

Key Points
Most substitutions of R338 increase the specific activity of human or canine FIX and the Padua substitution is one of the most active.
These data suggest an evolutionary pressure to limit, rather than maximize, factor IX activity.
Abstract
The high-specific-activity factor IX (FIX) variant Padua (R338L) is the most promising transgene for hemophilia B (HB) gene therapy. Although R338 is strongly conserved in mammalian evolution, amino acid substitutions at this position are underrepresented in HB databases. We therefore undertook a complete 20 amino acid scan and determined the specific activity of human (h) and canine (c) FIX variants with every amino acid substituted at position 338. Notably, we observe that hFIX-R338L is the most active variant and cFIX-R338L is sevenfold higher than wild-type (WT) cFIX. This is consistent with the previous identification of hFIX-R338L as a cause of a rare X-linked thrombophilia risk factor. Moreover, WT hFIX and cFIX are some of the least active variants. We confirmed the increased specific activity relative to FIX-WT in vivo of a new variant, cFIX-R338I, after gene therapy in an HB dog. Last, we screened 232 pediatric subjects with thromboembolic disease without identifying F9 R338 variants. Together these observations suggest a surprising evolutionary pressure to limit FIX activity with WT FIX rather than maximize FIX activity.
Visual Abstract

Introduction
The hyperactive factor IX (FIX) variant R338L (FIX Padua) has emerged as an attractive transgene for hemophilia B (HB) gene therapy.1 We initially identified this variant as the cause of a rare X-linked thrombophilia, which was associated with an eightfold increase in FIX specific activity compared with wild type (WT).2 The high specific activity of FIX-R338L has successfully been used to enhance the potency of gene therapy vectors for mice, dogs, and humans, which has helped address vector-dose–dependent safety limitations.1,3,4
Before our identification of FIX-R338L, 2 earlier studies investigated amino acid substitutions at the R338 position in FIX. In an analysis of HB-causing mutations focusing on CpG dinucleotides that are hot spots for base pair changes, Bottema et al5 recognized that R338 was unique within FIX as an evolutionary conserved amino acid where the predicted most likely missense variant (R338Q) did not cause HB, although a nonsense variant did (Table 1). The authors speculated that missense variants at this position might cause only a very mild hemophilia or even thrombophilia.5 Subsequently, Chang et al,6 in an alanine-screening study of FIX, identified FIX-R338A as having a threefold increased specific activity compared with FIX-WT in vitro, which was subsequently confirmed in vivo after gene transfer in HB mice by Schuettrumpf et al.7
Table 1.
Predicted and observed R338 variants (X:139561835-7)
| Codon | Amino acid | Type of DNA change | Phenotype | Frequency* | References |
|---|---|---|---|---|---|
| gga | G | Transversion | Predicted to be mild hyperactive/wild type | This work | |
| tga | Stop | Transition | Severe HB | 9.46746E-6 | 38 |
| aga | R | Transversion | Synonymous | 5.45625E-5 | |
| cga | R | Wild type | |||
| caa | Q | Transition | FIX Shanghai | 23 | |
| cta | L | Transversion | FIX Padua | 2 | |
| cca | P | Transversion | Mild HB | 5 |
*Variant table hF9 ENSG00000101981.
These disparate observations suggested the hypothesis that the evolutionary conservation of R338 in mammalian FIX was to constrain, rather than optimize, FIX activity. To test this hypothesis, we investigated the effect of all amino acid substitutions at 338 position in FIX orthologs from the 2 most studied mammalian species: human (h) and canine (c). Canine HB is a naturally occurring large animal model that faithfully recapitulates the spontaneous bleeding phenotype of the clinical disease and has predicted doses for a number of new therapeutics for HB.8-10 Our results confirm our hypothesis, inform on the evolutionary pressures on mammalian FIX, and may advise bioengineering strategies for new HB therapeutics.
Materials and methods
Molecular biology and cellular studies
Amino acid substitutions were introduced into hFIX-WT or cFIX-WT cDNA containing plasmids with QuikChange II site-directed mutagenesis kit (Agilent) per the manufacturer’s instructions and verified by sequencing. Confluent human embryonic kidney (HEK) 293, HepG2, Huh7, or SKHep cells in 6-well plates were transfected with 10 µg plasmid DNA with Lipofectamine 2000 (ThermoFischer). After 1 day, media was changed to expression media consisting of Dulbecco's modified eagle medium (Nutrient Mixture F-12) augmented with 10 µg/mL vitamin K; after an additional day, conditioned media were collected, and expressed FIX was assayed.
