

Abstract
A diverse series of hemozoin-inhibiting quinolines, benzamides, triarylimidazoles, quinazolines, benzimidazoles, benzoxazoles and benzothiazoles have been found to lead to exchangeable heme in cultured Plasmodium falciparum (NF54) that ranged over an order of magnitude at the IC50. Surprisingly, less active compounds often exhibited higher levels of exchangeable heme than more active ones. Quantities of intracellular inhibitor measured using the inoculum effect exhibited a linear correlation with exchangeable heme, suggesting formation of heme-inhibitor complexes in the parasite. In an effort to confirm this, the presence of a Br atom in one of the benzimidazole derivatives was exploited to image its distribution in the parasite using electron spectroscopic imaging of Br, an element not naturally abundant in cells. This showed that the compound colocalized with iron, consistent with its presence as a heme complex. Direct evidence for this complex was then obtained using confocal Raman microscopy. Exchangeable heme and inhibitor were found to increase with decreased rate of killing, suggesting that slow-acting compounds have more time to build up exchangeable heme complexes. Lastly, some, but not all compounds evidently cause pro-oxidant effects since their activity could be attenuated with N-acetylcysteine and potentiated with t-butyl hydroperoxide. Collectively, these findings suggest that hemozoin inhibitors act as complexes with free heme, each with its own unique activity.
Keywords: hemozoin, antimalarial, electron spectroscopic imaging, Raman microscopy, ROS
Graphical Abstract
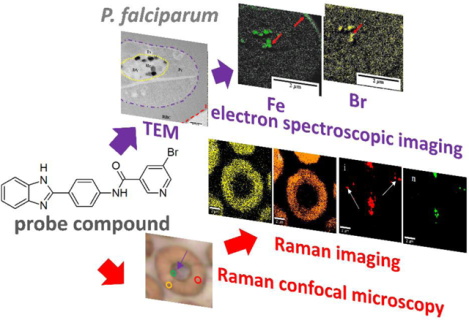
The graphic illustrates the use of a probe compound to show colocalization of Fe and the probe compound Br in Plasmodium falciparum using electron microscopy and identification of various compounds in the parasite by confocal Raman microscopy
Hemozoin microcrystals are a striking feature in the digestive vacuole (DV) of malaria parasites. They are formed following digestion of host hemoglobin, sequestering ≈95% of the released heme in a non-toxic insoluble form.1 Inhibition of hemozoin formation has been implicated in the mechanism of action of chloroquine as well as amodiaquine and piperaquine, currently used in artemisinin combination therapy (ACT).2–6 Studies have shown that chloroquine decreases formation of hemozoin, increases exchangeable heme, disrupts the hemozoin crystal lattice and redistributes heme into the parasite cytoplasm. The dose-dependent increase in exchangeable heme coincides with decreased parasite survival.7 Similar behavior has been shown with other clinical and experimental hemozoin inhibiting compounds.6, 8–15
It has been hypothesized that inhibition of hemozoin formation causes a build-up of heme, killing the parasite via pro-oxidant activity, membrane disruption or osmotic pressure.16–19 Surprisingly, levels of exchangeable heme (all heme other than undigested hemoglobin and hemozoin) at the IC50 values of a range of hemozoin inhibitors are remarkably variable.6, 8–15 To better understand this mechanism, we have investigated a diverse range of hemozoin inhibiting scaffolds (Figure 1).
Figure 1.
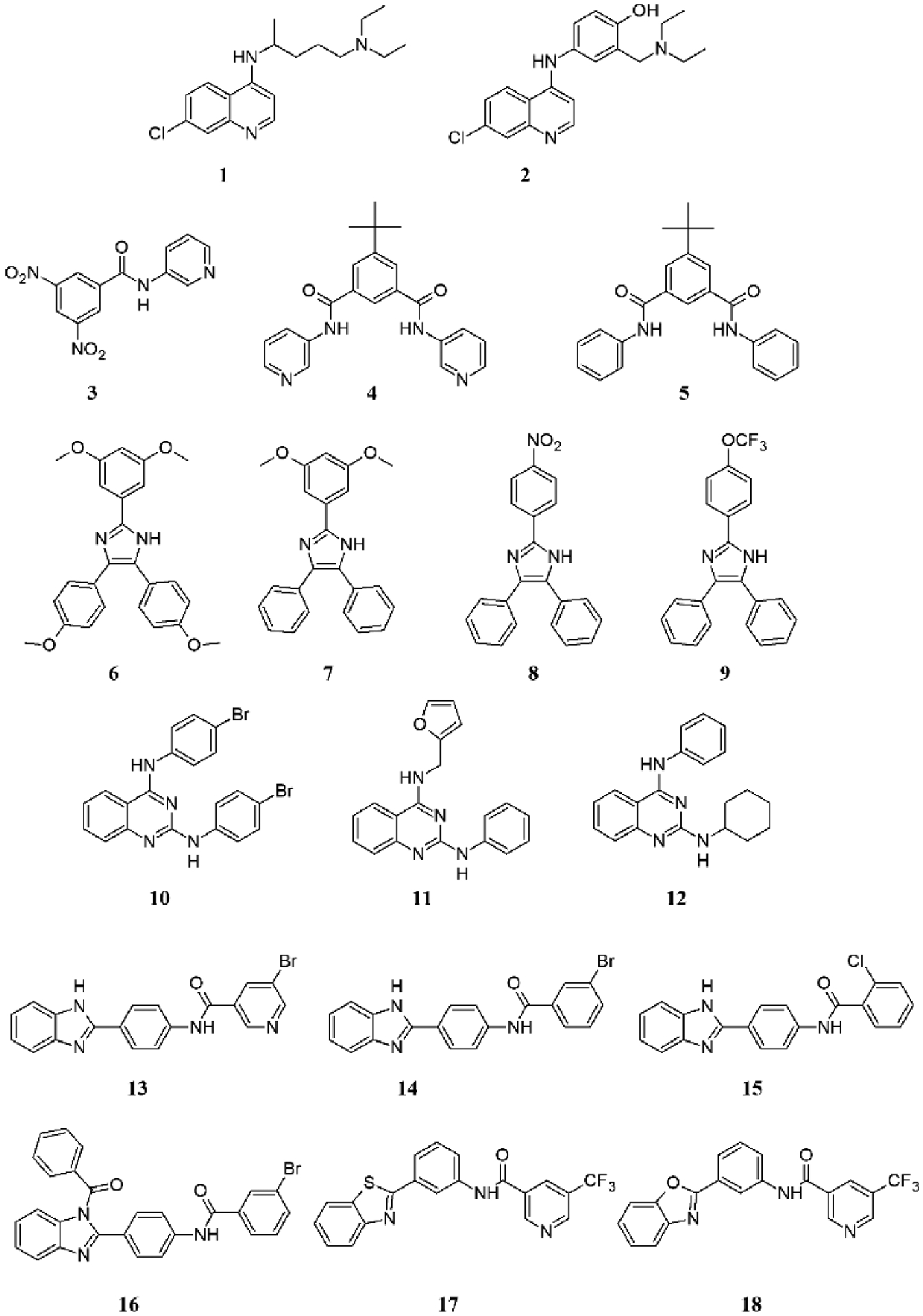
Compounds investigated in this study falling into seven scaffolds: quinolines (chloroquine 1 and amodiaquine 2), benzamides (3 – 5), triarylimidazoles (6 – 9), quinazolines (10 – 12), benzimidazoles (13 – 16), benzothiazole (17) and benzoxazole (18).
Here we show that the amounts of intracellular exchangeable heme and intracellular test compound coincide. Using electron spectroscopic imaging, we demonstrate that a derivative bearing a Br atom (compound 13) co-localizes with iron, suggesting the presence of a heme complex in the DV, a finding strongly supported by confocal Raman microscopy. Higher levels of exchangeable heme are associated with a decreased rate of killing, while heme-related pro-oxidant activity is involved in the antiparasitic activity of some but not all compounds. Based on these findings, we propose a comprehensive model for the antimalarial mechanism of action of hemozoin inhibitors.
Results
Chloroquine and amodiaquine are commercially available, while the syntheses of compounds 3 – 9 and 13 – 16 have been previously reported.9–11 The quinazolines 10 – 12 and benzazoles 17 and 18 have not previously been reported. Compounds 3 – 18 are all based on scaffolds previously discovered in a high throughput screening campaign for β-hematin inhibitors.20 The quinazolines were synthesized in three steps from benzoylene urea (Scheme 1a). This involved refluxing with a large excess of POCl3 to produce 2,4-dichloroquinazoline, followed by a regioselective SNAr reaction with an appropriate amine to produce a 2-chloroquinazoline derivative followed by a second SNAr reaction to yield the final products. The benzazoles were also prepared in three steps, this time starting with an SNAc reaction between 2-aminophenol or 2-aminothiophenol and 3-aminobenzoic acid to produce an azole intermediate which was then reacted with an acyl halide to afford the product (Scheme 1b). The purity of all final products was confirmed to be >95% by HPLC.
Scheme 1.
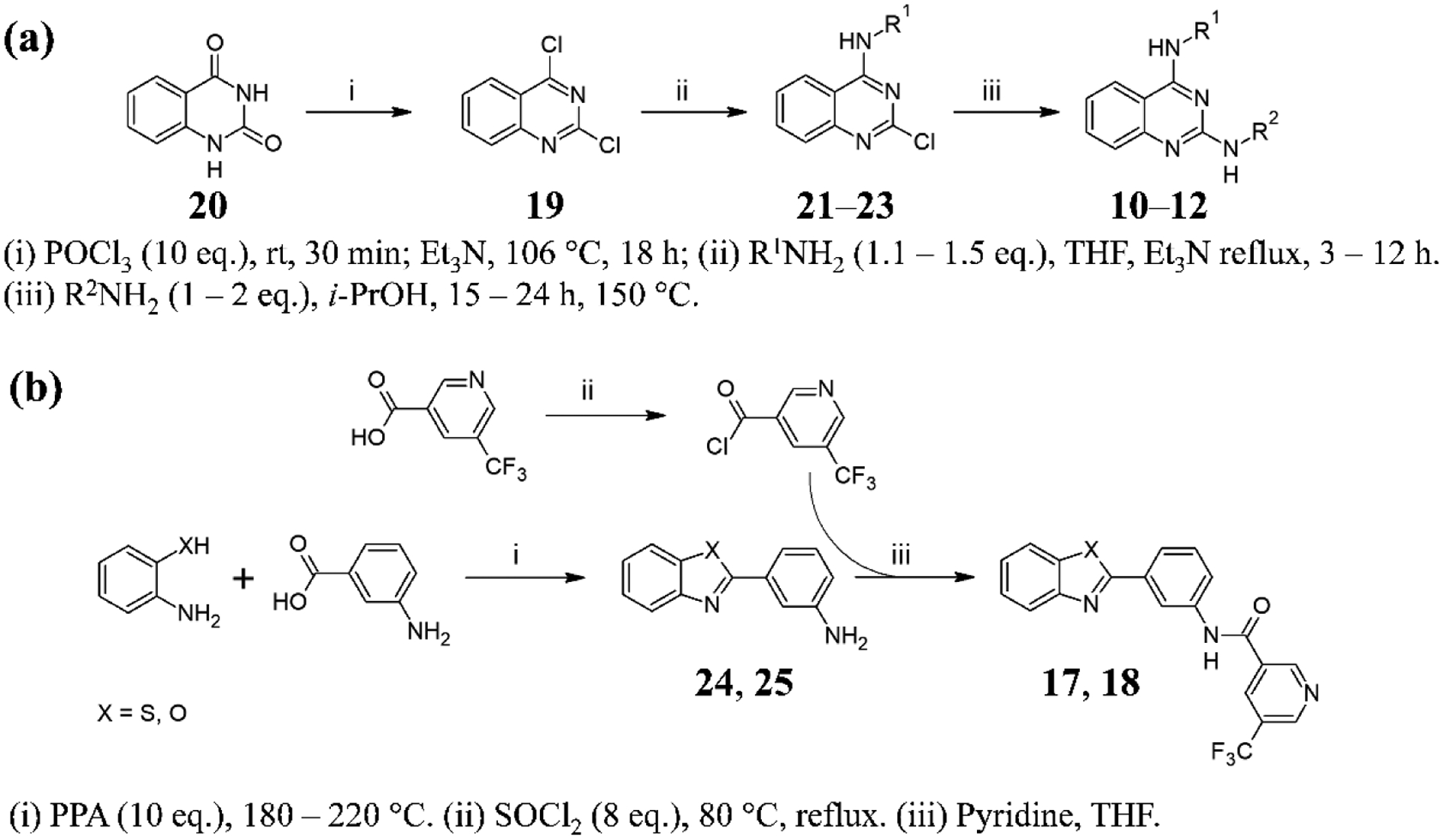
Synthesis of quinazolines (a) and benzoxazoles and benzothiazoles (b).
