

Key Points
A split GFP minigene accurately models endogenous posttranscriptional processing of hematopoietic transcription factor RUNX1.
The RNA-binding proteins HNRNPA1 and KHDRBS1 antagonistically regulate RUNX1a isoform generation by alternative polyadenylation.
Abstract
The proper balance of hematopoietic stem cell (HSC) self-renewal and differentiation is critical for normal hematopoiesis and is disrupted in hematologic malignancy. Among regulators of HSC fate, transcription factors have a well-defined central role, and mutations promote malignant transformation. More recently, studies have illuminated the importance of posttranscriptional regulation by RNA-binding proteins (RBPs) in hematopoiesis and leukemia development. However, the RBPs involved and the breadth of regulation are only beginning to be elucidated. Furthermore, the intersection between posttranscriptional regulation and hematopoietic transcription factor function is poorly understood. Here, we studied the posttranscriptional regulation of RUNX1, a key hematopoietic transcription factor. Alternative polyadenylation (APA) of RUNX1 produces functionally antagonistic protein isoforms (RUNX1a vs RUNX1b/c) that mediate HSC self-renewal vs differentiation, an RNA-processing event that is dysregulated in malignancy. Consequently, RBPs that regulate this event directly contribute to healthy and aberrant hematopoiesis. We modeled RUNX1 APA using a split GFP minigene reporter and confirmed the sensitivity of our model to detect changes in RNA processing. We used this reporter in a clustered regularly interspaced short palindromic repeats (CRISPR) screen consisting of single guide RNAs exclusively targeting RBPs and uncovered HNRNPA1 and KHDRBS1 as antagonistic regulators of RUNX1a isoform generation. Overall, our study provides mechanistic insight into the posttranscriptional regulation of a key hematopoietic transcription factor and identifies RBPs that may have widespread and important functions in hematopoiesis.
Visual Abstract

Introduction
Hematopoiesis is dependent on the proper balance between hematopoietic stem cell (HSC) self-renewal and differentiation. Perturbation in either direction is the basis of various hematologic malignancies. Among molecular mechanisms governing HSC fate, there is growing interest in the role of posttranscriptional regulation by RNA-binding proteins (RBPs), ignited by the identification of common splice factor mutations in leukemia and myelodysplastic syndrome (MDS).1-4 In addition to splicing factors, RBPs that regulate RNA methylation,5 editing,6 and translation7 have been described to impact HSC self-renewal and differentiation. However, only a small subset of all RBPs have been implicated in hematopoietic regulation and leukemia development. As such, the mechanistic impact and breadth of RBP involvement in hematopoiesis is only beginning to be understood.
By contrast, transcription factors have well-defined roles in regulating proper hematopoiesis, and mutations are common in hematologic malignancies. Interestingly, posttranscriptional regulation of various hematopoietic transcription factors (GATA1,8 IKZF1,9 RUNX1,10 SCL,11 and TEL/ETV612) produces functionally distinct isoforms, highlighting the intersection between these 2 regulatory processes. Consequently, elucidating the posttranscriptional mechanism of a key hematopoietic transcription factor will illuminate additional RBPs with a critical role in hematopoiesis.
RUNX1 is a hematopoietic transcription factor that plays a direct role in regulating HSC fate through the antagonistic action of its 3 major protein isoforms.13-15 The long RUNX1b/c isoforms use alternative promoters and differ by 27 amino acids at their N terminus.13,16 They both contain the runt homology domain for DNA binding and heterodimerization with CBFβ and the downstream transcriptional regulatory domain, which recruits essential cofactors.17 RUNX1a is a C-terminally truncated RUNX1 isoform that is generated by alternative polyadenylation (APA)13,18; it retains the runt homology domain but lacks the transcriptional regulatory domain.14 This structural difference confers enhanced DNA binding14,19 and divergent effects on target gene transcription compared with RUNX1b/c.14,20,21 RUNX1a overexpression expands functional HSCs in vitro and in vivo,10,21-23 retards hematopoietic differentiation,14,22 and enhances engraftment of murine bone marrow cells following transplantation.10,21,22 Conversely, RUNX1b/c induce HSC quiescence,24 promote differentiation,14,25 and abrogate engraftment of transplanted murine bone marrow cells.10
In a healthy hematopoietic system, RUNX1b/c are the dominantly expressed isoforms, whereas RUNX1a represents a minor fraction of the isoform pool.10,13 RUNX1a is further restricted to immature hematopoietic stem and progenitor cells (HSPCs),10 suggesting that RUNX1 APA is dynamically regulated during differentiation. Despite this restricted expression, short isoforms of RUNX1 are conserved across species,26-28 playing an important role in healthy HSC pool maintenance.29 Because RUNX1a mediates HSC expansion, overexpression can be leukemogenic.20 Indeed, RUNX1a is overexpressed in some patients with acute myeloid leukemia (AML),14,20 acute lymphoblastic leukemia,20 or MDS.30 Importantly, overexpression is achieved by a change in the relative ratio of RUNX1a/total RUNX1 transcript, linking posttranscriptional mechanisms to aberrant transcription factor function.14,30
Collectively, these observations support the hypothesis that posttranscriptional regulators of RUNX1 isoform generation contribute to HSC biology and protect against leukemia development. So far, the RBPs responsible for this regulation and the cis-acting elements to which they bind are unknown. Here, we devised a fluorescent minigene model that accurately recapitulates RUNX1 isoform generation and used our construct that monitors RUNX1a formation to perform an RBP clustered regularly interspaced short palindromic repeats (CRISPR) screen. We learned that RUNX1 proximal poly(A) site usage and, therefore, RUNX1a formation, is repressed primarily as a result of splicing factor RBPs, not core APA machinery. We identified HNRNPA1 as a potent repressor of RUNX1a formation throughout hematopoiesis, with a putative binding site in alternative terminal exon 7a. Conversely, KHDRBS1 is an activator of RUNX1a production in HSPCs, exhibiting declining expression throughout hematopoietic differentiation. Overall, our study highlights the intersection between posttranscriptional regulation and transcription factor function, while uncovering RBPs that play essential and previously underappreciated roles in normal and aberrant hematopoiesis.
Methods
Please see supplemental Methods for a complete description of all methods.
Split GFP minigene cloning
The split GFP vector was kindly provided by Zefeng Wang (University of North Carolina at Chapel Hill).31,32 RUNX1 exon 7a and 7b reporters were made by amplifying genomic DNA from KG-1a cells (exon 7a: 5′-GTTTTCACGTGACCCAGCAC-3′/5′-GGGACCTAGCATCTCCCTGA-3′; exon 7b: 5′-CTTGGGAGAGAATTCGCCTTA-3′/5′-TGGAACCAGTCCTCCATGGA-3′) and inserting the resulting fragment into the reporter by HindIII/KpnI digestion.
CRISPR screening and analysis
A total of 13 million MDS-L dual fluorescent reporter cells was infected with virus produced from the RBP single guide RNA (sgRNA) library33 (#141438; Addgene) in duplicate at a multiplicity of infection ∼0.3 using the transduction protocol described in supplemental Methods. Four million cells were collected following 72 hours of puromycin selection (day 0) and at the 3-week end point (day 21). At this time, GFP low and high cells relative to mCherry were sorted on a FACSAria II. Genomic DNA was extracted from each cell population using a DNeasy Blood and Tissue Kit (#69504; Qiagen). Integrated sgRNA sequences were amplified using polymerase chain reaction (PCR) and Herculase II Fusion DNA Polymerase (#600679; Agilent Technologies) with the following primers:
forward (equimolar mixture of the following):
5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCTAGTGGAAAGGACGAAACACCG-3′
5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGCTAGTGGAAAGGACGAAACACCG-3′
5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTGCCAGTGGAAAGGACGAAACACCG-3′
5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGAAGTCCGTGGAAAGGACGAAACACCG-3′;
reverse: 5′-TGGAAAAGATAGCTGGATCCTGGCTGGGGAGAGGG-3′.
PCR products were purified using agarose gel electrophoresis and then Nextera XT Indexes (#15032353; Illumina) were added to the amplicons using NEB Taq DNA Polymerase (M0273L). Libraries were pooled and sequenced on an Illumina HiSeq 4000. β scores representing the cumulative sgRNA enrichment per gene in GFP high and low populations were calculated using MAGeCK maximum-likelihood estimation.34 Enrichment of individual sgRNAs was determined by MAGeCK robust rank aggregation.34
Results
RUNX1 poly(A) site strength minimally contributes to suppression of the RUNX1a isoform
RNA-processing events are regulated by core machinery and unique RBPs that confer cellular context specificity. Therefore, we first assessed the likelihood that RUNX1 APA is regulated solely by core polyadenylation machinery. Previous studies regarding global poly(A) site usage revealed that genes tend to terminate at distal sites because proximal poly(A) sites generally contain weaker cis-acting elements than do their distal counterparts and recruit core machinery less efficiently.35,36 In these cases, usage of a weaker site is mostly dependent upon the cellular concentration of core polyadenylation machinery.37 Because RUNX1a isoform generation is due to polyadenylation at a proximal poly(A) site, we examined whether minimal endogenous RUNX1a formation can be attributed to a weak poly(A) site. To this end, we profiled endogenous RUNX1 poly(A) site usage by performing 3′READS38 of sorted common myeloid progenitors (CMPs) (CD34+/CD38+/CD123+/CD45RA−) from leukapheresis products obtained from 3 healthy donors (supplemental Figure 1A). Among 4 previously annotated major poly(A) sites,13 we observed that poly(A) site #1, which results in RUNX1a formation, was the second most used site behind only distal site #4 (Figure 1A-B; supplemental Figure 1B). Next, we compared the sequences of core polyadenylation cis elements for poly(A) sites #1 and #4 (supplemental Figure 2). Interestingly, both poly(A) sites have the canonical AAUAAA poly(A) hexamer, predictive of a strong poly(A) site.35,39,40 Both also have an upstream UGUA motif that enhances core machinery recruitment41 and downstream G/U-rich elements containing UU dinucleotides that enhance cleavage efficiency.42,43 Together, RUNX1 poly(A) site #1 has strong, not weak, cis elements. Finally, we tested the cleavage efficiency of both sites using a tandem poly(A) reporter system and RNase protection assays44 (supplemental Figure 3A). Although poly(A) site #4 had stronger cleavage than did poly(A) site #1, cleavage at poly(A) site #1 occurred in 40% of transcripts when present in tandem with a strong downstream synthetic poly(A) site (Figure 1C; supplemental Figure 3B-C). Altogether, these data support the conclusion that RUNX1a formation is not limited primarily as a result of weak polyadenylation cis elements and poor cleavage efficiency. Although core machinery may play a role, we hypothesize that context-specific RBPs regulate isoform expression of this crucial hematopoietic transcription factor.
