

Abstract
Janus kinases (JAKs) are non-receptor tyrosine kinases that are essential components of the JAK-STAT signaling pathway. Associated aberrant signaling is responsible for many forms of cancer and disorders of the immune system. The present focus is on the discovery of molecules that may regulate the activity of JAK2 by selective binding to the JAK2 pseudokinase domain, JH2. Specifically, the Val617Phe mutation in JH2 stimulates the activity of the adjacent kinase domain (JH1) resulting in myeloproliferative disorders. Starting from a non-selective screening hit, we have achieved the goal of discovering molecules which preferentially bind to the ATP binding site in JH2 instead of JH1. We report the design and synthesis of the compounds and binding results for the JH1, JH2, and JH2 V617F domains, as well as five crystal structures for JH2 complexes. Testing with a selective and non-selective JH2 binder on the autophosphorylation of wild-type and V617F JAK2 is also contrasted.
Keywords: Janus Kinase 2, JAK2, JAK2 JH2, Pseudokinase, V617F, Structure-Based Drug Design, Fluorescence Polarization, Protein Crystallography
INTRODUCTION
JAK1, JAK2, JAK3, and TYK2 are members of the Janus family of non-receptor tyrosine kinases, which are activated by and mediate the cellular responses induced by binding of a variety of cytokines to specific cytokine receptors.1 Cytokine-induced activation of the JAK-STAT signaling pathway and other intracellular pathways play important roles in the control of cell proliferation, hematopoiesis, and immune functions. In addition to a canonical tyrosine kinase domain (JH1) located in the C-terminal region, JAK proteins contain a pseudokinase domain designated JH2. Though JH2 domains have an ATP-binding site, they show little or no catalytic activity. However, JH2 domains do have a regulatory function for the JH1 kinase activity such that mutations in JH2 can cause hyperactivation leading to numerous diseases and cancer.2 In particular, the single point-mutation Val617Phe (V617F) in JAK2 JH2 is responsible for the majority of myeloproliferative disorders including polycythemia vera, myelofibrosis, and essential thrombocythemia.3,4 Undesirable side effects such as anemia and thrombocytopenia are associated with inhibition of the wild-type kinase activity of the JH1 domain of JAK2.2 Thus, selective inhibition of V617F JAK2 could be therapeutically valuable. Interestingly, mutations of residues that eliminate ATP-binding by JAK2 JH2 were found to enhance the autophosphorylation of wild-type JAK2 by 2–5 fold, while they generally reduced the activity of V617F JAK2 by 5–15 fold.5 An exception was L579F JAK2 mutation, which does not remove ATP binding, and showed reduced effect on V617F JAK2 activation. This work has raised the possibility of selective reversal of the activating effect of the V617F mutation by blocking ATP binding to JAK2 JH2.5,6 To test this hypothesis, our first goal was to discover small molecules that can bind strongly to the ATP binding site of JAK2 JH2, but that bind at most weakly to the ATP binding site of JAK2 JH1. The next step is testing the effects on the autophosphorylation activity of full-length wild-type and V617F JAK2.
Towards these ends, in seeking molecules that bind to the JH2 ATP-site, we reported results of a high-throughput screen that yielded the known pan-CDK and pan-JAK inhibitor JNJ7706621 (1, Figure 1)7,8 as the strongest JH2 binder in a fluorescence polarization (FP) assay.9 Subsequent analysis by isothermal titration calorimetry (ITC) yielded binding constants Kd of 106 nM and 31 nM with JAK2 JH2 and JH1, respectively, showing an undesirable greater than three-fold preference for binding JH1.9 It may be noted that these values are lower than an earlier Kd report of 220 nM for 1 with a JH1-JH2 construct of JAK2 in a competition binding assay.8 Crystal structures for 1 with JAK2 JH2 and JH1 were also determined, as illustrated in Figure 2.9 The binding sites are similar in the hinge regions, Glu627-Phe628-Val629 for JH2, and Glu930-Tyr931-Leu932 for JH1; the diaminotriazole fragment of 1 engages in three hydrogen bonds with the backbone in both cases. In addition, for JAK2 JH2 there are hydrogen bonds between the amino group of 1 and the sidechain oxygen of the gatekeeper, Gln626, and between the carbonyl group of 1 and Lys581. The strong binding of 1 to both JAK2 JH1 and JH2 is consistent with the similar binding modes in the hinge regions and the limited contact of the difluorophenyl substituent. The molecule is U-shaped with the aryl group directed out of the binding site towards the solvent.
Figure 1.

Compound 1 is the known pan-CDK and pan-JAK kinase inhibitor JNJ7706621. Compounds 2 – 15 are JAK2 JH2-binding molecules reported in this work.
Figure 2.
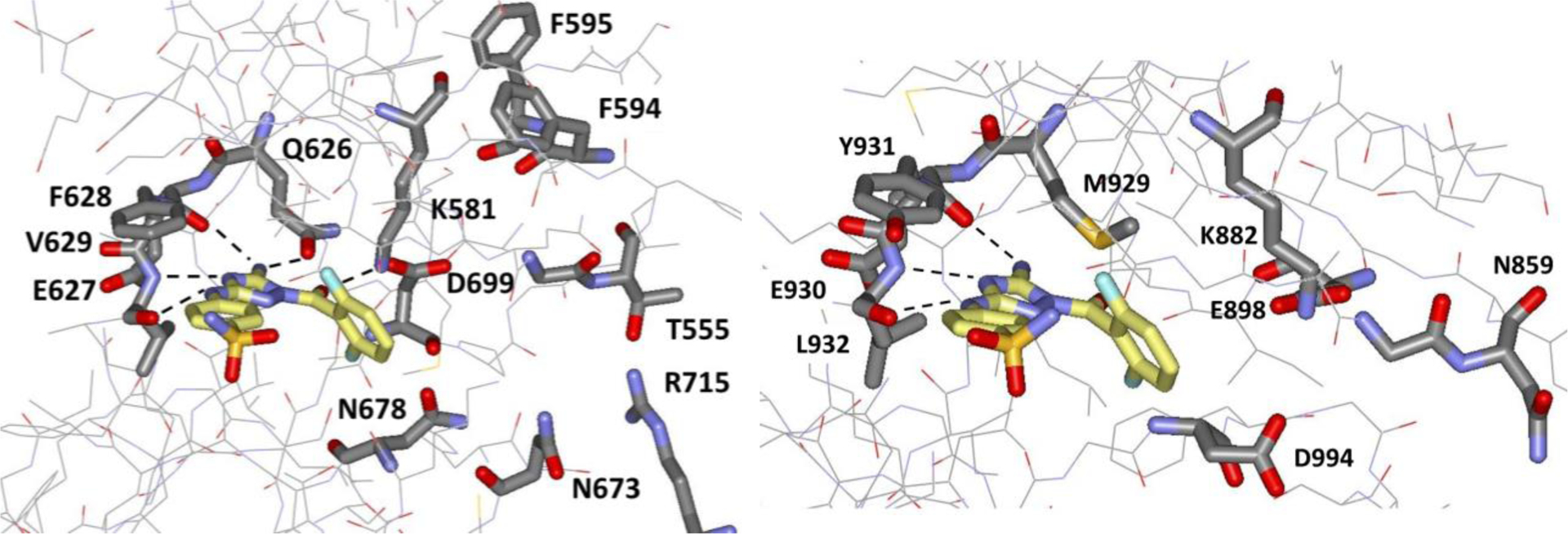
(Left) 1 bound to JAK2 JH2 (PDB ID 5USZ); (Right) 1 bound to JAK2 JH1 (PDB ID 5USY). Hydrogen bonds with rON < 3.6 Å are indicated with dashed lines. Selective binding to JAK2 JH2 is sought by growing the ligands towards the vacant region near T555 and R715.
In order to seek molecules that may selectively bind to the ATP site in JAK2 JH2 and avoid inhibition of the kinase activity of wildtype (WT) JAK2, the immediate goal was to discover small molecules whose affinities are at least as strong as 1 for JAK2 JH2, while showing much diminished affinity for JAK2 JH1. For the present series, 1 was chosen as the starting point with retention of the 3,5-diamino-1,2,4-triazole substructure. In viewing Figure 2, it is apparent that the desired selectivity needs to exploit differences in the vacant eastern part of the binding clefts heading towards Thr555 and Arg715 in JAK2 JH2. However, this is the region where the triphosphate portion of ATP is bound with the terminal phosphate group in a hydrogen-bond cluster with Thr555, Asn678, and Arg715, as shown in Figure 3.10 Thus, it is a very polar region with the concomitant targeting difficulties owing to the competition between an envisioned polar substituent being well-solvated in water and in the polar binding site. Nevertheless, the challenge was taken on with the notion that precise fit of a relatively rigid, polar substituent ending in an anionic mimic of the terminal phosphate might be successful. Competition with ATP is not expected to be problematic since its binding is relatively weak with binding constants of 1.3 μM for both WT JAK2 JH2 and the V617F mutant.5 It should also be noted that the principal structural changes for JAK2 V617F occur in this vicinity.10 Val617 is located above Phe595 in Figure 3 and upon mutation, Phe617-Phe595-Phe594 form an edge-face-edge stack with downward displacement of Phe594 towards the terminal phosphate in the ATP-bound structures.10 Phe595 has been shown to be indispensable for the constitutive activity of JAK2 V617F; mutation to non-aromatic residues significantly decreases the activity.11 Thus, exploration of alternative ligands that occupy the ATP site could reasonably be expected to affect the activity of JAK2 V617F, e.g., by reversing the displacement of F594.
Figure 3.

Rendering from the crystal structure for ATP bound to JAK2 JH2 (PDB ID 4FVQ).
RESULTS AND DISCUSSION
Design and Assays.
For thoroughness, molecular design emphasized inspection of hundreds of trial structures for JAK2 JH2 complexes built with the BOMB (Biochemical and Organic Model Builder) program12 and energy-minimized with MCPRO13 using the OPLS-AA/M force field for proteins14 and OPLS/CM1A for ligands.15 Free energy perturbation (FEP) calculations were also carried out in some cases to better predict potential differences in free energies of binding for analogues;12 these calculations included ca. 2000 explicit water molecules and extensive sampling of configurations for the JAK2 JH2 protein, ligands and water. The principal assays measured binding constants with JAK2 JH2, V617F JH2, and JH1 via fluorescence polarization (FP)16 using a fluorescein-conjugate of 1 as the tracer.17 The assay followed the previous description17 with minor changes to the buffer composition, as fully described in the Experimental section. For several compounds, microscale thermophoresis was carried out as an additional approach for obtaining Kd values. The present measurements like the prior ITC and FP assays were carried out at pH 8.0, which provided more consistent results than current tests at pH 7.0 and 8.5.
For the FP assays with JAK2 JH2, 1 was used as a control compound so multiple Kd results were obtained yielding an average value of 0.456 ± 0.124 μM (Table 1). The result from the MST measurements, 0.489 ± 0.084 μM, was notably consistent given the differences in the utilized buffers, as detailed in the SI. These values are somewhat lower than the previous FP result of 0.80 ± 0.05 μM,17 and they are significantly higher than the ITC result of 0.106 μM,9 and of a recent report of 0.094 μM from intrinsic tryptophan fluorescence.18 We have also assayed a second compound, BI-D1870, that was tested in reference 18; our Kd of 2.0 ± 0.2 μM is again several-fold higher than the 0.62 ± 0.04 μM in the prior report.18 Clearly alternative binding assays are yielding somewhat different results, which likely reflects differences in the assay conditions and preparation of the protein. Thus, it is advisable to make comparisons relative to a common control, e.g., compound 1.
Table 1.
Binding Affinity for JAK2 JH2 (Kd, μM) from the FP Assay
| Compd | Kd (μM)a |
|---|---|
| 1 | 0.456 ± 0.124 |
| 2 | 16.7 ± 5.4 |
| 4 | 4.7 ± 0.8 |
| 5 | 47.8 ± 9.2 |
| 6 | 30.5 ± 3.6 |
| 7 | 12.3 ± 0.6 |
| 8 | 1.9 ± 0.1 |
| 9 | 0.643 ± 0.019 |
| 10 | 0.571 ± 0.034 |
| 11 | 4.0 ± 0.3 |
| 12 | 0.346 ± 0.034 |
| 13 | 0.439 ± 0.064 |
| 14 | 7.0 ± 0.1 |
| 15 | 0.374 ± 0.013 |
Kd data from quadruplicate measurements in three independent assays. Mean ± SEM.
Synthesis of New Compounds.
The new compounds reported here are numbered 2 – 15 in Figure 1; complete synthetic details and characterization data are reported in the Experimental section. However, the general approach can be illustrated with the synthetic route for 12 in Scheme 1. The key coupling step is the regioselective acylation of a 1H-[1,2,4]triazole-3,5-diamine7 with a phenylcarbamate (step e in Scheme 1). Of course, obstacles arose. Indeed, for the coupling, the original intention was to employ a common procedure for urea formation, the reaction of two amines with triphosgene in the presence of triethylamine. This strategy afforded compound 3 in ~10% yield, but failed for more complex ureas, even when attempted in several different solvents (DCM, THF, dioxane, DMSO) and at various temperatures (r.t., 80 °C, 130 °C). The problems appeared to be associated with the poor solubility in organic solvents for the polar diaminotriazole precursor and the presence of its tautomeric 2-H form, which leads to the undesirable and difficult-to-separate, regioisomeric 2-H byproduct. In search of a more efficient route, phenyl carbamates were investigated as alternative carbonyl sources.19 This approach proved to be effective in generating the desired products when dioxane was used as the solvent, but the yields were not optimal since the problems with the diaminotriazole precursor remained. Better results were obtained by increasing the reaction temperature from 80 to 110 °C, extending the reaction time, and diluting the mixture from 1.0 to 0.5 M to improve dissolution of the diaminotriazole. Finally, the use of DCM:MeCN as the solvent system for the chromatography allowed for the successful purification of ester precursor of 12.
Scheme 1. Synthesis of Compound 12.a.