The simplest explanation for the activity of hemozoin inhibitors is that they cause a build-up of free heme which directly kills the parasite. Exchangeable heme levels should then be the same at the parasite growth IC50 of each compound. While the IC50 values of different compounds could vary because of the different extents of accumulation in the DV through pH trapping and different hemozoin inhibiting strengths, the toxicity of heme to the parasite should remain constant. In previous studies, we have shown that the level of exchangeable heme at the IC50 varies over several fold,6, 8–14 making this hypothesis untenable. It is tempting to suggest that these compounds act on a target other than heme, with adventitious inhibition of hemozoin formation causing a build-up of exchangeable heme with no role in parasite killing. This argument is also untenable, however, since no match would then be expected between the dose-dependent increase in exchangeable heme and decrease in parasite survival for a range of compounds. For example, a compound that inhibited hemozoin formation more strongly than its drug target would build up exchangeable heme levels at doses below the IC50, while one that inhibited the drug target more strongly than hemozoin formation would only exhibit rising exchangeable heme levels at doses well above the IC50. This is not what we have previously observed.8–10 Rather, the decrease in survival is closely linked to the increase in exchangeable heme. This seems to leave one alternative; that inhibition of hemozoin formation causes a build-up of exchangeable heme in the form of a unique complex, with each complex having its own specific level of activity. The implication is that heme complexes with drugs or test compounds should be observable in the parasite with variable levels of heme at the IC50 of each complex.
The β-hematin and parasite growth inhibition IC50 values for all compounds are presented in Table S1. We have reported most of these previously, but quinazolines 10 – 12, benzothiazole 17 and benzoxazole 18 are new compounds. While we have reported levels of exchangeable heme upon treatment with test compounds previously for compounds 3 – 9 and 13,9–11 we have now also measured these for the remaining ten compounds. There is a wide range of exchangeable heme levels at the respective IC50 of each compound with no definite trend relating exchangeable heme and parasite IC50. A counterintuitive tendency for higher exchangeable heme levels to be associated with higher IC50 is evident, especially if chloroquine, amodiaquine and compounds 6 and 15 are disregarded (Figure 2a). Not only do these results confirm recent observations of variable levels of exchangeable heme at the IC50, but they also illustrate a tendency for higher levels to be associated with weaker activity. This surprising result further invalidates the hypothesis that exchangeable heme alone is responsible for parasite death.
Figure 2.
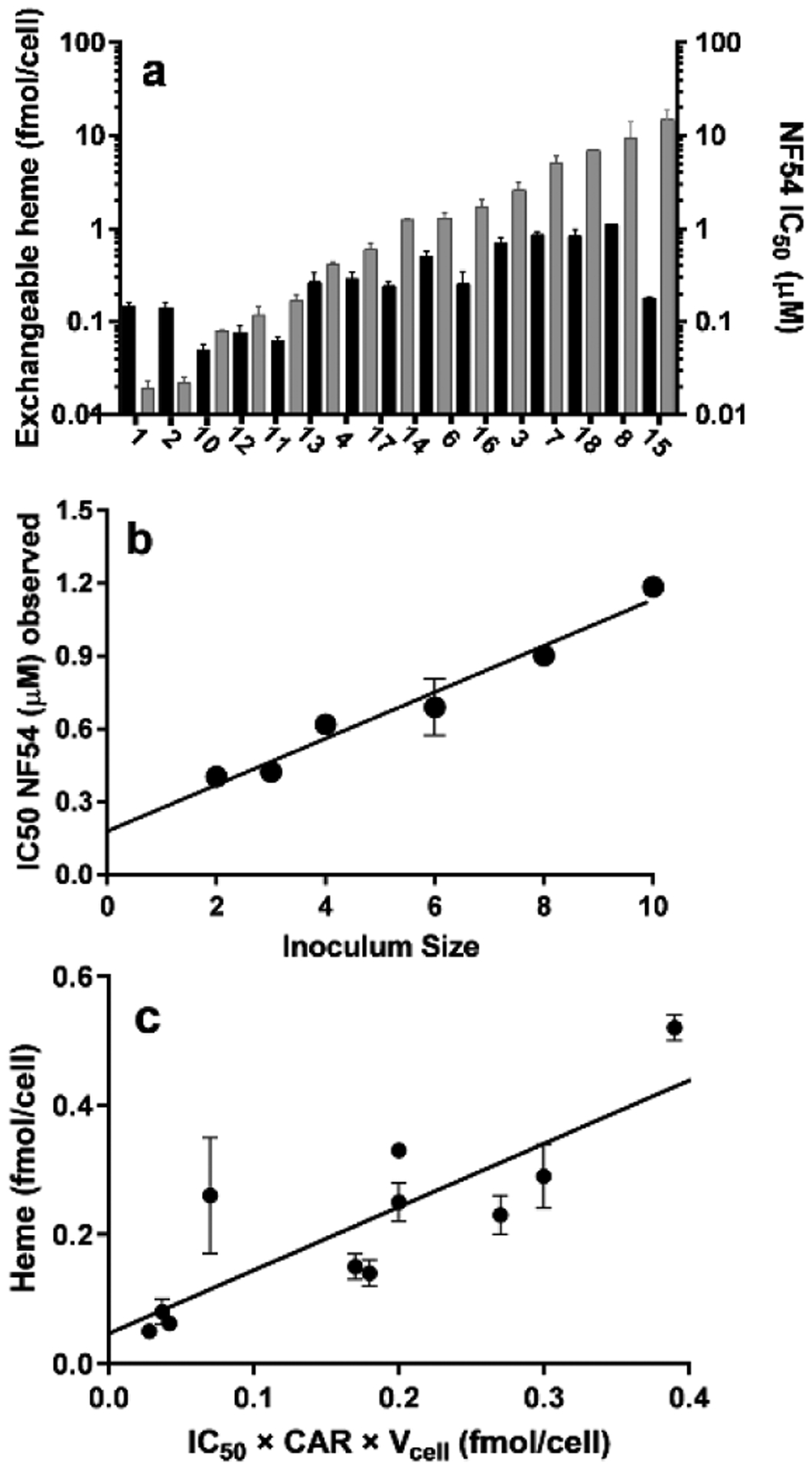
Comparison of levels of exchangeable heme, parasite growth inhibition and compound accumulation in NF54 P. falciparum. (a) Exchangeable heme levels at the IC50 (black bars, left axis) and the corresponding IC50 values of each compound (grey bars, right axis) showing no direct correlation. Compounds are arranged in order of increasing IC50. (b) Inoculum effect for compound 13, r2 = 0.95. (c) A plot of exchangeable heme versus amount of intracellular test compound at the IC50. Slope 1.0 ± 0.2, intercept 0.05 ± 0.04, r2 = 0.70, P=0.0013, P value (runs test) = 0.61.
Next, we measured amounts of selected active compounds accumulated in the parasite to establish whether they stoichiometrically correlate with levels of exchangeable heme as might be expected if they act as complexes. To this end, we measured the accumulation of eleven of the active compounds in the parasite by using the inoculum effect (Figure 2b and Table S2). These consisted of all compounds with parasite growth IC50 values ≤ 2 μM. A direct 1:1 correlation was found between amounts of exchangeable heme and active compound in NF54 parasites (Figure 2c), with a linear correlation exhibiting a slope of 1 and intercept of 0. Runs tests of the data showed no deviation from linearity.
The statistical correlation between the amount of test compound accumulated in the parasite and exchangeable heme over seven scaffolds with a wide range of inhibitory activities suggests they are present as complexes. Several compounds (10, 13, 14, 16 and 17) contain either F or Br atoms that do not occur naturally at high concentrations in cells and can be exploited as labels for electron spectroscopic imaging (ESI) based on electron energy loss spectroscopy (EELS). The EELS K edge of F is close to the L2 and L3 edges of Fe making it unsuitable for elemental mapping in this Fe rich system,21 ruling out mapping of compound 17. Compound 10 was unsuitable because exchangeable heme at the IC50 represents only a very small fraction of the total heme in the parasite. This left compounds 13, 14 and 16. Compound 13 was finally chosen for further investigation because it was the most active against the parasite and produced fairly high levels of exchangeable heme, making up ≈25% of total heme iron at the IC50. In untreated trophozoites, the distribution of Fe was readily observed using ESI and was almost exclusively confined to hemozoin (Figure 3a and b). At the Br edge, no signal was observed (Figure 3c), confirming no significant overlap in the EELS of Fe and Br and no measurable Br in the untreated cell. When parasites were treated with 13, ESI images of the cryofixed parasitized red blood cells at the Br edge readily permitted the distribution of the compound to be visualized (Figure 3f). Comparison with the Fe distribution (Figure 3d) showed almost perfect co-localization of Fe and high concentration regions of Br (Figure 3e). This region of co-localization coincided with hemozoin and in view of the lack of any other strong Fe signals, also the region occupied by most of the exchangeable heme. This co-localization supports the hypothesis of a complex between exchangeable heme and compound 13 and likely association of the compound with hemozoin as well.
Figure 3.

Distribition of Fe and Br in untreated P. falciparum infected red blood cells and cells treated with compound 13. (a) Electron spectroscopic imaging using EELS showing distribution of Fe (green) and (c) Br in an untreated parasite with TEM image in (b). A parasite treated with 13 is shown in (d) to (f) with the Fe map in (d), Br map (yellow) in (f) and manual overlay in (e).
A confocal Raman microscope with 532 nm laser was used to further investigate parasites treated with 13. Figure 4a shows average Raman spectra obtained from various parts of an infected RBC treated with compound 13 that was obtained using the built-in true component analysis tool in Witec Suite FIVE software from large area scan spectra obtained separately from 10 – 15 cells. Extracted spectra were averaged and produced distinct spectra of oxyhemoglobin, deoxyhemoglobin and hemozoin following demixing. An additional spectrum obtained near the hemozoin and not seen in untreated parasites was flagged as a possible complex between heme and compound 13. This spectrum differed from other heme species, including hematin and hemin and from compound 13 alone, but resembled a synthetically prepared complex of hematin and compound 13 (Figure 4b). This visual similarity was confirmed using principal components analysis (PCA). For this purpose, second derivatives of Raman spectra normalized to the strongest peak were used and only second derivative peaks lying outside 95th percentile of the mean were considered. For each representative part of the infected RBC, 150 such spectra were recorded from multiple cells and the dimensionality of these data reduced to two using PCA which clustered similar spectra (Figure 5a and b). As a statistical measure of separation of these clusters, we computed the centroid of each cluster and projected the data onto a line coincident with the vector joining the cluster centroids and applied a Welch’s t-test. This showed that the spectrum of the putative heme-13 complex was statistically distinct from all other heme species (oxy- and deoxyhemoglobin, hemozoin, hemin, hematin and β-hematin) as well as from the spectrum of pure compound 13 (P < 0.0001) but was statistically indistinguishable from the synthetically prepared complex (P = 0.8043, Table S3). A unique peak was observed in the synthetic complex at 1080 cm−1 as well as in the putative complex in the cell. This likely involves a shift in the δas(=CH2) vinyl mode in the porphyrin which occurs at 1090 cm−1 in hemozoin and oxyhemoglobin and at 1099 cm−1 in deoxyhemoglobin. This peak was subsequently used for Raman imaging of the complex in the cell.
Figure 4.
