, this group was enriched in ASXL1-mutated patients compared with the low-risk one (Table 2).
Table 2.
Clinical and molecular characteristics of patients included in our dataset, classified according to our gene expression–based model
| Variable | Low-risk (n = 60) | High-risk (n = 54) | P |
|---|---|---|---|
| OS, median (95% CI), y | 6.93 (5.56-NA) | 3.26 (2.68-3.81) | 1.78E-07 |
| Males | 32 (53.3) | 28 (51.9) | 1.00E+00 |
| Disease | |||
| Pre-PMF | 25 (41.7) | 10 (18.5) | |
| Overt PMF | 20 (33.3) | 17 (31.5) | |
| PET-MF | 11 (18.3) | 15 (27.8) | |
| PPV-MF | 4 (6.7) | 12 (22.2) | 1.17E-02 |
| Age | |||
| Median (range), y | 61.4 (32.1-81) | 69.95 (31.1-85.6) | 5.73E-05 |
| >65 y | 18 (30) | 35 (64.8) | 3.15E-04 |
| Hb | |||
| Median (range), g/dL | 12.1 (6.5-16.6) | 11.15 (5.2-14.4) | 1.89E-03 |
| <10 g/dL | 6 (10) | 16 (29.6) | 9.44E-03 |
| Leukocytes | |||
| Median (range), ×109/L | 8.00 (2.3-38.7) | 14.2 (3.15-104) | 3.98E-06 |
| >25 × 109/L | 1 (1.7) | 18 (34) | 3.80E-06 |
| Platelets | |||
| Median (range), ×109/L | 386 (22-1568) | 226 (20-1271) | 6.85E-03 |
| <100 × 109/L | 4 (7) | 10 (18.9) | 8.62E-02 |
| Circulating blasts ≥1% | 8 (14.3) | 16 (34.8) | 1.94E-02 |
| Circulating blasts ≥2% | 5 (8.9) | 12 (26.1) | 3.13E-02 |
| BM fibrosis grade ≥2 | 33 (55.9) | 36 (76.6) | 3.97E-02 |
| Constitutional symptoms | 10 (16.7) | 18 (34.6) | 4.78E-02 |
| Splenomegaly | 34 (56.7) | 45 (88.2) | 2.93E-04 |
| Driver mutation | |||
| JAK2 V617F | 22 (36.7) | 32 (59.3) | 2.38E-02 |
| JAK2 ex12 | 1 (1.7) | 0 | — |
| CALR unspecified | 1 (1.7) | 0 | — |
| CALR type 1 | 14 (23.3) | 7 (13) | 2.26E-01 |
| CALR type 2 | 6 (10) | 2 (3.7) | 2.77E-01 |
| MPL | 8 (13.3) | 3 (5.6) | 2.11E-01 |
| Triple negative | 8 (13.3) | 10 (18.5) | 6.08E-01 |
| JAK2 V617F | |||
| Heterozygous | 14 (66.7) | 7 (22.6) | |
| Homozygous | 7 (33.3) | 24 (77.4) | 3.41E-03 |
| CALR type 1 absence | 46 (76.7) | 47 (87) | 2.26E-01 |
| ASXL1 mutation (n evaluable, total = 85) | 46 | 39 | |
| n (%) | 9 (19.6) | 23 (59) | 2.79E-04 |
| EZH2 mutation (n evaluable, total = 82) | 48 | 34 | |
| n (%) | 3 (6.2) | 3 (8.8) | 6.88E-01 |
| SRSF2 mutation (n evaluable, total = 81) | 47 | 34 | |
| n (%) | 4 (8.5) | 5 (14.7) | 4.81E-01 |
| IDH1/2 mutation (n evaluable, total = 81) | 47 | 34 | |
| n (%) | 3 (6.4) | 1 (2.9) | 6.36E-01 |
| HMR (n evaluable, total = 86) | 48 | 38 | |
| n (%) | 14 (29.2) | 25 (65.8) | 1.02E-03 |
| ≥2 | 4 (8.3) | 7 (18.4) | 2.03E-01 |
| DIPSS (n evaluable, total = 107) | 57 | 50 | |
| Low | 23 (40.4) | 7 (14) | |
| Intermediate-1 | 24 (42.1) | 17 (34) | |
| Intermediate-2 | 8 (14) | 16 (32) | |
| High | 2 (3.5) | 10 (20) | 6.00E-04 |
| MIPSS70 (n evaluable, total = 73) | 41 | 32 | |
| Low | 12 (29.3) | 1 (3.1) | |
| Intermediate | 22 (53.7) | 11 (34.4) | |
| High | 7 (17.1) | 20 (62.5) | 1.01E-04 |
| MYSEC-PM (n evaluable, total = 38) | 14 | 24 | |
| Low | 5 (35.7) | 4 (16.7) | |
| Intermediate-1 | 5 (35.7) | 11 (45.8) | |
| Intermediate-2 | 3 (21.4) | 4 (16.7) | |
| High | 1 (7.1) | 5 (20.8) | 4.36E-01 |
| Progression to leukemia | 4 (6.7) | 9 (16.7) | 1.40E-01 |
| Death | 15 (25) | 34 (63) | 6.01E-05 |
Unless otherwise noted, data are n (%). Significant P values (<.05) are highlighted in bold.
Figure 2.