aReagents and conditions: (a) Boc2O, Et3N, iPrOH, r.t., 14 h; (b) 10 mol % Pd(OAc)2, 20 mol % JohnPhos, Cs2CO3, dioxane, 110 °C, 19 h; (c) TFA, DCM, 0 °C to r.t., 30 min; (d) NaHCO3, H2O/THF, 0 °C, 1h; (e) Et3N, dioxane, 110 °C, 50 min; (f) DBN, LiBr, MeCN, 2 vol % H2O, r.t., 68h.
In several cases preparation of the carbamates also proved challenging. For 12, the original plan to prepare the 2-aryloxazole (step c in Scheme 1) was to utilize regioselective, palladium-catalyzed direct arylation of the commercially available ethyl oxazole-4-carboxylate with unprotected 4-iodo aniline, under the conditions described by Verrier et al.20 However, the suggested conditions (5 mol % Pd(OAc)2, 10 mol% JohnPhos) proved inefficient, affording the desired product in single-digit yields. Consequently, it was decided to protect the aniline and repeat the arylation with increased catalyst and ligand loading (10 mol % Pd(OAc)2, 20 mol % JohnPhos). This approach allowed access to the 2-anilinyloxazole in a viable yield (30%). And, finally, hydrolysis of the ester precursor of 12 in the last step required care. Standard conditions using strong, nucleophilic bases led to decomposition of the urea. Fortunately, heteroatoms were present in the α or β-position of most ester precursors, which allowed use of the mild ester hydrolysis method introduced by Mattsson et al.21 In this transformation, lithium coordination to the carbonyl group and the neighboring heteroatom increases the electrophilicity and thus selectivity of the ester toward nucleophilic attack, allowing hydrolysis by water at room temperature. However, long reaction times were required due to the low solubility of the compounds in the reaction media.
Lead Optimization.
An initial question that was addressed was the importance of the carbonyl group in 1 for binding to JAK2 JH2. Though the carbonyl oxygen atom participates in a hydrogen bond with the ammonium group of Lys581 (rNO = 3.16 Å), it is in repulsive contact, 3.06 Å, with the sidechain carbonyl oxygen atom of Gln626 (Figure 2). Thus, 2, the desoxy analog of 1, was prepared; it was found to yield a much-reduced affinity for JAK2 JH2 with a Kd of 16.7 μM in the FP assay (Table 1). Apparently, the hydrogen bond with the charged ammonium group more than offsets the repulsion with Gln626 and/or the reduced torsional flexibility of 1 compared to 2 is beneficial.
Having established the desirability of the carbonyl group, the next step was to append a substituent that should project eastwardly past Lys581 towards Thr555 rather than outward from the binding site as for 1. Though several alternatives were considered, a secondary amide linkage modeled well, especially with an N-phenyl substituent that might form a cation-π interaction with Lys581. This notion led to the synthesis of 3 and 4, which only differ by the p-anilinyl substituent being cyano or sulfamyl. In general, we have observed little difference in binding affinity for these alternatives, but the lesser solubility of the cyano analogs sometimes led to assay difficulties as for 3. The FP result for the N-phenylamide 4 with a 3-fold improvement over 2 to 4.7 μM was encouraging in view of the fact that the phenyl ring is being placed in a polar environment. It was also possible to obtain crystal structures for the complexes of both 3 and 4 with JAK2 JH2 at 1.94-Å and 1.90-Å resolution, respectively. The structures clearly reveal the desired eastwardly projection and the cation-π interaction with Lys581, as illustrated in Figure 4 for 4. The hydrogen-bond length between the amide carbonyl oxygen of 4 and the ammonium nitrogen atom of Lys581 is 2.85 Å, and the distances between the ammonium nitrogen and the carbon atoms of the phenyl ring are as short as 3.92 Å for the ipso carbon atom and 3.77 Å for the nearer ortho carbon atom, while the hydrogen bonding in the hinge region is essentially the same for 1, 3 and 4.
Figure 4.
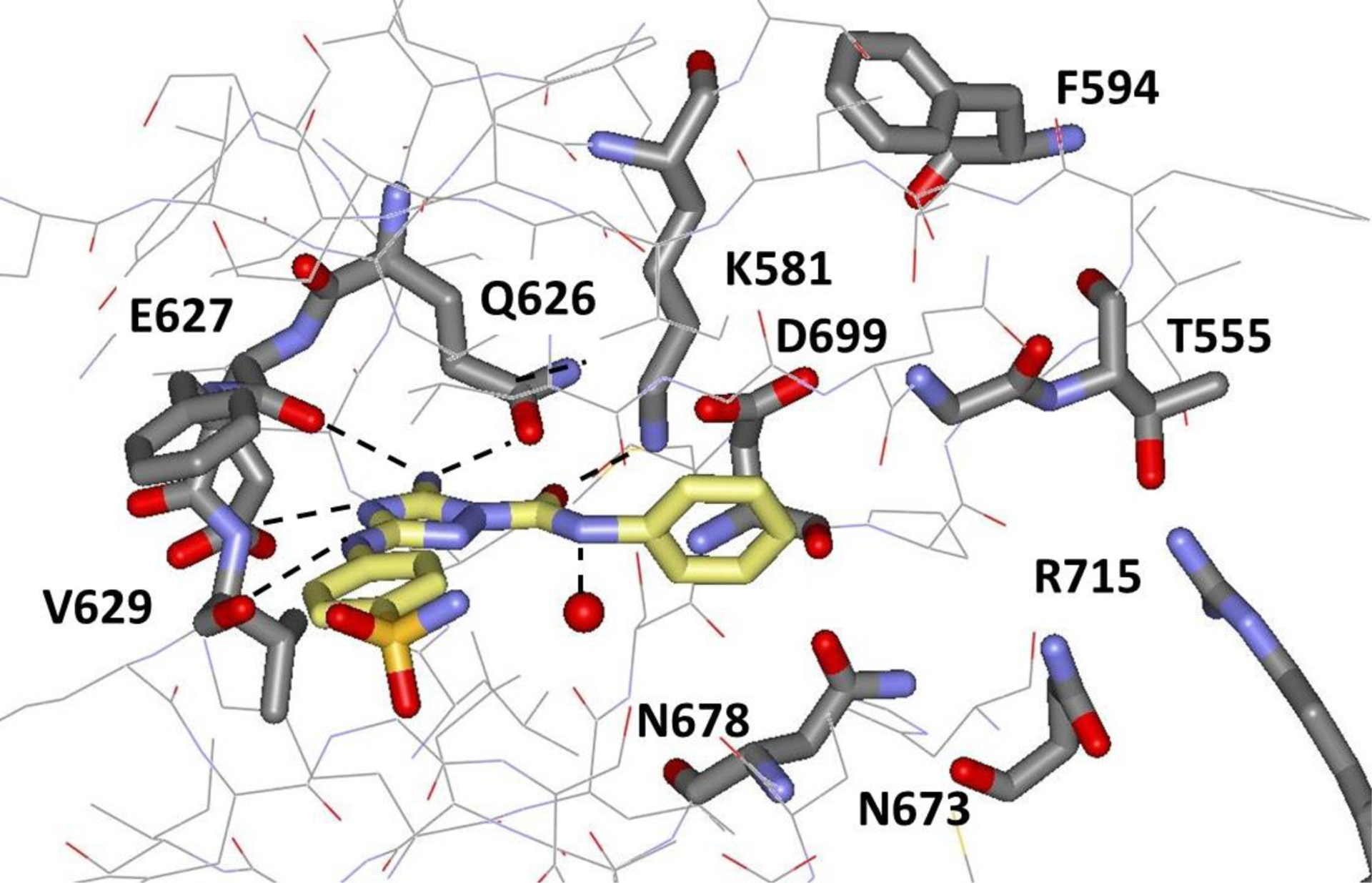
Rendering from the 1.90-Å crystal structure for 4 bound to JAK2 JH2 (PDB ID 6OBB).
To expand the structure-activity data, 5, the tertiary methylamide corresponding to 4 was checked. Since there is a water molecule hydrogen-bonded to the amide NH in 4 (rNO = 3.11 Å, Figure 4), it might seem likely that 4 and 5 would have similar Kd values. However, a conformational issue should be noted for acetanilides. From small-molecule crystallography and quantum chemical calculations, it is known that acetanilide prefers the Z-conformer, while N-methylacetanilide prefers the E-conformer.22,23 We confirmed this with DFT B3LYP/6-31G(d) geometry optimizations and subsequent single-point energy evaluations at the MP2/6-311+G(d,p) level.24 The B3LYP and MP2 calculations favor the Z-conformer for acetanilide by 3.19 and 1.29 kcal/mol, respectively, while the E-conformer is preferred by 3.87 and 3.39 kcal/mol for N-methylacetanilide (Scheme 2). Assuming this preference carries over to 4 and 5, there would be a significant conformational penalty for 5 to achieve the Z-conformation that is needed for the cation-π interaction with Lys581. As listed in Table 2, FEP calculations were also performed for the conversion of Z-5 to Z-4; when adjusted for the conformational penalty, weaker binding is expected for 5. This was borne out by the measured Kd of 47.8 μM for 5 in Table 1.
Scheme 2.

Computed Conformational Preferences (kcal/mol) for Acetanilides
Table 2.
Differences in Free Energies of Binding with JAK2 JH2 from FEP Calculations
| A → B | Transformation | ΔGb (kcal/mol)a |
|---|---|---|
| 5 → 4 | (Z)-O=CNMePh → O=CNHPh | 1.47 ± 0.54 |
| 6 → 4 | 2-pyridinyl → phenyl | −0.43 ± 0.59 |
| 7 → 4 | 3-pyridinyl → phenyl | −1.44 ± 0.45 |
| 4-Pyrb → 4 | 4-pyridinyl → phenyl | −0.64 ± 0.16 |
4-pyridinyl analogue of 6 or 7.
Pyridinyl replacements 6 and 7 for the phenyl group were also considered. Initial structure building with BOMB indicated that constructive interaction of the pyridinyl nitrogen atoms with Lys581 might be possible with N-N contacts of 3.32 Å for 6 (Figure 5) and 3.72 Å for 7. Energy minimizations with the force-field find a more favorable interaction with the protein by 3.0 kcal/mol for 6 than 7; however, this is offset by a 3.4 kcal/mol conformational penalty for placement of the pyridine nitrogen atom syn rather than anti to the carbonyl oxygen in 6, which is needed for the hydrogen bond with Lys581. B3LYP/6-31G(d) optimizations for the corresponding syn and anti conformers of N-(pyridine-2-yl)acetamide indicate an even higher conformational penalty to achieve the syn conformer of 6, 8.98 kcal/mol.24 For both 6 and 7, the azine ring is now basically in-plane with the amide group, which replaces the cation-π interaction found for 4 with Lys581 (Figure 4) with a hydrogen bond (Figure 5). Results of FEP calculations were also obtained for conversion of 6 and 7 to 4 with the predictions that the pyridinyl analogs would be weaker binders for JAK2 JH2 (Table 2). The FEP calculations for 6 considered multiple starting conformers; both conformers with the pyridine nitrogen syn or anti to the carbonyl group were stable and the results indicated that the syn conformer is favored by 0.7 ± 0.2 kcal/mol. The compounds were synthesized and the Kd results of 30.5 (6) and 12.3 (7) μM confirmed the weaker binding than for 4 (Table 1). These relatively simple changes illustrate well the challenges in making even qualitatively correct predictions for differences in protein-ligand binding owing to the complexities of the inter- and intra-molecular energetics and solvation.
Figure 5.