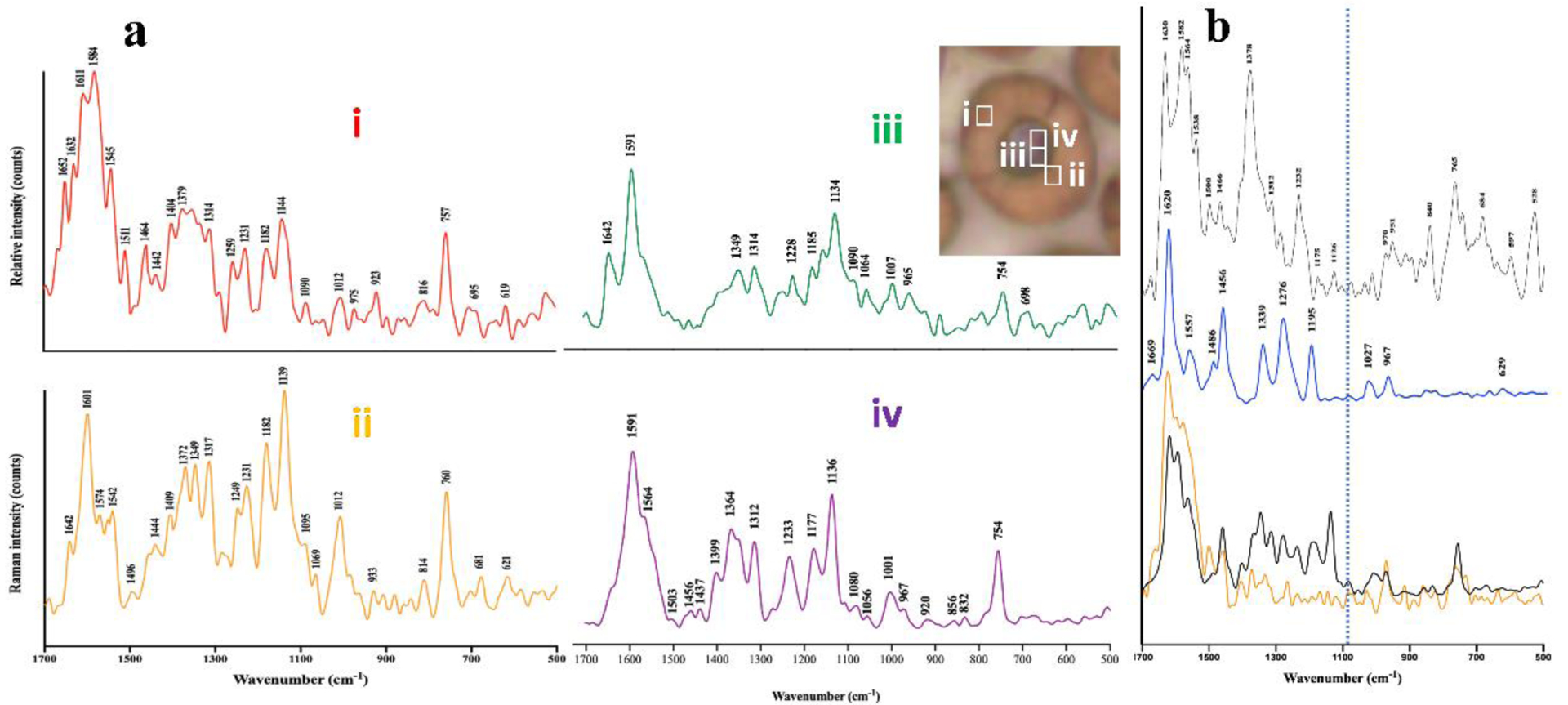
Raman spectra obtained from infected RBCs treated with 13 and compared with pure hemin, compound 13 and a complex of 13 with hemin in 40% aqueous DMSO in the presence of a 9-fold excess of compound 13. (a) Typical sampling sections where individual species were dominant are illustrated in the inset by 1 μm2 boxes which are ≈ 5× the lateral resolution of the instrument. Spectra represent (i) oxyhemoglobin; (ii) deoxyhemoglobin; (iii) hemozoin and (iv) putative heme-13 complex. (b) Raman spectra of hemin (top), 13 (middle), 10:1 mixture of hemin and compound 13 in 40% DMSO (bottom in orange) and spectrum iv from (a) added to the spectrum of compound 13 at a ratio of 9:1 (bottom in black). The vertical dotted line marks 1080 cm−1, where a unique peak occurs in the complex. Peak positions in the bottom spectra are very similar. Differences in relative intensities may reflect the fact that the synthetic complex was recorded in a precipitate obtained from an aqueous DMSO solution, whereas the parasite species represents a solid formed in the cell which may exhibit different heme-heme interactions.
Figure 5.
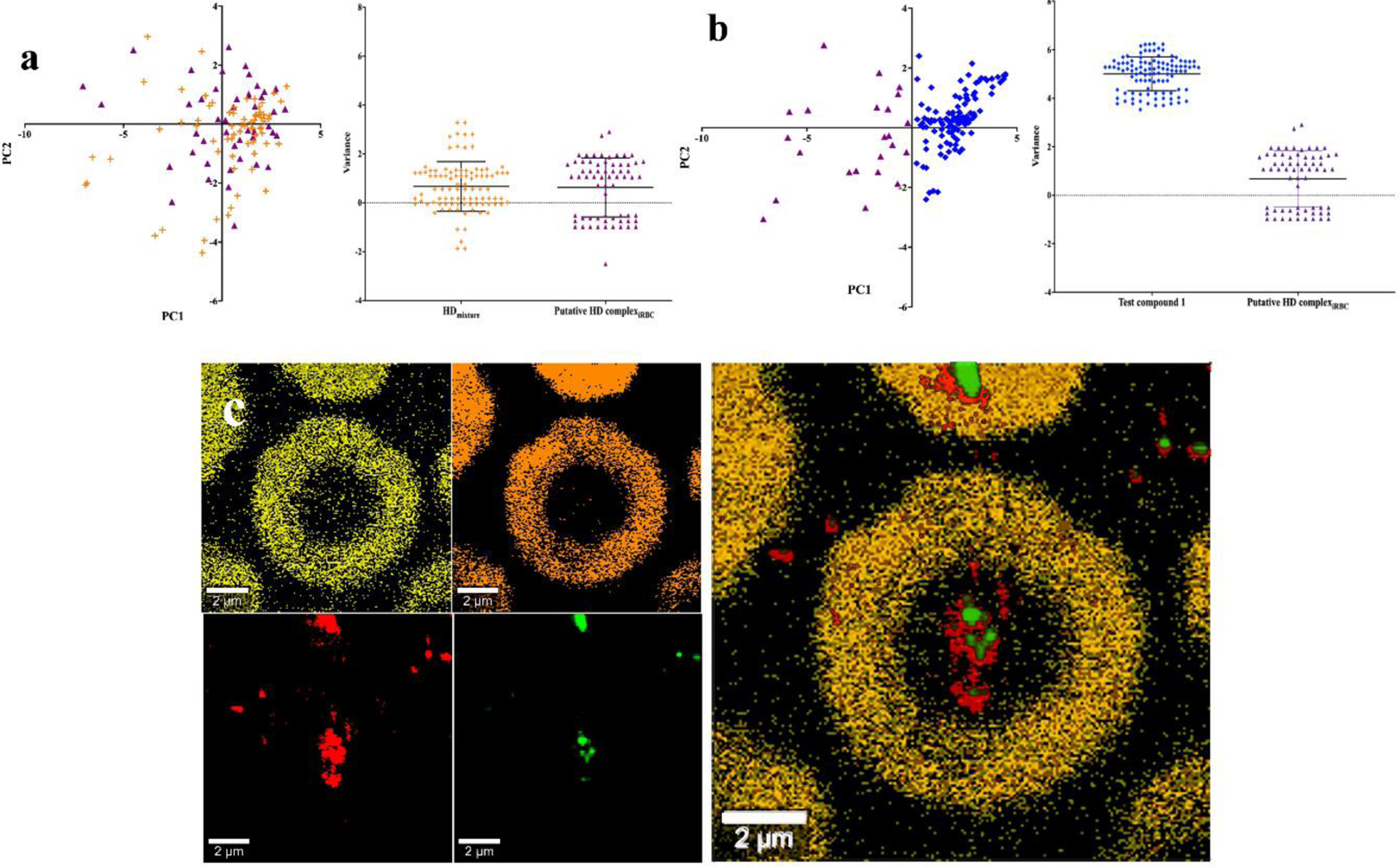
PCA analysis of Raman spectra and Raman imaging of P. falciparum infected RBCs treated with compound 13. (a) PCA scores plot of a solution of hemin with excess compound 13 (orange crosses) and spectra obtained from the parasite (purple triangles) used to constitute the average shown in Figure 5a spectrum iv (left panel) and scatter plots along the line coinciding with the vector joining the centroids of the two series in the PCA plot (right panel). (b) PCA scores plot for compound 13 alone (blue diamonds) and spectra obtained from the parasite (purple triangles, left panel) and scatter plots along the line coinciding with the vector joining the centroids of the two series in the PCA plot (right panel). (c) Raman imaging of a parasitized RBC obtained using unmixed spectra based on the average spectra as shown in Figure 5a as components with application of a filter tool: oxyhemoglobin (yellow), deoxyhemoglobin (orange), hemozoin (red) and heme-13 complex (green) imaged at 1090, 1642, 754 and 1080 cm−1 respectively (left and central panels) and an overlay of images (right panel).
Raman imaging was used to establish the distribution of the complex in the cell (Figure 5c). A spectral unmixing protocol like that described above used to collect single scan spectra was applied, but the single scan spectra previously identified were used as reference spectra. Use of a filter viewer tool then improved the quality of the images by reducing background noise. This procedure produced an image at 1080 cm−1 using the spectrum of the complex as basis spectrum that was totally absent in the untreated sample and in images based on any of the other component spectra. This image could thus be ascribed to the complex alone and was in close agreement with the evidence from the EELS data, showing that the complex co-localized with hemozoin (See also Figure S1 and S2).
The evidence for heme-inhibitor complexes in the parasite presented above supports the hypothesis that such complexes are responsible for parasite killing. One explanation for the variability in exchangeable heme relates to speed of killing. A heme-inhibitor complex that kills the parasite slowly or later in the lifecycle would permit more hemoglobin digestion, resulting in more heme available to form a complex with the inhibitor than if it kills early in the lifecycle. To explore this, we inoculated ring-stage parasitized RBCs with chloroquine, amodiaquine and compounds 3, 6, 13, and 18, with the test compound washed out after 4, 8, 12, 24 or 48 h. Parasite growth was measured at 72 h post-inoculation using flow cytometry with SYBR Green I staining (Figure S3). It was evident that some compounds kill parasites much earlier in the lifecycle than others (Figure 6a), with an overall increase in exchangeable heme as the rate of killing decreases as measured from the ratio of IC50 with 24 h washout to that without washout, both measured at 72 h (Figure 6b).
Figure 6.