OS and LFS. (A) Kaplan-Maier analysis demonstrated a statistically significant difference in terms of OS between the high-risk and low-risk groups, as identified by gene expression–based classifier. (B) The high-risk group showed an inferior LFS compared with the low-risk group.
Next, we studied the distribution of clinical variables included in contemporary prognostic models and observed that high-risk classification correlated with the presence of clinical markers of inferior survival. Indeed, patient age at sample collection was higher in the high-risk group, which was also characterized by inferior Hb levels and platelet counts, superior white blood cell counts, increased incidence of splenomegaly, circulating blasts > 1% or 2%, BM fibrosis grade ≥ 2, and constitutional symptoms (Table 2). Even if 9 of 13 patients who developed secondary AML clustered within the high-risk group, this difference failed to reach statistical significance (Table 2). Nevertheless, survival analysis revealed that LFS was reduced significantly in the high-risk group (P value = 1.9E-2, log-rank test) (Figure 2B). Collectively, these results demonstrated that belonging to the high-risk group represents a risk factor for survival (HR, 4.736; 95% confidence interval [CI], 2.5-8.9; P value = 1.48E-6, Wald test) and for leukemic transformation (HR, 3.976; 95% CI, 1.2-13.6; P value = 2.75E-2, Wald test). Of particular interest, this is also true considering samples stratified according to diagnosis. Indeed, high-risk patients displayed inferior OS when PMF and SMF samples were considered separately (supplemental Figure 2).
Comparison with contemporary prognostic model
Next, we looked at the correlation between GEP-based and prognostic model classifications. The low-risk group was significantly enriched in patients belonging to DIPSS Low and Intermediate-1 categories (P value = 6.00E-4, χ2 test), whereas the majority of patients classified as DIPSS Intermediate-2 (16/24 [66.7%]) or High (10/12 [83.3%]) risk clustered within the gene expression–defined high-risk group (Table 2). Interestingly, DIPSS Intermediate-1 and Intermediate-2 patients belonging to the high-risk group, according to our classification, showed a significantly inferior survival compared with those identified as low-risk (P values = 5.07E-4 and 2.53E-2, respectively, log-rank test) (Figure 3A), and Cox-regression analysis confirmed that high-risk classification represented a risk factor for inferior survival in DIPSS Intermediate-1 and Intermediate-2 groups (Figure 3B). High-risk classification retained its significance in multivariate analysis when evaluated in the context of DIPSS classification (P value = 5.69E-5, Wald test) (Figure 3C), as well as when considering risk factors included in the DIPSS prognostic model (P value = 4.96E-3, Wald test) (supplemental Table 4). With regard to SMF, we found that the distinction between high-risk and low-risk patients in terms of survival reached statistical significance for MYSEC-PM lower-risk categories (Low plus Intermediate-1), whereas it approached significance for higher-risk classes (Intermediate-2 plus High) (P values = 1.39E-2 and 7.26E-2, respectively, log-rank test) (supplemental Figure 3).