Clotting and FIX assays
The whole blood clotting time, FIX inhibitor titer, FIX activity and antigen levels, and FIX inhibitor titers were performed as previously described.11-13 FIX activity was determined with an activated partial thromboplastin time–based clotting assay using the TriniCLOT silica activated partial thromboplastin time reagent (TCoag). FIX antigen was determined using either hFIX or cFIX commercial enzyme-linked immunosorbent assay (Affinity Biologics). Commercial recombinant hFIX-WT (Benefix; Pfizer) produced in Chinese hamster ovary cells or recombinant cFIX-WT produced in HEK293 cells in our laboratory were used as standards for the transfections, whereas dilutions of pooled normal canine plasma were used as a standard for the in vivo dog experiments.
Evaluation of F9 R338 variants in pediatric thrombophilia cohort
The Institutional Review Board at Children’s Hospital of Philadelphia (CHOP) approved this study (08-005949). Samples from CHOP patients with thrombotic disease received consent, and blood was obtained in conjunction with clinical laboratory evaluation, typically at an outpatient hematology appointment. Screening for F9 R338 variants was performed as described previously.2 DNA was isolated with Qiagen’s QIAamp DNA Blood Mini Kit per the manufacturer’s instructions. Exon 8 of F9 was amplified by polymerase chain reaction assay using previously published forward (5′-GCCAATTAGGTCAGTGGTCC-3′) and reverse (5′-GATTAGTTAGTGAGAGGCCCTG-3′) primers. Changes at the R338 codon (cga) were screened for by digestion of the polymerase chain reaction product with TaqI endonuclease.
HB dog experiments
Recombinant adeno associated virus (AAV) vector derived from serotype 6 (AAV6) was produced by triple transfection using an expression cassette containing the cytomegalovirus promoter/enhancer.14,15 Vector was administered via transvenular delivery to an isolated limb as described previously.7,12 The Institutional Animal Care and Use Committees at CHOP and University of North Carolina at Chapel Hill approved all animal experiments. Complete blood counts, serum chemistries, and liver and kidney function were serially monitored for systemic toxicitity.12
Data and statistical analysis
Graphical production and statistical analysis were performed with the OriginPro Software package. The Tukey multiple comparison test was used to compare the means of the specific activities of the FIX variants. The frequency of FIX variants was based on the variant table of F9 (ENSG00000101981).
Results
R338 of FIX is conserved in mammalian evolution (Figure 1A) and is located on the periphery of the protease domain (Figure 1B). Based on previous identifications of gain-of-function variants with amino acid substitutions at FIX R3382,6,7 and the observation that missense variants at this position are underrepresent as causative mutations in HB,5 we hypothesized that selective pressures conserved R338 in mammalian FIX to limit, rather than maximize, FIX activity.
Figure 1.

Amino acid sequences surrounding R338 of FIX orthologs and FIXa structure. (A) R338 is indicated. Red-hued amino acids are evolutionary conserved, whereas positions highlighted by blue background are identical. (B) Ribbon rendering of partial structure of FIXa-WT (Protein Data Bank code: 1RFN) showing the protease domain in the standard orientation. R338 is highlighted in blue. The amino acids of catalytic triad (H221, D269, S365) that form the enzymatic activation site are shown in green.
Most amino acid substitutions for R338 increase the activity of hFIX
To test this hypothesis, we evaluated the specific activity of hFIX variants with all encoded amino acid substituted for R338 (Figure 2A). We initially used HEK293 cells to express the FIX variants; HEK293 cells are frequently used to study FIX,6,16,17 as well as manufacturing commercial FIX products for HB.18 In this 20 amino acid scan, we find the hFIX-R338L, the previously identified thrombophilia causing Padua variant,2 has the highest specific activity. hFIX-WT is one of the least active variants with more than two thirds of the substitutions at R338 increasing the specific activity of hFIX. Interestingly, hFIX-R338K, which is evolutionary conserved at the equivalent position in nonmammalian vertebrate FIX orthologs (Figure 1), also has low specific activity compared with most of the other FIX variants.
Figure 2.