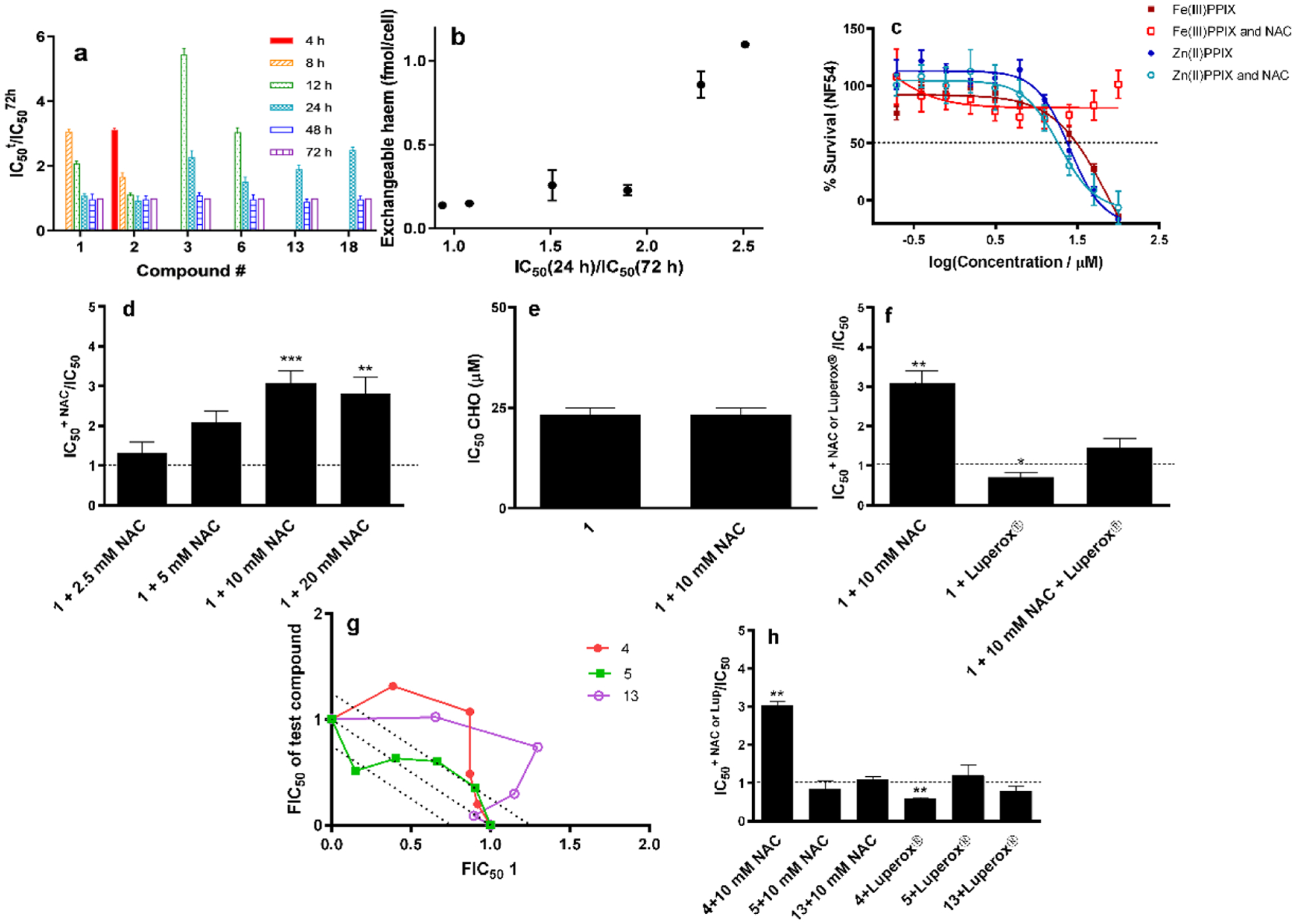
Speed and basis of killing of selected compounds. (a) IC50 values for test compounds washed out at various times (IC50t) relative to that with no washout (IC5072h). Where no bar is shown, there was no inhibition of parasite growth after that length of exposure. (b) Trend of increasing exchangeable heme levels as a function of prolonged duration of killing. (c) Effects of externally introduced Fe(III)PPIX and Zn(II)PPIX on parasite survival with and without NAC. (d) Effects of varying concentration of NAC on the relative IC50 of chloroquine (1). (e) Lack of effect of NAC on toxicity of chloroquine (1) in CHO cells. (f) Protective effect of NAC and potentiating effect of Luperox® on parasites in the presence of chloroquine (1). NAC was found to abrogate the effect of Luperox®. (g) Isobolograms showing strong antagonism between compound 4 & chloroquine (1) and compound 13 & chloroquine (1) but an additive effect between the non-hemozoin inhibitor 5 & chloroquine (1). Dotted line represents one standard deviation about the line. (h) Protective effect of NAC on the effects of compound 4 and potentiating effect of Luperox®, but lack of effect of either NAC or Luperox® on the activities of compounds 5 and 13. Statistical significance, calculated using a two-tailed t-test (error bars showing 95% CI) is expressed using asterisks. * P < 0.05, ** P < 0.01, *** P < 0.001.
Finally, we investigated the basis of inhibitor toxicity to the parasite, of several inhibitors. It has long been known that hemin is toxic to parasites,16 probably arising from pro-oxidant effects of the Fe center of hemin.17 To investigate this, we first incubated parasitized RBCs with various doses of hemin, determining an IC50 of 31 ± 7 μM (Figure 6c). The reductant and glutathione precursor N-acetylcysteine (NAC) at 10 mM, a concentration that did not interfere with the assay, protected the parasites from hemin up to 100 μM, the highest concentration that could be tested. This is consistent with a pro-oxidant effect of hemin. Interestingly, zinc(II) protoporphyrin IX (Zn(II)PPIX) also inhibited parasite growth, but consistent with the redox inactivity of Zn(II) was unaffected by 10 mM NAC, showing that this metalloporphyrin acted by a different mechanism (Figure 6c). The IC50 of Zn(II)PPIX was slightly lower than hemin (24 ± 4 μM) and we speculate that this may involve deactivation of crucial hemoproteins in the parasite by replacement of heme in their active sites with catalytically inactive Zn(II)PPIX.
Chloroquine is the most studied hemozoin inhibitor, so we used it as a benchmark for studying oxidative effects. Consistent with a prooxidant effect of chloroquine, the presence of 10 mM NAC caused a significant increase in its IC50. This increase was dependent on the concentration of NAC up to 10 mM, but above this concentration, levelled off, probably because higher concentrations of NAC started to interfere with the parasite survival assay (Figure 6d). In the case of mammalian (CHO) cells, where chloroquine is far less toxic, NAC had no effect at 10 mM (Figure 6e). This shows that chloroquine alone has no evident pro-oxidant effects and that it is the presence of heme that is responsible for the effect in the parasite. To further, explore whether chloroquine has an oxidative mechanism of action in the parasite, we introduced t-butyl hydroperoxide (Luperox®) at a dose found to have no effect on parasite growth (50 μM). This significantly potentiated the activity of chloroquine. This pro-oxidant effect of Luperox® could be completely abrogated by 10 mM NAC (Figure 6f). These findings are consistent with the hypothesis that chloroquine kills the parasite through the pro-oxidant effects of heme.
For comparison with this benchmark compound, we constructed isobolograms of chloroquine in combination with two hemozoin inhibitors (4 and 13) and a close analogue of 4 (compound 5) that is not a β-hematin inhibitor and was also found not to inhibit parasite hemozoin formation but displayed moderate growth inhibition activity. For the first two combinations, the isobolograms were strongly antagonistic, consistent with compounds 4 and 13 competing with chloroquine for intracellular heme, while for the last combination, the relationship was merely additive consistent with independent targets (Figure 6g). Finally, we investigated the effects of NAC and Luperox® on compounds 4, 5 and 13 (Figure 6h). As expected, the parasite was protected from compound 4 by NAC, while Luperox® potentiated its effect. There was no effect of either NAC or Luperox® on compound 5, consistent with a mechanism not involving hemozoin inhibition. Unexpectedly, compound 13 was also unaffected by either NAC or Luperox® despite the antagonistic relationship with chloroquine and the strong evidence of complex formation with heme presented earlier. This shows that not all heme-inhibitor complexes act in the same way. Since heme-13 apparently does not kill the parasite via prooxidant effects, it must kill it in some other way, possibly involving inhibition of a protein target in the DV.
Discussion
From the data collated in Figure 2a, it is evident that the amount of exchangeable heme at the parasite growth inhibition IC50 values across the diverse scaffolds investigated in this study is not constant. Rather, it covers an order of magnitude range from 0.05 to 0.52 fmol/cell (Table S2). This implies that free heme on its own cannot be responsible for inhibition of parasite growth in all cases, invalidating the supposition that all hemozoin-inhibiting antimalarials kill the parasite merely by causing a build-up of free heme alone.
The linear relationship between the amount of exchangeable heme and amount of accumulated active compound at the parasite inhibition IC50 shown in Figure 2c strongly suggests that the exchangeable heme is present as a complex with the inhibitor in each case. There has been only one previous report of direct spectroscopic evidence for a complex between chloroquine and heme in malaria parasites based on photoacoustic spectroscopy.22 While Raman microscopy has provided evidence of effects on the spectra of parasitized RBCs and the heme species therein,23 changes in heme spectra are quite subtle and direct evidence for a complex has proven elusive. It is clear from Figure 2a and c that the amount of complex present in the case of chloroquine is low (Table S2), making its detection in the presence of a large hemozoin background extremely challenging. An advantage of using a wide range of scaffolds was the presence of compounds that produce substantial quantities of heme complex and which contain substituents that could be exploited for imaging. Taking advantage of the Br atom in compound 13, it was possible to demonstrate co-localization of this compound with Fe, strongly indicating the presence of a complex with heme. That this is in fact a complex between compound 13 and heme was demonstrated by Raman spectroscopy. This observation supports the contention that the direct linear relationship between exchangeable heme and inhibitor amount in the parasite is a result of complex formation. It is therefore likely that hemozoin inhibitors either act as complexes or that their activity is strongly influenced by complex formation. Interestingly, a very recent soft X-ray fluorescence imaging study of P. falciparum treated with bromoquine, an analogue of chloroquine with a 7-Br, rather than 7-Cl substituent has shown that this compound also co-localizes with hemozoin, but is found in the DV membrane as well, which the authors interpreted as evidence of its presence as a heme-bromoquine complex.24
The level of exchangeable heme attained with different compounds can evidently be traced to the speed of killing, with slow killing compounds leading to high levels of exchangeable heme and fast killing compounds resulting in only low levels of exchangeable heme. A possible explanation for this is that slow acting complexes allow the parasite to grow further into the trophozoite stage relative to fast acting compounds, with consequent increased digestion of hemoglobin, resulting in more heme being available to bind to the test compound. This would account for the trend seen in Figure 6b.
Several authors have suggested that the inhibition of hemozoin formation by chloroquine and other quinolines results in increased reactive oxygen species (ROS) owing to the pro-oxidant effect of free heme.17, 25–26 This hypothesis is consistent with the observation that the activities of chloroquine and compound 4 are significantly reduced with the use of NAC which is expected to raise the concentration of cytosolic glutathione protecting the cell from ROS, while also being able to act directly as an antioxidant. Further support for the ROS hypothesis is the fact that the activities of these compounds were enhanced by the pro-oxidant Luperox®. A notable point, however, is that not all hemozoin inhibitors seem to act in this way, since neither NAC nor Luperox® had any effect on the activity of compound 13. Thus, the killing mechanism of these compounds is not always the same. Interestingly, despite the difference in parasite killing mechanism between chloroquine and compound 13, they nonetheless have an antagonistic relationship as revealed in the isobologram study. This is to be expected if the first step in the antiparasitic process is inhibition of hemozoin formation in both cases.
Taking account of all the above findings, we propose a hypothesis that can integrate these observations. This is presented in summary in Figure 7. It involves: (i) accumulation of the charged protonated form of the compound (D) in the DV via pH trapping; (ii) inhibition of hemozoin (HZ) formation by this protonated compound resulting in increased free Fe(III) heme (H) in the DV; (iii) precipitation of free Fe(III) heme, which is poorly soluble at DV pH. It has been reported that the solubility of hemin at pH 5 is 0.1 μM,27 with the result that the overwhelming majority of the observed free heme in the DV is in the solid state; (iv) the compound forms a complex with free heme (HD) in solution; (v) the complex is also likely to be relatively insoluble in this environment. The observed exchangeable heme is a combination of mainly precipitated Fe(III) heme and the complex, itself probably largely solid; (vi) The dissolved complex may inhibit enzymes or transporters in the DV or; (vii) it may diffuse or be transported out of the DV into the cytoplasm where the concentration of compound D is much lower, driving the equilibrium towards dissociation; (viii) Finally, soluble free heme in the cytoplasm may generate ROS, killing the parasite.
Figure 7.