Figure 3.
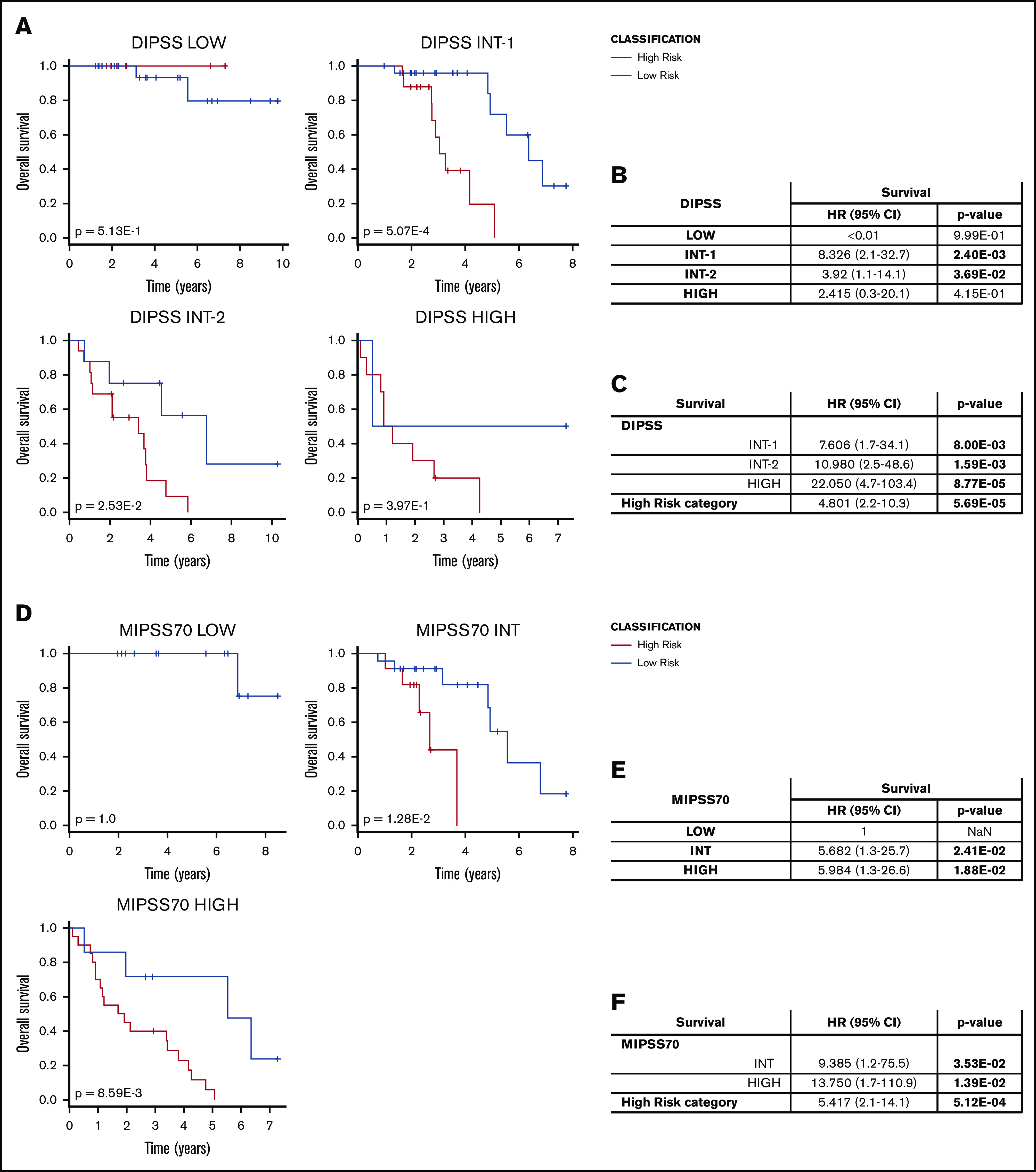
Comparison with DIPSS and MIPSS70 classification. (A) Survival curves for samples stratified according to DIPSS and classified in the high-risk and low-risk groups using our gene expression–based model. P values were calculated using the log-rank test. (B) HR for death comparing the high-risk and low-risk groups in the context of DIPSS classification. (C) Results of multivariate analyses. (D-F) The same analysis was performed for patients classified using the MIPSS70 scoring system. (D) Survival analysis. HR for death of samples stratified according to MIPSS70 (E) and using multivariate analyses (F).