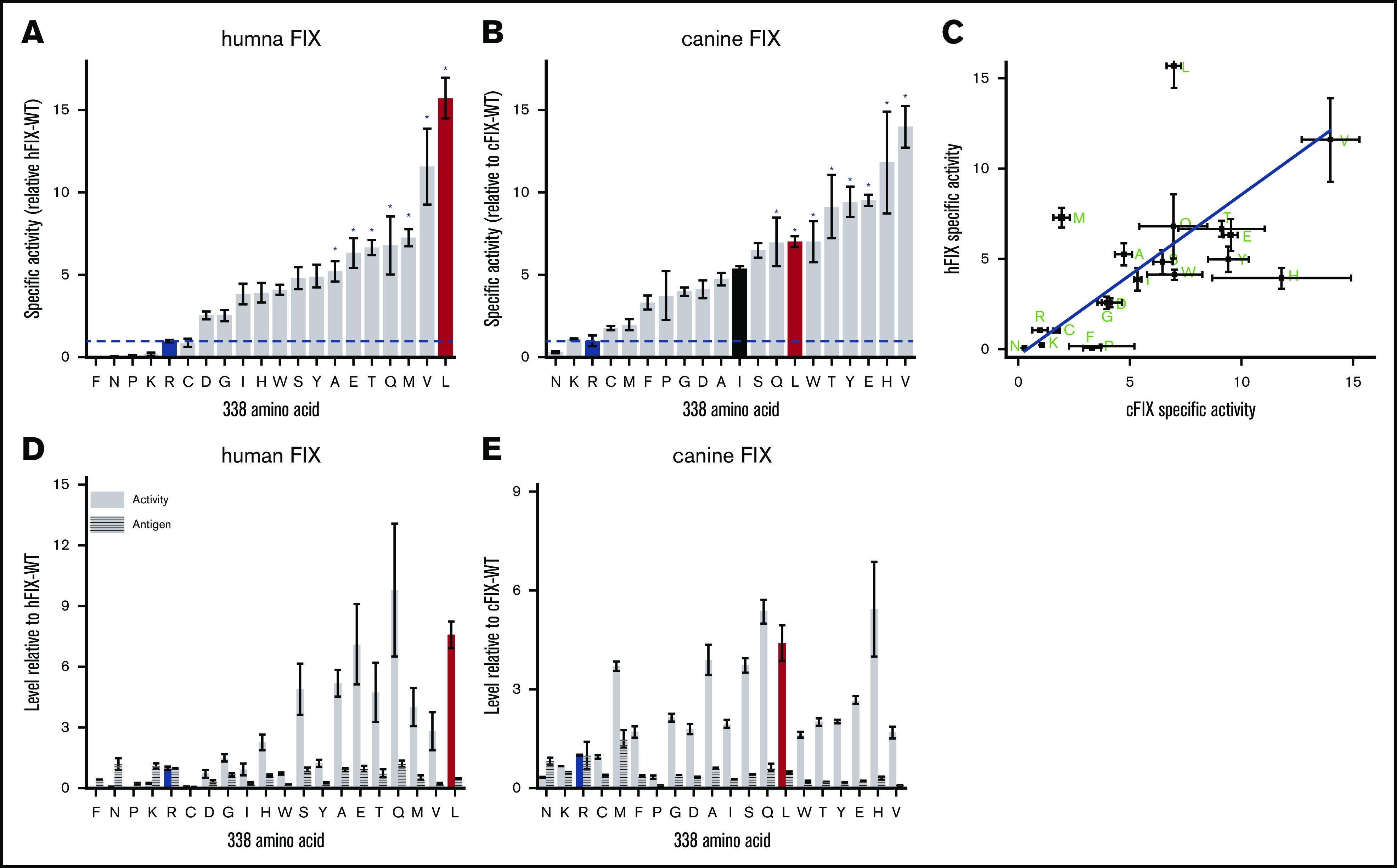
Amino acid screen at FIX position 338. Specific activity of hFIX variants (A) and cFIX-variants (B) relative to hFIX-WT (R338) and cFIX-WT (R338), respectively. FIX-WT (R338) is highlighted in in blue, and R338L (Padua) is highlighted in red. Variants expressed in HEK-293 cells. cFIX-R338I is highlighted in black in 2B. *Statistically significant difference in specific activity from FIX-WT at the .05 level. (C) Scatter plot of specific activities of hFIX and cFIX R338 substituted variants (green letters). Line is linear correlation (y = (0.9 ± 0.2)x – (0.5 ± 0.2); Pearson’s r = .62). Total activity and total antigen hFIX variants (D) and cFIX variants (E) relative to hFIX-WT (R338) and cFIX-WT (R338), respectively. Bars or square points represent the mean specific activity of 3 to 8 independent transient transfections, and errors bars represent ± standard error of the mean.
Most of the hFIX variants display similar antigen levels (less than twofold difference) compared with hFIX-WT; as such, there is an increase in the total FIX activity relative to hFIX-WT for most variants (Figure 2D). The variants hFIX-R338L, hFIX-R338Q, and hFIX-R338E have the highest total activity. The variant hFIX-R338W has a decreased antigen level but a fourfold increase in specific activity, consistent with previous descriptions of this variant.17 The only known HB-causing variant, hFIX-R338P, has decreased specific activity, total activity, and antigen consistent with the clinical description.19 These results highlight that hFIX-WT is not optimized for specific or total FIX activity, which supports our hypothesis that R338 in mammalian FIX was conserved through evolution to constrain, rather than increase, FIX activity.