Figure 1.
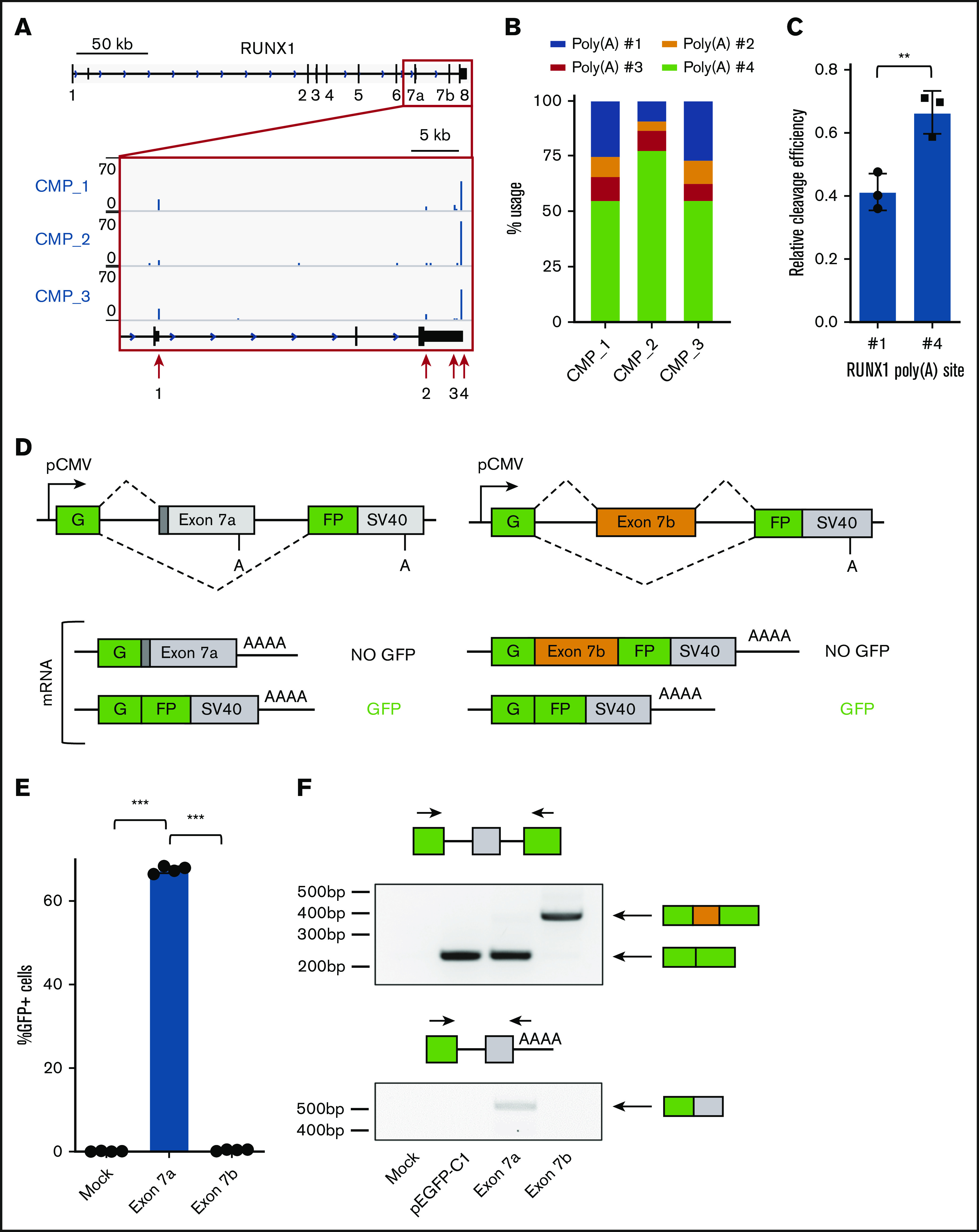
A split GFP minigene model recapitulates RUNX1 isoform generation. (A) Genome browser tracks depicting sequencing reads in the RUNX1 gene obtained from 3′READS of sorted common myeloid progenitors (CMP). The full RUNX1 genomic structure is shown above and the red boxed region is expanded below. The locations of the 4 major poly(A) sites are marked with red arrows. (B) Usage of the 4 major RUNX1 poly(A) sites, calculated from 3′READS analysis of sorted CMPs. (C) Quantification of RUNX1 poly(A) site #1 or #4 usage relative to a common synthetic poly(A) site via RNase protection assay. Data are mean ± standard deviation (SD) of 3 independent experiments. (D) Schematic diagram of the split GFP minigene constructs containing RUNX1 exon 7a (left panels) and RUNX1 exon 7b (right panels). Poly(A) sites are shown, as well as possible messenger RNA products. (E) Percentage of GFP+ cells, as measured by flow cytometry of KG-1a cells nucleofected with the RUNX1 exon 7a or exon 7b minigene construct. Data are mean ± SD of 4 independent experiments. (F) RT-PCR analysis of RNA extracted from KG-1a cells nucleofected with the RUNX1 minigene constructs, a GFP vector positive control, or mock-nucleofected negative control. Primer sets used for analysis are shown above the corresponding agarose gel image. **P < .01, 2-tailed Student t test; ***P < .001, 1-way analysis of variance (ANOVA) with a post hoc Tukey’s test.
A split GFP minigene model recapitulates RUNX1 isoform generation
Next, we modeled RUNX1a formation by including more of its genomic context, capturing regions that bind accessory RBPs and regulate context-specific isoform generation. RUNX1 isoform generation is mediated by a particular type of coding sequence APA in which proximal poly(A) site usage is coupled with splicing of an alternative terminal exon.45 For RUNX1a formation, exon 6 of the RUNX1 gene splices exon 7a and terminates at poly(A) site #1 (Figure 1A). RUNX1b/c are formed by skipping exon 7a, splicing exon 7b, and terminating at 1 of 3 poly(A) sites in exon 8.13 Therefore, we devised a minigene model that accounts for splicing and polyadenylation of RUNX1 alternative terminal exon 7a. We cloned exon 7a with ∼500 bp of flanking intron between the 2 exons of a split GFP reporter31,32 (Figure 1D, left panel). Because RUNX1a is globally suppressed in hematopoiesis, we expect that exon 7a will be skipped and GFP will be detected in cells expressing the transgene. We also generated an analogous construct containing constitutive exon 7b, which is spliced instead of exon 7a to produce the major RUNX1b/c isoforms (Figure 1D, right panel). Unlike the exon 7a minigene construct, we expect that exon 7b will be spliced between the 2 GFP exons, and GFP will not be produced.
To select an appropriate cell line in which to test our reporters, we profiled endogenous RUNX1 isoform levels in 8 leukemia cell lines (supplemental Figure 4). We selected KG-1a cells, with high relative RUNX1a expression, reasoning that we should be able to detect exon 7a usage of our reporter, despite its global suppression in blood cells. Upon nucleofection into KG-1a cells, the exon 7a minigene produced a strong GFP signal, whereas the exon 7b construct produced no detectable GFP (Figure 1E). We confirmed that GFP fluorescence reflected the expected splice products by performing reverse transcription PCR (RT-PCR) (Figure 1F, upper panel). Additionally, we observed low levels of exon 7a spliced and polyadenylated product from cells expressing the exon 7a minigene, effectively modeling low RUNX1a isoform generation (Figure 1F, lower panel). The opposite processing of these minigenes recapitulates endogenous RUNX1 posttranscriptional regulation. Consequently, these constructs are suitable for further study of cis-acting elements and RBPs that differentiate these RNA-processing events.