Modeled structure illustrating the potential hydrogen bonding between Lys581 and the amide carbonyl oxygen atom and the pyridinyl nitrogen atom for 2-pyridinyl analogues of 3 and 4 such as 6.
The next step in the design was to consider substituents in the para position of the phenyl ring of 4 that would extend further towards Thr555 and Arg715 (Figure 4). In view of the placement of the terminal phosphate of ATP in this region (Figure 3), substituents terminating in an anionic group seemed desirable. Modeling was performed for various carboxyalkyl and carboxyalkoxy alternatives with the conclusion that carboxymethoxy should be particularly promising; the OCH2 fragment should be in-plane with the phenyl ring, as in anisole, and the carboxylate group should be extended to form hydrogen bonds with Thr555. For comparison, both carboxyethyl and carboxyethoxy were expected to incorporate a gauche dihedral angle and be less extended. Thus, the carboxymethoxy analogues 9 and 10 were synthesized and assayed along with 8, the methyl-ester precursor of 10. The results were gratifying with a ca. 8-fold boost in binding strength with JAK2 JH2 for 9 (0.64 μM) and 10 (0.57 μM) compared to 4. The corresponding result for the ester 8 (1.9 μM) confirmed the importance of an anionic terminus for the substituent, though it is not obvious intrinsically that a carboxylate group would yield any improvement in binding since it would be well hydrated unbound in water. 11, the meta-substituted isomer of 9 was also synthesized and showed reduced binding with a Kd of 4.0 μM (Table 1). The modeling in this case predicted that the substituent would likely be directed into the solvent from the upper meta position in Figure 4.
It was also possible to obtain crystal structures for the complexes of 9 and 10 with JAK2 JH2 at resolutions of 2.06 Å and 1.71 Å, respectively. As shown in Figure 6, the carboxylate group of 10 is extended, as expected, with a COCC dihedral angle of 170°. It is positioned similarly to the terminal phosphate group of ATP in Figure 3, and it forms hydrogen bonds with the backbone NH and side-chain OH of Thr555 with lengths of 3.06 and 2.83 Å. As expanded upon in Figure 7, the carboxylate group also participates in an extensive network of hydrogen bonds with localized water molecules in the crystal structure including three hydrogen bonds between the carboxylate oxygen atoms and water molecules.
Figure 6.
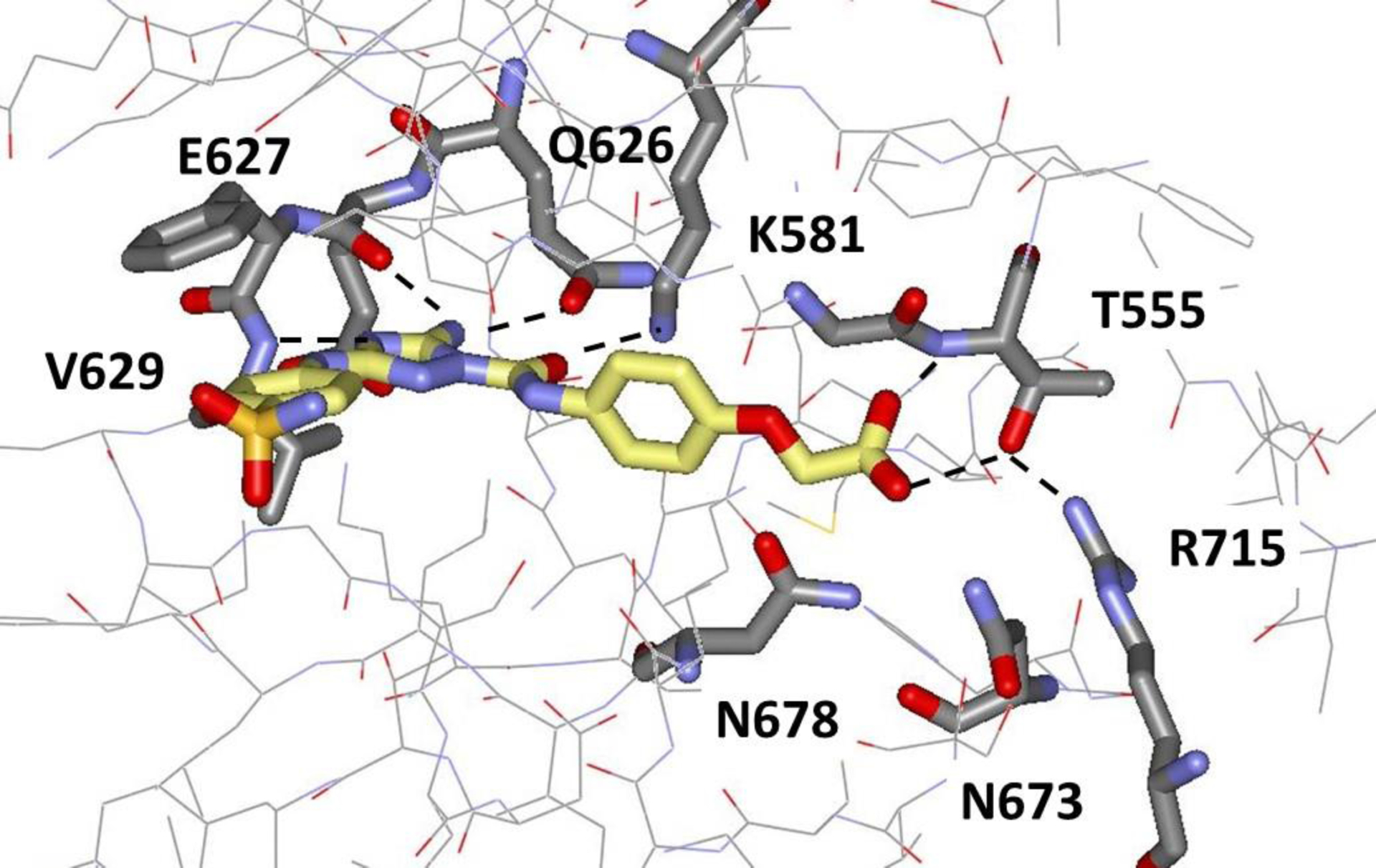
Rendering from the 1.71-Å crystal structure for 10 bound to JAK2 JH2 (PDB ID 6OBF).
Figure 7.
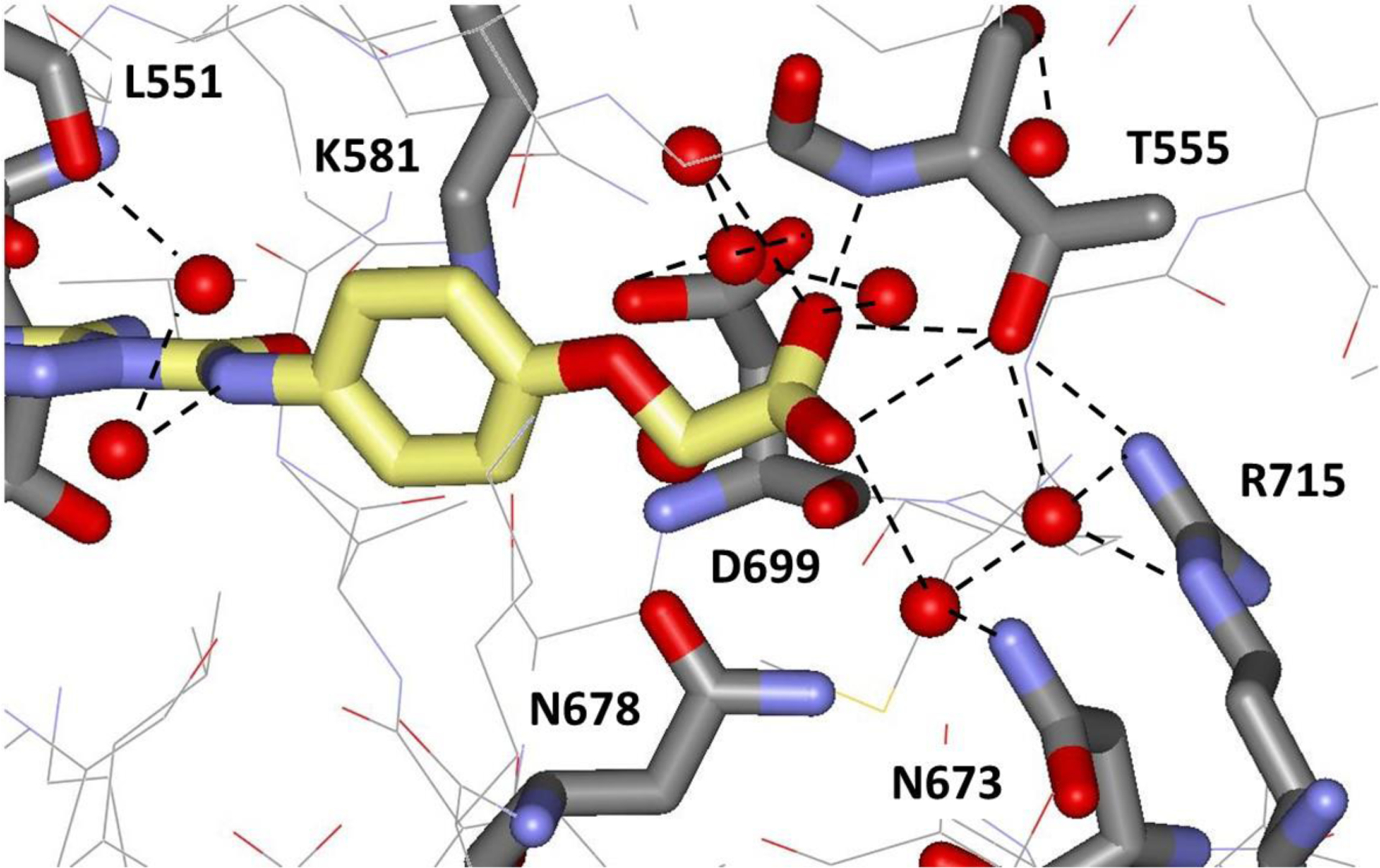
Illustration of the hydrogen-bonding in the vicinity of the carboxylate group of 10 from the 1.71-Å crystal structure with JAK2 JH2 (PDB ID 6OBF). Red spheres represent oxygen atoms of localized water molecules.
In viewing the crystal structure for 10, it was felt that a 1-Å additional extension of the carboxylate group towards Arg715 to form a salt bridge would likely be beneficial for both binding and selectivity. Addition of a methylene group was not expected to be optimal based on the modeling noted above. Instead, replacement of the methoxy linking group with a five-membered heterocycle yielded computed structures that delivered the desired contact. The added rigidity of the heterocycle was also viewed as a desirable feature, though there were concerns about unfavorable disruption of the hydrogen-bonding networks (Figure 7). More than 30 alternative structures were built with BOMB and visually inspected; our preference was for heterocycles with multiple hydrogen-bonding sites. The syntheses brought new challenges; however, several illustrative compounds were prepared starting with the oxazole-4-carboxylic acid 12 (Scheme 1). The FP assay result of 0.346 ± 0.034 μM showed the validity of the design and yielded the first compound that is a clearly stronger binder for JAK2 JH2 than JNJ7706621 (1). Again, we were fortunate to obtain a crystal structure for this complex at a resolution of 2.03 Å (Figure 8). The shortened contact with Arg715 was achieved with an O…N distance of 3.56 Å, while maintaining the hydrogen bonds with the NH (2.98 Å) and side-chain OH (2.70 and 3.58 Å) of Thr555. There are again three water molecules hydrogen-bonded to the carboxylate group with O…O separations of 2.28, 2.82, and 3.09 Å. The water molecule with the 2.28-Å contact is part of a striking triangle of hydrogen-bonded water molecules that bridges between the carboxylate groups of the ligand and Asp699. The central water molecule in the cluster is also hydrogen-bonded to the nitrogen atom of the oxazole at a distance of 2.79 Å. Furthermore, the cluster is in van der Waals contact with a meta carbon atom of Phe594 with separations of 3.37, 3.91, and 4.27 Å. Thus, this interesting water trimer is providing a carefully arranged molecular cushion between the ligand and JAK2 JH2 residues Asp699 and Phe594.
Figure 8.
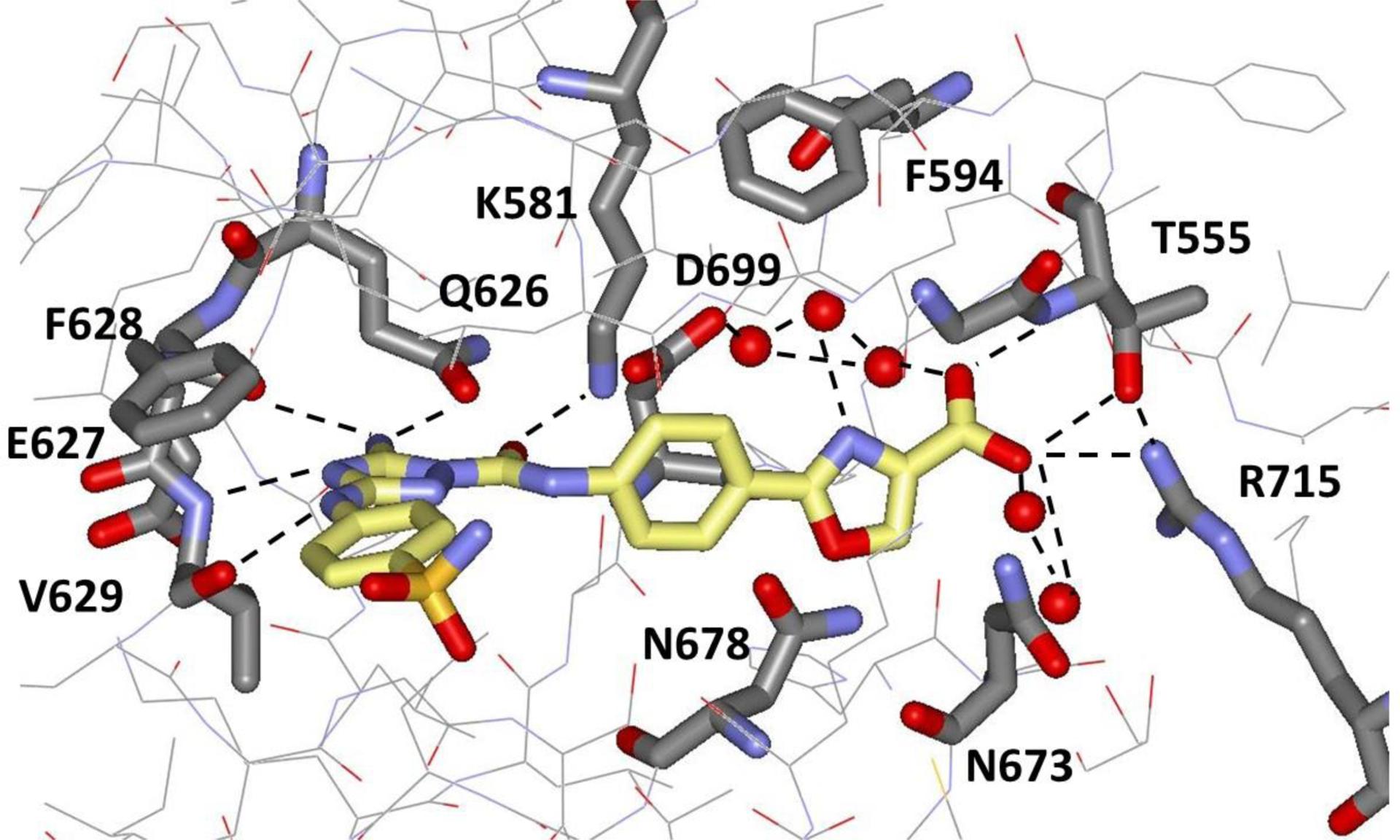
Rendering from the 2.03-Å crystal structure for 12 bound to JAK2 JH2 (PDB ID 6OCC). Red spheres represent oxygen atoms of localized water molecules.
Among additional five-membered heterocycles as linking groups, the furan (13) and 1,3,4-oxadiazole (14) analogues of 12 were synthesized. These were expected to be less robust binders for JAK2 JH2 than the oxazole 12. First, a furan-type oxygen atom is a weak hydrogen-bond acceptor and its replacement of the oxazole nitrogen atom in Figure 8 would weaken the link to the water cluster. Furthermore, furan and thiophene are the most lipophilic five-membered heteroaromatic molecules with octanol/water log P values of 1.34 and 1.81 versus, for example, 0.12 for oxazole, 0.08 for isoxazole, and 0.44 for thiazole.25 Finally, the oxazole oxygen atom in the crystal structure for 12 (Figure 8) does not participate in a hydrogen bond and, in fact, there is no room for a neighboring water molecule. Thus, if the oxadiazole ring in 14 is oriented as for 12 in Figure 8, there would be a significant penalty for desolvation of one of the nitrogen atoms. The qualitative expectations were borne out by the measured Kd values of 0.44 and 7.0 μM for 13 and 14 (Table 1).
From viewing the crystal structure for 10 (Figures 6 and 7) and modeling with BOMB, the (E)-cinnamic acid analog 15 was also suggested with the vinyl group replacing the OCH2 linker. This compound was synthesized and turned out to be a somewhat better binder than 10 with a Kd of 0.37 μM. The improvement can be attributed to the greater rigidity of 15 stemming from removal of one torsional degree of freedom. It is clear from the modeling of 15 and the crystal structures of ATP, 10, and 12 bound to the JH2 domain in Figures 3, 6, and 8 that the terminal carboxylate group in the designed compounds overlaps closely with the terminal phosphate of ATP; in all cases hydrogen bonds are formed between these groups and both the hydroxyl group and backbone NH of Thr555.
JH2/JH1 Selectivity.
The next step was to study the selectivity of the reference compound 1 and the potent new compounds 10, 12 and 15 towards wild-type JAK2 JH1 and JH2, and the V617F JH2 variant. Kd results were obtained in all cases via fluorescence polarization using the same tracer.17 As listed in Table 3, the pan-JAK binder JNJ7706621 (1) shows no significant selectivity in the FP assays towards the three proteins, which is in accord with prior reports and expectations from the crystallography (Figure 2).8,9,18 However, as designed, compound 10 exhibits strong (ca. 75-fold) selectivity for binding the wild-type or V617F JH2 pseudokinase domain over the JH1 kinase domain. For the oxazole 12 and cinnamic acid 15 there are also ca. 20-fold preferences for binding wild-type JH2 over the kinase domain, and there are again modest differences in binding for wild-type JH2 and the V617F variant.
Table 3.
Binding Affinities (Kd, μM) from the FP and MST Assays for JAK2 Domainsa
| JH1 | JH2 | JH2 | V617F JH2 | V617F JH2 | |
|---|---|---|---|---|---|
| Cmpd | FP | FP | MST | FP | MST |
| 1 | 0.67 ± 0.18 | 0.46 ± 0.12 | 0.49 ± 0.08 | 0.60 ± 0.09 | 0.82 ± 0.12 |
| 10 | 42.3 ± 2.3 | 0.57 ± 0.03 | 0.31 ± 0.03 | 0.54 ± 0.13 | 0.40 ± 0.05 |
| 12 | 6.6 ± 0.9 | 0.35 ± 0.03 | 0.29 ± 0.07 | 0.48 ± 0.14 | 0.43 ± 0.03 |
| 15 | 6.0 ± 1.0 | 0.37 ± 0.01 | 0.20 ± 0.05 | 0.18 ± 0.08 | 0.23 ± 0.03 |
In addition, eight Kd values were determined using microscale thermophoresis.26 Optimization of this assay was possible for the wild-type JH2 and JH2 V617F domains, but an adequate signal-to-noise ratio could not be obtained for JH1. MST was found to give similar Kd values as FP (Table 3), though some differences could be expected in view of the variations in optimized buffer compositions. With MST, compounds 10 and 12 are found to have identical Kd values within the error limits for JAK2 JH2 near 0.30 μM, which improves on the 0.49 μM result for 1. The binding constants for JAK2 JH2 and the V617F variant are again close. Finally, compound 15 is found with MST to be the most tenacious binder with Kd values at the 200-nM level for both the JH2 and V617F JH2 domains.
At this point the primary goals were achieved. Compounds were discovered that show good selectivity for binding to the JAK2 JH2 domain in preference to the JH1 kinase domain and that were at least as potent as the non-selective JNJ7706621 (1), which has previously been reported from alternative binding measurements to give Kd values of 0.106 and 0.094 μM.9,18
Results with Full-Length Wild-Type and V617F JAK2.
The next step was to test compounds 1 and 10 for their influence on the autophosphorylation of wild-type and V617F JAK2 mutant. These compounds were chosen to contrast the non-selective JH1/JH2 binder 1 with the JH2-selective 10. The expectation was that 1 would be an inhibitor of the JH1-based kinase activity for both the WT and variant proteins, while the outcome with 10 was unclear. Though the mutational studies suggested that displacement of ATP from the JH2 domain by selective binders could deactivate V617F JAK2,5 the JH2 binders may also be viewed as potential ATP surrogates leading to no effect. Since activation of kinase activity with some mutations was also observed, this outcome is also possible with the present compounds.
As detailed in the Experimental Section, full-length cDNAs encoding human WT and V617F JAK2 (residues 1–1132) were amplified by PCR, cloned into an expression plasmid, and the JAK2 constructs were expressed in HEK293T cells grown at 37 °C. The transfected cells were lysed and the JAK2 proteins were immunoprecipitated from the supernatant and used to measure kinase activity by treatment with [γ−32P]ATP in the presence or absence of increasing concentrations of 1 and 10. The autophosphorylation reaction was stopped after 15 min at 30 °C and the extent of phosphorylation was gauged by autoradiography. Details of the protocols were worked out with compound 1 to establish good signal-to-noise and reproducibility in triplicate experiments.
Though 1 has previously been established to bind to the JH1 and JH2 domains of JAK2,8,9,17,18 its status as an inhibitor has not been determined until now. As shown in Figure 9A, we do find that 1 is an inhibitor of the autophosphorylation of both WT and V617F JAK2. There are steady, dose-dependent decreases in phosphorylation in the presence of 1. It was possible to quantify the gel bands from the phosphor autoradiography to yield IC50 values of 2.96 ± 0.41 μM for 1 with WT JAK2 and 11.05 ± 3.21 μM with V617F JAK2. These results seem consistent with the Kd of 0.67 μM for 1 with the JH1 kinase domain of JAK2 (Table 3).
Figure 9.

