

Dear Editors
1.
Malignant fibrous histiocytoma (MFH) is a rare neoplasm that was classified in the 1970s as pleomorphic soft tissue sarcoma for which a definable line of differentiation has not been found.1
At present, these lesions are considered to be a variant of high‐grade sarcoma. MFH tends to develop in the skin in the form of small, slow‐growing nodules, although its behaviour is more aggressive when the lesions are located in deep‐lying tissues.2 It affects males more frequently and mostly occurs at the extremity or the retroperitoneum and pelvis.1, 2 The lesions are usually diagnosed in advanced stages, and despite currently proposed therapies such as radiotherapy or chemotherapy, the patient's prognosis is usually poor, with a tendency of local recurrence and systemic metastasis in relation to the histological grade and size of the tumour.1, 2, 3, 4, 5 We report a case of a 86‐year‐old male with a history of multiple basal cell carcinomas and actinic keratosis, who presented at our unit in March 2012 with a nodular, asymptomatic lesion of the scalp that appeared about 2 years earlier. Clinically, the lesion presented as a red, mobile, nodule, easily bleeding, with a “tomato sea” appearance, which measured 4 cm in diameter (Figure 1 ). The biopsy showed the presence of a stromal malignant tumour, with no cellular differentiation and giants cells. MRI and CT scan revealed no evidence of bone and lymph node invasion. We performed wide local excision (2 cm surgical margins) and reconstruction with full‐thickness skin graft, harvested from the left arm. The patient was discharged the same day of the operation without any complications. Histopathological report showed a solid, everted, and ulcerated neoplasm with the proliferation of atypical stromal elements, numerous atypical and roundish mitosis, eosinophilic cytoplasm, nucleus polymorph and numerous giant cells (Figure 2A,B). Immuno‐histochemical study showed a positive test to CD68 and negative test to CD10, CD34, aktina, desmin, melan‐A, HMB‐45, and S‐100 (Figure 2C). From these findings, the diagnosis of MFH was made, confirming free surgical margins. The radiotherapist and oncologist proposed no treatment, and a strict follow up of the patient and lesion was recommended. A repeat MRI and CT scan every year was advised, and at 6‐year follow up, the patient showed no local recurrence of the tumour, and he had no more tumour lesions on the skin with satisfactory cosmetic outcome of the skin graft (Figure 3 ).
Figure 1.
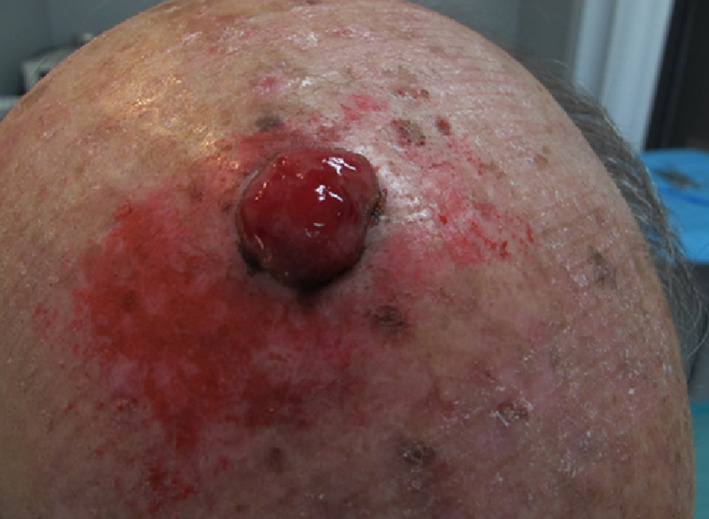
Preoperative presentation of the lesion on the scalp [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 2.
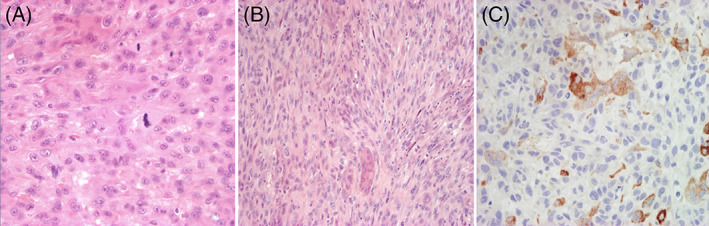
A, Proliferation of atypical stromal elements, numerous atypical and roundish mitosis, eosinophilic cytoplasm, nucleus polymorph, with numerous giant cells (H and E, ×40); B, Spindle cells arranged in fascicles and showing a storiform pattern, with numerous mitoses and multinucleated giant cells (H and E, ×20); C, Positive test to CD68 and negative test to CD10, CD34, aktina, desmin, melan‐A, HMB‐45, and S‐100 (Immunohistochemical test) [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 3.