Chimeric minigenes reveal locations of critical cis-acting elements regulating RUNX1 exon 7a and 7b inclusion
To assess whether the disparity in RUNX1 exon usage is generally defined by exons or flanking intronic regions, we subcloned 2 series of chimeric minigene constructs. We first modified the split GFP reporter that contained exon 7a by replacing the upstream (bAa), downstream (aAb), or both (bAb) flanking introns with the analogous introns that typically surround exon 7b (Figure 2A). Cells nucleofected with all 4 exon 7a minigene constructs still produced GFP, as shown by flow cytometry (Figure 2B) and RT-PCR (Figure 2C, upper panel). This observation suggests that major suppressive cis elements are located within exon 7a. Interestingly, we noticed consistent differences in the GFP mean fluorescent intensity (MFI) of cells expressing each of the 4 constructs (Figure 2D). To test whether these differences meaningfully predict changes in inclusion of exon 7a, we performed quantitative RT-PCR (RT-qPCR) to quantify the relative amount of polyadenylated exon 7a product generated by each minigene construct (Figure 2E). Indeed, decreased GFP MFI (bAa) correlated with increased exon 7a inclusion, whereas increased GFP MFI (aAb and bAb) correlated with exon 7a exclusion. The former observation (bAa) indicates a minor suppressive cis element in the upstream intron of exon 7a. The latter (aAb and bAb) result from ablation of exon 7a polyadenylation by removal of the G/U-rich downstream element (Figure 2C, lower panel). Overall, GFP MFI precisely detected changes in exon 7a inclusion of the minigene reporter.
Figure 2.
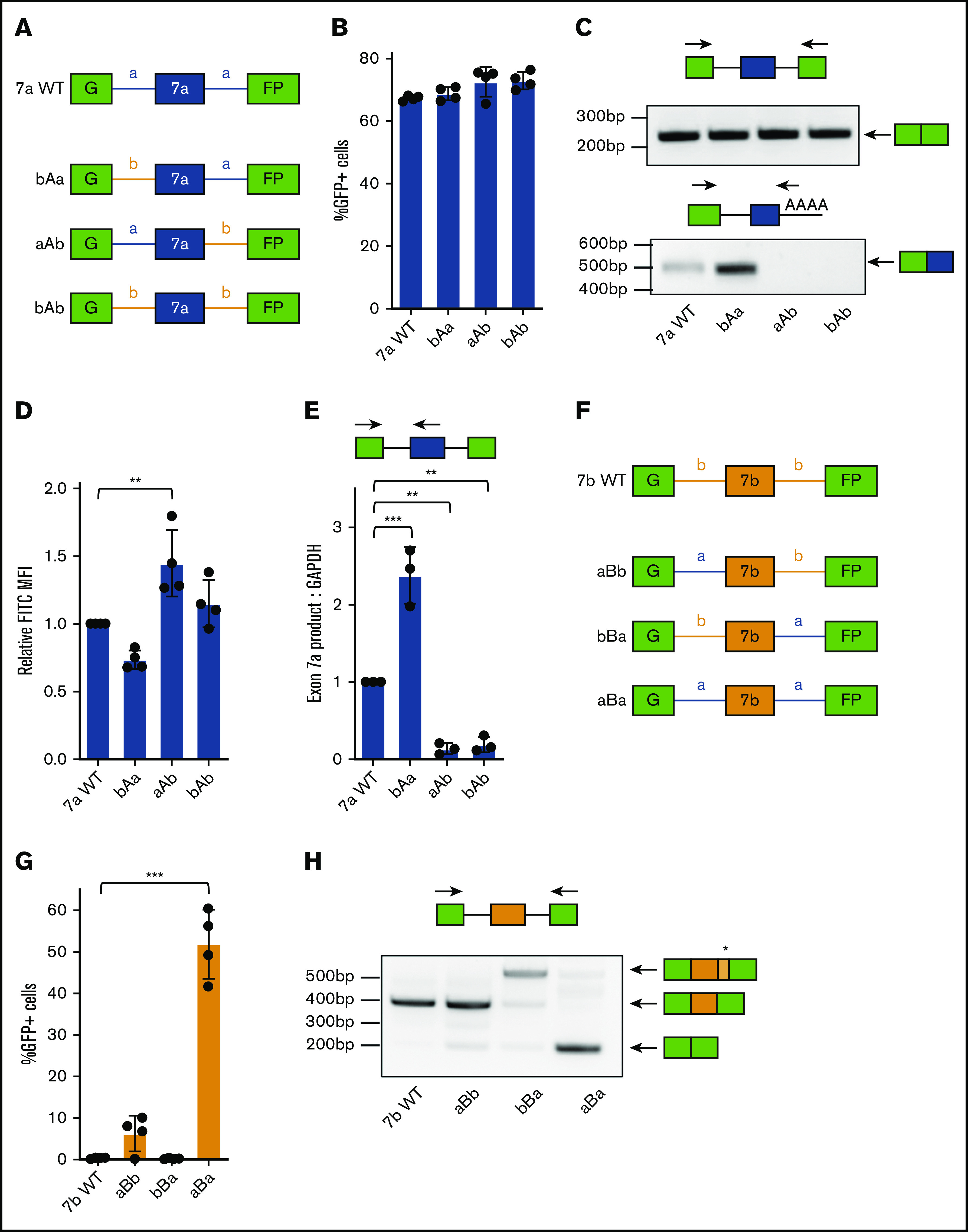
Chimeric minigenes reveal locations of critical cis-acting elements regulating RUNX1 exon 7a and 7b inclusion. (A) Schematic diagram of the RUNX1 exon 7a chimeric split GFP minigene constructs. Changes to the intronic context are indicated by label and color: introns normally flanking exon 7a are blue and are labeled “a’; introns normally flanking exon 7b are yellow and are labeled “b.” (B) Percentage of GFP+ cells, as measured by flow cytometry, in KG-1a cells nucleofected with the indicated RUNX1 exon 7a chimeric minigene constructs. Data are mean ± SD of 4 independent experiments. The data are not significantly different by 1-way analysis of variance (ANOVA). (C) RT-PCR analysis of RNA extracted from KG-1a cells nucleofected with the RUNX1 exon 7a chimeric minigene constructs. Primer sets used for analysis are shown above the corresponding agarose gel image. (D) Fluorescein isothiocyanate (FITC) MFI measured by flow cytometric analysis of KG-1a cells nucleofected with the indicated RUNX1 exon 7a chimeric minigene constructs. FITC MFI is normalized to KG-1a cells nucleofected with the exon 7a wild-type (WT) minigene construct. Data are mean ± SD of 4 independent experiments. (E) RT-qPCR analysis of exon7a product mRNA normalized to GAPDH mRNA from KG-1a cells nucleofected with the respective constructs. mRNA levels were normalized to that of the RUNX1 exon 7a WT control. Data are mean ± SD of 3 independent experiments. (F) Schematic diagram of the RUNX1 exon 7b chimeric split GFP minigene constructs. Changes to the intronic context are indicated by label and color: introns normally flanking exon 7a are blue and are labeled “a”; introns normally flanking exon 7b are yellow and are labeled “b.” (G) Percentage of GFP+ cells, as measured by flow cytometry, of KG-1a cells nucleofected with the indicated RUNX1 exon 7b chimeric minigene constructs. Data are mean ± SD of 4 independent experiments. (H) RT-PCR analysis of RNA extracted from KG-1a cells nucleofected with the indicated RUNX1 exon 7b chimeric minigene constructs. The asterisk represents the presence of a cryptic splice site that results in a higher molecular weight product. **P < .01, ***P < .001, 1-way ANOVA with post hoc Tukey’s test.
We next generated a set of minigene constructs that contained constitutive exon 7b. We replaced the upstream (aBb), downstream (bBa), or both (aBa) flanking introns with the analogous introns that typically flank exon 7a (Figure 2F). When nucleofected into cells, the wild-type construct did not produce GFP, and replacing only the downstream intron (bBa) had no effect on GFP production. However, replacing the upstream intron (aBb) produced a slight increase in GFP+ cells, and replacing both introns (aBa) produced a dramatic increase in GFP+ cells (Figure 2G). RT-PCR analysis revealed a striking reversal from complete inclusion of exon 7b in the wild-type context to nearly complete exclusion when both introns were replaced (Figure 2H). This observation indicates that the exon 7b flanking introns may contain enhancer cis elements, the exon 7a flanking introns may contain suppressive cis elements, or both hypotheses may be true. Ultimately, the constitutive nature of exon 7b is disrupted by modifying its intronic context, in contrast to exon 7a, which was modestly affected by alterations to its flanking introns.
A CRISPR RBP screen uncovers putative regulators of RUNX1a production
Motivated by the unique regulation of exon 7a, we next wanted to identify specific RBPs that regulate exon 7a inclusion and, therefore, RUNX1a formation. We performed a CRISPR/Cas9 screen using a library composed of sgRNAs exclusively targeting RBPs.33 This library consists of 10 sgRNAs targeting each of 1078 RBPs, 628 positive control sgRNAs targeting essential genes, 1058 negative control nontargeting sgRNAs, and 12 sgRNAs targeting fluorescent proteins. Because the exon 7a split GFP minigene reporter was sensitive in detecting bidirectional changes in exon inclusion (Figure 2D), we modified this reporter for screening. To account for RBPs that may affect transcription, messenger RNA (mRNA) stability, export, localization, and translation of the reporter independent of changes in inclusion of exon 7a, we added mCherry and a P2A peptide directly upstream of the split GFP (Figure 3A). Using this bicistronic model, RBPs that regulate reporter expression will influence both fluorescent proteins, and the fluorescence ratio will remain unchanged. Conversely, RBPs that regulate RUNX1 exon 7a processing will alter the ratio of GFP/mCherry fluorescence. We selected MDS-L cells as a diploid RUNX1a intermediate-expressing cell line (supplemental Figure 4A) and generated a stable clonal line expressing the dual fluorescent minigene reporter. As expected, these cells displayed a strong correlation between mCherry and GFP fluorescence (Figure 3B).