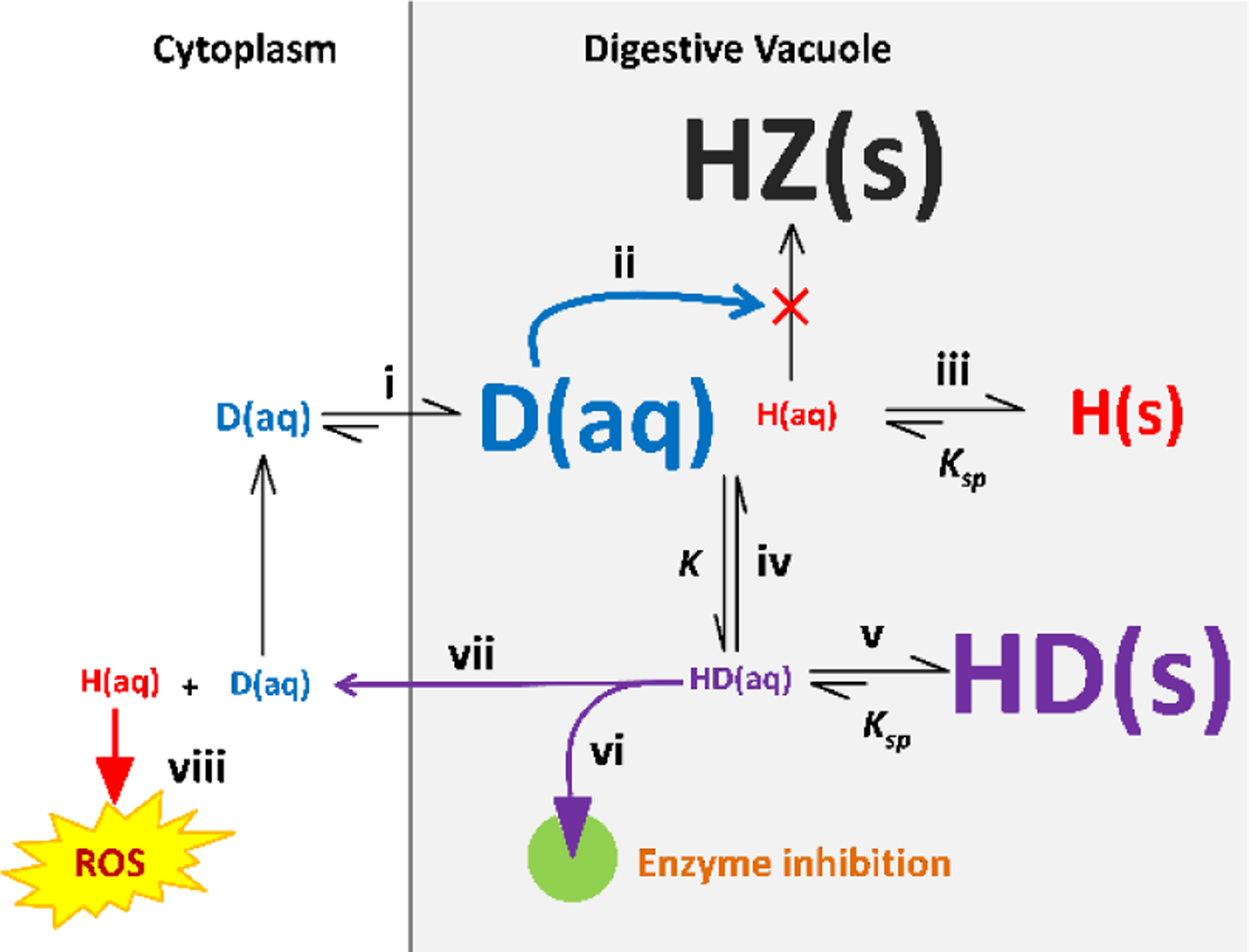
Model of the factors involved in the activity of hemozoin inhibitors. Protonation states and charges are omitted for the sake of simplicity. Refer to the main text for the numbered steps. Abbreviations: D, drug; H, heme; HZ, hemozoin.
In terms of this hypothesis, if the compound delivers heme to the cytoplasm, increased glutathione produced by the introduction of NAC would protect the parasite from the effects of ROS, while the pro-oxidant Luperox® would exacerbate them. On the other hand, if the complex inhibits a target in the DV, neither NAC nor Luperox® would have any effect on its activity. This would explain the differential effects of NAC and Luperox® on chloroquine and compound 4 on the one hand versus compound 13 on the other. In the case of a compound that delivers free heme to the cytoplasm, the solubility of the complex would determine the rate of delivery. For a very insoluble complex, a relatively long time would be needed for heme to reach toxic levels, and consequently hemoglobin digestion would continue for a longer time, with consequent build-up of a larger quantity of exchangeable heme in the form of precipitated complex. Conversely, for a more soluble complex, delivery of heme to the cytoplasm would be much faster, with consequent faster killing of the parasite and less time to build-up the solid complex in the DV. Differential rates of transport of complexes out of the DV by transporters would have a similar effect. In the case of a complex that inhibits a target in the DV, the time to appearance of the target would determine the speed of killing. Once again, if the target were expressed early, the killing speed would be fast, with little time to build-up the precipitated complex and vice versa in the case of a target that appears late. This can explain the relationship between speed of killing and levels of exchangeable heme. Key factors governing the concentrations of Fe(III) heme complexes are likely to be the solubility products Ksp) of Fe(III) heme and its complex as well as the equilibrium constant for formation of the complex (K). The availability of free Fe(III) heme for complexation is determined by inhibition of hemozoin formation. This is dependent on the strength of inhibition of hemozoin formation and degree of accumulation of the inhibitor in the DV by pH trapping. This accumulation will also influence the position of the equilibrium formation of the Fe(III) heme complex. Inhibition of hemozoin formation is the key initiating step in the mechanism. Thus, even when compounds have different subsequent effects, they will show an antagonistic relationship as seen for chloroquine and compound 13. This complicated process, involving an interplay of multiple factors, would explain why no simple relationship between antiparasitic activity and β-hematin inhibition activity, or levels of exchangeable heme has been found.
Methods
Synthesis
All chemicals and solvents were bought from Sigma-Aldrich or Fluka and unless otherwise specified, were used as received. Tetrahydrofuran (THF) and dichloromethane (DCM) were freshly distilled under N2, dried over Na wire with benzophenone and P4O10 respectively. Reactions were monitored by TLC using pre-coated silica-gel 60 F254 (0.2 mm) mounted on aluminum-backed plates, commercially available from Merck. Compounds were detected using a UV light with an absorption of 254 nm and stained for visible detection using either iodine filings, ninhydrin or anisaldehyde. Purification procedures involved the use of flash column chromatography and single or double solvent recrystallization techniques, using a Biotage Isolera 4 EXP and analytical reagent grade solvents respectively. Characterization procedures comprised: the determination of the melting points of the compounds using a Reichert-Jung Thermovar hot stage microscope; IR spectra recorded on a Bruker Tensor 27 FT-IR spectrometer; NMR spectra recorded on Bruker Ultrashield 400 Plus (for 1H, 13Cstan, HSQC and COSY). The NMR spec solvents used were deuterated acetone ((CD3)2CO), deuterated dimethyl sulfoxide ((CD3)2S=O) and deuterated chloroform (CDCl3) with reference peaks occurring at 2.05, 2.50 and 7.26 ppm in the 1H NMR and 29.84, 39.52 and 77.16 ppm in the 13C NMR spectra respectively. All chemical shifts are stated in ppm and coupling constants in Hz; HPLC was conducted using a Agilent 1220 LC system V; HRMS (ESI) was conducted at the University of Stellenbosch, Central Analytical facilities (CAF) using a Waters Synapt G2 instrument.
2,4-dichloroquinazoline (19)
Et3N (1.4 mL, 10 mmol) was added to a suspension of benzoylene urea 20 (0.82 g, 5.1 mmol) in POCl3 (9.2 mL, 51 mmol) under inert conditions and set to reflux at 106 °C for approximately 18 h. The reaction was monitored using TLC (MeOH/DCM, 1:99, Rf = 0.85) until the presence of the starting material diminished, indicating the completion of the reaction. After completion, the reaction mixture was diluted with DCM (80 mL) and poured over crushed ice, after which the slurry was neutralized using a saturated solution of Na2CO3 (approx. 30 mL) and allowed to stir for 1 h. The aqueous layer was extracted several times with DCM, after which all the DCM fractions were pooled, dried over sodium sulfate (Na2SO4) and the organic fraction concentrated under vacuum. The crude material was purified using flash column chromatography (MeOH/DCM 1:99, Rf = 0.85), followed by recrystallisation from boiling MeOH to obtain compound 19 as a white solid (0.87 g, 87%), m.p. 117–118 °C (Lit:28 118–120 °C). 1H NMR (400 MHz, Acetone-d6) δH 8.33 (1H, ddd, J 8.4, 1.4, 0.7 Hz), 8.16 (1H, td, J 8.4, 1.4 Hz), 8.01 (1H, ddd, J 8.4, 1.4, 0.7 Hz), 7.90 (1H, td, J 8.4, 1.4 Hz); 13C NMR (101 MHz, Acetone-d6) δC 164.5 (C-q), 155.5 (C-q), 153.4 (C-q), 137.5, 130.7, 128.7, 126.8, 123.2 (C-q); HRMS (ESI) m/z: Found 198.9822. Calculated 198.9830, C8H5Cl2N2 [M+H]+; HPLC purity: 99%.
General Procedure for the synthesis of 2-chloro-quinazolin-4-amine intermediates (21–23).
The following general procedure for the synthesis of intermediates (21–23) was followed unless otherwise stated in the specific intermediate experimental details.
The 2-chloro-quinazolin-4-amine intermediates were synthesized on a 0.50–1.0 g scale by reacting the preferred amine derivatives (1.1–1.7 eq) with dichloroquinazoline (19, 1.0 eq) and Et3N (1.5–2 eq) in THF (10–15 mL) for 3–12 h at 60 °C.
TLC was used to monitor the reaction progress in combination with UV (254 nm) absorption and staining using iodine, ninhydrin or anisaldehyde.
Upon reaction completion, the reaction vessel was allowed to cool to room temperature before the reaction contents were diluted in EtOAc and DCM (50:50), after which the contents were neutralized with a saturated solution of Na2CO3 and left to stir for several minutes. The aqueous and organic layers were separated and the former washed multiple times with DCM, after which the DCM fractions were pooled and concentrated under high vacuum. Chromatography of the residue followed by recrystallization from boiling MeOH or DCM, produced solid products that were characterized and evaluated for analytical purity.
N-(4-Bromophenyl)-2-chloroquinazolin-4-amine (21)
4-Bromoaniline (0.50 g, 2.9 mmol, 1.4 eq) and Et3N (0.51 mL, 3.5 mmol, 1.7 eq) were added to a suspension of 19 (0.40 g, 2.0 mmol) for the synthesis of 21. Compound 21 was purified by recrystallization from boiling MeOH and obtained as a white solid (0.51 g, 75%), m.p. 167–168 °C, Rf = 0.3 (EtOAc/hexane, 30:70). 1H NMR (400 MHz, DMSO-d6) δH 10.21 (1H, s), 8.55 (1H, ddd, J 8.4, 1.2, 0.5 Hz), 7.89 (1H, td, J 8.4, 1.2 Hz), 7.82 – 7.76 (2H, m), 7.72 (1H, ddd, J 8.4, 1.2, 0.5 Hz), 7.65 (1H, td, J 8.4, 1.2 Hz), 7.63 – 7.59 (2H, m); 13C NMR (101 MHz, DMSO-d6) δC 159.2 (C-q), 155.9 (C-q), 150.8 (C-q), 137.6 (C-q), 134.1, 131.4, 126.9, 126.6, 124.6, 123.4, 116.5 (C-q), 113.7 (C-q); HRMS (ESI) m/z: Found 333.9713. Calculated 333.9747, C14H10BrClN3 [M+H]+; HPLC purity: 99%.
2-Chloro-N-(furan-2-ylmethyl) quinazolin-4-amine (22)
Furfurylamine (0.16 mL, 1.8 mmol, 1.1 eq) and Et3N (0.36 mL, 2.5 mmol, 1.5 eq) were added to a suspension of 19 (0.34 g, 1.7 mmol). Compound 21 was purified by recrystallization from boiling MeOH and obtained as a white solid (0.33 g, 50%), m.p. 158–159 °C, Rf = 0.2 (EtOAc/hexane, 20:80). 1H NMR (400 MHz, DMSO-d6) δH 9.18 (1H, t, J 5.5 Hz), 8.30 (1H, ddd, J 8.4, 1.2, 0.5 Hz), 7.81 (1H, td, J 8.4, 1.2 Hz), 7.63 (1H, ddd, J 8.4, 1.2, 0.5 Hz), 7.60 (1H, dd, J 1.8, 0.9 Hz), 7.54 (1H, td, J 8.4, 1.2 Hz), 6.42 (1H, dd, J 3.2, 1.8 Hz), 6.37 (1H, dd, J 3.2, 0.9 Hz), 4.74 (2H, d, J 5.5 Hz); 13C NMR (101 MHz, DMSO-d6) δC 160.9 (C-q), 156.7 (C-q), 151.2 (C-q), 150.3 (C-q), 142.2, 133.7, 126.6, 126.2, 123.1, 113.4 (C-q), 110.5, 107.7, 37.3; HRMS (ESI) m/z: Found 260.0538. Calculated 260.0591, C13H11ClN3O [M+H]+; HPLC purity: 99%.
2-Chloro-N-phenylquinazolin-4-amine (23)
Aniline (0.26 mL, 2.8 mmol, 1.1 eq) and Et3N (0.53 mL, 3.8 mmol, 1.5 eq) were added to a suspension of 19 (0.50 g, 2.5 mmol). Compound 23 was purified by recrystallization from boiling DCM and obtained as a white solid (0.57 g, 89%), m.p. 187–188 °C, Rf = 0.3 (EtOAc/hexane, 30:70). 1H NMR (400 MHz, DMSO-d6) δH 10.16 (1H, s), 8.57 (1H, ddd, J 8.4, 1.2, 0.5 Hz), 7.87 (1H, td, J 8.4, 1.2 Hz), 7.82 – 7.75 (2H, m), 7.71 (1H, ddd, J 8.4, 1.2, 0.5 Hz), 7.64 (1H, td, J 8.4, 1.2 Hz), 7.48 – 7.39 (2H, m), 7.22 – 7.18 (1H, m); 13C NMR (101 MHz, DMSO-d6) δC 159.4 (C-q), 156.2 (C-q), 150.8 (C-q), 138.2 (C-q), 134.0, 128.6, 126.8, 126.5, 124.7, 123.4, 122.9, 113.7 (C-q); HRMS (ESI) m/z: Found 256.0633. Calculated 256.0642, C14H11ClN3 [M+H]+; HPLC purity: 100%.