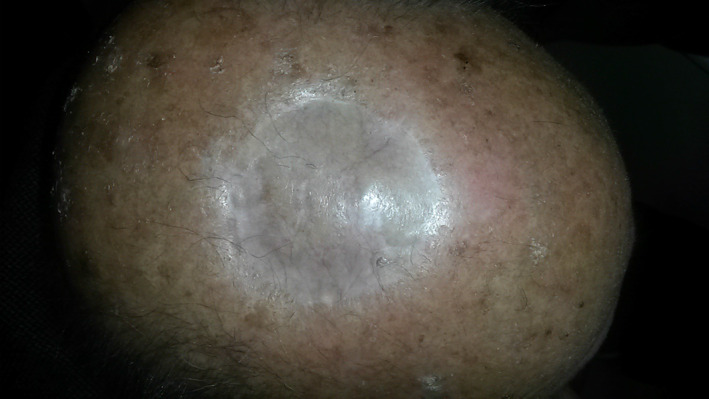
At 5 years of follow up, the patient shows no local recurrence of the tumour, with satisfactory cosmetic outcome of the skin graft [Colour figure can be viewed at wileyonlinelibrary.com]
MFH is a high‐grade sarcoma that was first described by O'Brien and Stout in 1964 and is currently classified as undifferentiated pleomorphic sarcoma.1
The age of presentation ranges between 50 and 70 years, and two‐thirds occur in men. It mostly affects the extremity or the retroperitoneum and pelvis. The region of the head and neck is affected in 3% of cases, the nasosinusal tract being the most common location.1, 2
It originates from pluripotent mesenchymal cells with the ability to differentiate into histiocytes, fibroblasts and myofibroblasts of the muscle or muscle fascia. The aetiology of these tumours is unknown, although, as in all sarcomas, previous radiotherapy in the area may induce its occurrence. There are five histological subtypes: storiform‐pleomorphic, myxoid, giant cell, inflammatory, and angiomatoid. Pleomorphic storiform pattern is the most common, found in two‐thirds of cases. In this variety, a large pleomorphic cell population with high nuclear atypia and mitosis is observed, together with cells with fusiform morphology that adopt a “spoke wheel” or storiform pattern.3
It can present as a cutaneous lesion in the form of a primary tumour or as metastasis from MFH at other sites.3 Approximately two‐thirds of the tumours are located within the skeletal muscle, with fewer than 10% confined to the subcutis. It usually presents as a slow‐growing tumorous lesion, although there is no clinically characteristic presentation that allows for differentiation from other sarcomas3. Definitive diagnosis of MFH relies on histological studies, while immunohistochemistry helps establish the differential diagnosis with other entities with similar morphological patterns.
This tumour does not display features on monocytes or macrophages but, rather, fibroblasts. Reliable histiocytic markers such as EMA, Leu‐3, Leu‐M3, and CD68 have not been identified in this tumour, while fibroblast‐associated antigens can be identified on the surface of the cells. The focal immunoreactivity for intermediate filaments such as keratin or desmin in an otherwise MFH alone is not sufficient to alter the diagnosis.5 Actually, the use of a panel including S‐100, cytokeratin stains, and smooth muscle actin is helpful in differentiating MFH from desmoplastic melanoma, spindle cell squamous cell carcinoma, and leiomyosarcoma. The CD74 has been suggested as a marker to help distinguish MFH from atypical fibroxanthoma.4 The lesions are usually diagnosed in advanced stages, and despite currently proposed therapies, such as radiotherapy or chemotherapy, the patient's prognosis is usually poor with a tendency of local recurrence and systemic metastasis in relation to the histological grade and size of the tumour. Tumour size and depth have been identified as the main prognostic factors.1, 2, 3, 4
Successful treatment and management depends on the achievement of local control; in fact, the relapse rate is 16%–52% and is fundamentally determined by the presence of tumour‐positive surgical resection margins. Therefore, initial aggressive surgical management is indicated in order to obtain complete excision with a negative margin. Radiotherapy is considered postoperatively, although some authors include MFH as a radio‐induced tumour. Chemotherapy appears to be of scant utility. Distant metastases appear in one‐third of cases, involving mainly lung, bone, and liver. Lymph node involvement is less common.1, 2, 3, 4
Postoperative follow up using MRI or PET is recommended, and annual check‐ups should be performed up to 5 years after definitive therapy.
As per our extensive search of the literature and the unique presentation of our patient, we can confidently say that this is, in most likelihood, the first ever reported case of MFH of scalp origin. Despite its infrequency, we emphasise the need of including MFH as one of the rare differential diagnoses of scalp tumours, comprising the sarcomas subtypes and other lesions, to establish an accurate and early diagnosis in order to perform prompt treatment and improve the tumour prognosis.
ACKNOWLEDGEMENTS
The authors wish to thank Dr Julie‐Ann Smith for revising the manuscript. None of the authors have a financial interest in any of the products, devices or drugs mentioned in this manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
REFERENCES
- 1. Weiss SW, Enzinger FM. Malignant fibrous histiocytoma: an analysis of 200 cases. Cancer. 1978. Jun;41(6):2250‐2266. [DOI] [PubMed] [Google Scholar]
- 2. Gibbs J, Huang P, Lee R, et al. Malignant fibrous histiocytoma: an institutional review. Cancer Invest. 2001;19:23‐27. [DOI] [PubMed] [Google Scholar]
- 3. Weiss SW, Goldblum JR. Malignant fibrohistiocytic tumors. In: Gibson LE, ed. Enzinger and Weiss's Soft Tissue Tumors. 4th ed. St. Louis, MO: CV Mosby; 2001:535‐569. [Google Scholar]
- 4. Henderson MT, Hollmig ST. Malignant fibrous histiocytoma: changing perceptions and management challenges. J Am Acad Dermatol. 2012;67:1335‐1341. [DOI] [PubMed] [Google Scholar]
- 5. Lazova R, Moynes R, May D, Scott G. LN‐2 (CD74): a market to distinguish atypical fibroxanthoma from malignant fibrous histiocytoma. Cancer. 1997;79:2115‐2124. [DOI] [PubMed] [Google Scholar]