Figure 3.
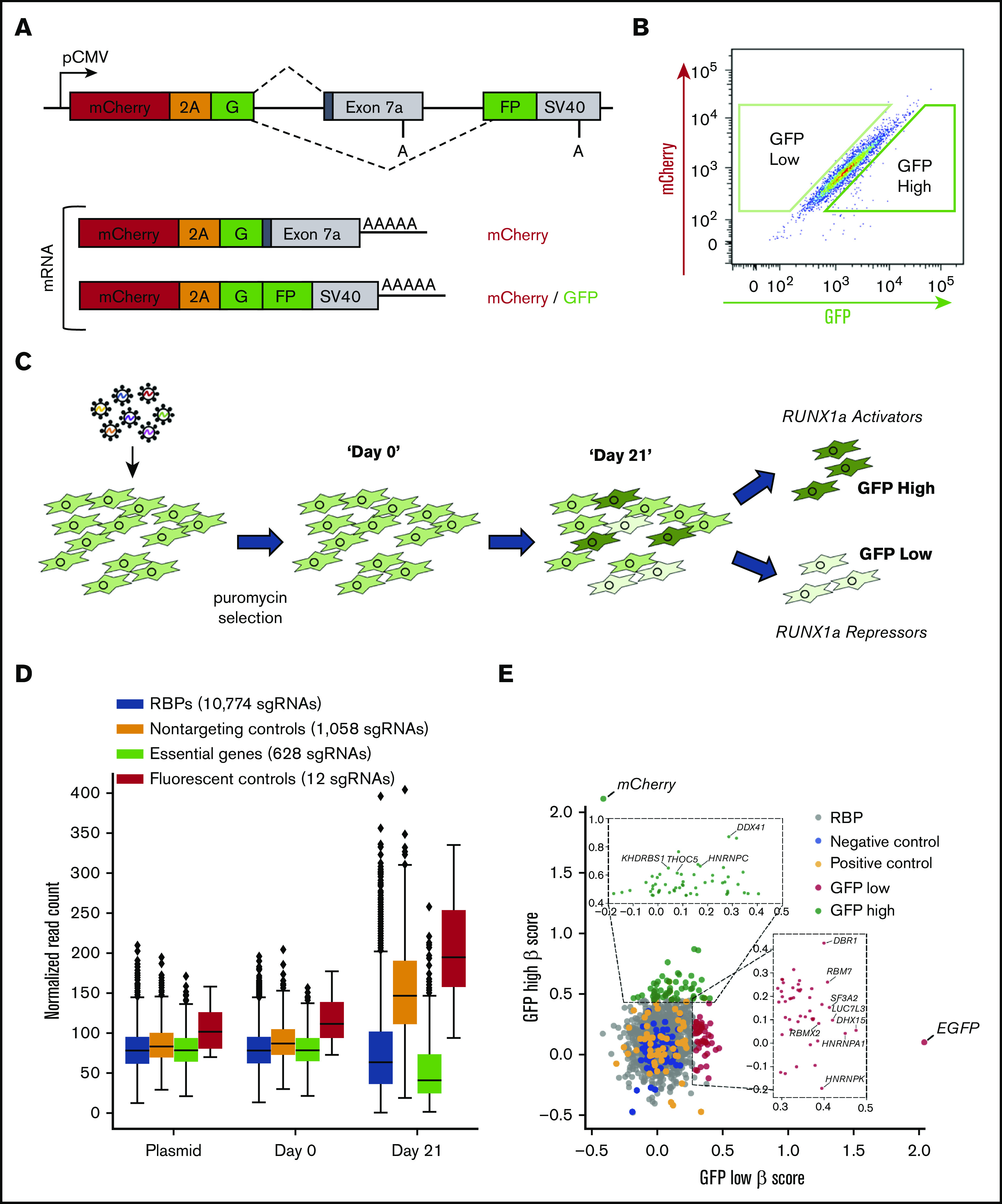
A CRISPR RBP screen reveals regulators of RUNX1a isoform formation. (A) Schematic of the bicistronic dual fluorescent minigene reporter used for the CRISPR screen. The RUNX1 exon 7a minigene reporter was modified by subcloning mCherry and a P2A peptide upstream of the first GFP exon. Poly(A) sites are shown. (B) Representative flow cytometry plot of the stable clonal MDS-L cell line containing the dual fluorescent reporter from panel A. Boxed regions represent the gating of GFP high and low cell populations that were sorted at the end point of the CRISPR screen. (C) Schematic diagram of the CRISPR screen. MDS-L cells stably expressing the dual fluorescent RUNX1 exon 7a minigene construct were infected with lentiviral particles containing the RBP sgRNA library. Bulk cell populations were collected at day 0 and day 21. GFP high and low cells were sorted as depicted in panel B on day 21. (D) Box and whisker plots depicting sgRNA representation in the indicated cell populations and the plasmid library used for lentiviral production. Comparing plasmid vs day 0 using a 2-sided Student t test, all sgRNA populations are insignificant with the exception of essential sgRNAs (0.001526493), which were modestly depleted during the 72 hours of puromycin selection between transduction and our day 0 time point. Comparing day 0 with day 21, all sgRNA populations are significant. P < .001 for all comparisons with the exception of fluorescent controls (P < .01). (E) Plot depicting the β -scores of sgRNA enrichment in GFP high and low populations. The insets show all significantly enriched RBPs for both cell populations (P < .05). Splicing-related RBPs with P < .01 that were included in the secondary validation are labeled. Two sgRNAs targeting mCherry and 3 targeting EGFP were present in the sgRNA library and served as the positive controls for enrichment in the sorted GFP high and low cell populations, respectively. These controls were most significantly enriched in their respective populations and are labeled in the plot.
We infected this reporter cell line with the lentiviral sgRNA library and selected for transduced cells with puromycin (Figure 3C). We collected a population of cells 3 days posttransduction to confirm representation of the sgRNA library (day 0) (supplemental Figure 5A) and after 3 weeks (day 21) to allow adequate time for editing of target RBP genomic sequences. At day 21, positive control sgRNAs were significantly depleted from the pool (approximately twofold) along with those targeting essential RBPs (Figure 3D). We also sorted GFP low and GFP high cells relative to mCherry (Figure 3B) to identify sgRNAs targeting putative RUNX1a repressors and activators, respectively. We used MAGeCK34 to calculate β scores describing the level of enrichment of multiple sgRNAs targeting each RBP in these subpopulations. Altogether, we identified 47 putative repressor RBPs and 55 activator RBPs from the 1078 RBPs screened (P < .05) (Figure 3E). We focused on RBPs with P < .01, which displayed a high proportion of enriched sgRNAs per RBP (supplemental Figure 5B). Among the 10 most significant putative RBP repressors, 8 carry the “RNA splicing” (GO:0008380) and/or “Regulation of mRNA splicing” (GO:0043484) Gene Ontology annotation; none are annotated for “mRNA 3′-end processing” (GO:0031124) (supplemental Table 1). This observation suggests potent splicing regulation of alternative terminal exon 7a and supports our previous conclusion that APA core machinery plays a minor role in repressing proximal poly(A) site usage (Figure 1A-C).
HNRNPA1 is a potent repressor of RUNX1a formation
To validate putative RUNX1a repressors, we performed a secondary screen using pooled small interfering RNAs (siRNAs) as a different method of gene knockdown to confidently identify RBP regulators of RUNX1 isoform generation. We knocked down the 8 splicing-related RBPs and measured relative endogenous RUNX1a mRNA to total RUNX1 transcript by RT-qPCR (supplemental Figure 6A). From this secondary screen, we identified HNRNPA1 as a repressor of RUNX1a isoform generation. Next, we knocked down HNRNPA1 using stably expressed short hairpin RNAs (shRNAs). In the K562 and MDS-L leukemia cell lines, HNRNPA1 knockdown (Figure 4A) led to a significant increase in relative RUNX1a mRNA to total RUNX1 mRNA (Figure 4B), with a concurrent decrease in RUNX1b/c transcript (Figure 4C). In MDS-L cells, HNRNPA1 knockdown also led to the clear detection of RUNX1a protein, which was difficult to detect in the parental cell line (Figure 4D). We further validated this regulation in primary human CD34+ HSPCs. HNRNPA1 knockdown in HSPCs (Figure 4E) led to a significant increase in relative RUNX1a mRNA (Figure 4F) and an obvious increase in RUNX1a protein that coincided with a modest decrease in RUNX1b protein (Figure 4G).
Figure 4.
