

Abstract
Luspatercept (Reblozyl) was recently approved for treating patients with transfusion-dependent lower-risk myelodysplastic syndrome (MDS) with ring sideroblasts (RS) and/or SF3B1 mutation who were not eligible for erythropoiesis-stimulating agents (ESAs) or patients for whom those agents failed. Luspatercept acts as an activin receptor type IIB fusion protein ligand trap that targets the altered transforming growth factor beta pathway in MDS, which is associated with impaired terminal erythroid maturation. Treatment with luspatercept results in decreased SMAD signaling, which enables erythroid maturation by means of late-stage erythroblast differentiation and thus improves anemia. ESAs, the current standard first-line therapeutic option for anemic lower-risk patients with MDS, also improve red cell parameters mainly by expanding proliferation of early erythroid progenitor cells. However, erythropoietin (EPO) and its receptor (EPO-R) are also required for survival of late-stage definitive erythroid cells, and they play an essential role in promoting proliferation, survival, and appropriate timing of terminal maturation of primitive erythroid precursors. Thus, luspatercept joins the mechanism of ESAs in promoting erythroid maturation. Especially in the subgroup of MDS patients with RS, luspatercept showed high clinical activity for the treatment of anemia in the phase 2 (PACE-MDS) trial and subsequently in the phase 3 (MEDALIST) trial, which resulted in approval by both the US Food and Drug Administration and the European Medicines Agency in April 2020. Additional studies are needed to better understand the mechanism of action and pharmacodynamics of this novel agent in MDS.
Visual Abstract

Introduction
Luspatercept (Reblozyl) is a specific activin receptor fusion protein that acts as a ligand trap to neutralize negative regulators of late-stage erythropoiesis.1-3 Luspatercept was recently approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for treating anemia that results from transfusion-dependent (TD) low- to intermediate-risk myelodysplastic syndromes (MDS) with ring sideroblasts (RS) and/or SF3B1 mutation or myelodysplastic/myeloproliferative neoplasms with RS and thrombocytosis (MDS/MPN-RS-T). Thus, it can now be prescribed in the second-line setting after erythropoiesis-stimulating agents (ESAs) have failed or for patients who are not eligible because of high serum erythropoietin (sEPO) levels.
In the double-blind phase 3 BELIEVE trial (NCT02604433), luspatercept was associated with significant reductions in transfusion burden in adult patients with TD beta thalassemia. On the basis of these data, FDA (2019) and EMA (2020) approved luspatercept also for treating anemia in adult patients with beta thalassemia who required regular transfusions of red blood cells (RBCs).4 According to the initial results of an open-label phase 2 study (NCT03194542), luspatercept also showed promising efficacy in patients with myelofibrosis-associated anemia.5 Thus, a phase 3 randomized study of luspatercept combined with ruxolitinib is under way.
In this review, we will first focus on the evolution of luspatercept from its initial development for treating bone disorders to anemia associated with MDS to its preclinical and clinical development. We will provide directions for additional studies, especially those concerning the activity of luspatercept in patients who do not have RS (currently being evaluated in the ongoing COMMANDS trial) and those concerning the potential of drug combinations. After a short road to approval, luspatercept now fills a long-standing gap by providing a much needed additional treatment option for a large subset of patients with lower-risk MDS (LR-MDS).
Luspatercept in LR-MDS
Most patients with MDS belong to the lower-risk categories, defined by an International Prognostic Scoring System (IPSS) score of low or intermediate-1 risk and a revised IPSS (IPSS-R) score of ≤3.5 points.6 On the basis of IPSS risk stratification, estimated median survival of patients with low-risk MDS who receive supportive care only is 5 to 7 years; median survival for patients with intermediate-1 risk is ∼3 to 5 years.3 It should also be taken into account that many LR-MDS patients die as a result of non-MDS causes.
Especially in LR-MDS, therapeutic decision-making is a challenge because there are only a few options currently available, and treatment is mainly aimed at improving cytopenias to prevent complications such as bleeding and severe infections, decreasing transfusion burden, and improving quality of life.3,7-9 Anemia remains the most frequent cytopenia in these patients, causing major symptoms such as shortness of breath, loss of energy, tachycardia, and dizziness with a significant impact on the quality of life.10-12 Moreover, chronic anemia is associated with multiple secondary complications like worsening of coronary heart or pulmonary disease and an increased tendency to fall.1,3,13
The majority of patients become RBC-TD during the course of their disease, which leads to significant iron overload, with an additional dose-dependent negative impact on survival and a substantial financial burden.3,14 Moreover, the severity of anemia and RBC transfusion dependency correlate with inferior outcomes in MDS.15
Especially for patients who do not have del(5q), single-agent ESAs (ie, recombinant EPO or darbepoetin) represent the first therapeutic step and are the mainstay of therapy in patients with anemia with or without (low) transfusion burden, as long as endogenous EPO levels are <500 U/L, preferably <200 U/L.16,17 Overall response rates are 20% to 40%, but in the small group of RBC-TD patients or with serum EPO levels >200 U/L, responses are lower.16,18 Approximately 80% of LR-MDS patients who are eligible for treatment with ESAs have endogenous EPO levels <200 IU/L, whereas only ∼10% exceed 500 IU/L.19 Most responses to ESAs occur within 3 months of treatment, with a median duration of about 15 to 18 months.3,19 Patients with MDS-RS have a shorter median duration of response to ESAs than those who do not have RS.1,16,19 ESAs especially target early stages of erythropoiesis by inhibiting apoptosis and stimulating erythropoietin-responsive erythroid precursor proliferation, but they are also capable of promoting erythroid maturation.20
In eligible patients who do not respond to single-agent ESAs, the response in as many as 20% of them may be rescued by the addition of granulocyte colony-stimulating factor (G-CSF).21 Treatment with G-CSF is of particular benefit to patients with RS who may display a shorter duration of response to ESAs than patients without RS.21
If treatment with ESAs fails, treatment options are limited but may include lenalidomide (currently approved for del(5q)-positive patients only),22 hypomethylating agents (HMAs) (approved by FDA, but not by EMA, in LR-MDS),23 and new experimental agents available within clinical trials, or supportive care only with regular RBC transfusions.3 In a randomized phase 3 trial comparing lenalidomide monotherapy with lenalidomide plus ESAs in ESA-resistant RBC-TD (≥4 units over 8 weeks) LR-MDS patients, the overall RBC transfusion independent (RBC-TI) rate at ≥8 weeks was 13.8% in the lenalidomide arm vs 24.2% in the lenalidomide plus ESA arm.24
The choice for second-line treatment should always consider various individual characteristics such as biologic, cytogenetic, and molecular data, as well as frailty and comorbidities. In patients with del(5q) disease and the need for continuous transfusion support, lenalidomide is the treatment of choice, and it results in erythroid responses in ∼70% of patients25-27 with a median response duration of 2 years.22 Some patients remain transfusion free for extended periods, even after discontinuing lenalidomide.25 However, RBC-TI rates are only 25% with a median duration of response of <1 year in patients who do not have del(5q) MDS.28
Over the last few years, altered signaling of the transforming growth factor beta (TGF-β) pathway in bone marrow precursor cells has been revealed as a potential therapeutic target in MDS patients. Luspatercept (Figure 1), now approved by FDA and EMA, interferes with the TGF-β pathway and represents a promising new treatment option for MDS-RS+ and/or SF3B1 mutation for whom ESAs have failed (Figure 2). Given the limited number of approved and effective treatments, new treatment strategies are needed for patients who do not have del(5q) LR-MDS and who are dependent on RBC transfusions.1,3,29,30
Figure 1.