General Procedure for the synthesis of 2,4-diaminoquinazoline (10–12)
The quinazoline-2,4-diamines were synthesized on a 0.05 – 0.30 g scale by reacting the required 2-chloro-quinazolin-4-amine intermediate (1 eq) with the desired amine compound (1 – 2 eq) in a sealed tube overnight at a temperature of 150 °C in isopropanol (2–5 mL). TLC was used to monitor the reactions in combination with UV (254 nm) absorption and staining using iodine, ninhydrin or anisaldehyde.
Upon reaction completion, the reaction contents were allowed to cool to ambient temperature before being diluted with EtOAc, after which they were neutralized with a solution of NaOH (1 M, 3–4 mL), which was allowed to stir for several minutes. The aqueous and organic layers were separated and the former washed multiple times with EtOAc, after which the EtOAc fractions were pooled and concentrated under high vacuum. Chromatography of the residue, followed by recrystallization from boiling MeOH or DCM, produced solid products.
N2, N4-Bis(4-bromophenyl)quinazoline-2,4-diamine (10)
4-Bromoaniline (0.17 g, 1.0 mmol, 1.3 eq) was reacted with compound 21 (0.25 g, 0.75 mmol) to produce 10, which was purified by recrystallization from boiling MeOH and obtained as a light-brown solid (0.29 g, 82%), m.p. 183–184 °C, Rf = 0.6 (EtOAc/hexane, 50:50). 1H NMR (400 MHz, DMSO-d6) δH 9.69 (1H, s), 9.31 (1H, s), 8.38 (1H, dd, J 7.5, 1.2 Hz), 7.92 – 7.88 (4H, m), 7.69 (1H, td, J 7.5, 1.2 Hz), 7.57 (2H, d, J 8.9 Hz), 7.52 (1H, dd, J 7.5, 1.2 Hz), 7.41 (2H, d, J 8.9 Hz), 7.31 (1H, td, J 7.5, 1.2 Hz); 13C NMR (101 MHz, DMSO-d6) δC 158.2 (C-q), 155.9 (C-q), 151.4 (C-q), 140.5 (C-q), 138.8 (C-q), 133.1, 131.2, 130.9, 125.7, 124.4, 123.1, 122.2, 120.7, 115.2 (C-q), 112.1 (C-q), 111.8 (C-q); HRMS (ESI) m/z: Found 468.9663. Calculated 468.9663, C20H15Br2N4 [M+H]+; HPLC purity: 98%.
N4-(Furan-2-ylmethyl)-N2-phenylquinazoline-2,4-diamine (11)
Phenylamine (0.11 mL, 1.2 mmol, 1.2 eq) was reacted with compound 22 (0.26 g, 1.0 mmol). Compound 11 was purified by recrystallization from boiling MeOH and obtained as a white solid (0.11 g, 36%) m.p. 103–104 °C, Rf = 0.4 (EtOAc/hexane, 30:70). 1H NMR (400 MHz, DMSO-d6) δH 9.10 (1H, s), 8.64 (1H, t, J 5.6 Hz), 8.12 (1H, dd, J 8.4, 1.0 Hz), 7.86 (2H, dd, J 8.6, 1.0 Hz), 7.62 (1H, td, J 8.4, 1.0 Hz), 7.59 (1H, dd, J 1.8, 0.8 Hz), 7.43 (1H, dd, J 8.4, 1.0 Hz), 7.29 – 7.23 (2H, m), 7.19 (1H, td, J 8.4, 1.0 Hz), 6.95 – 6.86 (1H, m), 6.40 (1H, dd, J 3.2, 1.8 Hz), 6.36 (1H, dd, J 3.2, 0.8 Hz), 4.78 (2H, d, J 5.6 Hz); 13C NMR (101 MHz, DMSO-d6) δC 159.8 (C-q), 156.4 (C-q), 152.3 (C-q), 150.4 (C-q), 141.9, 141.0 (C-q), 132.8, 128.2, 124.8, 122.8, 121.6, 120.8, 118.9, 111.5 (C-q), 110.4, 107.1, 37.2; HRMS (ESI) m/z: Found 317.1393. Calculated 317.1402, C19H17N4O [M+H]+; HPLC purity: 97%.
N2-Cyclohexyl-N4-phenylquinazoline-2,4-diamine (12)
Cyclohexylamine (0.11 mL, 0.89 mmol, 1.2 eq) was reacted with compound 23 (0.20 g, 0.78 mmol) to form 12, which was purified by flash chromatography, Rf = 0.3 (EtOAc/Hexane, 30:70) as a white solid (0.18 g, 72%), m.p. 133–134 °C. 1H NMR (400 MHz, DMSO-d6) δH 9.35 (1H, s), 8.28 (1H, dd, J 8.0, 1.2 Hz), 7.96 (2H, d, J 7.9 Hz), 7.54 (1H, td, J 8.0, 1.2 Hz), 7.35 (2H, t, J 7.9 Hz), 7.30 (1H, dd, J 8.0, 1.2 Hz), 7.15 – 7.03 (2H, m), 6.57 (1H, s), 3.79 (1H, s), 1.94 – 1.29 (8H, m), 1.61 – 1.58 (1H, m), 1.21 – 1.17 (1H, m); 13C NMR (101 MHz, DMSO-d6) δC 158.1 (C-q), 158.0 (C-q), 152.4 (C-q), 139.8 (C-q), 132.5, 128.2, 124.6, 123.0, 122.8, 121.5, 120.1, 110.9 (C-q), 49.2, 32.8, 25.4, 25.0; HRMS (ESI) m/z: Found 319.1917. Calculated 319.1923, C20H23N4 [M+H]+; HPLC purity: 99%.
3-(benzo[d]thiazol-2-yl)aniline (24)
3-Aminobenzoic acid (2.500 g, 18.637 mmol) was added to 2-aminothiophenol (2 mL, 18.637 mmol) and polyphosphoric acid (PPA) (25.00 g, 10 eq.). The mixture was continually stirred whilst heated at 180 °C until completion (3 h) as confirmed by TLC (5:95 MeOH:DCM, product Rf = 0.67). After completion, the reaction mixture was cooled and poured into ice cold Na2CO3 solution (400 mL, 10%) whilst stirring in order to neutralize residual PPA. The resulting mixture was filtered and washed with cold water (5 mL). The filtrate was retained and dried under vacuum at 60 °C to produce crude 24. The product was recrystallized in boiling MeOH and water to afford pure 24 (3.623 g, 16.028 mmol, 86%) as a green solid, m.p. 138–139 °C. IR (ATR) ν/cm−1: 3440, 3323 (NH str.), 3062 (Ar-H str.), 1627 (C=N str.), 1604 (NH bend), 761 (m. disub. benzene); 1H NMR (400 MHz, DMSO-d6) δH 8.02 (d, J 7.9 Hz, 1H), 8.02 (d, J 8.1 Hz, 1H), 7.52 (t, J 7.2 Hz, 1H), 7.43 (t, J 8.0 Hz 1H), 7.38 (m, 1H), 7.19 (m, 2H), 6.77 (d, J 6.6 Hz, 1H), 5.44 (br. s, 2H); 13C NMR (101 MHz, DMSO-d6) δC 168.3 (Cq), 153.7 (Cq), 149.4 (Cq), 134.4 (Cq), 133.6 (Cq), 129.9, 126.6, 125.4, 122.5, 117.0, 114.9, 112.1. EI-MS m/z: Found 226. Calculated 226, C13H10N2S [M]+. HPLC purity: 100%.
3-(benzo[d]oxazol-2-yl)aniline (25)
3-Aminobenzoic acid (2.500 g, 18.229 mmol) was added to 2-aminophenol (1.989g, 18.229 mmol) and PPA (25 g, 10 eq.). The mixture was continually stirred whilst heated at 180 °C until completion (3 h) as confirmed by TLC (5:95 MeOH:DCM, product Rf = 0.63). After completion, the reaction mixture was cooled and poured into ice cold Na2CO3 solution (400 mL, 10%) whilst stirring in order to neutralize residual PPA. The resulting mixture was filtered and washed with cold water (5 mL). The filtrate was retained and dried under vacuum at 60 °C to produce crude 25. The product was recrystallized in boiling MeOH and water to afford pure 25 (3.411 g, 16.225 mmol, 89%) as a red solid. m.p. 179–180 °C (Lit.:29 178 °C). IR (ATR) ν/cm−1: 3458, 3305 (NH str.), 3091 (Ar-H str.), 1625 (C=N str.), 1550 (NH bend), 746 (m. disub. benzene); 1H NMR (400 MHz, DMSO-d6) δH 7.76 (m, 2H), 7.46 (t, J 1.8 Hz, 1H), 7.40 (m, 2H), 7.35 (m, 1H), 7.23 (t, J 7.8 Hz, 1H), 6.81 (ddd, J 8.0, 2.3, 1.0, 1H), 5.45 (br. S, 2H); 13C NMR (101 MHz, DMSO-d6) δC 163.1 (Cq), 150.2 (Cq), 149.5 (Cq), 141.7 (Cq), 129.8, 127.0 (Cq), 125.3, 124.8, 119.7, 117.4, 114.7, 112.2, 110.8. EI-MS m/z: Found 210. Calculated 210, C13H10N2O [M]+. HPLC purity: 96%.
Acid chloride formation and amide bond formation
To 5-(trifluoromethyl)pyridine-3-carboxylic acid (2 mmol) thionyl chloride (SOCl2) (8 eq.) was added and the reaction mixture heated under reflux at 80 °C until completion. TLC was performed to monitor the reaction using 10:90 MeOH/DCM and stained with I2. After completion residual thionyl chloride was removed under pressure and the product dried under vacuum.
The acid chloride was dissolved in THF and added to the required intermediate (24 or 25) (1 mmol) dissolved in pyridine (2 mL) under an inert atmosphere until completion. Reactions were maintained at −40 °C using an acetonitrile/liquid N2 bath. Reaction progress was monitored using TLC 5:95 MeOH/ DCM and stained using ninhydrin. The starting material (amine) appeared dark pink and the product (amide) stained light pink. Once complete the reaction was quenched with MeOH and dried under vacuum to afford the appropriate amide. Following purification by column chromatography (MeOH: DCM, from 2:98 to 20:80) the product was recrystallized in boiling MeOH.
N-(3-(benzo[d]oxazol-2-yl)phenyl)-5-(trifluoromethyl)nicotinamide (17)
The product of the reaction between 24 (0.226 g, 1 mmol) and the acid chloride (2 mmol) dissolved in THF (2 mL) was purified by column chromatography (5:95 MeOH: DCM, product Rf = 0.56) and the appropriate fractions recrystallized to afford pure 17 (0.295 g, 0.739 mmol, 74%) as a brown solid. m.p. 212–213 °C. IR (ATR) ν/cm−1 3302 (NH str.), 3053 (Ar-H str.), 1650 (C=O str.), 1656 (C=N str.), 1139 (C-F str.), 710 (m. disub. benzene); 1H NMR (400 MHz, DMSO-d6) δH 10.84 (s, 1H), 9.43 (d, J 2.0 Hz, 1H), 9.20 (m, 1H), 8.75 (m, 1H), 8.62 (t, J 1.8 Hz, 1H), 8.16 (d, J 7.3 Hz, 1H), 8.08 (d, J 7.5 Hz, 1H), 8.03 (ddd, J 8.2 Hz, 2.1 Hz, 0.9 Hz, 1H), 7.86 (dt, J 8.1 Hz, 1.2 Hz, 1H), 7.60 (m, 1H), 7.56 (m, 1H), 7.48 (m, 1H); 13C NMR (400 MHz, DMSO-d6) δC 166.9 (Cq), 162.7, 153.4 (Cq), 152.7, 148.6 (Cq), 139.4 (Cq), 134.4 (Cq),133.3, 132.6 (Cq), 132.5 (Cq), 130.3 (Cq), 129.9, 126.6, 125.6, 125.0, 122.9, 122.8, 122.8, 122.3, 118.6. EI-MS m/z: Found 399. Calculated 399 [M]+ C20H12F3N3OS. HPLC purity: 99%.