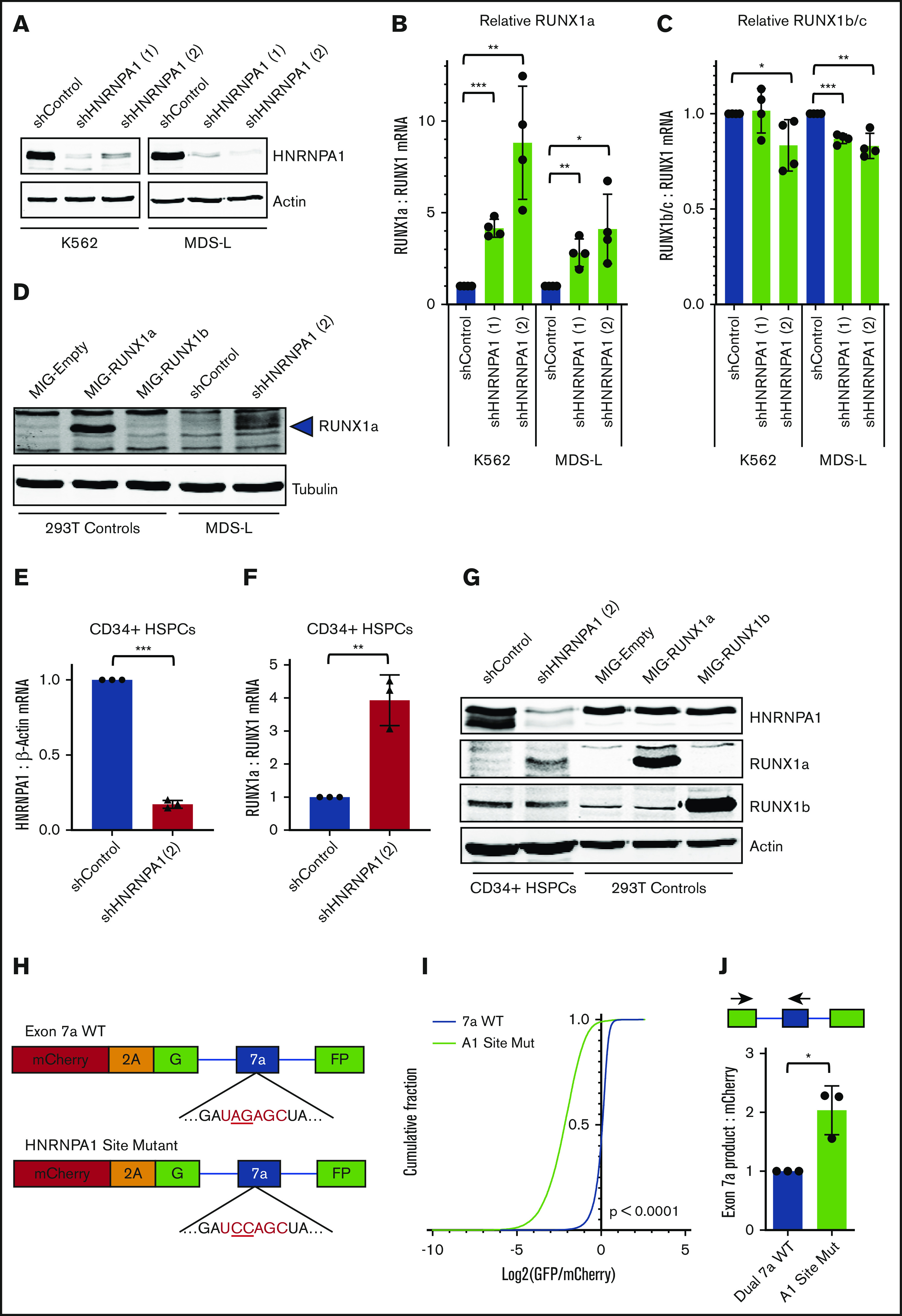
HNRNPA1 represses RUNX1a isoform formation. (A) Western blot showing HNRNPA1 and actin (loading control) protein in K562 and MDS-L cells 6 days following transduction with control shRNAs or shRNAs targeting HNRNPA1. (B) RT-qPCR analysis of RUNX1a mRNA normalized to total RUNX1 mRNA upon HNRNPA1 knockdown in K562 and MDS-L cells. mRNA levels were normalized to the respective control for each cell line. Data are mean ± SD of 4 independent experiments. *P < .05, **P < .01, ***P < .001, 2-tailed Student t test. (C) RT-qPCR analysis of RUNX1b/c mRNA normalized to total RUNX1 mRNA upon HNRNPA1 knockdown in K562 and MDS-L cells. mRNA levels were normalized to the respective control for each cell line. Data are mean ± SD of 4 independent experiments. *P < .05, **P < .01, ***P < .001, 2-tailed Student t test. (D) Western blot showing RUNX1a and tubulin (loading control) protein. The first 3 lanes contain protein from 293T cells transfected with empty vector, vector containing untagged RUNX1a cDNA, or vector containing RUNX1b cDNA. Lane 2 is a positive control for RUNX1a protein (blue arrowhead). The last 2 lanes contain protein from MDS-L cells 6 days following transduction with shControl lentivirus or shHNRNPA1 (2) lentivirus. Signal above the main RUNX1a band could be due to posttranslational modifications of the RUNX1 protein or usage of distal promoter P1. (E) RT-qPCR analysis of HNRNPA1 mRNA normalized to total β-Actin mRNA upon HNRNPA1 knockdown in primary CD34+ HSPCs. mRNA levels were normalized to cells transduced with control shRNAs. Data are mean ± SD of 3 independent experiments. ***P < .001, 2-tailed Student t test. (F) RT-qPCR analysis of RUNX1a mRNA normalized to total RUNX1 mRNA upon HNRNPA1 knockdown in primary CD34+ HSPCs. mRNA levels were normalized to cells transduced with control shRNAs. Data are mean ± SD of 3 independent experiments. **P < .01, 2-tailed Student t test. (G) Western blot showing HNRNPA1, RUNX1a, RUNX1b, and actin (loading control) protein. The first 2 lanes contain protein from CD34+ HSPCs 6 days following transduction with control shRNA lentivirus or shHNRNPA1 (2) lentivirus. The last 3 lanes contain protein from 293T cells transfected with empty vector, vector containing untagged RUNX1a cDNA, or vector containing RUNX1b cDNA. Quantification of RUNX1a and RUNX1b protein was performed by densitometry and normalized to actin protein. Compared to HSPCs transduced with control shRNAs, HNRNPA1 knockdown cells have 4.5 times more RUNX1a protein and 0.78 times RUNX1b protein. (H) Schematic diagram of the dual fluorescent RUNX1 exon 7a wild-type (WT) and HNRNPA1 site mutant constructs. The underlined “AG” nucleotides in the WT construct were mutated to “CC.” (I) Cumulative distribution function plot showing the log2 ratio of GFP/mCherry in individual KG-1a cells nucleofected with the exon 7a WT construct or HNRNPA1 site mutant construct. Data are from 1 representative experiment of 3 independent experiments. ***P < .001, Kolmogorov-Smirnov test. (J) RT-qPCR analysis of exon 7a product mRNA normalized to mCherry mRNA from KG-1a cells nucleofected with the respective HNRNPA1 site mutant constructs. mRNA levels were normalized to that of the RUNX1 exon 7a WT control. Data are mean ± SD of 3 independent experiments. *P < .05, 2-tailed Student t test.
Because HNRNPA1 is abundantly expressed in hematopoietic cells, we also generated HNRNPA1-knockout MDS-L cells via CRISPR/Cas9. We validated 4 unique HNRNPA1-knockout clones and 2 heterozygous clones by western blot (supplemental Figure 7A) and sequencing of individual alleles (supplemental Figure 7B). HNRNPA1 protein reduction conferred a dose-dependent increase in relative RUNX1a mRNA (supplemental Figure 7C) and RUNX1a protein (supplemental Figure 7D), further implicating HNRNPA1 as a potent repressor of RUNX1a formation.
Mutation of a predicted HNRNPA1 binding site in exon 7a enhances its splicing and polyadenylation
We next addressed whether HNRNPA1 likely represses RUNX1a production through direct binding to exon 7a or its adjacent introns. A consensus binding motif for HNRNPA1 was previously deduced using SELEX,46 HITS-CLIP,47 and iCLIP48 techniques. Based on our cis element studies, we hypothesized that suppressive elements are located within exon 7a (Figure 2A-E). Indeed, we identified a putative HNRNPA1 binding motif (UAGAGC) in the 3′ region of exon 7a. To disrupt HNRNPA1 binding, we mutated the essential “AG” dinucleotide to cytosines (UAGAGC → UCCAGC) in the dual fluorescent minigene49,50 (Figure 4H). We observed a significantly lower GFP/mCherry MFI ratio in cells expressing the mutated construct compared with cells expressing the wild-type construct (Figure 4I). In agreement with this shift, the construct containing the mutated HNRNPA1 binding site also produced significantly more exon 7a polyadenylated product (Figure 4J). We concluded that HNRNPA1 likely binds to the consensus motif in RUNX1 exon 7a to repress splicing and polyadenylation.
Additional regulators contribute to the proper balance of RUNX1 isoforms
HNRNPA1 is abundantly expressed throughout hematopoiesis, playing a pivotal role in repressing RUNX1a isoform production. However, its absence does not confer dominance of the RUNX1a isoform over RUNX1b/c. There must be additional RBPs that contribute to this repression and others that activate exon 7a inclusion, maintaining the low levels of RUNX1a necessary for HSC pool maintenance.
In our CRISPR screen interrogating RBPs, we also sorted GFP high cells that contained sgRNAs targeting putative RUNX1a activators (Figure 3B-C). To validate endogenous RUNX1a activators, we again performed a secondary siRNA screen of RBPs with documented roles in splicing (supplemental Figure 6B; supplemental Table 1). From this secondary screen, we identified HNRNPC and KHDRBS1 as putative activators of RUNX1a formation (Figure 5A-B; supplemental Figure 6B). Of these 2 RBPs, overexpression of KHDRBS1 in K562 cells led to a significant increase in relative RUNX1a mRNA to total RUNX1 mRNA (Figure 5C-D). Next, we validated this regulation in primary HSPCs. Indeed, knockdown of KHDRBS1 (Figure 5E) led to a significant decrease in relative RUNX1a mRNA (Figure 5F). Finally, we examined the effect of KHDRBS1 overexpression combined with HNRNPA1 knockdown and observed further increased RUNX1a transcript production (Figure 5G-H). These results highlight the combinatorial role of multiple RBPs in maintaining isoform pools of critical transcription factors, such as RUNX1.