More recently, outcome prediction in MF was improved by the inclusion of molecular information in prognostic models. Because of the lack of cytogenetics data for most of the samples, we decided to compare our classification using the MIPSS70 model.12 Most MIPSS70 High-risk patients clustered within the high-risk group, whereas the low-risk group contained higher frequencies of Low and Intermediate samples (P value = 1.01E-4, χ2 test) (Table 2). Nevertheless, gene expression–based classification distinguished high-risk patients from low-risk ones with different survival within MIPSS70 Intermediate and High categories (P values = 1.28E-2 and 8.59E-3, respectively, log-rank test) (Figure 3D). The high-risk classification represented a risk factor for inferior survival for patients belonging to these prognostic categories (Figure 3E), also retaining its significance in multivariate analyses when considering MIPSS70 classification (P value = 5.12E-4, Wald test) (Figure 3F) or factors included in the MIPSS70 model (P value = 1.12E-4, Wald test) (supplemental Table 5).
Taken together, these results demonstrated that a gene expression–based classifier might also identify groups of patients characterized by different outcomes in the context of contemporary prognostic models.
Discussion
MF is a complex hematologic disorder arising from the mutation of hematopoietic stem cells. Excessive proliferation of cells from the neoplastic clones gives rise to granulocyte and megakaryocyte hyperplasia, whereas the altered interaction between hematopoietic and stromal cells in the BM microenvironment leads to the development of the fibrosis that is the hallmark of the disease.27 PMF originates from the acquisition of somatic driver mutations in JAK2, MPL, or CALR genes by hematopoietic stem/progenitor cells, even if almost 5% of patients do not harbor any of these and, therefore, are considered triple negative.7 Several other mutations might be present in PMF patients; among them, those occurring in EZH2, ASXL1, SRSF2, and IDH1/2 genes are termed HMR because of their negative impact on OS and LFS.28
MF can be primary or secondary to PV and ET; moreover, a prefibrotic stage and overt PMF must be distinguished within PMF, depending on the degree of BM fibrosis, because they represent distinct entities. As demonstrated by clinical features of patients included in our data set, in agreement with literature findings, PET-MF was characterized by increased platelet counts,20,26 whereas pre-PMF displayed higher Hb levels and platelet counts and a lower frequency of splenomegaly.24,25 Increased Hb was also evident in PPV-MF, which was also characterized by an increased leukocyte count and the presence of a JAK2V617F mutation in all patients.20,26 Despite these differences, MF cases are currently managed in the same way.29 To study whether gene expression might provide clinical and prognostic information at any time during the disease course, we included samples from diagnosis and during follow-up in our dataset and correlated GEP with patient features at those time points. Many prognostic models were developed in the last decade, allowing risk stratification in MF patients based on clinical features (ie, IPSS, DIPSS), as well as on molecular and cytogenetics characteristics (ie, DIPSS-plus, MIPSS70, MIPSS70+v2.0, GIPSS).12,13,30-32 These models were developed for prognostication in PMF but are also applied in SMF cases, even if a specific model was recently developed (MYSEC-PM).10 More recently, Grinfeld et al emphasized the impact of driver mutations on patient prognosis by defining a classification scheme for MPNs and related disorders based on the type of genomic alterations harbored by patients.3
Because clinical decision making in MF is mainly influenced by survival prediction, it is fundamental to develop easy-to-use models that can identify patients who are at the highest risk of death. Gene expression analysis was recently adopted to predict the risk of recurrence in breast cancer,33 but gene expression signatures were also developed to predict survival in hematopoietic malignancies, such as AML (ie, 17-gene leukemia stem cell score [LSC17]).15 Therefore, we decided to study the GEP of MF granulocytes to evaluate its impact on disease phenotype and patient outcome. We focused on granulocytes because they represent an easily accessible myeloid cell population that belongs to the neoplastic clone, unlike peripheral blood mononuclear cells that also contain lymphoid elements.34