Most amino acid substitutions for R338 increase the activity of cFIX
To further evaluate this hypothesis, we completed a similar amino acid scan at position R338 in cFIX expressed in HEK293 cells (Figure 2B,E). Although the ordering of the variants is not identical between hFIX and cFIX, there is a linear correlation of the specific activities (Figure 2C). Moreover, the same qualitative pattern is apparent with most amino acid substitutions increasing the specific activity and total activity relative to cFIX-WT. Homology modeling of the hFIX and cFIX variants suggest overall similar structures, although there are larger differences between the species than within the amino acid substituted variants (supplemental Figure 1). As with hFIX, cFIX-WT, cFIX-R338N, and cFIX-R338K are the least active variants. These results indicate that R338 in cFIX limits FIX activity rather than maximizes it, which further supports the hypothesis that R338 was likely evolutionarily selected to limit FIX activity.
Amino acid substitutions for R338 increase the activity of hFIX in multiple human cell lines
To certify that our results were not specific to the HEK293 cell line, we expressed the hFIX variants in 3 different human cell lines able to efficiently express functional recombinant FIX: HepG2,20 Huh7,21 and SK-Hep22; all 3 cell lines are derived from liver tumors and have been ascribed similar properties to hepatocytes.20-22 In all 3 human cell lines, most amino acids substitutions at R338 in hFIX increase the specific activity and total activity compared with hFIX-WT (Figure 3A-F). hFIX-R338L consistently has the highest or near-highest specific activity across all cell lines. We also observe that in addition to L, the E, Q, A, S, M, and H substituted variants also mostly have high specific activity and total activity compared with FIX-WT. Conversely, hFIX-R338P, the HB causative missense variant, consistently has low specific activity and total activity; hFIX-WT, hFIX-R338N, and hFIX-R338K also consistently have low specific activity and total activity. These qualitative groupings of high and low specific and total activity across cell lines are illustrated in Figure 3G-H, respectively. For clarity, we also grouped hFIX-R338G, hFIX-R338C, hFIX-R338Y, hFIX-R338I, and hFIX-R338D in an intermediate group, whereas we note that hFIX-R338F, hFIX-R338W, hFIX-R338V, and hFIX-R338T had variable specific activity across the cell lines evaluated. We observe that most amino acid substitutions at R338 increase the specific activity and total activity of hFIX-WT across human cell lines, further supporting the hypothesis that R338 was evolutionarily favored to restrain FIX activity.
Figure 3.

Specific activity, total activity, and total antigen of hFIX variants expressed in different cell lines. Specific activity (A,C,E) and total activity and antigen (B,D,F) FIX variants expressed in the specified liver cell line. Bars or square points represent the mean specific activity of ≥3 independent transient transfections, and errors bars represent ± standard error of the mean. For clarity, variants are grouped as (1) high specific activity and total activity across all cell lines (L, E, Q, A, S, M, H; red and pink, respectively), (2) low specific activity and total activity across all cell lines (R, K, N, P; blue), (3) medium specific activity and total activity across all cell lines (C, Y, G, I, D; black), and (4) variable specific activity and total activity across all cell lines (F, W, V, T; green). Summary of specific activity (G) and total activity (H) across cell lines with each letter represents the average of ≥3 independent transient transfections.
Comparison of FIX expressed in HEK293 cells and in vivo studies
To determine the in vivo relevance of our in vitro studies, we compared the FIX activity and antigen expressed in HEK293 cells against the clinical description of a range of FIX variants (supplemental Table 1) including FIX Padua (R338L), the FIX Malmo polymorphism (A148T), and 4 HB missense mutations with a range of FIX antigen levels. In supplemental Figure 2, we observe that these FIX variants expressed from HEK293 recapitulate both the total activity and antigen level of the clinical description. These data further support the biological relevance of our in vitro results.
Likewise, we observe that our in vitro cFIX results (Figure 2) are consistent with cFIX expressed after AAV gene therapy (Table 2). Here, we compare the cFIX-specific activity of cFIX-R338I with our previous published experience with cFIX-WT (R338)12 and cFIX-R338L.14 We used that same AAV expression cassette (supplemental Figure 3A), vector dose (3 × 1012 vg/kg), delivery method (transvenular delivery to an isolated limb), and HB dog model.12,14 Delivery of AAV6-cFIX-R338I resulted in increased cFIX activity and antigen levels (supplemental Figure 3B) with plateau levels of 0.95 ± 0.01% and 0.22 ± 0.05% normal, respectively. The cFIX specific activity was 4.4, which is intermediate between cFIX-WT and cFIX-R338L (Table 2) and consistent with our in vitro data (Figure 2B). As expected with these FIX activity levels, the whole blood clotting time decreased from the HB range (supplemental Figure 3C). These observations support the relevance of our in vitro results.
Table 2.