Figure 5.
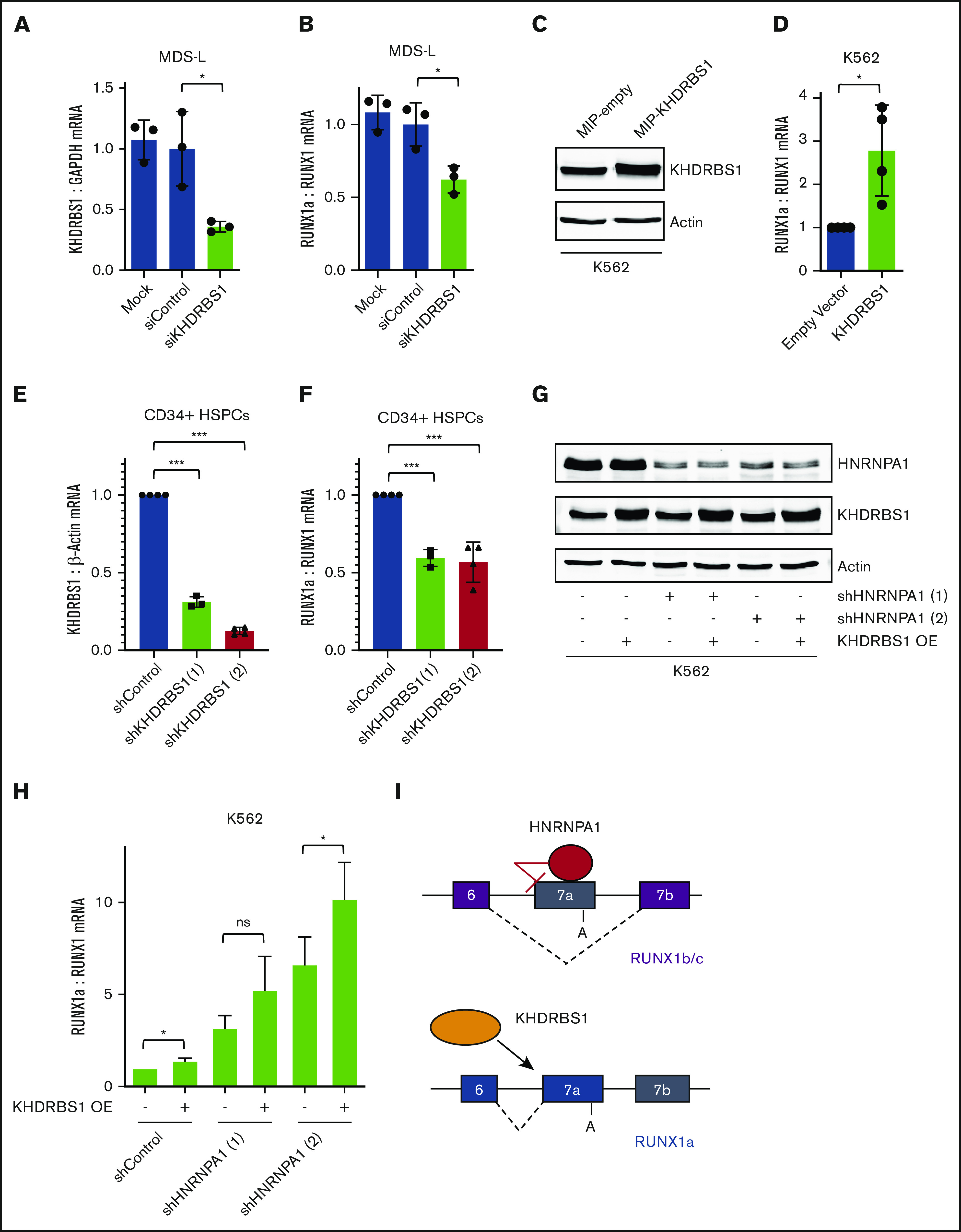
KHDRBS1 activates RUNX1a isoform formation. (A) RT-qPCR analysis of KHDRBS1 mRNA normalized to GAPDH mRNA upon siRNA knockdown of KHDRBS1 in MDS-L cells. mRNA levels were normalized to cells treated with control siRNAs. Data are mean ± SD of 3 independent experiments. *P < .05, 2-tailed Student t test. (B) RT-qPCR analysis of RUNX1a mRNA normalized to total RUNX1 mRNA upon siRNA knockdown of KHDRBS1 in MDS-L cells. mRNA levels were normalized to cells treated with control siRNAs. Data are mean ± SD of 3 independent experiments. *P < .05, 2-tailed Student t test. (C) Western blot showing overexpressed KHDRBS1 protein in K562 cells following transduction with retrovirus expressing KHDRBS1 cDNA. Actin protein is the loading control. (D) RT-qPCR analysis of RUNX1a mRNA normalized to total RUNX1 in K562 cells following transduction with retrovirus expressing KHDRBS1 cDNA. mRNA levels were normalized to cells transduced with an empty vector. Data are mean ± SD of 4 independent experiments. *P < .05, 2-tailed Student t test. (E) RT-qPCR analysis of KHDRBS1 mRNA normalized to β-Actin mRNA upon shRNA knockdown of KHDRBS1 in primary CD34+ HSPCs. mRNA levels were normalized to cells transduced with control shRNAs. Data are mean ± SD of 3 (shRNA 1) or 4 (shRNA 2) independent experiments. ***P < .001, 2-tailed Student t test. (F) RT-qPCR analysis of RUNX1a mRNA normalized to total RUNX1 mRNA upon shRNA knockdown of KHDRBS1 in primary CD34+ HSPCs. mRNA levels were normalized to cells transduced with control shRNAs. Data are mean ± SD of 3 (shRNA 1) or 4 (shRNA 2) independent experiments. ***P < .001, 2-tailed Student t test. (G) Western blot showing HNRNPA1, KHDRBS1, and actin (loading control) proteins in K562 cells with the indicated combinations of shRNA-mediated HNRNPA1 knockdown and KHDRBS1 cDNA retroviral overexpression. (H) RT-qPCR analysis of RUNX1a mRNA normalized to total RUNX1 in K562 cells with the indicated combinations of shRNA-mediated HNRNPA1 knockdown and KHDRBS1 cDNA retroviral overexpression. mRNA levels were normalized to cells transduced with control shRNAs and empty vector. Data are mean ± standard error of the mean of 5 independent experiments. *P < .05, paired 2-tailed Student t test. (I) Diagram depicting the antagonistic regulation of RUNX1 APA by HNRNPA1 and KHDRBS1. ns, not significant.
Discussion
Because transcription factors have well-defined roles in healthy hematopoiesis and leukemogenesis, studying the posttranscriptional processing of a critical hematopoietic transcription factor is a suitable strategy for identifying functionally important RBPs in hematology. Here, we studied APA of RUNX1, a posttranscriptional event that produces antagonistic isoforms mediating HSC dynamics and is dysregulated in malignancy. By studying this event, we uncovered 2 RBPs, HNRNPA1 and KHDRBS1, with previously unappreciated roles in proper and aberrant hematopoiesis. Additionally, our dual fluorescent model and screening approach can be adapted to probe RBP regulation of other critical posttranscriptional events. Finally, our study represents one of few reports on the role of APA in normal hematopoiesis and assigns a role for splicing regulators to alternative terminal exon APA.
To study RBP-RNA interactions that impact RUNX1 isoform generation, we modeled RUNX1 posttranscriptional processing using a split GFP minigene.31,32 This fluorescent reporter recapitulated the endogenous disparity in the expression of RUNX1a (alternative terminal exon 7a) and RUNX1b/c (constitutive exon 7b) in hematopoiesis10 and was sensitive enough to detect small changes in exon inclusion. When converted to a dual fluorescent bicistronic reporter and paired with a CRISPR sgRNA library targeting RBPs,33 we identified key posttranscriptional regulators of RUNX1 function. Certainly, our strategy can be readily adapted to probe regulation of RNA-processing events beyond this current study.
Among the screened RBPs, we identified HNRNPA1 as a potent repressor of RUNX1a formation with a presumptive binding site in the 3′ untranslated region of alternative terminal exon 7a (Figure 5I). Considering the global suppression of RUNX1a in hematopoiesis, the discovery of HNRNPA1 as a major player in exon 7a repression is not surprising. HNRNPA1 has a broadly documented repressive role on exon usage and is one of the most abundantly expressed nuclear proteins.51 Indeed, HNRNPA1 is highly expressed throughout hematopoiesis, maintaining a RUNX1 isoform pool that favors RUNX1b/c (supplemental Figure 8A).52 Expression remains high in HSPCs and, therefore, cannot fully explain RUNX1a upregulation in these cells. Expression is also lower in differentiated myeloid cells,53 suggesting the presence of additional repressors.
HNRNPA1 is overexpressed in AML53 (supplemental Figure 8B) and BCR-ABL+ chronic myeloid leukemia patients,54 where it contributes to leukemic phenotypes.55 Consequently, decreased HNRNPA1 expression is not a pervasive mechanism for RUNX1a upregulation in leukemia. An intriguing alternative possibility is that posttranslational modification of HNRNPA1, independent of changes in expression, can impact target gene splicing.56 So far, we cannot exclude the importance of HNRNPA1 posttranslational modifications on the regulation of RUNX1 isoform generation.