N-(3-(benzo[d]oxazol-2-yl)phenyl)-5-(trifluoromethyl)nicotinamide (18)
The product of the reaction between 25 (0.210 g, 1 mmol) and the acid chloride (2 mmol) dissolved in THF (6 mL) and DMF (0.1 mL) was purified by column chromatography (5:95 MeOH:DCM, product Rf = 0.67). The appropriate fractions were recrystallized in boiling MeOH to afford pure 18 (0.198 g, 0.518 mmol, 52 %) as a cream solid, m.p. 229–230 °C. IR (ATR) ν/cm−1 3259 (NH str.), 3051 (Ar-H str.), 1649 (C=O str.), 1635 (C=N str.), 1128 (C-F str.), 736 (m. disub. benzene); 1H NMR (400 MHz, DMSO-d6) δH 10.83 (s, 1H), 9.41 (d, J 1.6 Hz, 1H), 9.18 (d, J 0.9 Hz, 1H), 8.73 (d, J 1.6 Hz, 2H), 8.02 (m, 1H), 7.96 (m, 1H), 7.80 (m, 2H), 7.62 (t, J 8.0 Hz, 1H), 7.42 (m, 2H); 13C NMR (400 MHz, DMSO-d6) δC 163.3, 162.5 (Cq), 153.3, 150.7 (Cq), 149.2 (Cq), 142.0 (Cq), 139.9 (Cq), 133.2, 130.8 (Cq), 130.4, 127.4 (Cq), 126.1, 125.4, 124.7, 123.9, 123.4, 120.4, 119.4, 111.4. EI-MS m/z: Found 383. Calculated 383 [M]+ C20H12F3N3O2. HPLC purity: 97%.
β-Hematin inhibition, parasite culturing and cellular heme measurements
The detergent mediated assay for β-hematin inhibition was based on that developed by Carter et al.30 modified to use pyridine to detect unconverted hematin as previously described,31 with modifications reported in detail elsewhere.32
Parasites were cultured according to the method of Trager and Jensen with minor modifications.33 Parasite growth inhibition experiments were carried out using previously reported methods. For most experiments the parasite lactate dehydrogenase assay was used to measure parasite growth,34 but for the experiments investigating speed of killing and the effects of NAC and Luperox® a SYBR Green I assay was used.35–36
Measurement of cellular hemoglobin, exchangeable heme and hemozoin was performed as previously described in detail8.
Cellular accumulation ratios
The inoculum effect analysis was carried out using a method similar to that previously reported.4 IC50 values measured using a modified version of the pLDH assay were determined in triplicate at inoculum sizes (IS) ranging from 1 to 5 and fractional volumes (Vf) of parasitized erythrocytes from 0.0001 to 0.001. The relationship was extrapolated via linear regression to give an absolute IC50 (IC50 at infinite parasite dilution) from the y-intercept in Graph Pad Prism v7.0. Experimental CAR values were calculated from a linear plot of IC50 values plotted as a function of their corresponding Vf in accordance with equation 1.
| (1) |
where and hematocrit × %parasitemia is the IS. The total amount of intracellular test compound was then calculated using equation 2:
| (2) |
Where Vcell is the average volume of an infected RBC in fL (84.3 ± 5.7 fL)37 and IC50 (4) the 50% inhibitory concentration in μM at IS=4, the standard parasite culturing conditions used for cellular heme measurements.
Electron microscopy
TEM and EELS experiments were carried out as described previously1, 7 except for the use of polysaccharide gel encapsulation and high pressure freezing. A 4% agarose gel solution was prepared using a modified microwave method previously described.38 The fixed PRBCs were washed twice with 1 mL PBS before the samples were suspended in an equal volume of the 4% agarose slurry. The suspension was allowed to solidify before adding 50 μL of PBS to avoid sample dehydration.
Agarose-fixed PRBC samples were high pressure frozen and embedded using a modified method previously described by Yamada et al.39–41 Briefly, the agarose-fixed PRBC samples were cut into thin sections (80–100 nm) using a diamond knife and sandwiched between two single-holed copper grids under a light microscope. Samples were rapidly frozen with liquid nitrogen at a pressure of 2,100 bar to a temperature of −81°C within 2 min using a Leica EM High Pressure Freezer 100. High pressure frozen samples were transferred to a Leica EM AFS embedding system after removing the copper grids from the Leica sample carrier. Samples were embedded at −80 °C in acetone for 24 h. After 24 h, acetone was replaced with OsO4 to maintain lipids and proteins within the sample for another 24 h. The samples were left in the Leica EM AFS UC7 embedding system in low viscosity Spurr’s resin for 5–7 days while the temperature was gradually increased to RT. Samples were removed and imaged using TEM with EELS.
Raman microscopy
Slides for confocal Raman microscopy were prepared as follows: a drop of clear nail polish was smeared evenly onto a clean glass microscope slide to create a thin film. Pre-cut aluminum foil (1.5 × 3.5 cm) strips were carefully placed on the film using thin tipped tweezers. The aluminum foil strips were then carefully pressed, using nitrile gloves, to create a smooth surface.
Trophozoite cultures were mixed and decanted into four 15 ml centrifuge tubes (250 μl). The decanted trophozoite cultures were left to stand for 10 min at rt. before mixing and gently smearing 2 μl onto an aluminum foil covered glass microscope slide. Glutaraldehyde fixation was avoided, since this is known to oxidize hemoglobin to methemoglobin. The sample slide was left to air dry at rt. for 15 min before analysis.
Samples for Raman imaging of the synthetically prepared material were prepared as follows: Solid aggregates of β-hematin crystals were gently broken up in a glass beaker before placing on an aluminum foil covered microscope glass slide for analysis; a 2 μL drop of 1 mM 13 in DMSO was dried onto an aluminum foil covered microscope glass slide using an incandescent lamp at RT before further analysis; a 2 μL drop of 1 mM solutions of hematin or hemin were dried onto separate aluminum foil covered microscope glass slides using an incandescent lamp at RT before further analysis; a 1:10 (v/v) mixture of hemin and 13 was prepared by titrating 5 mL of a 1 mM solution of hemin into 50 mL solution of 1 mM of compound 13 in a 100 mL glass beaker. DMSO was added to a final volume of 100 mL. The container was covered with parafilm and the solution was gently heated in a water bath (60 °C) for 30 min and cooled to RT. Roughly 2 μL of the mixture was dried, using an incandescent lamp, onto an aluminum foil covered glass microscope slide for further analysis.
Raman spectra were collected using a confocal Raman microspectrometer alpha300 R (WITec GmbH, Germany) equipped with a CCD camera cooled to −60 °C and three air-calibrated objective lenses (10×, 50× and 100×). All spectra were collected using the 100× objective lens. This Raman microspectrometer uses a green Nd:YAG laser with excitation wavelength of 532 nm and a laser spot of 1–2 μm with a resolution of 1–2 cm−1 that was set at a default grating of 2050. The Raman microspectrometer was controlled using WITec Control FIVE v5.0.0 software. Spectra were recorded in the iron porphyrin dominated spectral region of 1700–500 cm−1.
Single scan Raman spectra were recorded on uninfected RBCs, untreated and treated infected RBCs and synthetic samples. For cells, an average of 30 single scan Raman spectra from selected regions were collected from each of 10 randomly selected cells. For synthetic complexes, the same number of single scan Raman spectra were obtained from 5 randomly selected regions of the slide. Before performing scans, the oscillator was adjusted and calibrated to the aluminum foil covered microscope glass slide to correct for any background signals. This was also done to reduce the comic ray effect which can cause false-positive peaks. All experiments were carried out in triplicate and each single scan Raman spectrum was collected using a total of 100 accumulations at an integration time of 0.05 s. The spectra obtained from cells, including RBCs, were collected with an optimal laser power of 0.1 mW to maximize the signal-to-noise ratio, while avoiding hemolysis and photo-damage of cells. Furthermore, cells were allowed to rest for intervals of 10 min before each scan. Synthetic complexes were analyzed using a laser power of 1 mW.
All single scan spectra were further processed using WITec Project FIVE v5.0.0.40 software. This involved cosmic ray correction (filter size 4; dynamic factor 50), smoothing using a Savitzky-Golay algorithm (9 smoothing points; derivative 0), averaging (spectral 9; spatial 0) and subtraction of background (shape size 100; noise factor 4).
Large-scale image Raman spectra were also recorded for cells. Every large-scale image Raman spectrum has a corresponding image where each pixel corresponds to a single spectrum. An average of 15 lines per image and 15 points per line for each large-scale image Raman spectrum were recorded for an average collection time of about 35 min. Since there is a risk of photo-damage to cells with this laser exposure time, the integration times and laser power were set as low as possible to obtain acceptable signal to noise ratios, namely 0.05 s and 0.1 mW. For synthetic materials an integration time of 0.1 s at laser power 1 mW was used. These spectra were corrected for cosmic rays, smoothed, averaged and background subtracted using the same parameters described above for single scan Raman spectra.
For PCA analysis, spectra were normalized by subtracting the minimum relative intensity count (cmin) from each data point (ci), divided by the relative intensity of the most intense peak (cmax). Next, the normalized spectra were converted to their corresponding second derivative spectra, thus defining peak positions based on their curvature rather than intensity. The data points (± ai) at each peak position of the second derivative spectra were filtered based on whether they were above or below the 95th percentile (positive +p0.05 or negative −p0.05) to eliminate noise.
All resulting replicate peak positions for pairs of heme components analyzed (e.g. oxyhemoglobin versus hemozoin) were transferred to Molegro Data Modeler Executive software. This software was used to generate the eigenvectors (PC1 and PC2) and corresponding eigenvalues.
A simplified approach was taken to test the statistical significance of separation of clusters in each PCA plot for pairs of heme components. In brief, the eigenvalues for PC1 and PC2 for a given pair of Fe(III)PPIX species were used to calculate the centroids and (a = I or II) given by equations 3 and 4:
| 3 |
| 4 |
where na is the number of points in cluster a. The points were then shifted so that one of the clusters had a centroid at (0,0) by applying equations 5 and 6.
| 5 |
| 6 |
The new position of centroid II in polar coordinates is then given by equations 7 and 8
| 7 |
| 8 |
where
| 9 |
and
| 10 |
Finally, rotation of the axes through angle θ resulted in a new set of axes in which both centroids lay on the new x-axis with no separation of the clusters along the y-direction and maximum separation along x. The positions of points along the new x-axis were given by equation 11
| 11 |
The statistical significance of the separations of the means and was evaluated using a Welch’s t-test which allows for unequal variances in the data.
Speed of killing measurements
For speed of killing measurements an early ring-stage sorbitol synchronized parasite culture at 2% parasitemia and 2% hematocrit was prepared in complete medium and 200 μL was added to each well of a 96-well flat-bottom plate. For each compound five concentrations were prepared in complete medium from the original 2 mg/mL stock. 20 μL of these individual concentrations were added to the plate such that the final volume in each well was 220 μL and the final concentration in each well corresponded to a multiple of the IC50 of each compound (0.5×, 1×, 1.5×, 2× and 3×). Each assay plate had at least eight drug-free control wells. The assay plates were incubated at 37 °C in a gassing chamber with 3% O2, 4% CO2 and N2. Compound wash-out steps were performed by washing off the compound containing medium and replacing it with new complete medium at the time points shown in Figure S3. The assay plates were then left to incubate for 72 h. After 72 h, the plates were removed from the gassing chamber, the medium aspirated and the pellet washed with 0.22 μm filtered phosphate buffered saline (1×PBS). Following this, 100 μL of freshly prepared SYBR green I in 0.22 μm filtered PBS (1 μL SYBR green I: 9 mL PBS) was added to each well of the 96-well plate. This was left to incubate for at least 30 min in the dark at RT. After incubation the plates were resuspended and read on a BD AccuriTM C6 Plus flow cytometer with C-sampler software. For each sample measurement 20,000 events were collected. The acquired data was analyzed using FlowJo software version 10.5.3. A gating method was employed. The uninfected and infected red blood cells in each sample were characterized using forward (FSC) and side scatter (SSC), as well as the fluorescence intensity (FL1) parameter to determine SYBR green I positive cells.