Summary of results of transvenular delivery of AAV6-cFIX at 3 × 1012 vg/kg gene transfer to skeletal muscle of severe hemophilia B dogs
| HB dog | cFIX variant | Plateau cFIX expression* | Spontaneous bleeds/mo | |||
|---|---|---|---|---|---|---|
| Activity, % | Antigen, % | Specific activity | Before | After | ||
| M13† | WT (R338) | 5.2 ± 3 | 5.5 ± 0.8 | 1.0 | 0/12 | |
| M20† | WT (R338) | 6.2 ± 3 | 4.1 ± 0.2 | 1.5 | 0/12 | |
| N40 | R338I | 0.95 ± 0.01 | 0.22 ± 0.05 | 4.4 | 6/45 | 2/9 |
| M55‡ | R338L | 8.7 ± 3 | 1.4 ± 0.3 | 7.4 | 0/7 | 0/101 |
| M59‡ | R338L | 3.5 ± 1 | 0.52 ± 0.2 | 7.7 | 0/6 | 0/101 |
| N07‡ | R338L | 5.7 ± 2 | 0.60 ± 0.3 | 10.6 | 2/6 | 0/79 |
Screening pediatric thrombosis patients for FIX R338 substitutions
Our hypothesis that the evolutionary conservation of R338 in mammalian FIX was to constrain, rather than optimize, FIX activity, suggests that high-specific activity FIX variants may be a prothrombotic risk factor. Consistent with this concept is FIX-R338L (Padua) being initially identified as a strong thrombophilia risk factor, because this substitution is the most active (Figures 2 and 3). Recently, hFIX-R338Q has also been implicated as an X-linked thrombophilia, termed FIX Shanghai.23 The specific activity in the clinical description of hFIX-R338Q was fivefold higher than hFIX-WT, consistent with our studies. Table 1 shows that potential consequences of single-base pair changes in the R338 codon and their measured frequencies.
Based on our results with multiple amino acid substitutions at R338 resulting in hyperactive FIX variants, it is possible that other substitutions at FIX R338 may be eventually identified as thrombotic risk factors. To evaluate this hypothesis, we screened 232 pediatric thrombosis patients for missense variants at FIX R338. Patient characteristics of this cohort are shown in supplemental Table 2. This is the first assessment FIX R338 substitutions in pediatric subjects. Subjects were recruited mostly during hematology outpatient visits and are therefore probably enriched for clinical cases that required ongoing management or follow-up; they skew older and have a higher prevalence of inherited thrombophilias compared with thrombosis patients identified through administration databases24 but are similar to other studies with similar recruitment.25
However, none of the 333 X-chromosomes screened had FIX R338 substitutions. These results extend previous reports of FIX R338 substitutions not being identified in 1214 cumulative subjects including 767 adult patients with venous thromboembolism and 447 adult controls (supplemental Table 3).2,26-28
Discussion
Herein, we demonstrate that most amino acid substitutions at R338 in hFIX or cFIX increase the specific activity compared with WT. We found that hFIX-R338L is the most active hFIX variant across multiple cell lines, which supports its ongoing use in current gene therapy clinical trials for HB.1,3,4 These data also resolve the apparent paradox why R338 is strictly conserved during mammalian evolution, but missense mutations at this hot spot are underrepresented as causes of HB. Our results support our hypothesis that the evolutionary conservation of R338 was likely to limit, rather than optimize, FIX activity. We speculate that FIX-WT has evolved to have a finely calibrated procoagulant activity with too much activity leading to thrombosis and too little activity leading to bleeding, as exhibited by hFIX-R338L and hFIX-R338P, respectively.
Our results are consistent with previous descriptions of these FIX variants. We observe the specific activity of hFIX-R338A is fivefold higher than FIX-WT, consistent with previous in vitro6,29,30 and in vivo gene therapy studies.7,31 Likewise, hFIX-R338Q has intermediate specific activity between hFIX-WT and hFIX-R338L, as has been observed previously in vitro32 and in vivo gene therapy studies.23,31 hFIX-R338E also has increased specific activity compared with hFIX-WT, as has been reported.33 Combined, the consistency of our results with clinical descriptions,2,19 biochemical characterization of purified recombinant FIX,6,29,30,34 and in vivo gene transfer experiments17 all supports the validity of these results.
We find that R338 substitutions (L, E, Q, A, S, M, H) in hFIX and cFIX have the highest total activity in both orthologs. Interestingly, amino acids in this high activity group occur in the analogous position (170 by chymotrypsin numbering) in other cofactor-dependent serine proteases involved in coagulation: Q for FVII; L for FX; and E for activated protein C. We speculate that limiting the activity of FIX may have been evolutionarily more important than for these other proteases because of the positioning of FIX at the top of the coagulation cascade where small increases in FIX activity are amplified to large increases in fibrin formation.