Putative mechanism of action of luspatercept, a TGF-β superfamily ligand trap, to improve ineffective erythropoiesis. Baso, basophilic erythroblast; BFU-E, burst-forming unit–erythroid; CFU-E, colony-forming unit–erythroid; Erythro, erythrocyte; m, mature; OrthoC, orthochromatic erythroblast; p, primitive; PolyC, polychromatic erythroblast; ProEbl, proerythroblast; Reti, reticulocyte. Professional illustration by Patrick Lane, ScEYEnce Studios.
Figure 2.
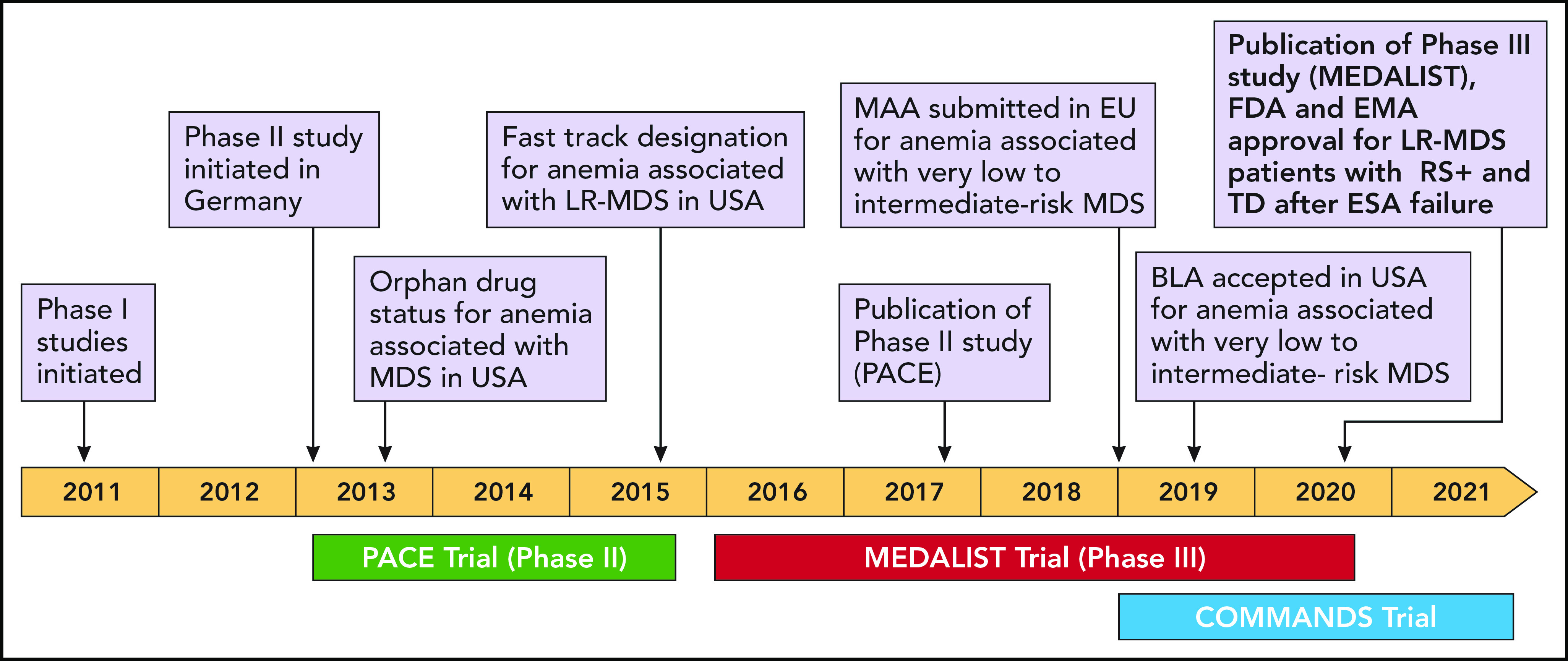
Drug development milestones. BLA, Biologics License Application; EU, European Union; MAA, Marketing Authorization Application; USA, United States of America. Professional illustration by Patrick Lane, ScEYEnce Studios.
Ineffective erythropoiesis in MDS
Ineffective erythropoiesis is the hallmark of LR-MDS, and it is evident by morphologically proven erythroid dysplasia and subsequent anemia. Under healthy conditions, early-stage erythropoiesis includes proliferation of hematopoietic stem cells and subsequent differentiation of those cells into erythroid progenitors; later-stage erythropoiesis is characterized by terminal erythroid maturation into enucleated RBCs.31
New erythrocytes are constantly produced in the bone marrow niche, and they consist of endothelial cells of the vascular system, osteoblasts, stromal cells, hematopoietic cells, and the extracellular matrix.32 In the niche, a complex direct cell-cell contact between the hematopoietic cells and cell adhesion molecules, growth factors, and cytokines is established.32,33 The earliest erythroid progenitors are responsive to several cytokines, including insulin-like growth factor 1 (IGF-1), interleukin-3 (IL-3), granulocyte-macrophage CSF (GM-CSF), along with EPO and stem cell factor, which are required for optimal development and terminal differentiation of erythroid cells.32 At subsequent stages, the stem cell factor acts synergistically with EPO in the proliferation and expansion of the developing erythroid progenitors; erythroid cells at the terminal stages of differentiation have shed their nucleus, endoplasmic reticulum, and mitochondria and are no longer able to proliferate.32
In MDS, impaired erythropoiesis in both early-stage and terminal erythroid differentiation is mainly explained by an inflammatory milieu in the microenvironment as well as defects that include intrinsic clonal disease features. Several mutations affecting the epigenetic modifiers (eg, TET2) or RNA splicing factors (eg, U2AF1) have been linked to NLRP3 inflammasome activation and enhanced innate immune signaling. Thus, inflammatory cytokines are increased in the serum and bone marrow of patients with MDS, establishing a platform for accelerated pyroptotic cell death.34,35 Impaired terminal erythroid differentiation in MDS, as a consequence of the altered cellular and molecular crosstalk within the bone marrow niche including TGF-β signaling, transforms the niche into a proleukemic environment.