In vitro [γ−32P]ATP kinase activity assay of immunoprecipitated full-length JAK2. Autoradiography of autophosphorylated JAK2 wild-type and V617F mutant exposed to different concentrations of (A) 1, and (B) 10. Shown is representative data from experiments performed in triplicate.
In contrast, with the JH2-selective 10 higher concentrations are needed to see clear effects (Figure 9B). Inhibition of WT JAK2 is apparent above ca. 10 μM, and an IC50 of 46.53 ± 4.74 μM could be assigned. The weak inhibition of the kinase activity is consistent with the Kd of 42 μM that was found for 10 with JAK2 JH1 (Table 3). Thus, it is expected that 10 is binding to the JH2 domain at low concentrations, but it has negligible effects on the kinase kinetics until significant binding to the ATP-site in the JH1 domain sets in at concentrations above 10 μM. For V617F JAK2, the effects of 10 are further diminished with little inhibition apparent below 25 μM, with an IC50 in the 100 – 200 μM range. Although 10 is expected to be binding to the ATP-site in the pseudokinase JH2 domain at low concentrations (Table 3), it is not affecting the kinase kinetics. This result contrasts the expectations from the mutational studies,5 though it is consistent with an alternative view of JH2-selective 10 as an ATP surrogate. The present finding for the one compound 10 does not imply that all JH2-selective binders will yield qualitatively similar results. If sufficient structural variety in JH2-binders can be explored, there will be a range of structural effects on the JH2/JH1 interface that can be expected to provide alternative outcomes.
CONCLUSION
Janus kinases are intracellular, non-receptor tyrosine kinases that are activated by cytokine binding to cytokine receptors resulting in stimulation of the JAK-STAT signaling pathway. Aberrant activation of this signaling pathway is associated with numerous immune disorders and cancer. The present focus is on selective reduction of the hyperactivation that occurs with the V617F mutation in the JH2 domain of JAK2 kinase, which is the primary source of myeloproliferative neoplasms. The initial goal has been to discover small molecules that selectively bind to the ATP binding site in the JH2 pseudokinase domain of JAK2 in preference to the ATP binding site in the JH1 kinase domain. Starting from the non-selective, multi-kinase inhibitor JNJ7706621 (1), new compounds were systematically designed, synthesized, and assayed that ultimately yielded compounds such as 10, 12, and 15, which bind more strongly to JAK2 JH2 than 1 and that show a substantial preference for binding JAK2 JH2 over JH1. The design was particularly challenging owing to the polarity of the ATP binding sites in the area which is amenable to differentiation of binding to JAK2 JH1 and JH2, and which is proximal to the region affected structurally by the V617F mutation. The ability to obtain multiple crystal structures for complexes with JAK2 JH2 was most valuable in monitoring the progress of the designs, and the ability to perform fluorescence polarization assays for binding to all three component proteins, JAK2 JH1, JH2, and V617F, was essential. Initial further exploration of 1 and 10 was pursued by examining the effects of the compounds on autophosphorylation of full-length JAK2 and V617F JAK2. 1 was established to be a low-μM inhibitor of both enzymes, while 10 showed little effects on the kinase activity below concentrations of ca. 25 μM. The interpretation is that, in contrast to expectations from mutational studies,5 10 binds well to the ATP-site of the pseudokinase JH2 domain and performs much the same function as ATP. However, it may also be expected that alternative JH2 ligands may perturb the proximal JH1/JH2 interface in different manners and lead to possible deactivation or activation of the oncogenic V617F JAK2.
EXPERIMENTAL SECTION
Generation of Recombinant Baculoviruses.
The isolated JH1 and JH2 domains of human JAK2 were expressed in baculovirus-infected Sf9 insect cells and purified similarly to the previously reported procedure.9,17 The two JH2 domain constructs contain residues 536–812 (with either mutations W659A, W777A, F794H, or mutations W777A, F794H, V617F), followed by a C-terminal thrombin cleavage site and 6×His-tag. The reported JH1 domain construct included an N-terminal 6×His-tag, followed by a TEV cleavage site and residues 840–1132. Recombinant bacmid and baculoviruses were generated using the Bac-to-Bac baculovirus expression system (Invitrogene). DH10Bac competent cells were transformed with recombinant pFastBac plasmid containing the gene of interest to generate the recombinant expression bacmid. P1, P2 and P3 baculovirus stocks were produced according to the manufacturer’s instructions. Sf9 cells were grown in HyClone SFX-Insect cell culture media (GE Healthcare) at 27 °C.
JAK2 Protein Expression and Purification.
Sf9 cells were grown in HyClone SFX-Insect cell culture media to a density of 2.5–4.0×106 cells/mL, followed by transfection with P3 baculovirus stock. After incubation for 48 h at 27°C, cells were harvested and separated from the supernatant by centrifugation (4000 rpm, 30 mins). Purification of the JH1 and JH2 domains of JAK2 was performed in an identical manner. Cell pellets were resuspended in lysis buffer composed of 20 mM Tris pH 8.0, 500 mM NaCl, 20% glycerol, 0.25 mM TCEP, and cOmplete EDTA free protease inhibitor (Roche). Cells were lysed by sonication, followed by pressure homogenization using an Emulsiflux cell disruptor (Avestin). Lysate was separated from cell debris by centrifugation (45 min, 16,500 rpm). Ni-NTA agarose beads (Qiagen) were added in batch mode, and incubated for 2 h at 4°C. Beads were washed with lysis buffer containing 10 mM imidazole, and JAK2 protein was eluted with lysis buffer containing 200 mM imidazole. The eluate was dialyzed overnight at 4°C using a MWCO 3.5 kDa Slide-A-Lyzer dialysis cassette (Thermo Fisher Scientific) against a low salt dialysis buffer composed of 20 mM Tris pH 8.0, 25 mM NaCl, 20% glycerol, and 0.25 mM TCEP. The dialysis product was filtered through a 0.45 μm membrane and loaded onto a pre-equilibrated Mono Q HR 16/19 column (GE Healthcare) linked to an ÄKTA pure protein purification system (GE Healthcare). Protein was eluted applying a linear gradient starting with dialysis buffer and ending with dialysis buffer containing 500 mM NaCl. JAK2 fractions were pooled and applied to a Superdex 75 10/300 (GE Healthcare) pre-equilibrated with a buffer composed of 20 mM Tris pH 8.0, 100 mM NaCl, 10% glycerol, and 1.0 mM TCEP. Purified protein was aliquoted, flash-frozen in liquid nitrogen, and stored at −80°C.
Competitive FP assay.17
In a flat black bottom 96 well plate (Corning), 200 μL of FP buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 20% Glycerol, 0.5 mM TCEP, 0.01% Tween 20) were added to column 1 (blank), 150 μL to column 2, and 140 μL to columns 3–12. 10 μL of 2.96 μM of JAK2-JH2 WT (3.52 μM for JAK2-JH2-VF, and 6.93 μM for JAK2-JH1), were added to columns 3–12, followed by the addition of 2 μL of DMSO to columns 1–3. 2 μL of inhibitor in DMSO at different concentrations were added from column 4 to 12. 50 μL of 24 nM of tracer were added to columns 2–12. Fluorescence polarization was measured at λexc = 485 ± 20 nm, λem = 535 ± 25 nm for 1 hour. Experiments were carried out in quadruplicate with three independent experiments. Data were analyzed by a least-squares non-linear fit, generated using Prism 7 in order to determine the compound’s IC50. Kd values for each inhibitor were calculated using the following equation based on the IC50, Kd of the tracer (), total (Lt) and bound (Lb) tracer, as well as total protein concentration (Pt).27
Microscale Thermophoresis Assay.
MST measurements were performed with a Monolith NT.115Pico device (NanoTemper Technologies). JH2 domain protein (triple mutant W659A, W777A, F794H, or triple mutant W777A, F794H, V617F) was fluorescently labeled with the His-tag labeling kit RED-tris-NTA 2nd generation. All dilutions were prepared with a buffer composed of 20 mM HEPES pH 8.0, 150 mM NaCl, 5% glycerol, and 0.05% Tween. Protein was labeled by incubating a mixture of 150 nM protein and 50 nM dye for 30 min at ambient temperature. MST measurements were performed with protein and dye concentrations adjusted to 30 nM and 10 nM, respectively. The serial dilution of 1 and 10 ranged from 160 μM to 0.00488 μM, for 12 from 20 μM to 0.00488 μM, and for 15 from 20 to 0.00061 μM. All measurement samples contained a constant DMSO concentration of 3%. Measurements were performed with standard capillaries (mutant W659A, W777A, F794H) or premium capillaries (mutant W777A, F794H, V617F), medium MST power, and 5% excitation power at ambient temperature. All measurements were performed in triplicate, and were analyzed with the MO.Affinity Analysis software (NanoTemper). All dose-response curves are displayed in Figure S2.
Protein Crystallization.
Crystals of JAK2 JH2 were grown by hanging drop vapor diffusion at 4 °C. Crystals were prepared by adding 1 μL of protein (6 mg/mL) in a solution composed of 20 mM Tris pH 8.0, 100 mM NaCl, 10% glycerol, and 1 mM TCEP to 1 μL of reservoir solution composed of 0.1 M Tris pH 8.0, 0.2 M sodium acetate, 12–24% PEG4000, and 1 mM TCEP. Crystallization was induced by streak seeding, and crystals grew to full size within a week. Complexes of JH2 with small-molecule ligands were prepared by ligand soaking. Therefore, crystals were transferred into a solution of 0.1 M Tris pH 8.0, 0.2 M sodium acetate, 22% PEG4000, 1 mM TCEP, 8% DMSO, and 4 mM of the respective small-molecule ligand. After 24 h of incubation, crystals were briefly exposed to a cryobuffer (0.1 M Tris pH 8.0, 0.2 M sodium acetate, 22% PEG4000, 1 mM TCEP, 8% DMSO, 4 mM of the respective small-molecule ligand, and 20% glycerol), and flash-frozen in liquid nitrogen.
Collection of X-ray Diffraction Data and Data Processing.
Crystal structures for complexes of five of the new compounds (3, 4, 9, 10, 12) with JAK2 JH2 were obtained at 1.7 – 2.1 Å resolution. All datasets were collected at 100 K. Datasets of JH2 in complex with 4, 10, and 12 were collected in-house on a Rigaku MicroMax-007HF X-ray generator (Cu rotating anode; λͅ=1.54 Å) with a Dectris Pilatus 200K detector. The dataset of JH2 in complex with 3 was collected at the Advanced Photon Source (APS; Argonne, Illinois, USA) on beamline 24-ID-E with a Dectris Eiger 16M detector (λͅ=0.97918 Å). The dataset of JH2 in complex with 9 was collected at the APS beamline 24-ID-C with a Dectris Pilatus 6M detector (λͅ=0.97910 Å). Datasets of 4, 10, and 12 were indexed, integrated, and scaled with HKL2000.28 Datasets of 3 and 9 were indexed, integrated, and scaled with XDS.29 Diffraction data and refinement statistics are listed in Table S1. Refinement procedures are also provided along with omit maps in Figure S3.
Cloning and Expression of Full-Length JAK2.
Full-length cDNA encoding human JAK2 (residues 1–1132) wild-type (NP_001309123) and V617F mutant with C-terminal FLAG-Tag (DYKDDDDK) were amplified by PCR, and subcloned into a modified pOptiVec expression vector (Invitrogen). JAK2 constructs were expressed in HEK293T cells grown at 37 °C at 5% CO2 in DMEM (Gibco) supplemented with 10% (v/v) FBS (Gibco) and 1% (v/v) penicillin-streptomycin (Gibco). HEK-293T cells were transiently transfected using Lipofectamin 2000 (Invitrogen) according to manufacturer’s instructions. 36 h post-transfection, cells were washed twice with ice-cold PBS, and lysed with ice-cold lysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 10% (v/v) glycerol, 1% (v/v) Triton-X 100, 1 mM EDTA, 1 mM EGTA, 25 mM NaF, 1.5 mM MgCl2, 1.0 mM Na3VO4, Roche complete mini EDTA-free protease inhibitor cocktail mixture). The lysate was centrifuged (20 min, 13000×g), and the lysate supernatant was flash-frozen in liquid nitrogen and stored at −80 °C.
Immunoprecipitation and In Vitro Kinase Assay.