It is difficult for us speculate on the role of antigen levels on the evolution of FIX, as the antigen levels of most variants is less than a twofold difference with WT, although exceptions include the HB-causing variant hFIX-R338P. Theoretically, there could be an antigen threshold below which the FIX could be meaningful depleted during normal hemostasis, despite normal FIX activity initially caused by high specific activity. This is unlikely the scenario for FIX-R338L based on our observation that expression of only 1% normal FIX activity after cFIX-R338L gene therapy was sufficient to prevent bleeding in a HB dog.15
We and others have recently demonstrated that the high specific activity of FIX-R338L requires FVIIIa cofactor activity.16,35 Additionally, very high FIX activity levels >700% normal also leads to defective fibrinolysis mediated through thrombin activatable fibrinolysis inhibitor activation.36 The observation that most amino acid substitutions at R338 of hFIX or cFIX increase the specific activity suggests that R338, at a molecular level, likely forms a suboptimal interaction with FVIIIa that is relieved by most substitutions.
Because hFIX-R338L is the most active human variant, it is not surprising that this substitution was first described as an X-linked thrombophilia risk factor.2 Recently, hFIX-R338Q was identified as the etiology of recurrent deep vein thrombosis despite anticoagulation, named FIX Shanghai.23 The FIX activity:antigen ratio in this subject and the specific activity of recombinant hFIX-R338Q protein was fivefold WT, consistent with our results. Likewise, hFIX-R338Q and hFIX-R338L have been directly compared in a limited study after AAV gene therapy in HB mice with hFIX-R338L displaying modestly higher levels compared with FIX activity levels,23,31 consistent with our results across different cell lines.
The variant hFIX-R338G is the only nonsynonymous single base-pair change that has not yet been clinically identified; our data suggest it would likely be asymptomatic because it has similar total activity and specific activity to hFIX-WT. Two substitutions (R338K and R338N) were consistently associated with decreased specific activity in both hFIX and cFIX. These loss-of-function substitutions likely have not been identified in HB patients because they would require 3 base pair changes, which is unlikely to spontaneously occur.
Our results are the largest screening of thrombosis subjects and the first of pediatric subjects for FIX R338 variant thrombophilias (supplemental Table 3). Given the cumulative negative screening of 767 thrombosis subjects and 3 positive cases of FIX Padua2 and Shanghai,23 we estimate an upper limit of prevalence among thrombosis subjects as 0.4%. This value is consistent with the observed frequencies of single base-pair changes at this codon and the approximately 50-fold enhancement in prevalence seen in thrombosis subjects with strong inherited thrombophilias.37 Notably, all 3 affected male patients developed venous thrombosis without any other identified risk factors. It is possible that the combination of FIX-R338L (Padua), and to a lesser extent FIX-R338Q (Shanghai), with other prothrombotic risk factors may not be compatible with life.
Our results confirm the hypothesis that R338 is suboptimal for FIX activity in at least 2 mammalian FIX orthologs. Amino acid substitutions at 8 additional positions in FIX have also been identified as increasing FIX specific activity,3 albeit to a lesser extent than R338L. This suggests that hFIX-WT (and cFIX-WT) at other amino acid positions is also not optimized for maximal FIX activity, and additional activity-enhancing amino acid substitutions may be located in the future. If such additional positions are identified, it is likely worthwhile to screen multiple amino acids because our data suggest that there may be a range of hyperactive variants. Thus, new hyperactive FIX variants may allow for further enhancements of protein- or gene-based therapy for HB. This approach also may be applicable to other proteins that have evolved to limit biological activity.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Sriram Krishnaswamy for helpful discussions; Julia Chau, Taylor V. Daniel, Benjamin J. Lee, Aaron F. McKeon-Fish, Lucas van Gorden, Maxwell Chappell, and Kevin B. Hayes for conducting experiments; James F. Jarrett III and Jian-Hua Liu for screening thrombosis subjects; and Zhiyuan Chen for homology modeling hFIX and cFIX.
B.J.S.-J. reports receiving support from the National Blood Foundation and National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (NHLBI) (grant 1K08HL140078). T.C.N. reports receiving support from the NIH/NHLBI (grant N01_75N92019D00041). V.R.A reports receiving support from the NIH/NHLBI (grants P01HL64190 and P01HL139420).
Footnotes
For data, please contact the corresponding author at arruda@email.chop.edu.