Using Cox-regression analysis, we identified survival-related transcripts that were used to construct a classifier based on the expression of 201 genes that could identify 2 groups of patients. High-risk patients identified by our model displayed an inferior OS and LFS compared with low-risk cases. Our results demonstrated that gene expression–based classification showed good agreement with contemporary prognostic models; indeed, the high-risk classification correlated with the presence of several detrimental features, such as advanced age, decreased Hb levels (<10 g/dL) and platelet count (<100 × 109/L), increased white blood cell count (>25 × 109/L), circulating blasts ≥ 1% and 2%, and the presence of constitutional symptoms and splenomegaly. Interestingly, in a recent report by Penna et al,35 the absence of the same detrimental features correlated with long survival (>20 years) in patients with PMF; this condition was similar to that observed in the low-risk group identified by gene expression signature. The frequency of patients with ≥1 HMR mutation was increased in the high-risk group, and the presence of these variants correlates with MF diagnosis and a more aggressive disease with inferior OS.3 Moreover, our findings underscored the impact of JAK2V617F allele burden on OS, because the high-risk group contained an increased frequency of homozygous patients, whereas heterozygous ones clustered within the low-risk group. These data are in agreement with those of Grinfeld et al, who demonstrated that patients with a JAK2V617F heterozygous disease have favorable outcomes.3
Our gene expression–based classifier was able to distinguish high-risk patients from low-risk ones within intermediate-risk classes. Indeed, in patients stratified according to DIPSS, belonging to the high-risk group represented a risk factor for inferior survival in the Intermediate-1 and Intermediate-2 categories. The high-risk group was characterized by significantly inferior OS compared with the low-risk group: 3.05 years and 3.42 years in the Intermediate-1 and Intermediate-2 classes, respectively. Likewise, within the MIPSS70 Intermediate group, high-risk patients displayed a median OS of 2.68 years, which was significantly lower compared with the low-risk group. Multivariable analysis demonstrated that gene expression–based classification might represent a risk factor for inferior survival independent from the DIPSS and MIPSS70 classifications, as well as from factors included in these models. Therefore, this suggests that gene expression analysis might provide additional information other than that included in contemporary prognostic models.
Overall, this study demonstrates the correlation between GEP and MF clinical features and provides the proof of concept that gene expression analysis should be considered to complement risk stratification in MF. To date, treatment algorithms suggest observation accompanied by palliative drug therapy for DIPSS Intermediate-1 patients, whereas ASCT and enrollment in clinical trials must be considered for patients belonging to Intermediate-2 and High-risk categories.6 Our results demonstrated that high-risk patients with an expected median survival < 5 years might also be identified within DIPSS Intermediate-1 and Intermediate-2 classes. The same is true when we consider Intermediate-risk patients based on MIPSS70 classification.
Overall, given the robustness of these analyses, we believe that these data may still be helpful to the hematological community in uncovering the possibility that GEPs might integrate contemporary prognostic models. Further studies might be able to prospectively validate these data in an independent cohort of MF cases, thus improving the identification of patients with expected inferior survival who can benefit from participation in clinical trials or ASCT.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by the Fondazione Associazione Italiana per la Ricerca sul Cancro (AIRC) under 5 per Mille 2018 - ID 21267 program and IG project ID 19818, the Italian Ministry of Health (project RF-2016-02362930), and the Ministry of Education, University and Research (project #2017WXR7ZT_005).