Biological background
TGF-β superfamily signaling plays an essential role in the regulation of hematopoiesis in the hematopoietic stem cell niche in the bone marrow (Figures 1 and 3), including the receptor ligands activin and growth differentiation factors (GDFs), through effects on cell inactivity, apoptosis, proliferation, differentiation, and migration.36,37 Under healthy conditions, TGF-β signaling acts as a myelosuppressive factor and inhibits erythroid differentiation by induction of apoptosis and cell cycle arrest in erythroblasts.38 As a consequence, erythroid maturation is mediated by parallel suppression of TGF-β signaling and stimulation by EPO.20 Thus, during MDS evolution, the TGF-β–mediated cellular and molecular crosstalk is consecutively altered and results in dyserythropoiesis.20
Figure 3.

Luspatercept presumed mechanism of action. Professional illustration by Patrick Lane, ScEYEnce Studios.
The TGF-β receptor ligands are polypeptide growth factors, including TGF-β, activins, bone morphogenetic proteins (BMPs), and growth differentiation factor 11 (GDF11), with an influence on several cell-intrinsic mechanisms including hematopoiesis (Figures 3 and 4). In the TGF-β superfamily, SMADs are important physiologic regulators of hematopoiesis. After ligand binding and receptor phosphorylation, the SMAD signaling pathway becomes activated and SMAD2/3 and SMAD1/5/8 mediate intracellular canonical signaling by ligand-receptor complexes.
Figure 4.

Regulators of erythropoiesis. EPO-R, EPO receptor. Professional illustration by Patrick Lane, ScEYEnce Studios.
In MDS, SMAD2/3 downstream mediators are constitutively activated and overexpressed in MDS CD34+ cells, which leads to impaired terminal erythroid differentiation and subsequent anemia as a result of an inhibitory effect on red-cell maturation.36,37,39 TGF-β, activins, GDF11, and GDF8 are SMAD2/3 pathway ligands that mediate inhibitory regulatory effects on multiple phases of erythropoiesis.37,40 Interestingly, pharmacologic inhibition of TGF-β receptors as well as SMAD2/3 inhibition by short hairpin RNA was able to enhance hematopoiesis in a variety of MDS subtypes in vitro.
The increased TGF-β signaling in patients with MDS is related to decreased expression of SMAD7, an important negative feedback regulator of superfamily signaling. The marked reduction of SMAD7 in MDS hematopoietic cells leads to the overactivation of SMAD2 signaling.36 GDF11, a negative regulator of late-stage erythrocyte development, is increased in MDS patients and is further increased in the course of the disease along with ineffective erythropoiesis, iron overload, and erythroid hyperplasia (Figures 3 and 4). In wild-type mice, chronic administration of GDF11 induced mild anemia and erythroid hyperplasia.
BMPs and many GDFs signal through SMAD1/5/8.36,41 Thus, the 2 branches of the canonical pathway (SMAD2/3 and SMAD1/5/8) seem to exert opposing effects, an imbalance of activity in 1 of the 2 branches potentially leading to the emergence of MDS.36,42
By sequestering SMAD2/3 pathway ligands, overactivated SMAD2/3 signaling is decreased by luspatercept, a novel recombinant fusion protein composed of modified Activin receptor type IIb (ActRIIB). The novel agent consists of the extracellular domain of the TGF-β receptor linked to the Fc domain of human immunoglobulin.1,2 By neutralizing TGF-β superfamily ligands before binding the receptor, SMAD signaling is decreased, which enables erythroid maturation by means of late-stage erythroblast differentiation which thus improves anemia.1,36,37
Commercial development of luspatercept
Luspatercept was initially developed by Acceleron Pharma and Celgene Corporation for treating anemia associated with MDS, myelofibrosis, and beta thalassaemia.43,44 Acceleron Pharma signed a sublicensing agreement with Celgene Corporation for the joint development, manufacturing, and commercialization of luspatercept for treating anemia in August 2011.43,44 From that date forward, Acceleron was in charge of conducting phase 1 and initial phase 2 trials, and Celgene was to conduct the remaining phase 2 and phase 3 trials (Table 1; Figure 2).45 .
Table 1.
Pharmacologic features of luspatercept
| Parameter | Details |
|---|---|
| Class | Antianemic drug, immunoglobulin Fc fragments, recombinant fusion protein |
| ATC code | WHO: B03XA06 (other antianemic preparations); EphMRA: B3X (other antianemic products) |
| Additional names | ACE-536, Reblozyl |
| Route of administration | Subcutaneously once every 21 days |
| Pharmacodynamics | SMAD2/3 ligand GDF11 and activating B signaling inhibition |
| Pharmacokinetics | Mean steady-state Cmax, 8.17 μg/mL; AUC, 126 days⋅μg/mL after administration at 1 mg/kg in patients with beta-thalassaemia |
| AEs | Frequent: headache, diarrhea, dizziness, bone pain, arthralgia, fatigue, abdominal pain; occasional: nausea, hyperuricemia, viral infection; rare: deep vein thrombosis, cerebrovascular accident |
Reprinted by permission from: Springer, Drugs. Markham, A. Luspatercept: First Approval. Drugs 80, 85-90 (2020). © 2020.43
ATC, Anatomical Therapeutic Chemical; AUC, area under the curve; EphMRA, European Pharmaceutical Market Research Association; WHO, World Health Organization.