JAK2 protein was immunoprecipitated from the lysate supernatant by adding anti-FLAG M2 antibody (Sigma-Aldrich, no. F1804) and protein G-PLUS agarose (Santa Cruz Biotechnology, no. sc-2002) followed by incubation overnight while rocking at 4°C. Immunoprecipitates were washed four times with wash buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 10% (v/v) glycerol, 0.1% (v/v) Triton-X 100, 1 mM EDTA, 1 mM EGTA, 25 mM NaF, 1.5 mM MgCl2, 1.0 mM Na3VO4, Roche complete mini EDTA-free protease inhibitor cocktail mixture), and once with kinase reaction buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 0.5 mM DTT, 5 mM MnCl2). The [γ−32P]ATP in vitro kinase activity assay was performed based on a previously published protocols.30,31 Washed immunoprecipitates were divided into equal parts, centrifuged, their residual solvent was removed, and the resulting pellets were resuspended in 25 μL kinase reaction buffer containing different concentrations of 1 or 10, followed by incubation for 1 h at 4°C. The autophosphorylation reaction of JAK2 was initiated by adding 25 μL of phosphorylation mixture consisting of kinase reaction buffer supplemented with 10 μM cold ATP, and 2 μCi (for JAK2 V617F) or 5 μCi (for JAK2 wild-type) of [γ−32P]ATP (EasyTides, PerkinElmer) per reaction. The mixture was allowed to react for 15 min at 30°C (within the linear range of kinase activity), and stopped by putting on ice and adding 18 μL of reducing Laemmli sample buffer (4×). Samples were heated at 95 °C for 5 min, and run by 7.5% SDS-PAGE. Gels were rocked in a solution of 10% glycerol, 20% ethanol for 30 min, dried with a vacuum drier, and autoradiographed using a phosphor imager. To calculate IC50 values, phosphor autoradiography was quantified using ImageJ,32 and curves were plotted with Prism 8.0 (GraphPad Software Inc., La Jolla, CA).
General Synthetic Methods.
All purchased compounds were used as received from vendors without further purification. Reactions were conducted under a nitrogen atmosphere and monitored by thin layer chromatography on Merck silica gel plates pre-coated with fluorescent indicator F254. Visualization of plates was achieved with UV light or potassium permanganate stain. Mass analysis of intermediates was done with an Agilent 6120 Quadrupole LC/MS instrument via electrospray ionization. Chromatographic purification was performed with a Teledyne ISCO CombiFlash automated system employing RediSep Normal Phase Silica (particle size: 35–70 μm; pore size: 60 Å) or RediSep Gold Normal Phase Silica (particle size: 20–40 μm; pore size: 60 Å) disposable cartridge columns. RediSep Gold C18 reusable columns (particle size: 20 – 40 μm spherical; pore size: 100 Å) were employed for reversed-phase chromatography. Preparative reverse phase HPLC was performed using a Shimadzu Prominence system equipped with LC-20AP pumps, CBM-20A Communications BUS module, SPD-20A UV/vis detector, SIL-10AP autosampler, FRC-10A fractions collector and a Waters SymmetryPrepTM C8, 19 × 300 mm column (particle size: 7 μm; pore size: 100 Å) with a gradient of 0.1 % formic acid in water and 0.1 % formic acid in acetonitrile as the mobile phase. 1H and 13C NMR spectra were recorded on Agilent DD2 400 MHz, DD2 500 MHz, or DD2 600 MHz instruments. The IR spectrum was recorded on Thermo Scientific Nicolet 6700 FT-IR spectrometer using a thin film of solid sample. HRMS analysis of final products was done on a Waters Xevo QTOF with a Z-spray electrospray ionization source. Purity of all assayed compounds was determined on a Shimadzu Prominence HPLC equipped with an Agilent Poroshell 120 SB-C18 2.7 μm column, using 0.1% TFA in water and 0.1% TFA in acetonitrile as the mobile phase. The compounds (2–4, 6–10, 12, 14, 15) were ≥ 95% pure by analytical HPLC, while the purities of three non-key compounds, 5, 11, and 13, were 94%, 91%, and 90%, respectively. The HPLC traces of all compounds showed no minor peaks above 3%. It is possible that the HPLC results may be affected by formation of dimers of the carboxylic acids or alternative protonation states. The systematic SAR data and multiple crystal structures reported here demonstrate that the reported binding data for this series is reflective of the compounds themselves. Spectra of all new compounds are provided in the SI.
Synthesis of Intermediates and Final Compounds.
Phenyl-N’-cyano-N-(4-sulfamoylphenyl)carbamimidate (16a).
Sulfanilamide (4.0 g, 23.2 mmol) was combined with diphenyl cyanocarbonimidate (5.5 g, 23.2 mmol) in anhydrous tetrahydrofuran (26 mL, 0.9 M). The reaction mixture was stirred at reflux for 24 h and afterwards was concentrated under reduced pressure. The residue was suspended in dichloromethane, filtered, washed with dichloromethane, and dried overnight to provide the title product as a pale white solid (6.2 g, 84% yield). 1H NMR (500 MHz, DMSO-d6) δ 11.12 (s, 1H), 7.83 (d, J = 8.5 Hz, 2H), 7.66 (d, J = 8.5 Hz, 2H), 7.46 (t, J = 7.8 Hz, 2H), 7.36 (s, 2H), 7.35 – 7.29 (m, 3H). 13C NMR (126 MHz, DMSO-d6) δ 151.43, 140.97, 139.12, 129.85, 127.39, 126.51, 126.39, 123.32, 120.79, 112.39. ESI-MS m/z: [M+H]+: 317.1.
Phenyl-N’-cyano-N-(4-cyanophenyl)carbamimidate (16b).
Sodium hydride (760 mg (60% dispersion in mineral oil), 19.0 mmol) was added at 0 °C to a mixture of 4-aminobenzonitrile (2.2 g, 19.0 mmol) in anhydrous tetrahydrofuran (65 mL, 0.3 M). The mixture was stirred at 60 °C for 45 min, followed by portion-wise addition of diphenyl cyanocarbonimidate (4.0 g, 17.0 mmol). The reaction was brought to reflux and allowed to run for 24 h. The crude mixture was quenched with methanol and solvent was evaporated under reduced pressure. Hot acetone was added until complete dissolution of the residue, at which point hexanes were added until the solution became cloudy. The precipitate was filtered to provide the title product as a white, fluffy solid (3.11 g, 70% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.21 (s, 1H), 7.86 (d, J = 8.1 Hz, 2H), 7.66 (d, J = 8.1 Hz, 2H), 7.46 (t, J = 7.6 Hz, 2H), 7.37 – 7.28 (m, 3H). 13C NMR (151 MHz, DMSO-d6) δ 157.71, 155.53, 154.16, 132.12, 128.84, 124.11, 123.67, 121.98, 121.78, 120.32, 100.73. ESI-MS m/z: [M+H]+: 263.1.
4-((5-amino-1H-1,2,4-triazol-3-yl)amino)benzenesulfonamide (17a).7
16a (5.0 g, 15.9 mmol) was suspended in anhydrous tetrahydrofuran (18 mL, 0.9 M) and hydrazine (17.5 mL (1 M in THF), 17.5 mmol) was added at 0 °C. The reaction mixture was warmed to room temperature and stirred at reflux for 7.5 h. The mixture was then filtered, washed three times with tetrahydrofuran, and dried overnight to afford the title product as white solid (3.4 g, 84.5% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.30 (s, 1H), 9.19 (s, 1H), 7.60 (q, J = 9.0 Hz, 4H), 7.03 (s, 2H), 5.95 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 157.22, 155.50, 145.46, 133.07, 126.72, 114.55. ESI-MS m/z: [M+H]+: 255.1.
4-((5-amino-1H-1,2,4-triazol-3-yl)amino)benzonitrile (17b).
16b (1.0 g, 3.8 mmol) was suspended in dry tetrahydrofuran (11 mL, 0.3 M) and combined with hydrazine (7.6 mL (1 M in THF), 7.6 mmol) at 0 °C. The reaction mixture was brought to reflux and stirred for 19 h, then was concentrated and purified by column chromatography (DCM/MeOH) to afford a yellow solid (461 mg, 61% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.38 (s, 1H), 9.40 (s, 1H), 7.60 (q, J = 8.9 Hz, 4H), 5.97 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 155.23, 147.97, 146.51, 133.04, 120.16, 115.49, 99.10. ESI-MS m/z: [M+H]+: 201.1.
Methyl 2-(4-nitrophenoxy)acetate (18a’).33
Para-nitrophenol (1.5 g, 10.8 mmol) was suspended in dry acetonitrile (50 mL, 0.2 M) and followed by addition of potassium carbonate (2.98 g, 21.6 mmol). The mixture was allowed to stir until homogenized, methyl-2-bromoacetate (1.0 mL, 10.8 mmol) was added, and stirring continued for 16.5 h at room temperature. The crude mixture was concentrated and diluted with ethyl acetate (50 mL), washed with water (2 × 20 mL) and brine (2 × 15 mL), and was dried over anhydrous sodium sulfate. Solvent was evaporated under vacuum to afford the title compound as a white solid (2.0 g, 89% yield). 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, J = 9.3 Hz, 2H), 7.17 (d, J = 9.3 Hz, 2H), 5.01 (s, 2H), 3.71 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.47, 162.78, 141.35, 125.78, 115.23, 65.02, 52.01. ESI-MS m/z: [M+H]+: 212.1.
Methyl 2-(4-aminophenoxy)acetate (18a).33
Pd/C (10% Pd basis, 565 mg) was added to a solution of 18a’ (1.85 g, 8.8 mmol) in 1:1 tetrahydrofuran:methanol (50 mL). The reaction stirred under hydrogen atmosphere for 20 h. The mixture was then filtered through celite and purified by column chromatography (Hexanes/EtOAc) to provide the title compound as an orange solid (1.3 g, 82% yield). 1H NMR (400 MHz, DMSO-d6) δ 6.64 (d, J = 8.8 Hz, 2H), 6.49 (d, J = 8.8 Hz, 2H), 4.65 (s, 2H), 4.59 (s, 2H), 3.67 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.71, 148.93, 143.08, 115.49, 114.75, 65.52, 51.60. ESI-MS m/z: [M+H]+: 182.1.
Methyl 2-(3-nitrophenoxy)acetate (18b’).34
Potassium carbonate (2.0 g, 14.5 mmol) was added to a mixture of m-nitrophenol (1.5 g, 10.8 mmol) in dry acetone (5 mL, 2 M). The mixture was allowed to stir until homogenized, and then methyl-2-bromoacetate (1.3 mL, 14.1 mmol) was added. The reaction refluxed for 5 h, then was cooled to room temperature and poured into water (25 mL). The precipitate was filtered and dried in a Buchner funnel under house vacuum overnight to provide the title compound as a pale white solid (2.2 g, 96% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.85 (dd, J = 8.1, 2.0 Hz, 1H), 7.73 (t, J = 2.3 Hz, 1H), 7.59 (t, J = 8.2 Hz, 1H), 7.45 (dd, J = 8.3, 2.5 Hz, 1H), 5.00 (s, 2H), 3.71 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.79, 158.15, 148.70, 130.79, 121.96, 116.22, 109.10, 65.01, 52.00. ESI-MS m/z: [M+H]+: 212.1.
Methyl 2-(3-aminophenoxy)acetate (18b).33
Pd/C (10% Pd basis, 565 mg) was added to a solution of 18b’ (1.85 g, 8.8 mmol) in 1:1 THF:MeOH (50 mL). The reaction stirred under hydrogen atmosphere for 20 h. The mixture was then filtered through celite and purified by column chromatography (Hexanes/EtOAc) to provide the title compound as a pale white solid (1.5 g, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ 6.90 (t, J = 8.0 Hz, 1H), 6.19 (d, J = 7.9 Hz, 1H), 6.11 (s, 1H), 6.05 (d, J = 8.1 Hz, 1H), 5.07 (s, 2H), 4.65 (s, 2H), 3.69 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 169.45, 158.62, 150.03, 129.58, 107.53, 101.65, 100.01, 64.31, 51.71. ESI-MS m/z: [M+H]+:182.1.
N-Boc-4-iodoaniline (18c”).35
Triethylamine (1.27 mL, 9.1 mmol) was added to a mixture of 4-iodoaniline (1.0 g, 4.6 mmol) and Boc anhydride (997 mg, 4.6 mmol) in isopropanol (23 mL, 0.2 M). The reaction was stirred at room temperature for 14 h and then concentrated under reduced pressure. The crude mixture was purified by column chromatography (Hexanes/EtOAc) to provide the title product as a pale yellow solid (908 mg, 62% yield). 1H NMR (400 MHz, Chloroform-d) δ 7.57 (d, J = 8.8 Hz, 2H), 7.14 (d, J = 8.7 Hz, 2H), 6.43 (s, 1H), 1.51 (s, 9H). 13C NMR (101 MHz, Chloroform-d) δ 152.55, 138.31, 137.96, 120.49, 85.86, 81.07, 28.45. HRMS (ESI): calc. for [IC6H4NHCO2H+H]+ 263.9521 found 263.9529.
Ethyl 2-(4-((tert-butoxycarbonyl)amino)phenyl)oxazole-4-carboxylate (18c’).20
Ethyl 4-oxazole carboxylate (200 mg, 1.4 mmol), was combined with 18c” (452 mg, 1.4 mmol), palladium acetate (32 mg, 0.14mmol), JohnPhos (85 mg, 0.28 mmol) and cesium carbonate (923 mg, 2.8 mmol) in dry dioxane (4 mL, 0.35 M). The reaction was sealed under N2 atm and heated at 110 °C for 19 h. The crude mixture was filtered through celite and purified by column chromatography (Hexanes/EtOAc) to provide the title product as an off-white solid (141 mg, 30% yield). 1H NMR (600 MHz, DMSO-d6) δ 9.76 (s, 1H), 8.86 (s, 1H), 7.91 (d, J = 8.7 Hz, 2H), 7.64 (d, J = 8.7 Hz, 2H), 4.31 (q, J = 7.1 Hz, 2H), 1.49 (s, 9H), 1.30 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 161.43, 160.79, 152.57, 145.16, 142.45, 133.55, 127.24, 119.52, 118.07, 79.69, 60.63, 28.07, 14.21. ESI-MS m/z: [M+H]+: 333.1.
Ethyl 2-(4-aminophenyl)oxazole-4-carboxylate (18c).
18c’ (101 mg, 0.3 mmol) was dissolved in anhydrous dichloromethane (0.8 mL, 0.38 M). The reaction mixture was cooled to 0 °C and trifluoroacetic acid (0.79 mL, 10.3 mmol) was added dropwise under vigorous stirring. The reaction was warmed to room temperature and stirred for 30 min. The pH was then adjusted to ~8 by adding sodium bicarbonate at 0 °C and the mixture was extracted with ethyl acetate. The combined organic phases were dried over anhydrous sodium sulfate, filtered, and concentrated to provide the product as a grey, shiny solid (71 mg, 100% yield). 1H NMR (400 MHz, Chloroform-d) δ 8.19 (s, 1H), 7.91 (d, J = 8.5 Hz, 2H), 6.71 (d, J = 8.4 Hz, 2H), 4.42 (q, J = 7.1 Hz, 2H), 3.98 (s, 2H), 1.40 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, Chloroform-d) δ 163.26, 161.83, 149.31, 142.92, 134.48, 128.76, 116.76, 114.70, 61.32, 14.51. ESI-MS m/z: [M+H]+: 233.1.
Ethyl 5-(4-aminophenyl)-1,3,4-oxadiazole-2-carboxylate (18d).36
Ethyl glyoxylate (1.98 mL (50% solution in THF), 10 mmol) in ethanol (100 mL, 0.1 M) was heated to reflux for 5 min. 4-aminobenzoic hydrazide (1.5 g, 10 mmol) was then added portion wise. After refluxing for 10–15 min, condensation was completed, as indicated by MS. The reaction mixture was concentrated under reduced pressure and the residue was dissolved in dimethylsulfoxide (50 mL). Potassium carbonate (4.2 g, 30 mmol) and iodine (3.0 g, 12 mmol) were added and then the reaction mixture was heated at 100 °C. At 40 min MS indicated completion of the cyclization and the reaction was cooled to room temperature, diluted with 5% Na2S2O3 (200 mL) and stirred for 30 min. The resulting suspension was extracted with ethyl acetate (4 × 100 mL), washed with brine, and dried over sodium sulfate. Purification by column chromatography (Hexanes/EtOAc) followed by recrystallization from hot methanol afforded the title product as a yellow solid (277 mg, 12% yield over 2 steps). 1H NMR (400 MHz, DMSO-d6) δ 7.71 (d, J = 8.7 Hz, 2H), 6.69 (d, J = 8.7 Hz, 2H), 6.13 (s, 2H), 4.42 (q, J = 7.1 Hz, 2H), 1.35 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz DMSO-d6) δ 166.34, 155.26, 154.21, 153.26, 128.84, 113.62, 108.32, 62.65, 13.92. ESI-MS m/z: [M+H]+: 234.1.