Footnotes
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE159514; private access token: qnkjcckqhhgtxsz).
Data sharing requests should be sent to Rossella Manfredini (rossella.manfredini@unimore.it).
Authorship
Contribution: S.R. performed GEP analyses and statistical analyses; S.C., A.G., and S.B. constructed the prediction model; R.Z., E.B., E.G., and C.C. performed granulocyte sorting; S.P., S.F., and S.M. performed total RNA purification; L.T., S. Sartini, and M.M. performed RNA quantification and quality control; S.M., L.T., and S. Sartini performed RNA quality analyses; P.G., C.M., F.G., A.P., D.P., E.R., S. Salmoiraghi, B.M., L.V., V.R., G.B., F.P., A.R., L.M., and M.C. selected and recruited patients and provided clinical data; and S.R., E.T., A.M.V., and R.M. designed the research and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The members of the Mynerva Project are listed in the supplemental Appendix.
Correspondence: Rossella Manfredini, Centre for Regenerative Medicine “Stefano Ferrari”, University of Modena and Reggio Emilia, via Gottardi n. 100, 41125 Modena, Italy; e-mail: rossella.manfredini@unimore.it.
References
- 1.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia [published correction appears in Blood. 2016;128(3):462-463]. Blood. 2016;127(20):2391-2405. [DOI] [PubMed] [Google Scholar]
- 2.Barosi G, Mesa RA, Thiele J, et al. ; International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) . Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. 2008;22(2):437-438. [DOI] [PubMed] [Google Scholar]
- 3.Grinfeld J, Nangalia J, Baxter EJ, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379(15):1416-1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marneth AE, Mullally A. The molecular genetics of myeloproliferative neoplasms. Cold Spring Harb Perspect Med. 2020;10(2):a034876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92(1):94-108. [DOI] [PubMed] [Google Scholar]
- 6.Tefferi A. Primary myelofibrosis: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91(12):1262-1271. [DOI] [PubMed] [Google Scholar]
- 7.Rumi E, Cazzola M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood. 2017;129(6):680-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masarova L, Bose P, Daver N, et al. Patients with post-essential thrombocythemia and post-polycythemia vera differ from patients with primary myelofibrosis. Leuk Res. 2017;59:110-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernández-Boluda JC, Pereira A, Gómez M, et al. ; Grupo Español de Enfermedades Mieloproliferativas Filadelfia Negativas . The International Prognostic Scoring System does not accurately discriminate different risk categories in patients with post-essential thrombocythemia and post-polycythemia vera myelofibrosis. Haematologica. 2014;99(4):e55-e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Passamonti F, Giorgino T, Mora B, et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia. 2017;31(12):2726-2731. [DOI] [PubMed] [Google Scholar]
- 11.Tefferi A. Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am J Hematol. 2018;93(12):1551-1560. [DOI] [PubMed] [Google Scholar]
- 12.Guglielmelli P, Lasho TL, Rotunno G, et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for transplantation-age patients with primary myelofibrosis. J Clin Oncol. 2018;36(4):310-318. [DOI] [PubMed] [Google Scholar]
- 13.Tefferi A, Guglielmelli P, Lasho TL, et al. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J Clin Oncol. 2018;36(17):1769-1770. [DOI] [PubMed] [Google Scholar]
- 14.Tefferi A, Guglielmelli P, Nicolosi M, et al. GIPSS: genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia. 2018;32(7):1631-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ng SW, Mitchell A, Kennedy JA, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540(7633):433-437. [DOI] [PubMed] [Google Scholar]