Pharmacokinetics and pharmacodynamics of luspatercept
When luspatercept is administered by subcutaneous (SC) injection once every 3 weeks in MDS patients, serum concentration reaches steady state after 3 doses; the absorption is not affected by the site of SC injection46 (Table 1). The accumulation ratio is ∼1.5, with a mean apparent distribution volume of 7.1 L (Table 1). Luspatercept is expected to be catabolized by general protein degradation in a wide range of body tissues. Mean half-life of luspatercept is ∼11 days, and the mean apparent total clearance was 0.44 L/day; the apparent volume of distribution and total clearance increase with increasing body weight.46 The initial dose of luspatercept is 1 mg/kg via SC injection once every 3 weeks; if there are no reductions in RBC transfusions after 2 doses (6 weeks), the dose should be increased to 1.33 mg/kg and after 2 consecutive doses, the dose should be increased up to a maximum of 1.75 mg/kg.1 Interestingly, mild to moderate hepatic or renal impairment, baseline serum EPO or albumin levels, degree of RBC transfusion burden, and co-administration of iron-chelating agents had no clinically significant effect on the pharmacokinetics of luspatercept.46
Preclinical studies of luspatercept
The activity of luspatercept has been assessed in various preclinical studies by using the murine analogs RAP-536, RAP-011, or the fully human fusion proteins.36 RAP-536, the murine analog of luspatercept, showed a measurable increase in erythrocyte count and reduced anemia in mouse models.37 Animals treated with RAP-536 displayed faster hematologic recovery after modeling acute blood loss, anemia from chronic kidney disease, or chemotherapy-induced anemia.36 Moreover, RAP-536 was tested in an MDS mouse model, which recapitulates ineffective erythropoiesis by transgenic expression of the NUP98/HOXD13 (NHD13) fusion protein. NHD13 mice also displayed ineffective hematopoiesis and inefficacious precursor maturation.36,47 After treatment with RAP-536, NHD13 mice presented enhanced erythropoiesis and improvement in anemia, consistent with reduced erythroid hyperplasia and enhancement of the abnormal myeloid:erythroid ratios in the bone marrow.37 In wild-type mice, experimental stimulation of SMAD2/3 signaling by administering GDF11 resulted in extenuated erythroid maturation and anemia.37 RAP-536 was more effective in stimulating red cell production in wild-type mice compared with single-ligand neutralizing antibodies against SMAD2/3 or a combination of antibodies against GDF8, GDF11, and activin B. These results indicate that multiple SMAD2/3 pathway ligands collaborate on suppression of erythropoiesis in vivo and that sequestration of multiple ligands is necessary to explain the robust stimulation of red cell production by RAP-536 (luspatercept) in normal and disease settings. Recent preliminary data show evidence that RAP-536 also has effects on the stromal niche compartment in the bone marrow microenvironment.48,49 Mesenchymal stem cells from MDS patients and age-adjusted healthy controls were treated with GDF11 with or without RAP-536, which reversed the GDF11–dependent SMAD2/3 activation and inhibition of SMAD4 levels. As a result, defects in the support properties for hematopoietic stem and progenitor cells were ameliorated, mainly mediated by restored SDF-1 levels. Thus, luspatercept has demonstrated a direct impact on the bone marrow microenvironment, which improves the disturbed functional capacities of mesenchymal stem cells associated with MDS (Figure 1).48,49
Luspatercept increased hematocrit, RBC counts, and hemoglobin concentration in a dose-dependent fashion in monkeys, mice, and rats.1,17,36 Recent data suggest that luspatercept increases RBC parameters by enhancing maturation of late-stage erythroblasts without distinctly modifying RBC lifespan.36 This is in contrast with EPO, which increases RBC parameters mainly by expanding proliferation of early erythroid progenitor cells.36,37 Nevertheless, EPO and its receptor EPO-R (Figure 4) are also required for survival of late-stage definitive erythroid progenitors.20 Mouse and human primitive erythroblasts cultured without EPO undergo accelerated maturation and apoptosis at later stages of maturation.20 Thus, EPO plays an essential role in promoting the proliferation, survival, and appropriate timing of terminal maturation of primitive erythroid precursors.20 Therefore, stating that luspatercept is the first and only in-class erythroid maturation agent is rather misleading. In addition, recent data suggest that after treatment initiation, luspatercept increases endogenous EPO levels, which trended downward over time during treatment and were lower in responders compared with nonresponders. Thus, EPO and luspatercept possibly share a common pathway in the final stage of maturation.
In mice, co-treatment with luspatercept and EPO resulted in a synergistic, robust increase in RBC parameters, probably because of the increased availability of early progenitors induced by EPO.37 A shift in cells to more mature stages revealed that treatment with a combination of luspatercept and EPO significantly increased maturation of basophilic erythroblasts compared with EPO alone.37 Moreover, mice pretreated with EPO showed fewer erythroid precursors in early phases after treatment with RAP-536 compared with vehicle, consistent with the concept that RAP-536 stimulates erythroid precursor maturation.37 Clinical studies of the combination of both agents may be prudent, given the synergism in mouse studies.
Clinical studies of luspatercept
Phase 1 study
Luspatercept was initially investigated within a phase 1 trial in healthy postmenopausal females. The trial evaluated increasing dose levels of luspatercept (n = 24), and 8 probands received placebo.50 Five of the 6 patients who received the highest dose (0.25 mg/kg) had a hemoglobin increase of ≥1.0 g/dL, in contrast to only 1 of 8 probands receiving placebo who met this hemoglobin threshold.50 Mean duration of response in probands was 14 days after 1 dose and 21 days after 2 consecutive doses. Treatment with luspatercept was generally safe, with no serious adverse events (AEs), and the occurrence of AEs was comparable between the placebo and the treatment group50 (Figure 2).
Phase 2 study
After the successful phase 1 clinical investigation, the phase 2, multicenter, open-label, dose-finding (PACE-MDS) study enrolled 58 patients (between 2013 and 2015) who had IPSS low-risk or intermediate-1–risk MDS or nonproliferative (white blood cell count <13 × 103/μL) chronic myelomonocytic leukemia within the German MDS Study Group (D-MDS).2 Eligible patients had anemia with or without RBC transfusion dependency. Patients were initially classified as having low transfusion burden (LTB), defined as baseline hemoglobin <10 g/dL and a requirement for <4 RBC units in the 8 weeks before random assignment or as having high transfusion burden (HTB), defined as requiring ≥4 RBC units in the 8 weeks before treatment initiation.2 Eligible patients received SC luspatercept once every 21 days at dose concentrations ranging from 0.125 mg/kg to 1.75 mg/kg of body weight for 5 doses (over a maximum of 12 weeks). The study had 2 stages: the first stage (base study) included 58 patients in the dose-finding and expansion cohorts; the second stage (extension study) enrolled 32 patients.2 Of the 58 patients, 19 had LTB (11 with no transfusions, and 8 with 1 to 3 RBC units transfused in the 8 weeks before treatment), and 39 had HTB (range, 4 to 18 RBC units over 8 weeks). Of the 32 patients in the extension study, 13 had LTB and 19 had HTB.2 In the base study, all patients received up to 5 doses (over a maximum of 12 weeks) of luspatercept treatment at 1 of 7 prespecified dose concentrations ranging from 0.125 mg/kg to 1.75 mg/kg (0.125, 0.25, 0.50, 0.75, 1.00, 1.33, and 1.75 mg/kg). Patients in the expansion cohort were treated with 1.0 mg/kg luspatercept; dose titration up to 1.75 mg/kg was allowed, and patients could be treated with luspatercept for a maximum of 5 years.2 Patients in the base study were assessed for response and safety after 12 weeks so they could be considered for enrollment into the extension study. After a review of the efficacy data by the sponsor and investigators for the dose-escalation cohorts, there was a clear dose-dependent efficacy response; dose concentrations of 0.125 to 0.5 mg/kg were deemed subtherapeutic and the higher dose concentrations of 0.75 to 1.75 mg/kg were deemed therapeutic (Figure 2; Table 2).2
Table 2.