General Procedure A: Synthesis of Phenyl Carbamates 19a, e-j.19
The appropriate aromatic amine (1.0 eq.) was suspended in a mixture of tetrahydrofuran (2 M) and water (1 M) containing sodium bicarbonate (1.2 eq. for all except 19j; 2.2 eq. for 19j). The mixture was cooled to 0 °C and a solution of phenyl chloroformate (1.05 eq.) in tetrahydrofuran (1 M) was slowly added to the reaction mixture. The reaction was stirred at 0 °C unless otherwise stated until completion as determined by TLC and MS. Then the reaction mixture was diluted with ethyl acetate (20 mL), washed with water (3 × 5 mL) and brine (2 × 5 mL), and dried over sodium sulfate. The solvent was evaporated under reduced pressure to afford the corresponding phenyl carbamate without further purification, unless otherwise stated.
Phenyl phenylcarbamate (19a).
Aniline (0.3 mL, 3.3 mmol) was stirred for 40 min with phenyl chloroformate (0.44 mL, 3.5 mmol) as described in General Procedure A, to afford the title compound as a white solid (700 mg, 99% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.21 (s, 1H), 7.51 (d, J = 7.7 Hz, 2H), 7.43 (t, J = 7.8 Hz, 2H), 7.32 (t, J = 7.8 Hz, 2H), 7.27 (d, J = 7.5 Hz, 1H), 7.23 (d, J = 8.4 Hz, 2H), 7.05 (t, J = 7.3 Hz, 1H).13C NMR (101 MHz, DMSO-d6) δ 151.68, 150.50, 138.61, 129.41, 128.87, 125.41, 122.96, 121.95, 118.44. ESI-MS m/z: [M+H]+: 214.1.
Phenyl pyridin-2-ylcarbamate (19c).
2-amino pyridine (311 mg, 3.3 mmol) was combined with phenyl chloroformate (3.1 mL, 24.4 mmol) in the presence of sodium bicarbonate (2.3 g, 27.8 mmol), using the solvent composition mentioned in General Procedure A. The reaction was stirred at 0 °C for 2–3 h and then was warmed to r.t. and stirred 45 h. The reaction was extracted as in Procedure A, and subsequently purified by column chromatography (Hexanes/EtOAc) to afford the title product as a white solid (94 mg, 13% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.71 (s, 1H), 8.33 – 8.31 (m, 1H), 7.83 – 7.77 (m, 2H), 7.46 – 7.41 (m, 2H), 7.29 – 7.25 (m, 1H), 7.24 – 7.20 (m, 2H), 7.12 – 7.08 (m, 1H). ESI-MS m/z: [M+H]+: 215.1.
Phenyl pyridin-3-ylcarbamate (19d).37
3-amino pyridine (941 mg, 10 mmol) was dissolved in mixture of acetonitrile (8 mL) and pyridine (0.9 mL, 11.0 mmol). The reaction mixture was cooled to 0 °C, phenyl chloroformate (1.25 mL, 10.0 mmol) was added, and the reaction was warmed to room temperature and stirred for 2.5 h. Upon quenching with water (40 mL), the title product precipitated out of the reaction mixture, and was washed with water and dried under vacuum to give a reddish-brown solid (1.7 g, 79% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.47 (s, 1H), 8.69 (d, J = 2.4 Hz, 1H), 8.27 (dd, J = 4.7, 1.3 Hz, 1H), 7.93 (d, J = 8.7 Hz, 1H), 7.44 (t, J = 7.9 Hz, 2H), 7.37 (dd, J = 8.3, 4.7 Hz, 1H), 7.31 – 7.21 (m, 3H).13C NMR (101 MHz, DMSO-d6) δ 151.87, 150.37, 144.04, 140.28, 135.42, 129.48, 125.64, 125.34, 123.74, 121.94. ESI-MS m/z: [M+H]+: 215.1.
Methyl 2-(4-((phenoxycarbonyl)amino)phenoxy)acetate (19e).
Amine 18a (600 mg, 3.3 mmol) reacted with phenyl chloroformate (0.44 mL, 3.5 mmol) for 40 min to afford a white, crystalline solid (919 mg, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 7.46 – 7.36 (m, 4H), 7.25 (t, J = 7.4 Hz, 1H), 7.21 (d, J = 7.6 Hz, 2H), 6.91 (d, J = 9.1 Hz, 2H), 4.75 (s, 2H), 3.69 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.30, 153.41, 151.81, 150.59, 132.26, 129.40, 125.34, 121.95, 119.95, 114.82, 64.80, 51.78. ESI-MS m/z: [M+H]+: 302.1.
Methyl 2-(3-((phenoxycarbonyl)amino)phenoxy)acetate (19f).
Amine 18b (600 mg, 3.3 mmol) reacted with phenyl chloroformate (0.44 mL, 3.5 mmol) for 40 min to afford a white solid (895 mg, 90% yield).1H NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H), 7.43 (t, J = 7.7 Hz, 2H), 7.29 – 7.19 (m, 3H), 7.18 – 7.10 (m, 2H), 6.78 – 6.72 (m, 1H), 6.61 (dd, J = 8.2, 2.3 Hz, 1H), 4.75 (s, 2H), 3.69 (s, 3H). ESI-MS m/z: [M+H]+: 302.1.
Ethyl 2-(4-((phenoxycarbonyl)amino)phenyl)oxazole-4-carboxylate (19g).
Amine 18c (71 mg, 0.31 mmol) reacted with phenyl chloroformate (42 mL, 0.33 mmol) for 1 h to provide the title product as a light brown solid (99 mg, 91% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.62 (s, 1H), 8.90 (s, 1H), 7.99 (d, J = 8.8 Hz, 2H), 7.70 (d, J = 8.7 Hz, 2H), 7.45 (t, J = 7.9 Hz, 2H), 7.30 – 7.24 (m, 3H), 4.31 (q, J = 7.1 Hz, 2H), 1.31 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 161.25, 160.75, 151.58, 150.33, 145.32, 141.51, 133.60, 129.49, 127.44, 125.64, 121.94, 120.44, 118.51, 60.64, 14.20. ESI-MS m/z: [M+H]+: 353.1.
Methyl 5-(4-((phenoxycarbonyl)amino)phenyl)furan-2-carboxylate (19h).
Methyl 5-(4-aminophenyl)furan-2-carboxylate (719 mg, 3.3 mmol) reacted with phenyl chloroformate (0.44 mL, 3.5 mmol) for 1 h to afford the title product as a cream white solid (1.09 g, 97% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.48 (s, 1H), 7.79 (d, J = 8.7 Hz, 2H), 7.63 (d, J = 8.6 Hz, 2H), 7.44 (t, J = 7.9 Hz, 2H), 7.40 (d, J = 3.6 Hz, 1H), 7.27 (t, J = 7.4 Hz, 1H), 7.25 (d, J = 7.7 Hz, 2H), 7.07 (d, J = 3.6 Hz, 1H), 3.83 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 158.31, 156.82, 151.62, 150.40, 142.51, 139.53, 129.46, 125.56, 125.42, 123.66, 121.96, 120.71, 118.66, 107.06, 51.76. ESI-MS m/z: [M+H]+: 338.1.
Ethyl 5-(4-((phenoxycarbonyl)amino)phenyl)-1,3,4-oxadiazole-2-carboxylate (19i).
Amine 18d (200 mg, 0.86 mmol) was reacted with phenyl chloroformate (115 mL, 0.91 mmol) for 50 min. The crude mixture was processed according to Procedure A. The product was further purified by column chromatography (Hexanes/EtOAc) to afford the title product as a yellow solid (117 mg, 39% yield). 1H NMR (600 MHz, DMSO-d6) δ 10.74 (s, 1H), 8.05 (d, J = 8.7 Hz, 2H), 7.77 (d, J = 8.7 Hz, 2H), 7.45 (t, J = 7.9 Hz, 2H), 7.28 (dd, J = 16.7, 7.9 Hz, 3H), 4.45 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 165.66, 156.61, 154.51, 152.01, 150.70, 143.31, 129.94, 128.82, 126.14, 122.36, 119.12, 117.08, 63.30, 14.33. ESI-MS m/z: [M+H]+: 354.4.
(E)-3-(4-((phenoxycarbonyl)amino)phenyl)acrylic acid (19j).
(E)-4-amino cinnamic acid (540 mg, 3.3 mmol) was reacted with phenyl chloroformate (0.44 mL, 3.5 mmol) in the presence of 2.2 eq. sodium bicarbonate (612 mg, 7.3 mmol), for 1 h. The reaction mixture was acidified to pH 4, followed by an extraction workup, as described in Procedure A. The product was further purified by column chromatography (DCM/MeCN) to afford the title compound as a white solid (500 mg, 53% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.29 (s, 1H), 10.47 (s, 1H), 7.66 (d, J = 8.7 Hz, 2H), 7.58 – 7.51 (m, 3H), 7.44 (t, J = 7.9 Hz, 2H), 7.27 (t, J = 7.6 Hz, 1H), 7.24 (d, J = 7.6 Hz, 2H), 6.43 (d, J = 16.0 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 167.73, 151.56, 150.37, 143.53, 140.51, 129.47, 129.21, 128.85, 125.58, 121.96, 118.32, 117.40. ESI-MS m/z: [M+H]+: 284.0.
4-((5-amino-1-(2,6-difluorobenzyl)-1H-1,2,4-triazol-3-yl)amino)benzenesulfonamide (2).
17a (150 mg, 0.6 mmol), 2-(bromomethyl)-1,3-difluorobenzene (75 μL, 0.6 mmol), and Cs2CO3 (192 mg, 0.6 mmol) were dissolved in dimethylformamide (2.4 mL, 0.25 M). The reaction was stirred 16 h at room temperature. The solvent was then evaporated and the residue purified by flash chromatography (DCM/MeOH) to afford 2 (54 mg, 24% yield). Purity 97%. 1H NMR (600 MHz, DMSO-d6) δ 9.24 (s, 1H), 7.56 (d, J = 8.9 Hz, 2H), 7.49 (d, J = 8.9 Hz, 2H), 7.46 – 7.40 (m, 1H), 7.12 (t, J = 7.9 Hz, 2H), 7.02 (s, 2H), 6.46 (s, 2H), 5.08 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 160.99 (dd, J = 248.7, 7.9 Hz), 156.55, 154.20, 145.26, 133.28, 130.64 (t, J = 10.3 Hz), 126.64, 114.56, 112.38 (t, J = 19.2 Hz), 111.62 (dd, J = 20.6, 4.7 Hz), 37.21 (t, J = 3.4 Hz). HRMS (ESI): calc. for [M+H]+ C15H15F2N6O2S 381.0945 found 381.0956.
General Procedure B: Synthesis of Ureas 3, 4, 6–8, 9’, 11’−14’, 15.
The appropriate phenyl carbamate (1.0 eq.), was dissolved in dry dioxane (0.4 M for 15, 0.5 M for 3, 8, 12’, 13’, and 14’; 1 M for 4, 6, 7, 9’, and 11’), triethylamine (1.0 eq. for all except 15; 2.0 eq. for 15) was added, and the mixture was heated at 90–100 °C for five minutes. In a separate vial, a mixture of 17a or 17b (1.0 eq.) and triethylamine (1.0 eq.) in dry dioxane (0.2 M for 15, 0.5 M for 3, 8, 12’, 13’, and 14’; 1 M for 4, 6, 7, 9’, and 11’) was sonicated at 80–100 °C for 5–10 min and was added to the reaction mixture dropwise. The reaction vial was sealed under N2 atmosphere and the mixture stirred for 1–3 h at 90–110 °C (90 °C for 11’; 100 °C for 3–4, 6–7, 9’, and 15; 110 °C for 8, 12’, 13’, and 14’).
Workup A (3, 8, 9’,11’ 14’).
The reaction mixture was diluted with ethyl acetate (50 mL), was washed with water (2 × 15 mL) and brine (1 × 15 mL). The organic layer was collected, dried over sodium sulfate and, concentrated under reduced pressure. The crude mixture was purified by column chromatography, (DCM/MeOH).
Workup B (7, 12’, 13’).
Solvent was evaporated under reduced pressure and the reaction mixture was purified directly by chromatography (DCM/MeOH).
Workup C (4, 6).
Solvent was evaporated under reduced pressure and the crude mixture was triturated with ethyl acetate (3 × 10 mL). The combined organic phases were purified by column chromatography (DCM/MeOH).
General Procedure C: Hydrolysis of Esters 8, 9’, 11’−14’.21
Esters 8, 9’, 11’−14’ were suspended in a mixture of acetonitrile with 2 vol % water. Base (DBN, 3.0 eq. for 8, 12’−13’; Et3N, 3.0 eq. for 9’, 11’, 14’) and lithium bromide (10 eq.) were then added and the reaction was allowed to run at r.t. for the indicated time.
Workup D (9, 11).
Solvent was evaporated and sat. NaHCO3 was added to the reaction residue. The mixture was washed with ethyl acetate and the aqueous phase was acidified to pH ~4. The precipitate was collected, washed with small amounts of ethyl acetate and water, and dried under vacuum to provide the desired product.
Workup E (10, 12–14).
Solvent was evaporated and a small amount of water was added to the residue. The pH was adjusted to ~4 and the mixture was kept at low temperature for 6–12 h. The mixture was centrifuged, and the precipitate was dried and purified by normal phase column chromatography (DCM/MeOH) for 10, 12, and 14, and by reverse phase column chromatography (H2O/MeCN) for 13. Compound 10 was further purified by triturating with methanol.
5-amino-3-((4-cyanophenyl)amino)-N-phenyl-1H-1,2,4-triazole-1-carboxamide (3).
19a (105 mg, 0.5 mmol) reacted with 17b (99 mg, 0.5 mmol) for 1.5 h to afford the title compound as a white, fluffy solid (45 mg, 29% yield). Purity: 99%. 1H NMR (600 MHz, DMSO-d6) δ 9.87 (s, 1H), 9.64 (s, 1H), 7.85 (d, J = 8.8 Hz, 2H), 7.67 (d, J = 8.8 Hz, 2H), 7.66 – 7.63 (m, 2H), 7.45 (s, 2H), 7.42 – 7.37 (m, 2H), 7.18 (tt, J = 7.3, 1.2 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 156.38, 155.91, 149.08, 145.05, 137.06, 133.12, 128.65, 124.51, 121.84, 119.83, 116.99, 101.05. HRMS (ESI): calc. for [M+H]+ C16H14N7O 320.1260 found 320.1245. [17b+H]+ 201.0882 was also observed due to in-source fragmentation.
5-amino-N-phenyl-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamide (4).
19a (59 mg, 0.3 mmol) reacted with 17a (70 mg, 0.3 mmol) for 1 h to afford the title compound as a white, fluffy solid (39 mg, 38% yield). Purity: 99%. 1H NMR (600 MHz, DMSO-d6) δ 9.68 (s, 1H), 9.59 (s, 1H), 7.82 (d, J = 8.9 Hz, 2H), 7.71 (d, J = 9.0 Hz, 2H), 7.65 (d, J = 8.7 Hz, 2H), 7.46 – 7.37 (m, 4H), 7.20 – 7.16 (m, 1H), 7.14 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 156.69, 155.90, 149.14, 143.90, 137.10, 134.88, 128.70, 126.84, 124.54, 121.87, 116.24. HRMS (ESI): calc. for [M+H]+ C15H16N7O3S 374.1035 found 374.1009. [17a+H]+ 255.0645 was also observed due to in-source fragmentation.
Phenyl 5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxylate (19b).
To a suspension of 17a (509 mg, 2.0 mmol) in tetrahydrofuran/water was added sodium bicarbonate (168 mg, 2.0 mmol). The mixture was cooled to 0 °C. Phenyl chloroformate (0.25 mL, 2.0 mmol) was added, followed by additional sodium bicarbonate (168 mg, 2.0 mmol). The reaction was stirred at 0 °C for 1 h and the crude mixture was processed according to Procedure A. The product was further purified by column chromatography (DCM/MeOH) to afford the title product as a pale white solid (581 mg, 78% yield). 1H NMR (600 MHz, DMSO-d6) δ 9.78 (s, 1H), 7.69 (s, 4H), 7.57 (s, 2H), 7.52 – 7.48 (m, 2H), 7.41 – 7.34 (m, 3H), 7.14 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 158.11, 157.01, 149.91, 148.58, 143.93, 135.04, 129.70, 126.80, 126.61, 121.87, 115.89. ESI-MS m/z: [M+H]+: 375.0.