- 16.Bullinger L, Döhner K, Bair E, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350(16):1605-1616. [DOI] [PubMed] [Google Scholar]
- 17.Pellagatti A, Benner A, Mills KI, et al. Identification of gene expression-based prognostic markers in the hematopoietic stem cells of patients with myelodysplastic syndromes. J Clin Oncol. 2013;31(28):3557-3564. [DOI] [PubMed] [Google Scholar]
- 18.Mills KI, Kohlmann A, Williams PM, et al. Microarray-based classifiers and prognosis models identify subgroups with distinct clinical outcomes and high risk of AML transformation of myelodysplastic syndrome. Blood. 2009;114(5):1063-1072. [DOI] [PubMed] [Google Scholar]
- 19.Shiozawa Y, Malcovati L, Gallì A, et al. Gene expression and risk of leukemic transformation in myelodysplasia [published correction appears in Blood. 2018;132(8):869-875]. Blood. 2017;130(24):2642-2653. [DOI] [PubMed] [Google Scholar]
- 20.Rotunno G, Pacilli A, Artusi V, et al. Epidemiology and clinical relevance of mutations in postpolycythemia vera and postessential thrombocythemia myelofibrosis: a study on 359 patients of the AGIMM group. Am J Hematol. 2016;91(7):681-686. [DOI] [PubMed] [Google Scholar]
- 21.Guglielmelli P, Rotunno G, Fanelli T, et al. Validation of the differential prognostic impact of type 1/type 1-like versus type 2/type 2-like CALR mutations in myelofibrosis. Blood Cancer J. 2015;5(10):e360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norfo R, Zini R, Pennucci V, et al. ; Associazione Italiana per la Ricerca sul Cancro Gruppo Italiano Malattie Mieloproliferative Investigators . miRNA-mRNA integrative analysis in primary myelofibrosis CD34+ cells: role of miR-155/JARID2 axis in abnormal megakaryopoiesis. Blood. 2014;124(13):e21-e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van ’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530-536. [DOI] [PubMed] [Google Scholar]
- 24.Guglielmelli P, Pacilli A, Rotunno G, et al. ; AGIMM Group . Presentation and outcome of patients with 2016 WHO diagnosis of prefibrotic and overt primary myelofibrosis. Blood. 2017;129(24):3227-3236. [DOI] [PubMed] [Google Scholar]
- 25.Mudireddy M, Shah S, Lasho T, et al. Prefibrotic versus overtly fibrotic primary myelofibrosis: clinical, cytogenetic, molecular and prognostic comparisons. Br J Haematol. 2018;182(4):594-597. [DOI] [PubMed] [Google Scholar]
- 26.Palandri F, Palumbo GA, Iurlo A, et al. Differences in presenting features, outcome and prognostic models in patients with primary myelofibrosis and post-polycythemia vera and/or post-essential thrombocythemia myelofibrosis treated with ruxolitinib. New perspective of the MYSEC-PM in a large multicenter study. Semin Hematol. 2018;55(4):248-255. [DOI] [PubMed] [Google Scholar]
- 27.Gangat N, Tefferi A. Myelofibrosis biology and contemporary management. Br J Haematol. 2020;191(2):152-170. [DOI] [PubMed] [Google Scholar]
- 28.Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861-1869. [DOI] [PubMed] [Google Scholar]
- 29.Tefferi A, Guglielmelli P, Pardanani A, Vannucchi AM. Myelofibrosis treatment algorithm 2018. Blood Cancer J. 2018;8(8):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895-2901. [DOI] [PubMed] [Google Scholar]
- 31.Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703-1708. [DOI] [PubMed] [Google Scholar]
- 32.Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392-397. [DOI] [PubMed] [Google Scholar]
- 33.Güler EN. Gene expression profiling in breast cancer and its effect on therapy selection in early-stage breast cancer. Eur J Breast Health. 2017;13(4):168-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mead AJ, Mullally A. Myeloproliferative neoplasm stem cells. Blood. 2017;129(12):1607-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Penna D, Lasho TL, Finke CM, et al. 20+ Years and alive with primary myelofibrosis: phenotypic signature of very long-lived patients. Am J Hematol. 2019;94(3):286-290. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.