Completed clinical trials of luspatercept in MDS
| Luspatercept trials | Patient population | End point | No. of erythroid responses | Dosing, mg/kg | |||
|---|---|---|---|---|---|---|---|
| Phase | Trial name | NCT no. | Luspatercept | Placebo | |||
| 1 | NCT01432717 | Postmenopausal, healthy women (age 45-75 years) | Mean hemoglobin change at day +15 | 24 | 8 | 0.0625-0.25 | |
| 2 | PACE-MDS | NCT01749514 | IPSS low or intermediate-1 risk, anemia with or without transfusion dependence | HI-E, RBC-TI ≥ 8 weeks | 32 (HI-E, 63%); 16 (RBC-TI, 38%) | — | 0.125-1.75 |
| 3 | MEDALIST | NCT02631070 | IPSS-R very low, low, or intermediate risk, ≥15% RS or ≥5% RS with SF3B1 mutation, R/R ESA or serum EPO >200 U/L, transfusion dependence (≥2 units once every 8 weeks) | HI-E, RBC-TI ≥ 8 weeks | 81 (HI-E, 53%); 58 (RBC-TI, 38%) | 9 (HI-E, 12%); 10 (RBC-TI, 13%) | 1.0-1.75 |
R/R, relapsed or refractory.
Median total duration of treatment of all patients across both the base and extension studies was 6.8 months (range, 2.0 to 19.8 months). Sixty-three percent of the patients (32 of 51) who received higher doses of luspatercept (0.75 to 1.75 mg/kg) achieved erythroid response (hematologic improvement in erythrocytes [HI-E]; defined according to the International Working Group [IWG] 2006 criteria51 as a reduction in RBC transfusions of ≥4 units per 8 weeks in patients with a baseline transfusion burden of ≥4 units per 8 weeks or as an increase in the hemoglobin level of ≥1.5 g/dL over a period of 8 weeks in patients with a baseline transfusion burden of <4 units per 8 weeks) vs 22% (2 of 9) receiving lower-dose concentrations (0.125 to 0.5 mg/kg).
The IWG 2018 criteria52 proposed a revision to the IWG 2006 standardized treatment response criteria for MDS patients. The revisions focused primarily on the more specific definition of the erythroid treatment response criteria (HI-E). The new IWG 2018 criteria recommended categorizing patients with no transfusion burden (0 to 2 RBC units within 16 weeks) at baseline, LTB (3 to 7 RBC units within 16 weeks), and HTB (≥8 RBC units within 16 weeks). The IWG 2018 criteria also suggested changing the threshold for treatment initiation from baseline hemoglobin <11 g/dL to hemoglobin levels <10 g/dL.52 During the first 16 weeks of treatment, erythroid response (HI-E) in patients who were not transfusion dependent was defined as achieving 2 consecutive measurements of hemoglobin >1.5 g/dL, and response in HTB or LTB patients was defined as achieving transfusion independence.1-54
Within the phase 2 study, RBC transfusion independence was achieved in 38% of patients (16 of 42). Of the 22 patients with previous RBC transfusions who carried over into the extension study, 11 patients (50%) were transfusion free for 8 weeks or longer.2 Median duration of RBC transfusion independence for these 11 patients was 15.3 months, and mean time to response was 1.1 months. Among patients treated with higher dose concentrations of luspatercept, 21 (62%) of 34 patients with previous ESA use achieved HI-E, and 11 (65%) of 17 patients with no previous ESA use achieved HI-E. Luspatercept showed similar activity regardless of previous ESA use and thus, previous ESA use was not an important predictor of response.2 For patients treated with higher dose concentrations of luspatercept and who had previously received lenalidomide, 5 (63%) of 8 achieved HI-E compared with 27 (63%) of 43 patients who had not previously received lenalidomide. Among patients treated with higher dose concentrations of luspatercept, 19 (76%) of 25 with baseline serum EPO levels <200 IU/L, 7 (58%) of 12 with baseline serum EPO levels of 200 IU/L to ≤500 IU/L, and 6 (43%) of 14 with baseline serum EPO levels >500 IU/L achieved HI-E. Thus, baseline endogenous EPO concentrations were predictive of response, but luspatercept was also effective in patients with higher endogenous EPO concentrations, which is associated with poor ESA response.
At screening, molecular analyses of commonly mutated genes in MDS were performed. Spliceosome mutations are common in MDS and are associated with RS-positive disease. SF3B1 mutation status was strongly associated with response: 24 (77%) of 31 SF3B1 mutation–positive patients achieved HI-E; all were also RS positive. By contrast, 6 (40%) of 15 SF3B1 mutation–negative patients achieved HI-E; 3 (50%) of these 6 responders were RS positive. Conversely, 3 (43%) of 7 RS-negative patients achieved HI-E.2 Thus, patients with the RS-positive phenotype or SF3B1 genotype or both seemed to display alterations in erythroid maturation, which in turn might render them more susceptible to SMAD2/3 inhibition by luspatercept compared with patients with other MDS subtypes. Given the activity of luspatercept in a variety of other hematologic diseases4,5,43 and in healthy volunteers, the mode of action may not be only causally connected to the presence or absence of spliceosome mutations.
To assess the association between baseline factors and IWG HI-E response, multivariable logistic regression analysis was performed. Both erythropoietin concentration (<100 IU/L vs ≥100 IU/L; P = .04) and SF3B1 mutation status (yes vs no; P = .01) had a significant effect. For RBC-TI patients, both endogenous EPO concentration (<500 IU/L vs ≥500 IU/L; P = .02) and iron chelation therapy use (yes vs no; P = .01) were significant predictors of response.2
The safety profile for luspatercept has been favorable, grade 3 AEs considered related to treatment were reported in only 3 patients (5%). Two of these treatment-related grade 3 AEs were reversible serious grade 3 events: 1 patient (2%) had myalgia and 1 patient (2%) had general physical health deterioration.2 No grade 4 AEs considered related to treatment were reported. Unlike certain other MDS-specific therapeutics, treatment-related cytopenias were not observed in patients receiving luspatercept. Instead, some patients with preexisting neutropenia had improved neutrophil counts.2
Phase 3 study
Because of the significant effectiveness and good tolerability of luspatercept in the phase 2 PACE-MDS study, the drug was subsequently investigated in MDS patients in the phase 3, randomized, double-blind, placebo-controlled MEDALIST trial.1 Eligible patients displayed TD anemia (≥2 units per 8 weeks during the 16 weeks before random assignment), an IPSS-R–defined very low, low, or intermediate risk if they were RS positive (with either ≥15% RS or ≥5% RS if an SF3B1 mutation was present and <5% bone marrow blasts) and had disease that was refractory to or unlikely to respond to ESA (endogenous EPO level of >200 U/L) (Figure 2; Table 2).