We also identified the RUNX1a activator KHDRBS1 (Figure 5I), belonging to the signal transduction and activation of RNA metabolism (STAR) family of RBPs, which link signal transduction to posttranscriptional regulation.57 KHDRBS1 and HNRNPA1 play cooperative and antagonistic roles in RNA-processing events.58-60 Our data support an antagonistic interaction on RUNX1 isoform generation.
Importantly, KHDRBS1 expression during healthy hematopoiesis and in leukemia supports a role for this RBP in fine-tuning the RUNX1 isoform ratio. KHDRBS1 is most highly expressed in HSPCs, with decreased expression upon differentiation (supplemental Figure 8C). Therefore, KHDRBS1 expression activates RUNX1a formation in HSPCs and might positively regulate HSC self-renewal. Consistent with this hypothesis, KHDRBS1 promotes neural progenitor cell self-renewal and knockdown induces differentiation, a system analogous to HSC regulation.61 Furthermore, KHDRBS1 plays an oncogenic role in various cancer types,62,63 including leukemia. KHDRBS1 expression is essential for MLL fusion–mediated leukemic transformation,64 and it is overexpressed in T-cell acute lymphoblastic leukemia65 and AML66 (supplemental Figure 8D). In these studies, the oncogenic role of KHDRBS1 is attributed to protein-protein interactions, not its RNA-binding ability. However, the posttranscriptional impact of KHDRBS1 in other cancer types is well documented.62,63 Our data demonstrate, for the first time, that KHDRBS1 regulates APA of a key hematopoietic transcription factor, thus making this RBP an attractive candidate for normal HSC regulation and dysregulation in leukemia.
Finally, our study highlights the importance of APA in hematopoiesis, a field of posttranscriptional regulation relatively understudied in hematology. Global APA trends across species and cellular contexts reveal a shift from proximal to distal poly(A) site usage during differentiation67-70 and the opposite trend during oncogenic transformation.71-73 In these respects, RUNX1 APA fits global trends: proximal poly(A) site #1 usage (RUNX1a) is highest in immature HSCs and decreases upon differentiation,10 and proximal poly(A) site #1 usage is elevated in hematologic malignancy.14,20,30 However, a closer analysis reveals unconventional APA regulation. Although proximal poly(A) sites tend to contain inherently weak polyadenylation cis elements,35,36 this is not the case for RUNX1. We saw that proximal poly(A) site #1 has higher endogenous usage than 2 of 3 distal sites, canonical cis-acting elements, and relatively strong cleavage. These observations support an evolutionarily conserved role for short RUNX1 isoforms in healthy hematopoiesis.26,27,29 Additionally, we implicated splicing RBPs, not APA machinery, in the regulation of alternative terminal exon polyadenylation. KHDRBS1 has recently been reported to regulate this unique APA category.61,74,75 Considering its expression in normal hematopoiesis and upregulation in leukemia, our finding warrants further study of the interaction between KHDRBS1 and APA in these contexts.
In summary, we uncovered RBPs involved in posttranscriptional regulation of RUNX1, a mechanism with implications for normal hematopoiesis and malignancy. Because of their newly assigned role in regulating RUNX1a formation, further studies on the global function of HNRNPA1 and KHDRBS1 in hematology are necessary.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the staff at the University of California, San Diego Human Embryonic Stem Cell Core Facility and Moores Cancer Center Flow Cytometry Core Facility for generous help with fluorescence activated cell sorting and the staff at the University of California, San Diego Institute for Genomic Medicine for assistance with next-generation sequencing.
This work was supported by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute grant F31HL131155 (A.G.D.) and NIH, National Institute of Diabetes and Digestive and Kidney Diseases grant R01DK098808 (D.-E.Z. and X.-D.F.). This work was also supported by Ruth. L. Kirschstein F31 National Research Service Award F31CA217173 (J.M.E.), the Cancer Systems Biology Training Program P50GM085764 and U54CA209891 (J.M.E.), and NIH, National Institute of General Medical Sciences grant HG004659 (G.W.Y.).
Footnotes
The 3′RNA sequencing data from healthy common myeloid progenitors and CRISPR RNA-binding protein screen results have been deposited in the Gene Expression Omnibus database (accession GSE145968). All DNA constructs cloned for this study will be deposited to Addgene for purchase or will be available by e-mail request to Dong-Er Zhang (dez@ucsd.edu).
Authorship
Contribution: A.G.D. and D.-E.Z. devised the study and designed the experimental strategies; A.G.D. and J.M.E. performed the research, collected the data, and analyzed the results; D.Z. prepared the 3′READS libraries; N.D.J. performed bioinformatic analyses pertaining to the 3′READS experiment; A.G.D. wrote the manuscript; J.M.E. edited the manuscript; X.-D.F., B.T., and G.W.Y. provided resources, imparted expertise, and critically reviewed the manuscript; and D.-E.Z. oversaw the study, supervised manuscript preparation, and secured funding to support the study.
Conflict-of-interest disclosure: G.W.Y. is a cofounder, a member of the Board of Directors, an equity holder, serves on the Scientific Advisory Boards, and is a paid consultant for Locana and Eclipse BioInnovations. G.W.Y. is also a member of the visiting faculty at the National University of Singapore; the terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. The remaining authors declare no competing financial interests.
Correspondence: Dong-Er Zhang, University of California, San Diego, 9500 Gilman Dr, Mail Stop 0815, La Jolla, CA 92037; e-mail: dez@ucsd.edu.
References
- 1.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64-69. [DOI] [PubMed] [Google Scholar]
- 2.Graubert TA, Shen D, Ding L, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2011;44(1):53-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ilagan JO, Ramakrishnan A, Hayes B, et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015;25(1):14-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim E, Ilagan JO, Liang Y, et al. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell. 2015;27(5):617-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vu LP, Pickering BF, Cheng Y, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.XuFeng R, Boyer MJ, Shen H, et al. ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proc Natl Acad Sci USA. 2009;106(42):17763-17768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park SM, Gönen M, Vu L, et al. Musashi2 sustains the mixed-lineage leukemia-driven stem cell regulatory program. J Clin Invest. 2015;125(3):1286-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Halsey C, Docherty M, McNeill M, et al. The GATA1s isoform is normally down-regulated during terminal haematopoietic differentiation and over-expression leads to failure to repress MYB, CCND2 and SKI during erythroid differentiation of K562 cells. J Hematol Oncol. 2012;5(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun L, Heerema N, Crotty L, et al. Expression of dominant-negative and mutant isoforms of the antileukemic transcription factor Ikaros in infant acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 1999;96(2):680-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsuzuki S, Hong D, Gupta R, Matsuo K, Seto M, Enver T. Isoform-specific potentiation of stem and progenitor cell engraftment by AML1/RUNX1. PLoS Med. 2007;4(5):e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calkhoven CF, Muller C, Martin R, et al. Translational control of SCL-isoform expression in hematopoietic lineage choice. Genes Dev. 2003;17(8):959-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sasaki K, Nakamura Y, Maki K, et al. Functional analysis of a dominant-negative DeltaETS TEL/ETV6 isoform. Biochem Biophys Res Commun. 2004;317(4):1128-1137. [DOI] [PubMed] [Google Scholar]
- 13.Miyoshi H, Ohira M, Shimizu K, et al. Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucleic Acids Res. 1995;23(14):2762-2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka T, Tanaka K, Ogawa S, et al. An acute myeloid leukemia gene, AML1, regulates hemopoietic myeloid cell differentiation and transcriptional activation antagonistically by two alternative spliced forms. EMBO J. 1995;14(2):341-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mevel R, Draper JE, Lie-A-Ling M, Kouskoff V, Lacaud G. RUNX transcription factors: orchestrators of development. Development. 2019;146(17):dev148296. [DOI] [PubMed] [Google Scholar]
- 16.Ghozi MC, Bernstein Y, Negreanu V, Levanon D, Groner Y. Expression of the human acute myeloid leukemia gene AML1 is regulated by two promoter regions. Proc Natl Acad Sci USA. 1996;93(5):1935-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lam K, Zhang DE. RUNX1 and RUNX1-ETO: roles in hematopoiesis and leukemogenesis. Front Biosci. 2012;17(1):1120-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci USA. 1991;88(23):10431-10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu TL, Goetz TL, Graves BJ, Speck NA. Auto-inhibition and partner proteins, core-binding factor beta (CBFbeta) and Ets-1, modulate DNA binding by CBFalpha2 (AML1). Mol Cell Biol. 2000;20(1):91-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Zhang Q, Zhang DE, et al. Overexpression of an isoform of AML1 in acute leukemia and its potential role in leukemogenesis. Leukemia. 2009;23(4):739-745. [DOI] [PubMed] [Google Scholar]
- 21.Guo H, Ma O, Speck NA, Friedman AD. Runx1 deletion or dominant inhibition reduces Cebpa transcription via conserved promoter and distal enhancer sites to favor monopoiesis over granulopoiesis. Blood. 2012;119(19):4408-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuzuki S, Seto M. Expansion of functionally defined mouse hematopoietic stem and progenitor cells by a short isoform of RUNX1/AML1. Blood. 2012;119(3):727-735. [DOI] [PubMed] [Google Scholar]