Isobolograms
Isobolograms were constructed according to the method of Fivelman et al.42 For each isobologram, two compounds were tested alone and in fixed concentration ratios of 1:4, 2:3, 3:2 and 4:1. All combinations were prepared on the day of the experiment and IC50 values were measured in triplicate using a modified version of the pLDH assay. Five IC50 values were determined for each compound for each isobologram. The IC50 values were used to determine the fractional inhibitory concentration (FIC) for each compound, for each combination according to equations 12 and 13:
| 12 |
Where IC50 (A+B) = 50% inhibitory concentration of compound A and B at a fixed concentration ratio and IC50 A = 50% inhibitory concentration of compound A alone
| 13 |
Where IC50 (B+A) = 50% inhibitory concentration of compound B and A at a fixed concentration ratio and IC50 B = 50% inhibitory concentration of compound B alone.
An isobologram was constructed for each combination by plotting FIC A as a function of FIC B.
Supplementary Material
Acknowledgments
We thank Miranda Waldron for assistance with Raman microscopy and Mohamed Jaffer for assistance with TEM. RO, KM and SJB thank the National Research Foundation of South Africa and JMC thanks CIDRI Africa for financial support. The research reported in this publication was supported by the United States National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01AI110329 and for the speed of killing studies R01AI143521. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Egan TJ; Combrinck JM; Egan J; Hearne GR; Marques HM; Ntenteni S; Sewell BT; Smith PJ; Taylor D; van Schalkwyk DA; Walden JC, Fate of haem iron in the malaria parasite Plasmodium falciparum. Biochem. J 2002, 365, 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Slater AFG; Cerami A, Inhibition by chloroquine of a novel haem polymerase enzyme activity in malaria trophozoites. Nature 1992, 355, 167–169. [DOI] [PubMed] [Google Scholar]
- 3.Egan TJ; Ross DC; Adams PA, Quinoline anti-malarial drugs inhibit spontaneous formation of b-haematin (malaria pigment). FEBS Lett. 1994, 352, 54–57. [DOI] [PubMed] [Google Scholar]
- 4.Hawley SR; Bray PG; Mungthin M; Atkinson JD; O’ Neill PM; Ward SA, Relationship between antimalarial drug activity, accumulation, and inhibition of heme polymerization in Plasmodium falciparum in vitro. Antimicr. Agents Chemother 1998, 42, 682–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dorn A; Vippagunta SR; Matile H; Jaquet C; Vennerstrom JL; Ridley RG, An assessment of drug-haematin binding as a mechanism for inhibition of haematin polymerisation by quinoline antimalarials. Biochem. Pharmacol 1998, 55, 727–736. [DOI] [PubMed] [Google Scholar]
- 6.Dinghra SK; Redhi D; Combrinck JM; Yeo T; Okombo J; Henrich PP; Cowell AN; Gupta P; Stegman ML; Hoke JM; Cooper RA; Winzeler E; Mok S; Egan TJ; Fidock DA, A variant PfCRT isoform can contribute to Plasmodium falciparum resistance to the first-line partner drug piperaquine. mBio 2017, 8, e00303–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Combrinck JM; Mabotha TE; Ncokazi KK; Ambele MA; Taylor D; Smith PJ; Hoppe HC; Egan TJ, Insights into the role of heme in the mechanism of action of antimalarials. ACS Chem. Biol 2013, 8, 133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Combrinck JM; Fong KY; Gibhard L; Smith PJ; Wright DW; Egan TJ, Optimization of a multi-well colorimetric assay to determine haem species in Plasmodium falciparum in the presence of anti-malarials. Malaria J. 2015, 14, e253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wicht KJ; Combrinck JM; Smith PJ; Hunter R; Egan TJ, Identification and SAR evaluation of hemozoin-inhibiting benzamides active against Plasmodium falciparum. J. Med. Chem 2016, 59, 6512–6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wicht KJ; Combrinck JM; Smith PJ; Hunter R; Egan TJ, Identification and mechanistic evaluation of hemozoin-inhibiting triarylimidazoles active against Plasmodium falciparum. ACS Med. Chem. Lett 2017, 8, 201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.L’abbate FP; Müller R; Openshaw R; Combrinck JM; de Villiers KA; Hunter R; Egan TJ, Hemozoin inhibiting 2-phenylbenzimidazoles active against malaria parasites. Eur. J. Med. Chem 2018, 159, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joshi MC; Okombo J; Nsumiwa S; Ndove J; Taylor D; Wiesner L; Hunter R; Chibale K; Egan TJ, 4‑Aminoquinoline antimalarials containing a benzylmethylpyridylmethylamine group are active against drug resistant Plasmodium falciparum and exhibit oral activity in mice J. Med. Chem 2017, 60, 10245–10256. [DOI] [PubMed] [Google Scholar]
- 13.Vanaerschot M; Lucatoni L; Li T; Combrinck JM; Ruecker A; Tiruppadiripuliyur SK; Rubiano K; Ferreira PE; Siciliano G; Gulati S; Henrich PP; Ng CL; Murithi JM; Corey VC; Duffy S; Lieberman OJ; Sinden RE; Alano P; Delves MJ; Sim KL; Winzeler EA; Egan TJ; Hoffman SL; Avery VM; Fidock DA, Identifying hexahydroquinolines as new antimalarial candidates with potent blood stage and transmission-blocking activity. Nat. Microbiol 2017, 2, 1403–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabryszewski S; Dhingra SK; Combrinck JM; Lewis IA; Callaghan PS; Hassett MR; Siriwardana A; Henrich PP; Lee AH; Gnädig NF; Musset L; Llinás M; Egan TJ; Roepe PD; Fidock DA, Evolution of fitness cost-neutral mutant PfCRT conferring P. falciparum 4-aminoquinoline drug resistance is accompanied by altered parasite metabolism and digestive vacuole physiology. PLoS Pathog. 2016, 12, e1005976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fong KY; Sandlin RD; Wright DW, Identification of b-hematin inhibitors in the MMV Malaria Box. Int. J. Parasitol. Drugs Drug Resist 2015, 5, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orjih AU; Banyal HS; Chevli R; Fitch CD, Hemin lyses malaria parasites. Science 1981, 214, 667–669. [DOI] [PubMed] [Google Scholar]
- 17.Sugioka Y; Suzuki M; Sugioka K; Nakano M, A ferriprotoporphyrin IX-chloroquine complex promotes membrane phospholipid peroxidation. A possible mechanism for antimalarial action. FEBS Lett. 1987, 223, 251–254. [DOI] [PubMed] [Google Scholar]
- 18.Schmitt TH; Frezzatti WA; Schreier S, Hemin-induced lipid membrane disorder and increased permeability: a molecular model for the mechanism of cell lysis. Arch. Biochem. Biophys 1993, 307, 96–103. [DOI] [PubMed] [Google Scholar]
- 19.Lew VL; Tiffert T; Ginsburg H, Excess hemoglobin digestion and the osmotic stability of Plasmodium falciparum-infected red blood cells. Blood 2003, 101, 4189–4194. [DOI] [PubMed] [Google Scholar]
- 20.Sandlin RD; Fong KY; Wicht KJ; Carrell HM; Egan TJ; Wright DW, Indentification of b-hematin inhibitors in a high-throughput screening effort reveals scaffolds with in vitro antimalarial activity. Int. J. Parasitol. Drugs Drug Resist 2014, 4, 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.EELS.info. www.eels.info/atlas (accessed 06/12/2020).
- 22.Balasubramanian D; Mohan Rao C; Panijpan B, The malaria parasite monitored by photoacoustic spectroscopy. Science 1984, 223, 828–830. [DOI] [PubMed] [Google Scholar]
- 23.Kozicki M; Creek DJ; Sexton A; Morahan BJ; Wesełucha-Birczyńska A; Wood BR, An attenuated total reflection (ATR) and Raman spectroscopic investigation into the effects of chloroquine on Plasmodium falciparum-infected red blood cells. Analyst 2015, 140, 2236–2246. [DOI] [PubMed] [Google Scholar]
- 24.Kapishnikov S; Staalsø T; Yang Y; Lee J; Pérez-Berná AJ; Pereiro E; Yang Y; Werner S; Guttmann P; Leiserowitz L; Als-Nielsen J, Mode of action of quinoline antimalarial drugs in red blood cells infected by Plasmodium falciparum revealed in vivo. Proc. Natl. Acad. Sci. USA 2019, 116, 22946–22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugioka Y; Suzuki M, The chemical basis for the ferriprotoporphyrin IX-chloroquine induced lipid peroxidation. Biochim. Biophys. Acta 1991, 1074, 19–24. [DOI] [PubMed] [Google Scholar]
- 26.Becker K; Tilley L; Vennerstrom JL; Roberts D; Rogerson S; Ginsburg H, Oxidative stress in malaria parasite-infected erythrocytes: host-parasite interactions. Int. J. Parasitol 2004, 34, 163–189. [DOI] [PubMed] [Google Scholar]
- 27.Morrison DB; Williams EFJ, The solubility and titration of hemin and ferrihemic acid. J. Biol. Chem 1941, 137, 461–473. [Google Scholar]
- 28.Mukherjee D; Mukhopadhyay A; Bhat KS; Shridhara AM; Rao KS, Synthesis, characterization and anticonvulsant activity of substituted 4-chloro-2-(4-piperazin-1-yl) quinazolines. Int. J. Pharm. Pharm. Sci 2014, 6, 567–571. [Google Scholar]
- 29.Stephens FF; Bower JD, The preparation of benzimidazoles and benzoxazoles from Schiff bases. Part 1. J. Chem. Soc 1949, 2971–2972. [Google Scholar]
- 30.Sandlin RD; Carter MD; Lee PJ; Auschwitz JM; Leed SE; Johnson JD; Wright DW, Use of the NP-40 detergent-mediated assay in discovery of inhibitors of b-hematin crystallization. Antimicr. Agents Chemother 2011, 55, 3363–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ncokazi KK; Egan TJ, A colorimetric high-throughput b-hematin inhibition screening assay for use in the search for antimalarial compounds. Anal. Biochem 2005, 338, 306–319. [DOI] [PubMed] [Google Scholar]
- 32.Lu W-J; Wicht KJ; Wang L; Imai K; Mei Z-W; Kaiser M; El Sayed I; Egan TJ; Inokuchi T, Synthesis and antimalarial testing of neocryptolepine analogues: Addition of ester function in SAR study of 2,11-disubstituted indolo [2,3-b]quinolines. Eur. J. Med. Chem 2013, 64, 498–511. [DOI] [PubMed] [Google Scholar]
- 33.Trager W; Jensen JB, Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [DOI] [PubMed] [Google Scholar]
- 34.Makler MT; Ries JM; Williams JA; Bancroft JE; Piper RC; Gibbins BL; Hinrichs DJ, Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am. J. Trop. Med. Hyg 1993, 48, 739–741. [DOI] [PubMed] [Google Scholar]
- 35.Smilkstein M; Swirilaijaroen N; Kelly JX; Wilairat P; Riscoe M, Simple and iInexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicr. Agents Chemother 2004, 48, 1803–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennett TN; Paguio M; Gligorijevic B; Seudieu C; Kosar AD; Davidson E; Roepe PD, Novel, rapid, and inexpensive cell-based quantification of antimalarial drug efficacy. Antimicr. Agents Chemother 2004, 48, 1807–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okafur UE; Tsoka-Gwegweni JM; Bibirigea A; Irimie A; Tomuleasa C, Parasitaemia and haematological changes in malaria-infected refugees in South Africa. S. Afr. Med. J 2016, 106, 413–416. [Google Scholar]
- 38.Koontz L, Agarose gel electrophoresis. Methods Enzymol 2013, 529, 35–45. [DOI] [PubMed] [Google Scholar]
- 39.Yamada H; Mitarai S; Chikamatsu K; Mizuno K; Yamaguchi M, Novel freeze-substitution electron microscopy provides new aspects of virulent Mycobacterium tuberculosis with visualization of the outer membrane and satisfying biosafety requirements. J. Micriobiol. Methods 2010, 80, 14–18. [DOI] [PubMed] [Google Scholar]
- 40.Yamada H; Chikamatsu K; Aono A; Mitarai S, Pre-fixation of virulent Mycobacterium tuberculosis with glutaraldehyde preserves exquisite ultrastructure on transmission electron microscopy through cryofixation and freeze-substitution with osmium-acetone at ultralow temperature. J. Micriobiol. Methods 2014, 96, 50–55. [DOI] [PubMed] [Google Scholar]
- 41.Yamada H; Yamaguchi M; Chikamatsu K; Aono A; Mitarai S, Structome analysis of virulent Mycobacterium tuberculosis, which survives with only 700 ribosomes per 0.1 fl of cytoplasm. PLoS One 2015, 10, e0117109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fivelman QL; Adagu IS; Warhurst DC, Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicr. Agents Chemother 2004, 48, 4097–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.