Between 2016 and 2017, 229 patients were enrolled at 65 sites in 11 countries and were double-blind randomized 2:1 to receive either luspatercept (n = 153) at a starting dose level of 1.0 mg/kg with titration up to 1.75 mg/kg if needed or SC placebo injection (n = 76) once every 3 weeks for at least 24 weeks.1 If new RBC transfusions became necessary after considering the patient as transfusion independent, dose adjustment to 1.33 mg/kg and then to 1.75 mg/kg was implemented. With regard to the IPSS-R categories, 10%, 72%, and 17% of the patients had an MDS defined as being very low risk, low risk, or intermediate risk, respectively. Baseline serum EPO levels were <100 U/L in 36%, 100 to <200 U/L in 24%, 200 to 500 U/L in 25%, and >500 U/L in 14% of the patients. At baseline, 29% had an RBC transfusion burden of <4 units per 8 weeks, 57% of patients required <6 RBC units per 8 weeks, and 43% of patients required at least 6 units per 8 weeks. Ninety-three percent of patients (138 of 148) in the luspatercept arm had an SF3B1 mutation compared with 86% (64 of 74) of those in the placebo group. Mutation profiles from bone marrow mononuclear cells were obtained at baseline, and they showed a similar distribution in the 2 groups. A total of 48% of patients had previous iron chelation therapy and 95% had previously received ESA treatment.
The primary end point was transfusion independence for 8 weeks or longer during weeks 1 through 24. The key secondary end point was transfusion independence for 12 weeks or longer, assessed during weeks 1 through 48 and during weeks 1 through 24. Other secondary end points included erythroid response (HI-E according to the IWG 2006 criteria51), longest duration of primary response, mean increase in hemoglobin levels of at least 1.0 g/dL, progression to acute myeloid leukemia, mean change in the serum ferritin level, and safety analyses.
The first response assessment was performed at week 25 when nonresponding patients discontinued receiving luspatercept or placebo and entered follow-up. Patients who had clinical benefit without evidence of disease progression (according to IWG 2006 criteria51) entered the extension phase and continued receiving luspatercept or placebo until they experienced disease progression or unacceptable toxicity or they had other reasons for being excluded from the study.1
Of 153 patients receiving luspatercept, 58 (37.9%) achieved the primary end point of RBC transfusion independence for at least 8 weeks compared with 10 (13.2%) of 76 patients receiving placebo (P < .0001). Forty-three (28.1%) of 153 patients receiving luspatercept achieved the key secondary end point of RBC transfusion independence for at least 12 weeks (weeks 1 to 24) compared with 6 (7.9%) of 76 receiving placebo (P = .0002). Moreover, in the luspatercept arm, HI-E was achieved in 81 (52.9%) of 153 patients vs 9 (11.8%) of 76 patients receiving placebo during the first 24 weeks. A total of 90 patients (59%) in the luspatercept group had an HI-E compared with 13 (17%) in the placebo group during weeks 1 through 48. The median duration of the longest single continuous period of response to luspatercept was 30.6 weeks.1
The longest single period of transfusion independence was a median 30.6 weeks in the luspatercept-treated group compared with 13.6 weeks in the placebo group. According to the baseline transfusion burden, RBC-TI rate in the luspatercept group was higher in patients with LTB. Eighty percent of patients (37 of 46) who received <4 RBC units per 8 weeks before treatment initiation achieved transfusion independence compared with 37% of patients (37 of 46) who received 4 to <6 units per 8 weeks and 9% of patients (6 of 66) who received at least 6 units per 8 weeks. In evaluable patients with hematologic improvement in platelet count (HI-P), 62.5% of those who received luspatercept and 33% of those who received placebo achieved HI-P.54 Hematologic improvement in neutrophil count (HI-N) was observed in 20% of luspatercept-treated patients compared with 10% in the placebo group.54
Interestingly, patients showed responses regardless of SF3B1 allelic burden and the total number of baseline somatic mutations. Moreover, no significant changes were observed in variant allele frequencies of somatic mutations during luspatercept treatment when comparing responders with nonresponders. Whether luspatercept has disease-modifying possibilities needs to be explored in future clinical trials. Interestingly, the analysis of neutrophil or platelet counts did not reveal any significant differences during treatment.
Again, luspatercept was associated with only low-grade toxicity. The most common treatment-associated AEs of any grade included fatigue, diarrhea, asthenia, nausea, and dizziness and were mostly grade 1 or 2. Thirty-one percent of patients (48 of 153) receiving luspatercept compared with 30% of patients (23 of 76) receiving placebo had at least 1 serious AE. The incidence of disease progression was low in both groups: 1 patient in each group progressed to higher-risk MDS (HR-MDS), and 3 patients in the luspatercept group and 1 patient who received placebo developed acute myeloid leukemia, consistent with the natural history of LR-MDS.1
Conclusion and future directions
After a long preclinical and clinical journey, the MEDALIST trial led to FDA approval (April 2020) and EMA approval (April 2020) of luspatercept for TD LR-MDS patients with RS and/or SF3B1 mutation after treatment with an ESA had failed (Figure 5). Hence, luspatercept will become the new treatment standard after ESA failure in RS-positive LR-MDS patients with TD anemia. The drug is now tested in LR-MDS patients without RS in the ongoing randomized phase 3 COMMANDS trial (NCT03682536), which evaluates the efficacy of luspatercept vs ESA as first-line therapy in ESA-naïve (serum EPO <500 U/L) LR-MDS patients (RS-positive and RS-negative) who require at least 2 RBC units every 8 weeks. The trial results will provide additional insights, whether outcomes of luspatercept-treated patients will be better than outcomes achieved with ESA and will determine the clinical activity in patients with other MDS subtypes without RS. Preclinical data are needed that will answer the still open questions of which exact TGF-β ligands are trapped by luspatercept and why patients with RS or SF3B1 mutation show higher response rates. The detection of further biomarkers that predict treatment response to luspatercept will help to preselect potential responders in subsequent studies.
Figure 5.

Historical overview of MDS-specific new drug registrations in the European Union and the United States. Professional illustration by Patrick Lane, ScEYEnce Studios.
Future clinical studies of combination treatments that evaluate the efficacy of luspatercept plus ESA, either as first-line therapy or in the relapsed/refractory setting, are necessary for verifying the promising preclinical synergistic effects of the 2 compounds in the clinical scenario. Further developments should also include clinical evaluation of the combination of luspatercept plus lenalidomide within an ongoing phase 1b/2 study (NCT04539236). The potential activity of luspatercept in patients with overlapping MDS and myeloproliferative neoplasms (MDS/MPN) as well as in more advanced HR disease is worth detailed clinical investigation. But there are still no data regarding the clinical activity of luspatercept after HMA failure. For patients who are receiving disease-modifying HMAs, the combination therapy with luspatercept in parallel could promote erythropoiesis and reduce transfusion burden in patients with HR-MDS.