- 23.Ran D, Shia WJ, Lo MC, et al. RUNX1a enhances hematopoietic lineage commitment from human embryonic stem cells and inducible pluripotent stem cells. Blood. 2013;121(15):2882-2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Challen GA, Goodell MA. Runx1 isoforms show differential expression patterns during hematopoietic development but have similar functional effects in adult hematopoietic stem cells. Exp Hematol. 2010;38(5):403-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goyama S, Schibler J, Cunningham L, et al. Transcription factor RUNX1 promotes survival of acute myeloid leukemia cells. J Clin Invest. 2013;123(9):3876-3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komeno Y, Yan M, Matsuura S, et al. Runx1 exon 6-related alternative splicing isoforms differentially regulate hematopoiesis in mice. Blood. 2014;123(24):3760-3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng CE, Osato M, Tay BH, Venkatesh B, Ito Y. cDNA cloning of Runx family genes from the pufferfish (Fugu rubripes). Gene. 2007;399(2):162-173. [DOI] [PubMed] [Google Scholar]
- 28.Tsuji K, Noda M. Identification and expression of a novel 3′ dentification and expression of a novel 3 pufferf. Biochem Biophys Res Commun. 2000;271(1):171-176. [DOI] [PubMed] [Google Scholar]
- 29.Osato M. An unsung runt 6e isoform for HSC expansion. Blood. 2014;123(24):3684-3686. [DOI] [PubMed] [Google Scholar]
- 30.Sakurai H, Harada Y, Ogata Y, et al. Overexpression of RUNX1 short isoform has an important role in the development of myelodysplastic/myeloproliferative neoplasms. Blood Adv. 2017;1(18):1382-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119(6):831-845. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Wang Z. Systematical identification of splicing regulatory cis-elements and cognate trans-factors. Methods. 2014;65(3):350-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wheeler EC, Vu AQ, Einstein JM, et al. Pooled CRISPR screens with imaging on microraft arrays reveals stress granule-regulatory factors. Nat Methods. 2020;17(6):636-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li W, Köster J, Xu H, et al. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015;16(1):281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beaudoing E, Freier S, Wyatt JR, Claverie JM, Gautheret D. Patterns of variant polyadenylation signal usage in human genes. Genome Res. 2000;10(7):1001-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian B, Hu J, Zhang H, Lutz CS. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005;33(1):201-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takagaki Y, Seipelt RL, Peterson ML, Manley JL. The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell. 1996;87(5):941-952. [DOI] [PubMed] [Google Scholar]
- 38.Hoque M, Ji Z, Zheng D, et al. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat Methods. 2013;10(2):133-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Proudfoot NJ, Brownlee GG. 3′ non-coding region sequences in eukaryotic messenger RNA. Nature. 1976;263(5574):211-214. [DOI] [PubMed] [Google Scholar]
- 40.Keller W, Bienroth S, Lang KM, Christofori G. Cleavage and polyadenylation factor CPF specifically interacts with the pre-mRNA 3′ processing signal AAUAAA. EMBO J. 1991;10(13):4241-4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown KM, Gilmartin GM. A mechanism for the regulation of pre-mRNA 3′ processing by human cleavage factor Im. Mol Cell. 2003;12(6):1467-1476. [DOI] [PubMed] [Google Scholar]
- 42.MacDonald CC, Wilusz J, Shenk T. The 64-kilodalton subunit of the CstF polyadenylation factor binds to pre-mRNAs downstream of the cleavage site and influences cleavage site location. Mol Cell Biol. 1994;14(10):6647-6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pérez Cañadillas JM, Varani G. Recognition of GU-rich polyadenylation regulatory elements by human CstF-64 protein. EMBO J. 2003;22(11):2821-2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hall-Pogar T, Zhang H, Tian B, Lutz CS. Alternative polyadenylation of cyclooxygenase-2. Nucleic Acids Res. 2005;33(8):2565-2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14(7):496-506. [DOI] [PubMed] [Google Scholar]
- 46.Burd CG, Dreyfuss G. RNA binding specificity of hnRNP A1: significance of hnRNP A1 high-affinity binding sites in pre-mRNA splicing. EMBO J. 1994;13(5):1197-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huelga SC, Vu AQ, Arnold JD, et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012;1(2):167-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bruun GH, Doktor TK, Borch-Jensen J, et al. Global identification of hnRNP A1 binding sites for SSO-based splicing modulation. BMC Biol. 2016;14(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morgan CE, Meagher JL, Levengood JD, et al. The first crystal structure of the UP1 domain of hnRNP A1 bound to RNA reveals a new look for an old RNA binding protein. J Mol Biol. 2015;427(20):3241-3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rollins C, Levengood JD, Rife BD, Salemi M, Tolbert BS. Thermodynamic and phylogenetic insights into hnRNP A1 recognition of the HIV-1 exon splicing silencer 3 element. Biochemistry. 2014;53(13):2172-2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jean-Philippe J, Paz S, Caputi M. hnRNP A1: the Swiss army knife of gene expression. Int J Mol Sci. 2013;14(9):18999-19024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bagger FO, Kinalis S, Rapin N. BloodSpot: a database of healthy and malignant haematopoiesis updated with purified and single cell mRNA sequencing profiles. Nucleic Acids Res. 2019;47(D1):D881-D885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song L, Lin HS, Gong JN, et al. microRNA-451-modulated hnRNP A1 takes a part in granulocytic differentiation regulation and acute myeloid leukemia. Oncotarget. 2017;8(33):55453-55466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iervolino A, Santilli G, Trotta R, et al. hnRNP A1 nucleocytoplasmic shuttling activity is required for normal myelopoiesis and BCR/ABL leukemogenesis. Mol Cell Biol. 2002;22(7):2255-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eiring AM, Neviani P, Santhanam R, et al. Identification of novel posttranscriptional targets of the BCR/ABL oncoprotein by ribonomics: requirement of E2F3 for BCR/ABL leukemogenesis. Blood. 2008;111(2):816-828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fang J, Bolanos LC, Choi K, et al. Ubiquitination of hnRNPA1 by TRAF6 links chronic innate immune signaling with myelodysplasia [published correction appears in Nat Immunol. 2017;18(4):474]. Nat Immunol. 2017;18(2):236-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vernet C, Artzt K. STAR, a gene family involved in signal transduction and activation of RNA. Trends Genet. 1997;13(12):479-484. [DOI] [PubMed] [Google Scholar]
- 58.Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol. 2007;176(7):929-939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pedrotti S, Bielli P, Paronetto MP, et al. The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J. 2010;29(7):1235-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh R, Valcárcel J. Building specificity with nonspecific RNA-binding proteins [published correction appears in Nat Struct Mol Biol. 2005;12(9):824]. Nat Struct Mol Biol. 2005;12(8):645-653. [DOI] [PubMed] [Google Scholar]
- 61.La Rosa P, Bielli P, Compagnucci C, et al. Sam68 promotes self-renewal and glycolytic metabolism in mouse neural progenitor cells by modulating Aldh1a3 pre-mRNA 3′-end processing. eLife. 2016;5:e20750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frisone P, Pradella D, Di Matteo A, Belloni E, Ghigna C, Paronetto MP. SAM68: signal transduction and RNA metabolism in human cancer. BioMed Res Int. 2015;2015:528954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bielli P, Busà R, Paronetto MP, Sette C. The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocr Relat Cancer. 2011;18(4):R91-R102. [DOI] [PubMed] [Google Scholar]
- 64.Cheung N, Chan LC, Thompson A, Cleary ML, So CW. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol. 2007;9(10):1208-1215. [DOI] [PubMed] [Google Scholar]
- 65.Wang Q, Li Y, Cheng J, et al. Sam68 affects cell proliferation and apoptosis of human adult T-acute lymphoblastic leukemia cells via AKT/mTOR signal pathway. Leuk Res. 2016;46:1-9. [DOI] [PubMed] [Google Scholar]
- 66.Benoit YD, Mitchell RR, Risueño RM, et al. Sam68 allows selective targeting of human cancer stem cells. Cell Chem Biol. 2017;24(7):833-844.e9. [DOI] [PubMed] [Google Scholar]
- 67.Ji Z, Lee JY, Pan Z, Jiang B, Tian B. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development [published correction appears in Proc Natl Acad Sci USA. 2009;106(23):9535]. Proc Natl Acad Sci USA. 2009;106(17):7028-7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hilgers V, Perry MW, Hendrix D, Stark A, Levine M, Haley B. Neural-specific elongation of 3′ UTRs during Drosophila development. Proc Natl Acad Sci USA. 2011;108(38):15864-15869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shepard PJ, Choi EA, Lu J, Flanagan LA, Hertel KJ, Shi Y. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA. 2011;17(4):761-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ulitsky I, Shkumatava A, Jan CH, et al. Extensive alternative polyadenylation during zebrafish development. Genome Res. 2012;22(10):2054-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138(4):673-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xia Z, Donehower LA, Cooper TA, et al. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat Commun. 2014;5(1):5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Singh P, Alley TL, Wright SM, et al. Global changes in processing of mRNA 3′ untranslated regions characterize clinically distinct cancer subtypes. Cancer Res. 2009;69(24):9422-9430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Naro C, Pellegrini L, Jolly A, et al. Functional Interaction between U1snRNP and Sam68 insures proper 3′ end pre-mRNA processing during germ cell differentiation. Cell Rep. 2019;26(11):2929-2941.e5. [DOI] [PubMed] [Google Scholar]
- 75.Iijima Y, Tanaka M, Suzuki S, et al. SAM68-specific splicing is required for proper selection of alternative 3′ UTR isoforms in the nervous system. iScience. 2019;22:318-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.