Authorship
Contribution: B.J.S.-J. designed and conducted experiments, analyzed and interpreted data, and drafted the manuscript; J.D.F. designed and conducted experiments and revised manuscript; L.J.R. provided clinical samples, guidance, and revised the manuscript; R.M.C. provided reagents, interpreted data, and revised the manuscript; T.C.N. and E.P.M. carried out canine experiments and revised the manuscript; V.R.A. designed experiments, interpreted data, provided guidance, and drafted the manuscript; and all authors approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Valder R. Arruda, The Children’s Hospital of Philadelphia, 3501 Civic Center Blvd, 5056 Colket Translational Research Center, Philadelphia, PA 19104; e-mail: arruda@email.chop.edu.
References
- 1.VandenDriessche T, Chuah MK. Hyperactive factor IX Padua: a game-changer for hemophilia gene therapy. Molec Therapy. 2018;26(1):14-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simioni P, Tormene D, Tognin G, et al. X-linked thrombophilia with a mutant factor IX (factor IX Padua). N Engl J Med. 2009;361(17):1671-1675. [DOI] [PubMed] [Google Scholar]
- 3.Samelson-Jones BJ, Arruda VR. Protein-engineered coagulation factors for hemophilia gene therapy. Mol Ther Methods Clin Dev. 2018;12:184-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.George LA, Sullivan SK, Giermasz A, et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N Engl J Med. 2017;377(23):2215-2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bottema CD, Ketterling RP, Vielhaber E, et al. The pattern of spontaneous germ-line mutation: relative rates of mutation at or near CpG dinucleotides in the factor IX gene. Hum Genet. 1993;91(5):496-503. [DOI] [PubMed] [Google Scholar]
- 6.Chang J, Jin J, Lollar P, et al. Changing residue 338 in human factor IX from arginine to alanine causes an increase in catalytic activity. J Biol Chem. 1998;273(20):12089-12094. [DOI] [PubMed] [Google Scholar]
- 7.Schuettrumpf J, Herzog RW, Schlachterman A, Kaufhold A, Stafford DW, Arruda VR. Factor IX variants improve gene therapy efficacy for hemophilia B. Blood. 2005;105(6):2316-2323. [DOI] [PubMed] [Google Scholar]
- 8.Nichols T, Whitford MH, Arruda VR, Stedman HH, Kay MA, High KA. Translational data from AAV-mediated gene therapy of hemophilia B in dogs. Hum Gene Ther Clin Dev. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nichols TC, Hough C, Agersø H, Ezban M, Lillicrap D. Canine models of inherited bleeding disorders in the development of coagulation assays, novel protein replacement and gene therapies. J Thromb Haemost. 2016;14(5):894-905. [DOI] [PubMed] [Google Scholar]
- 10.Sabatino DE, Nichols TC, Merricks E, Bellinger DA, Herzog RW, Monahan PE. Animal models of hemophilia. Prog Mol Biol Transl Sci. 2012;105:151-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haurigot V, Mingozzi F, Buchlis G, et al. Safety of AAV factor IX peripheral transvenular gene delivery to muscle in hemophilia B dogs. Molec Therapy. 2010;18(7):1318-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arruda VR, Stedman HH, Haurigot V, et al. Peripheral transvenular delivery of adeno-associated viral vectors to skeletal muscle as a novel therapy for hemophilia B. Blood. 2010;115(23):4678-4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crudele JM, Finn JD, Siner JI, et al. AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood. 2015;125(10):1553-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finn JD, Nichols TC, Svoronos N, et al. The efficacy and the risk of immunogenicity of FIX Padua (R338L) in hemophilia B dogs treated by AAV muscle gene therapy. Blood. 2012;120(23):4521-4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.French RA, Samelson-Jones BJ, Niemeyer GP, et al. Complete correction of hemophilia B phenotype by FIX-Padua skeletal muscle gene therapy in an inhibitor-prone dog model. Blood Adv. 2018;2(5):505-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samelson-Jones BJ, Finn JD, George LA, Camire RM, Arruda VR. Hyperactivity of factor IX Padua (R338L) depends on factor VIIIa cofactor activity. JCI Insight. 2019;5(14):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Branchini A, Ferrarese M, Campioni M, et al. Specific factor IX mRNA and protein features favor drug-induced readthrough over recurrent nonsense mutations. Blood. 2017;129(16):2303-2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCue J, Osborne D, Dumont J, et al. Validation of the manufacturing process used to produce long-acting recombinant factor IX Fc fusion protein. Haemophilia. 2014;20(4):e327-e335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rallapalli PM, Kemball-Cook G, Tuddenham EG, Gomez K, Perkins SJ. An interactive mutation database for human coagulation factor IX provides novel insights into the phenotypes and genetics of hemophilia B. J Thromb Haemost. 2013;11(7):1329-1340. [DOI] [PubMed] [Google Scholar]