Footnotes
Request data via e-mail to Uwe Platzbecker at uwe.platzbecker@medizin.uni-leipzig.de.
Authorship
Contribution: A.S.K., P.F., and U.P. wrote the manuscript and reviewed, edited, and approved the final manuscript.
Conflict-of-interest disclosure: A.S.K. received lecture fees from Novartis and Janssen Biotech. P.F. received consulting fees from Celgene. U.P. received grant support from Amgen; lecture fees, grant support, fees for serving on a steering committee, consulting fees, and travel support from Celgene; grant support from Janssen Biotech; grant support from Merck and Novartis; and lecture fees from Novartis.
Correspondence: Uwe Platzbecker, Department of Hematology, Cellular Therapy and Hemostaseology, Leipzig University Hospital, Liebigstr 22, 04103 Leipzig, Germany; e-mail: uwe.platzbecker@medizin.uni-leipzig.de.
References
- 1.Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in patients with lower-risk myelodysplastic syndromes. N Engl J Med. 2020;382(2):140-151. [DOI] [PubMed] [Google Scholar]
- 2.Platzbecker U, Germing U, Götze KS, et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017;18(10):1338-1347. [DOI] [PubMed] [Google Scholar]
- 3.Platzbecker U. Treatment of MDS. Blood. 2019;133(10):1096-1107. [DOI] [PubMed] [Google Scholar]
- 4.Cappellini MD, Viprakasit V, Taher AT, et al. ; BELIEVE Investigators . A phase 3 trial of luspatercept in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2020;382(13):1219-1231. [DOI] [PubMed] [Google Scholar]
- 5.Gerds AT, Vannucchi AM, Passamonti F, et al. A phase 2 study of luspatercept in patients with myelofibrosis-associated anemia [abstract]. Blood. 2019;134(suppl_1). Abstract 557. [Google Scholar]
- 6.Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montalban-Bravo G, Garcia-Manero G. Myelodysplastic syndromes: 2018 update on diagnosis, risk-stratification and management. Am J Hematol. 2018;93(1):129-147. [DOI] [PubMed] [Google Scholar]
- 8.Fenaux P, Adès L. How we treat lower-risk myelodysplastic syndromes. Blood. 2013;121(21):4280-4286. [DOI] [PubMed] [Google Scholar]
- 9.Malcovati L, Hellström-Lindberg E, Bowen D, et al. ; European Leukemia Net . Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122(17):2943-2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Efficace F, Gaidano G, Breccia M, et al. Prognostic value of self-reported fatigue on overall survival in patients with myelodysplastic syndromes: a multicentre, prospective, observational, cohort study. Lancet Oncol. 2015;16(15):1506-1514. [DOI] [PubMed] [Google Scholar]
- 11.Efficace F, Cottone F, Oswald LB, et al. The IPSS-R more accurately captures fatigue severity of newly diagnosed patients with myelodysplastic syndromes compared with the IPSS index. Leukemia. 2020;34(9):2451-2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stauder R, Yu G, Koinig KA, et al. Health-related quality of life in lower-risk MDS patients compared with age- and sex-matched reference populations: a European LeukemiaNet study. Leukemia. 2018;32(6):1380-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenberg PL, Stone RM, Al-Kali A, et al. Myelodysplastic syndromes, version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Cancer Netw. 2017;15(1):60-87. [DOI] [PubMed] [Google Scholar]
- 14.Angelucci E, Li J, Greenberg P, et al. ; TELESTO Study Investigators . Iron chelation in transfusion-dependent patients with low- to intermediate-1-risk myelodysplastic syndromes: A randomized trial. Ann Intern Med. 2020;172(8):513-522. [DOI] [PubMed] [Google Scholar]
- 15.Goldberg SL, Chen E, Sasane M, Paley C, Guo A, Laouri M. Economic impact on US Medicare of a new diagnosis of myelodysplastic syndromes and the incremental costs associated with blood transfusion need. Transfusion. 2012;52(10):2131-2138. [DOI] [PubMed] [Google Scholar]
- 16.Hellström-Lindberg E, Negrin R, Stein R, et al. Erythroid response to treatment with G-CSF plus erythropoietin for the anaemia of patients with myelodysplastic syndromes: proposal for a predictive model. Br J Haematol. 1997;99(2):344-351. [DOI] [PubMed] [Google Scholar]
- 17.Kubasch AS, Platzbecker U. Setting fire to ESA and EMA resistance: new targeted treatment options in lower risk myelodysplastic syndromes. Int J Mol Sci. 2019;20(16):3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hellström-Lindberg E, Gulbrandsen N, Lindberg G, et al. ; Scandinavian MDS Group . A validated decision model for treating the anaemia of myelodysplastic syndromes with erythropoietin + granulocyte colony-stimulating factor: significant effects on quality of life. Br J Haematol. 2003;120(6):1037-1046. [DOI] [PubMed] [Google Scholar]
- 19.Park S, Hamel JF, Toma A, et al. Outcome of lower-risk patients with myelodysplastic syndromes without 5q deletion after failure of erythropoiesis-stimulating agents. J Clin Oncol. 2017;35(14):1591-1597. [DOI] [PubMed] [Google Scholar]
- 20.Malik J, Kim AR, Tyre KA, Cherukuri AR, Palis J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica. 2013;98(11):1778-1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelaidi C, Park S, Sapena R, et al. Long-term outcome of anemic lower-risk myelodysplastic syndromes without 5q deletion refractory to or relapsing after erythropoiesis-stimulating agents. Leukemia. 2013;27(6):1283-1290. [DOI] [PubMed] [Google Scholar]
- 22.List A, Dewald G, Bennett J, et al. ; Myelodysplastic Syndrome-003 Study Investigators . Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355(14):1456-1465. [DOI] [PubMed] [Google Scholar]
- 23.Kubasch AS, Platzbecker U. The wolf of hypomethylating agent failure: what comes next? Haematologica. 2019;104(8):1505-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toma A, Kosmider O, Chevret S, et al. Lenalidomide with or without erythropoietin in transfusion-dependent erythropoiesis-stimulating agent-refractory lower-risk MDS without 5q deletion. Leukemia. 2016;30(4):897-905. [DOI] [PubMed] [Google Scholar]
- 25.Giagounidis AAN, Kulasekararaj A, Germing U, et al. Long-term transfusion independence in del(5q) MDS patients who discontinue lenalidomide. Leukemia. 2012;26(4):855-858. [DOI] [PubMed] [Google Scholar]