5-amino-N-methyl-N-phenyl-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamide (5).
N-methyl aniline (0.23 mL, 2.1 mmol) was dissolved in tetrahydrofuran (4.0 mL). Sodium hydride (51 mg, 2.1 mmol) was added at 0 °C and the mixture was allowed to stir for 1 h. Subsequently, this mixture was transferred dropwise to a separate vial containing 19b (400 mg, 1.1 mmol) in dioxane (2 mL) and the reaction was heated to reflux. After 20 h, solvent was evaporated and the crude mixture was purified by column chromatography (DCM/MeCN) to provide the product as a light brown solid (2 mg, Yield <1%). Purity: 94%. 1H NMR (500 MHz, DMSO-d6) δ 9.33 (s, 1H), 7.43 (dd, J = 13.3, 7.9 Hz, 4H), 7.27 (dd, J = 13.5, 7.5 Hz, 5H), 7.09 (s, 2H), 6.87 (d, J = 8.5 Hz, 2H), 3.36 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 156.93, 155.37, 151.42, 145.19, 143.66, 134.31, 129.00, 126.30, 126.08, 125.79, 115.34, 29.62. HRMS (ESI): calc. for [M+H]+ C16H18N7O3S 388.1192 found 388.1156.
5-amino-N-(pyridin-2-yl)-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamide (6).
19c (59 mg, 0.3 mmol) reacted with 17a (70 mg, 0.3 mmol) for 1 h, to afford the title compound as a white, fluffy solid (15 mg, 14% yield). Purity: 99%. 1H NMR (600 MHz, DMSO-d6) δ 9.76 (s, 1H), 9.42 (s, 1H), 8.41 (d, J = 4.1 Hz, 1H), 7.95 (d, J = 8.3 Hz, 1H), 7.92 – 7.88 (m, 1H), 7.75 – 7.70 (m, 4H), 7.52 (s, 2H), 7.25 – 7.21 (m, 1H), 7.15 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 156.89, 155.90, 150.13, 148.34, 143.79, 138.61, 135.13, 126.81, 120.34, 116.16, 113.66, 109.56. HRMS (ESI): calc. for [M+H]+ C14H15N8O3S 375.0988 found 375.0410. [17a+H]+ 255.0290 was also observed due to in-source fragmentation.
5-amino-N-(pyridin-3-yl)-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamide (7).
19d (59 mg, 0.3 mmol) was reacted with 17a (70 mg, 0.3 mmol) for 1 h to afford the title compound as a white, fluffy solid (20 mg, 20% yield). Purity: 100%. 1H NMR (600 MHz, DMSO-d6) δ 9.80 (s, 1H), 9.70 (s, 1H), 8.85 (d, J = 2.5 Hz, 1H), 8.38 (dd, J = 4.7, 1.4 Hz, 1H), 8.06 (ddd, J = 8.3, 2.6, 1.5 Hz, 1H), 7.84 – 7.80 (m, 2H), 7.72 – 7.69 (m, 2H), 7.47 – 7.42 (m, 3H), 7.15 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 156.81, 155.91, 149.28, 145.35, 143.80, 143.55, 134.91, 133.94, 129.16, 126.77, 123.45, 116.21. HRMS (ESI): calc. for [M+H]+ C14H15N8O3S 375.0988 found 375.0970. [17a+H]+ 255.0662 was also observed due to in-source fragmentation.
Methyl 2-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido) phenoxy)acetate (8).
19e (331 mg, 1.1 mmol) reacted with 17a (280 mg, 1.1 mmol) for 1.5 h. The reaction was processed using Workup A with modified column chromatography (DCM/MeCN), to afford the title compound as a white, fluffy solid (209 mg, 41% yield). Purity: 98%. 1H NMR (600 MHz, DMSO-d6) δ 9.66 (s, 1H), 9.53 (s, 1H), 7.81 (d, J = 8.8 Hz, 2H), 7.69 (d, J = 8.7 Hz, 2H), 7.52 (d, J = 9.0 Hz, 2H), 7.38 (s, 2H), 7.14 (s, 2H), 6.97 (d, J = 9.0 Hz, 2H), 4.81 (s, 2H), 3.71 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.26, 156.61, 155.80, 154.60, 149.31, 143.88, 134.79, 130.47, 126.77, 123.89, 116.16, 114.49, 64.77, 51.81. HRMS (ESI): calc. for [M+H]+ C18H20N7O6S 462.1196 found 462.1172. [17a+H]+ 255.0659 was also observed due to in-source fragmentation.
Methyl 2-(4-(5-amino-3-((4-cyanophenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenoxy) acetate (9’).
19e (151 mg, 0.5 mmol) reacted with 17b (100 mg, 0.5 mmol) for 35 min to afford the title compound as a white fluffy solid. (65 mg, 32% yield). 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H), 9.57 (s, 1H), 7.84 (d, J = 8.8 Hz, 2H), 7.65 (d, J = 8.9 Hz, 2H), 7.51 (d, J = 9.1 Hz, 2H), 7.40 (s, 2H), 6.97 (d, J = 9.1 Hz, 2H), 4.80 (s, 2H), 3.71 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.26, 156.34, 155.84, 154.62, 149.28, 145.07, 133.10, 130.45, 123.89, 119.85, 116.98, 114.49, 101.00, 64.76, 51.82. ESI-MS m/z: [M+H]+: 408.1.
2-(4-(5-amino-3-((4-cyanophenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenoxy)acetic acid (9).
9’ (14 mg, 35 μmol) was hydrolyzed according to the General procedure C (MeCN 0.2 M, Et3N, 11 h) and the reaction was processed using Workup D to afford the title compound as a white solid (10 mg, 70% yield). Purity: 97%. 1H NMR (600 MHz, DMSO-d6) δ 12.94 (bs, 1H), 9.86 (s, 1H), 9.57 (s, 1H), 7.85 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 7.51 (d, J = 8.7 Hz, 2H), 7.42 (s, 2H), 6.94 (d, J = 8.7 Hz, 2H), 4.68 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 170.25, 156.38, 155.88, 154.85, 149.33, 145.11, 133.14, 130.25, 123.91, 119.90, 117.03, 114.45, 101.04, 64.73. HRMS (ESI): calc. for [M+H]+ C18H16N7O4 394.1264 found 394.1285.
2-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenoxy) acetic acid (10).
8 (187 mg, 405 μmol) was hydrolyzed according to General procedure C (MeCN 0.03 M, DBN, 38 h) and the reaction was processed using Workup E to afford the title compound as a white solid (15.1 mg, 8% yield). Purity: 95%.1H NMR (600 MHz, DMSO-d6) δ 12.95 (bs, 1H), 9.66 (s, 1H), 9.52 (s, 1H), 7.81 (d, J = 8.8 Hz, 2H), 7.69 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 8.9 Hz, 2H), 7.38 (s, 2H), 7.14 (s, 2H), 6.93 (d, J = 8.9 Hz, 2H), 4.65 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 170.25, 156.60, 155.80, 154.89, 149.32, 143.89, 134.78, 130.17, 126.78, 123.85, 116.16, 114.40, 64.90. HRMS (ESI): calc. for [M+H]+ C17H18N7O6S 448.1039 found 448.1033. [17a+H]+ 255.0668 was also observed due to in-source fragmentation.
Methyl 2-(3-(5-amino-3-((4-cyanophenyl)amino)-1H-1,2,4-triazole-1-carboxamido) phenoxy) acetate (11’).
19f (151 mg, 0.5 mmol) was reacted with 17b (100 mg, 0.5 mmol) for 50 min to afford the title compound as a white fluffy solid. (59 mg, 29% yield). 1H NMR (600 MHz, DMSO-d6) δ 9.87 (s, 1H), 9.61 (s, 1H), 7.83 (d, J = 8.9 Hz, 2H), 7.67 (d, J = 9.0 Hz, 2H), 7.46 (s, 2H), 7.33 – 7.27 (m, 3H), 6.74 (dt, J = 7.0, 2.3 Hz, 1H), 4.80 (s, 2H), 3.72 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 169.14, 157.78, 156.40, 155.92, 148.93, 145.03, 138.35, 133.14, 129.50, 119.83, 117.00, 114.41, 110.05, 108.17, 101.08, 64.59, 51.87. ESI-MS m/z: [M+H]+: 408.1.
2-(3-(5-amino-3-((4-cyanophenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenoxy)acetic acid (11).
11’ (16 mg, 39 μmol) was hydrolyzed according to General Procedure C (MeCN 0.2 M, Et3N, 21 h) and the reaction was processed using Workup D to afford the title compound as a white solid (5 mg, 35% yield). Purity: 91%. 1H NMR (600 MHz, DMSO-d6) δ 12.99 (bs, 1H), 9.87 (s, 1H), 9.60 (s, 1H), 7.83 (d, J = 8.5 Hz, 2H), 7.67 (d, J = 8.7 Hz, 2H), 7.45 (s, 2H), 7.32 – 7.27 (m, 3H), 6.74 – 6.69 (m, 1H), 4.67 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 170.08, 157.99, 156.41, 155.92, 148.93, 145.05, 138.32, 133.16, 129.45, 119.84, 117.00, 114.17, 110.12, 108.01, 101.08, 64.53. HRMS (ESI): calc. for [M+H]+ C18H16N7O4 394.1264 found 394.1337.
Ethyl 2-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido) phenyl)oxazole-4-carboxylate (12’).
19g (60 mg, 0.2 mmol) reacted with 17a (43 mg, 0.2 mmol) for 50 min. The reaction was processed using Workup B with modified column chromatography (DCM/MeCN) to provide the title compound as a white solid. (21 mg, 25% yield). 1H NMR (600 MHz, DMSO-d6) δ 9.86 (s, 1H), 9.72 (s, 1H), 8.94 (s, 1H), 8.04 (d, J = 8.7 Hz, 2H), 7.93 (d, J = 8.7 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H), 7.72 (d, J = 8.9 Hz, 2H), 7.49 (s, 2H), 7.16 (s, 2H), 4.33 (q, J = 7.1 Hz, 2H), 1.32 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 161.16, 160.74, 156.77, 155.95, 148.87, 145.51, 143.79, 140.15, 134.96, 133.68, 127.03, 126.79, 121.57, 121.39, 116.26, 60.68, 14.21. ESI-MS m/z: [M+H]+: 513.1.
2-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl) oxazole-4-carboxylic acid (12).
12’ (14 mg, 27 μmol) was hydrolyzed according to General Procedure C (MeCN 0.14 M, DBN, 68 h). The reaction was processed using Workup E, and was further purified with HPLC to afford the title compound as a white solid (1.5 mg, 11% yield). Purity: 98%. 1H NMR (600 MHz, DMSO-d6) δ 9.82 (s, 1H), 9.72 (s, 1H), 8.33 (bs, 2H), 8.04 (bs, 2H), 7.88 (d, J = 7.6 Hz, 2H), 7.83 (d, J = 8.4 Hz, 2H), 7.72 (d, J = 8.4 Hz, 2H), 7.48 (s, 2H), 7.16 (s, 2H).13C NMR (151 MHz, DMSO-d6) δ 163.86, 163.23, 159.80, 156.77, 155.95, 148.91, 143.82, 139.44, 134.95, 126.81, 126.75, 122.65, 121.39, 116.26, 114.54. HRMS (ESI): calc. for [M+H]+ C19H17N8O6S 485.0992 found 485.0979. [17a+H]+ 255.0667 was also observed due to in-source fragmentation.
Methyl 5-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido) phenyl)furan-2-carboxylate (13’).
19h (86 mg, 0.26 mmol) was reacted with 17a (65 mg, 0.26 mmol) for 2.5 h, according to General Procedure B. The reaction was processed using Workup B with modified column chromatography (DCM/MeCN) to provide the title compound as a white solid (20 mg, 16% yield). 1H NMR (600 MHz, DMSO-d6) δ 9.76 (s, 1H), 9.70 (s, 1H), 7.87 – 7.81 (m, 6H), 7.72 (d, J = 8.7 Hz, 2H), 7.46 (s, 2H), 7.43 (d, J = 3.7 Hz, 1H), 7.17 – 7.13 (m, 3H), 3.85 (s, 3H). ESI-MS m/z: [M+H]+: 498.1.
5-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)furan-2-carboxylic acid (13).
13’ (20 mg, 40 μmol), in MeCN (0.035 M) with DBN, was hydrolyzed for 288 h (12 days) to afford the title product as a pale yellow solid (2 mg, 10% yield). Purity: 90%. 1H NMR (600 MHz, DMSO-d6) δ 9.70 (s, 2H), 7.83 (d, J = 8.6 Hz, 2H), 7.76 (s, 4H), 7.72 (d, J = 8.8 Hz, 2H), 7.46 (s, 2H), 7.15 (s, 2H), 6.91 (s, 1H), 6.86 (s, 1H). Carboxylic acid hydrogen is not observed. 13C NMR (151 MHz, DMSO-d6) δ 161.02, 156.71, 155.90, 148.96, 143.84, 137.01, 134.90, 126.81, 126.14, 124.70, 124.33, 121.76, 117.56, 116.24, 114.54, 106.81. HRMS (ESI): calc. for [M+H]+ C20H18N7O6S 484.1039 found 484.1027.
Ethyl 5-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido) phenyl)-1,3,4-oxadiazole-2-carboxylate (14’).
19i (90 mg, 0.26 mmol) was reacted with 17a (65 mg, 0.26 mmol) for 2.5 h. The reaction was processed using Workup A with modified column chromatography (DCM/MeCN) to provide the title product as a white solid (59 mg, 45% yield). 1H NMR (600 MHz, DMSO-d6) δ 9.96 (s, 1H), 9.72 (s, 1H), 8.11 (d, J = 8.7 Hz, 2H), 8.01 (d, J = 8.7 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H), 7.72 (d, J = 8.8 Hz, 2H), 7.50 (s, 2H), 7.16 (s, 2H), 4.46 (q, J = 7.1 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 165.21, 156.83, 156.31, 156.00, 154.11, 148.87, 143.79, 141.58, 135.00, 127.98, 126.82, 121.49, 117.82, 116.30, 62.94, 13.94. ESI-MS m/z: [M+H]+: 514.2.
5-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)-1,3,4-oxadiazole-2-carboxylic acid (14).
14’ (58 mg, 112 μmol) in MeCN (0.035 M) with Et3N, was hydrolyzed for 96 h (4 days). Additional purification with HPLC was needed to afford the product as a white solid (7 mg, 13% yield). Purity: 99%. 1H NMR (600 MHz, DMSO-d6) δ 9.90 (s, 1H), 9.72 (s, 1H), 9.33 (s, 1H), 8.06 (d, J = 8.7 Hz, 2H), 7.96 (d, J = 8.8 Hz, 2H), 7.83 (d, J = 8.8 Hz, 2H), 7.72 (d, J = 8.9 Hz, 2H), 7.49 (s, 2H), 7.16 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 163.43, 156.80, 155.97, 154.31, 148.89, 143.79, 140.76, 134.97, 127.42, 127.12, 126.79, 121.50, 118.65, 116.27. IR (solid, cm−1): 3291.7 (broad) [O-H stretch], 1724.8 [C=O stretch, −COOH]. HRMS (ESI): calc. for [M-CO2+H]+ C17H16N9O4S 442.1046 found 442.1042. [17a+H]+ 255.0678 was also observed due to in-source fragmentation.
(E)-3-(4-(5-amino-3-((4-sulfamoylphenyl)amino)-1H-1,2,4-triazole-1-carboxamido)phenyl)acrylic acid (15).
19j (100 mg, 0.35 mmol) was reacted with 17a (90 mg, 0.35 mmol) and 3.0 eq. triethylamine according to General Procedure B. After 1 h the reaction was stopped, solvent was evaporated, and the residue was triturated with methanol. Subsequently water was added, the mixture was acidified to ~ pH 4, and was kept at low temperature for 14 h. Afterwards the suspension was filtered, the solid was allowed to dry under vacuum, and was purified first by normal phase column chromatography (DCM/MeCN/MeOH) and last by HPLC, to afford the title compound as a white solid (8 mg, 5% Yield). Purity: 99%. 1H NMR (600 MHz, DMSO-d6) δ 9.73 (s, 1H), 9.70 (s, 1H), 7.82 (d, J = 8.8 Hz, 2H), 7.75 (d, J = 8.6 Hz, 2H), 7.71 (d, J = 8.6 Hz, 4H), 7.55 (d, J = 16.0 Hz, 1H), 7.46 (s, 2H), 7.15 (s, 2H), 6.49 (d, J = 16.0 Hz, 1H). Carboxylic acid hydrogen is not observed. 13C NMR (151 MHz, DMSO-d6) δ 167.93, 156.75, 155.93, 148.89, 143.83, 142.86, 138.94, 134.93, 130.28, 128.73, 126.82, 121.29, 118.82, 116.24. HRMS (ESI): calc. for [M+H]+ C18H18N7O5S 444.1090 found 444.1081.
Supplementary Material
Scheme 3. Synthesis of 17a-ba.