- 20.de Castilho Fernandes A, Fontes A, Gonsales N, et al. Stable and high-level production of recombinant Factor IX in human hepatic cell line. Biotechnol Appl Biochem. 2011;58(4):243-249. [DOI] [PubMed] [Google Scholar]
- 21.Enjolras N, Perot E, Le Quellec S, et al. In vivo efficacy of human recombinant factor IX produced by the human hepatoma cell line HuH-7. Haemophilia. 2015;21(4):e317-e321. [DOI] [PubMed] [Google Scholar]
- 22.de Sousa Bomfim A, Cristina Corrêa de Freitas M, Picanço-Castro V, et al. Human cell lines: a promising alternative for recombinant FIX production. Protein Expr Purif. 2016;121:149-156. [DOI] [PubMed] [Google Scholar]
- 23.Wu W, Xiao L, Wu X, et al. Factor IX alteration p.Arg338Gln (FIX Shanghai) potentiates FIX clotting activity and causes thrombosis. Haematologica. 2020;106(1):264-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raffini L, Huang YS, Witmer C, Feudtner C. Dramatic increase in venous thromboembolism in children’s hospitals in the United States from 2001 to 2007. Pediatrics. 2009;124(4):1001-1008. [DOI] [PubMed] [Google Scholar]
- 25.Ehrenforth S, Junker R, Koch HG, et al. ; Childhood Thrombophilia Study Group . Multicentre evaluation of combined prothrombotic defects associated with thrombophilia in childhood. Eur J Pediatr. 1999;158(suppl 3):S97-S104. [DOI] [PubMed] [Google Scholar]
- 26.Mazetto BM, Orsi FLA, Siqueira LH, de Mello TB, de Paula EV, Annichino-Bizzacchi JM. Prevalence of Factor IX-R338L (Factor IX Padua) in a cohort of patients with venous thromboembolism and mild elevation of factor IX levels. Thromb Res. 2010;126(2):e165. [DOI] [PubMed] [Google Scholar]
- 27.Koenderman JS, Bertina RM, Reitsma PH, de Visser MC. Factor IX-R338L (Factor IX Padua) screening in a Dutch population of sibpairs with early onset venous thromboembolism. Thromb Res. 2011;128(6):603. [DOI] [PubMed] [Google Scholar]
- 28.Campello E, Spiezia L, Bulato C, Gavasso S, Woodhams B, Simioni P. Factor IX activity/antigen ratio and the risk of first unprovoked venous thromboembolism. Thromb Haemost. 2013;109(4):755-756. [DOI] [PubMed] [Google Scholar]
- 29.Yuan QP, Walke EN, Sheehan JP. The factor IXa heparin-binding exosite is a cofactor interactive site: mechanism for antithrombin-independent inhibition of intrinsic tenase by heparin. Biochemistry. 2005;44(9):3615-3625. [DOI] [PubMed] [Google Scholar]
- 30.Kao CY, Yang SJ, Tao MH, Jeng YM, Yu IS, Lin SW. Incorporation of the factor IX Padua mutation into FIX-Triple improves clotting activity in vitro and in vivo. Thromb Haemost. 2013;110(2):244-256. [DOI] [PubMed] [Google Scholar]
- 31.Monahan PE, Sun J, Gui T, et al. Employing a gain-of-function factor IX variant R338L to advance the efficacy and safety of hemophilia B human gene therapy: preclinical evaluation supporting an ongoing adeno-associated virus clinical trial. Hum Gene Ther. 2015;26(2):69-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suwanmanee T, Hu G, Gui T, et al. Integration-deficient lentiviral vectors expressing codon-optimized R338L human FIX restore normal hemostasis in hemophilia B mice. Molec Therapy. 2014;22(3):567-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nichols TC, Levy H, Merricks EP, Raymer RA, Lee ML. Preclinical evaluation of a next-generation, subcutaneously administered, coagulation factor IX variant, dalcinonacog alfa. PLoS One. 2020;15(10):e0240896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buyue Y, Whinna HC, Sheehan JP. The heparin-binding exosite of factor IXa is a critical regulator of plasma thrombin generation and venous thrombosis. Blood. 2008;112(8):3234-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang H, Zögg T, Brandstetter H. Maturation of coagulation factor IX during Xase formation as deduced using factor VIII-derived peptides. FEBS Open Bio. 2019;9(8):1370-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ammollo CT, Semeraro F, Colucci M, Simioni P. Factor IX-Padua enhances the fibrinolytic resistance of plasma clots. Thromb Haemost. 2014;111(2):226-232. [DOI] [PubMed] [Google Scholar]
- 37.Middeldorp S. Inherited thrombophilia: a double-edged sword. Hematology Am Soc Hematol Educ Program. 2016;2016:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Belvini D, Salviato R, Radossi P, et al. ; AICE HB Study Group . Molecular genotyping of the Italian cohort of patients with hemophilia B. Haematologica. 2005;90(5):635-642. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.