- 26.Giagounidis A, Mufti GJ, Mittelman M, et al. Outcomes in RBC transfusion-dependent patients (Pts) with low-/intermediate (Int)-1-risk myelodysplastic syndromes (MDS) with isolated deletion 5q treated with lenalidomide (LEN): A subset analysis from the MDS-004 study [abstract]. Blood. 2013;122(21). Abstract 2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fenaux P, Giagounidis A, Selleslag D, et al. ; MDS-004 Lenalidomide del5q Study Group . A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with low-/intermediate-1-risk myelodysplastic syndromes with del5q. Blood. 2011;118(14):3765-3776. [DOI] [PubMed] [Google Scholar]
- 28.Santini V, Almeida A, Giagounidis A, et al. Randomized phase III study of lenalidomide versus placebo in RBC transfusion-dependent patients with lower-risk non-del(5q) myelodysplastic syndromes and ineligible for or refractory to erythropoiesis-stimulating agents. J Clin Oncol. 2016;34(25):2988-2996. [DOI] [PubMed] [Google Scholar]
- 29.Stauder R, Valent P, Theurl I. Anemia at older age: etiologies, clinical implications, and management. Blood. 2018;131(5):505-514. [DOI] [PubMed] [Google Scholar]
- 30.Adès L, Itzykson R, Fenaux P. Myelodysplastic syndromes. Lancet. 2014;383(9936):2239-2252. [DOI] [PubMed] [Google Scholar]
- 31.Valent P, Büsche G, Theurl I, et al. Normal and pathological erythropoiesis in adults: from gene regulation to targeted treatment concepts. Haematologica. 2018;103(10):1593-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dzierzak E, Philipsen S. Erythropoiesis: development and differentiation. Cold Spring Harb Perspect Med. 2013;3(4):a011601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Komrokji RS. Luspatercept in myelodysplastic syndromes: Who and when? Hematol Oncol Clin North Am. 2020;34(2):393-400. [DOI] [PubMed] [Google Scholar]
- 34.Basiorka AA, McGraw KL, Eksioglu EA, et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood. 2016;128(25):2960-2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winter S, Shoaie S, Kordasti S, Platzbecker U. Integrating the “immunome” in the stratification of myelodysplastic syndromes and future clinical trial design. J Clin Oncol. 2020;38(15):1723-1735. [DOI] [PubMed] [Google Scholar]
- 36.Verma A, Suragani RNVS, Aluri S, et al. Biological basis for efficacy of activin receptor ligand traps in myelodysplastic syndromes. J Clin Invest. 2020;130(2):582-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suragani RNVS, Cadena SM, Cawley SM, et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med. 2014;20(4):408-414. [DOI] [PubMed] [Google Scholar]
- 38.Zermati Y, Fichelson S, Valensi F, et al. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Exp Hematol. 2000;28(8):885-894. [DOI] [PubMed] [Google Scholar]
- 39.Zhou L, Nguyen AN, Sohal D, et al. Inhibition of the TGF-β receptor I kinase promotes hematopoiesis in MDS. Blood. 2008;112(8):3434-3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mullen AC, Wrana JL. TGF-β family signaling in embryonic and somatic stem-cell renewal and differentiation. Cold Spring Harb Perspect Biol. 2017;9(7):a022186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mueller TD, Nickel J. Promiscuity and specificity in BMP receptor activation. FEBS Lett. 2012;586(14):1846-1859. [DOI] [PubMed] [Google Scholar]
- 42.Ning J, Zhao Y, Ye Y, Yu J. Opposing roles and potential antagonistic mechanism between TGF-β and BMP pathways: Implications for cancer progression. EBioMedicine. 2019;41:702-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Markham A. Luspatercept: First approval. Drugs. 2020;80(1):85-90. [DOI] [PubMed] [Google Scholar]
- 44.BusinessWire.com . Celgene Corporation and Acceleron Pharma announce submission of luspatercept marketing authorization application to European Medicines Agency (EMA) for MDS and beta-thalassemia. 26 April 2019. https://www.businesswire.com/news/home/20190426005205/en/Celgene-Corporation-and-Acceleron-Pharma-Announce-Submission-of-Luspatercept-Marketing-Authorization-Application-to-the-European-Medicines-Agency-EMA-for-MDS-and-Beta-Thalassemia. Accessed 4 November 2020.
- 45.Biotech Fierce. Acceleron Pharma announces global collaboration with Celgene Corporation on ACE-536 Program. 3 August 2011. https://www.fiercebiotech.com/biotech/acceleron-pharma-announces-global-collaboration-celgene-corporation-on-ace-536-program. Accessed 4 November 2020.
- 46.Celgene Corporation . Highlights of prescribing information for REBLOZYL (luspatercept-aamt). Revised November 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761136lbl.pdf. Accessed 4 November 2020.
- 47.Lin YW, Slape C, Zhang Z, Aplan PD. NUP98-HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood. 2005;106(1):287-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wobus M, Mies A, Oelschlagel U, et al. Functional characterization of mesenchymal stromal cell-derived extracellular vesicles in myelodysplastic syndromes [abstract]. Oncol Res Treat. 2018;41(4):92. Abstract P291. [Google Scholar]
- 49.Wobus M, Mies A, Magno V, et al. Altered structure and function of mesenchymal stromal cell-derived extracellular matrix in MDS can be restored by luspatercept [abstract]. Blood. 2019;134(suppl_1). Abstract 1699. [Google Scholar]
- 50.Attie KM, Allison MJ, McClure T, et al. A phase 1 study of ACE-536, a regulator of erythroid differentiation, in healthy volunteers. Am J Hematol. 2014;89(7):766-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108(2):419-425. [DOI] [PubMed] [Google Scholar]
- 52.Platzbecker U, Fenaux P, Adès L, et al. Proposals for revised IWG 2018 hematological response criteria in patients with MDS included in clinical trials. Blood. 2019;133(10):1020-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Business Wire . Bristol-Myers Squibb completes acquisition of Celgene, creating a leading biopharma company. 20 November 2019. https://news.bms.com/news/details/2019/Bristol-Myers-Squibb-Completes-Acquisition-of-Celgene-Creating-a-Leading-Biopharma-Company/default.aspx. Accessed 4 November 2020.
- 54.Garcia-Manero G, Mufti GJ, Fenaux P, et al. Hematologic improvement-neutrophil and -platelet in the MEDALIST Trial: Multilineage data from a phase 3, randomized, double-blind, placebo-controlled study of luspatercept to treat anemia in patients with very low-, low-, or intermediate-risk myelodysplastic syndromes (MDS) with ring sideroblasts (RS) who require red blood cell (RBC) transfusions [abstract]. Blood. 2019;134(suppl_1). Abstract 4243. [Google Scholar]