aReagents and conditions: (a) THF, reflux, 24 h; (b) NaH, THF, reflux, 24 h; (c) Hydrazine, reflux, 8–19 h.
Scheme 4. Synthesis of 18a-ba.

aReagents and conditions: (a) K2CO3, MeCN, r.t., 16.5 h; (b) K2CO3, acetone, reflux, 5 h (c) H2, Pd/C, THF:MeOH, 20h.
Scheme 5. Synthesis of 18c.a.

aReagents and Conditions: (a) Boc2O, Et3N, iPrOH, r.t., 14 h; (b) 10 mol % Pd(OAc)2, 20 mol % JohnPhos, Cs2CO3, dioxane, 110 °C, 19 h; (c) TFA, DCM, 0 °C to r.t., 30 min.
Scheme 6. Synthesis of 18d.a.

aReagents and conditions: (a) EtOH, reflux, 10 min; (b) K2CO3, I2, DMSO, 100 °C, 40 min.
Scheme 7. Synthesis of phenyl carbamates 19a, c-ja.

aReagents and conditions: (a) NaHCO3, H2O/THF, 0 °C, 40 min-1 h; (b) NaHCO3, H2O/THF, 0 °C to r.t., 45h; (c) pyridine, MeCN, r.t., 2.5 h.
Scheme 8. Synthesis of 2.a.

aReagents and conditions: (a) Cs2CO3, DMF, r.t., 16 h.
Scheme 9. Synthesis of compounds 3–4, 6–15.a.

aReagents and conditions: (a) Et3N, dioxane, 90 °C–110 °C, 35 min-2.5 h.; (b) Base, LiBr, MeCN, 2 vol % H2O, r.t., 11–288 h.
Scheme 10. Synthesis of compound 5a.

aReagents and conditions: (a) NaHCO3, H2O/THF, 0 °C, 1 h; (b) NaH, THF/dioxane, reflux, 20 h.
ACKNOWLEDGMENTS
Gratitude is expressed to the U. S. National Institutes of Health (GM32136) for research support, to the Deutsche Forschungsgemeinschaft (DFG) for a postdoctoral fellowship to S. G. K. (KR 5228/1-1), and to the NIH for training support of D. E. P. (GM007324). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This research also used resources of the Yale Macromolecular X-ray Core Facility (NIH Grant 1S10OD018007-01).
ABBREVIATIONS USED
- ATP
adenosine triphosphate
- JAKs
Janus kinases
- JH1
kinase domain
- JH2
pseudokinase domain
- FEP
free-energy perturbation
- FP
fluorescence polarization
- MST
microscale thermophoresis
- STAT
signal transducer and activator of transcription
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00192.
1H and 13CNMR spectra for final compounds 2–15; IR spectrum for compound 14; details for the FP and MST assays; crystallographic data for complexes of 3, 4, 9, 10, and 12 with JAK2 JH2, which have been deposited in the RCSB Protein Data Bank with the PDB ID codes 6OAV, 6OBB, 6OBL, 6OBF, and 6OCC (PDF).
Molecular formula strings for compounds 1 – 15 (CSV).
The authors have filed patent applications for JAK2 binding agents.
REFERENCES
- (1).O’Shea JJ; Holland SM; Staudt LM JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med 2013, 368, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Leroy E; Constantinescu SN Rethinking JAK2 inhibition: towards novel strategies of more specific and versatile Janus kinase inhibition. Leukemia 2017, 31, 1023–1038. [DOI] [PubMed] [Google Scholar]
- (3).Baxter EJ; Scott LM; Campbell PJ; East C; Fourouclas N; Swanton S; Vassiliou GS; Bench AJ; Boyd EM; Curtin N; Scott MA; Erber WN; Green AR Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [DOI] [PubMed] [Google Scholar]
- (4).Levine RL; Wadleigh M; Cools J; Ebert BL; Wernig G; Huntly BJP; Boggon TJ; Wlodarska L; Clark JJ; Moore S; Adelsperger J; Koo S; Lee JC; Gabriel S; Mercher T; D’Andrea A; Frohling S; Dohner K; Marynen P; Vandenberghe P; Mesa RA; Tefferi A; Griffein JD; Eck MJ; Sellers WR; Meyerson M; Golub TR; Lee SJ; Gilliland DG Activating mutation in tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [DOI] [PubMed] [Google Scholar]
- (5).Hammaren HM; Ungureanu D; Grisouard J; Skoda RC; Hubbard SR; Silvennoinen O ATP binding to the pseudokinase domain of JAK2 is critical for pathogenic activation. Proc. Natl. Acad. Sci. USA 2015, 112, 4642–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kung JE; Jura N Prospects for pharmacological targeting of pseudokinases. Nature Rev. Drug Disc 2019, 18, 501–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lin RH; Connolly PJ; Huang SL; Wetter SK; Lu YH; Murray WV; Emanuel SL; Gruninger RH; Fuentes-Pesquera AR; Rugg CA; Middleton SA; Jolliffe LK 1-acyl-1H-[1,2,4]triazole-3,5-diamine analogues as novel and potent anticancer cyclin-dependent kinase inhibitors: synthesis and evaluation of biological activities. J. Med. Chem 2005, 48, 4208–4211. [DOI] [PubMed] [Google Scholar]
- (8).Karaman MW; Herrgard S; Treiber DK; Gallant P; Atteridge CE; Campbell BT; Chan KW; Ciceri P; Davis MI; Edeen PT; Faraoni R; Floyd M; Hunt JP; Lockhart DJ; Milanov ZV; Morrison MJ; Pallares G; Patel HK; Pritchard S; Wodicka LM; Zarrinkar PP A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol 2008, 26, 127–32. [DOI] [PubMed] [Google Scholar]
- (9).Puleo DE; Kucera K; Hammarén H; Ungureanu D; Newton AS; Silvennoinen O; Jorgensen WL; Schlessinger J Identification and characterization of JAK2 pseudokinase domain small molecule binders. ACS Med. Chem. Lett 2017, 8, 618–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bandaranayake RM; Ungureanu D; Shan Y; Shaw DE; Silvennoinen O; Hubbard SR Crystal structures of the JAK2 pseudokinase domain and the pathogenic mutant V617F. Nat. Struct. Mol. Biol 2012, 19, 754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dusa A; Mouton C; Pecquet C; Herman M; Constantinescu SN JAK2 V617F constitutive activation requires JH2 residue F595: a pseudokinase domain target for specific inhibitors. PLoS One 2010, 5, e11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jorgensen WL Efficient drug lead discovery and optimization. Acc. Chem. Res 2009, 42, 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jorgensen WL; Tirado-Rives J Molecular modeling of organic and biomolecular systems using BOSS and MCPRO. J. Comput. Chem 2005, 26, 1689–1700. [DOI] [PubMed] [Google Scholar]
- (14).Robertson MJ; Tirado-Rives J; Jorgensen WL Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comput 2015, 11, 3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jorgensen WL; Tirado-Rives J Potential energy functions for atomic-level simulations of organic and biomolecular systems. Proc. Nat. Acad. Sci. USA 2005, 102, 6665–6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lea WA; Simeonov A Fluorescence polarization assays in small molecule screening. Expert. Opin. Drug Discovery 2011, 6, 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Newton AS; Deiana L; Puleo DE; Cisneros JA; Cutrona KJ; Schlessinger J; Jorgensen WL JAK2 JH2 fluorescence polarization assay and crystal structures for complexes with three small molecules. ACS Med. Chem. Lett 2017, 8, 614–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).McNally R; Li Q; Li K; Dekker C; Vangrevelinghe E; Jones M; Chène P; Machauer R; Radimerski T; Eck MJ Discovery and structural characterization of ATP-site ligands for the wild-type and V617F-mutant JAK2 pseudokinase domain. ACS Chem. Biol 2019, 14, 587–593. [DOI] [PubMed] [Google Scholar]
- (19).Hron R; Jursic BS Preparation of substituted semicarbazides from corresponding amines and hydrazines via phenyl carbamates. Tetrahedron Lett 2014, 55, 1540–1543. [Google Scholar]
- (20).Verrier C; Martin T; Hoarau C; Marsais F Palladium-catalyzed direct (hetero) of ethyl oxazole-4-carboxylate: an efficient access to (hetero)aryloxazoles. J. Org. Chem 2008, 73, 7383–7386. [DOI] [PubMed] [Google Scholar]
- (21).Mattsson S; Dahlström M; Karlsson S A mild hydrolysis of esters mediated by lithium salts. Tetrahedron Lett 2007, 48, 2497–2499. [Google Scholar]
- (22).Schärfer C; Schulz-Gasch T; Ehrlich H-C; Guba W; Rarey M; Stahl M Torsion angle preferences in druglike chemical space: a comprehensive guide. J. Med. Chem 2013, 56, 2016–2028. [DOI] [PubMed] [Google Scholar]
- (23).Arjunan V; Santhanam R; Rani T; Rosi H; Mohan S Conformational, vibrational, NMR and DFT studies of N-methylacetanilide. Spectrochim. Acta A 2013, 104, 182–196. [DOI] [PubMed] [Google Scholar]
- (24).Gaussian 16, Revision A.03, Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian, Inc, Wallingford CT, 2016. [Google Scholar]
- (25).Hansch C; Leo A; Hoekman D “Exploring QSAR – hydrophobic, electronic, and steric constants”, American Chemical Society: Washington, 1995. [Google Scholar]
- (26).Jerabek-Willemsen M; André T; Wanner R; Roth HM; Duhr S; Baaske P; Breitsprecher D MicroScale thermophoresis: interaction analysis and beyond. J. Mol. Struct 2014, 1077, 101–113. [Google Scholar]
- (27).Rossi AM; Taylor CW Analysis of protein-ligand interactions by fluorescence polarization. Nat. Protoc 2011, 6, 365–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Otwinowski Z; Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (29).Kabsch W XDS. Acta Crystallogr. Sect. D Biol. Crystallogr 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Argetsinger LS; Stuckey JA; Robertson SA; Koleva RI; Cline JM; Marto JA; Myers MG; Carter-Su C Tyrosines 868, 966, and 972 in the kinase domain of JAK2 Are autophosphorylated and required for maximal JAK2 kinase activity. Mol. Endocrinol 2010, 24, 1062–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Prchal-Murphy M; Semper C; Lassnig C; Wallner B; Gausterer C; Teppner-Klymiuk I; Kobolak J; Müller S; Kolbe T; Karaghiosoff M; Dinnyés A; Rülicke T; Leitner NR; Strobl B; Müller M TYK2 kinase activity is required for functional type I interferon responses in vivo. PLoS One 2012, 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Schneider CA; Rashband WS; Eliceiri KW NIH Image to ImageJ: 25 Years of image analysis. Nature Methods 2012, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zidar N; Macut H; Tomašič T; Brvar M; Montalvão S; Tammela P; Solmajer T; Peterlin Mašič L; Ilaš J; Kikelj D N-phenyl-4,5-dibromopyrrolamides and N-phenylindolamides as ATP competitive DNA gyrase B inhibitors: design, synthesis, and evaluation. J. Med. Chem 2015, 58, 6179–6194. [DOI] [PubMed] [Google Scholar]
- (34).Argade A; Bhamidipati S; Li H; Carroll D; Clough J; Keim H; Sylvain C; Rossi AB; Coquilla C; Issakani SD; Masuda ES; Payan DG; Singh R Application of cultured human mast cells (CHMC) for the design and structure-activity relationship of IgE-mediated mast cell activation inhibitors. Bioorganic Med. Chem. Lett 2015, 25, 2117–2121. [DOI] [PubMed] [Google Scholar]
- (35).Kabro A; Lachance H; Marcoux-Archambault I; Perrier V; Doré V; Gros C; Masson V; Gregoire JM; Ausseil F; Cheishvili D; Laulan NB; St-Pierre Y; Szyf M; Arimondo PB; Gagnon A Preparation of phenylethylbenzamide derivatives as modulators of DNMT3 activity. Medchemcomm 2013, 4, 1562–1570. [Google Scholar]
- (36).Yu W; Huang G; Zhang Y; Liu H; Dong L; Yu X; Li Y; Chang J I2-mediated oxidative C-O bond formation for the synthesis of 1,3,4-oxadiazoles from aldehydes and hydrazides. J. Org. Chem 2013, 78, 10337–10343. [DOI] [PubMed] [Google Scholar]
- (37).Keith JM; Apodaca R; Tichenor M; Xiao W; Jones W; Pierce J; Seierstad M; Palmer J; Webb M; Karbarz M; Scott B; Wilson S; Luo L; Wennerholm M; Chang L; Brown S; Rizzolio M; Rynberg R; Chaplan S; Breitenbucher JG Aryl piperazinyl ureas as inhibitors of fatty acid amide hydrolase (FAAH) in rat, dog, and primate. ACS Med. Chem. Lett 2012, 3, 823–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.