

Abstract
Biofilm‐infected wounds are clinically challenging. Vascular endothelial growth factor and host defence S100A8/A9 are crucial for wound healing but may be suppressed by biofilms. The natural course of Pseudomonas aeruginosa biofilm infection was compared in central and peripheral zones of burn‐wounded, infection‐susceptible BALB/c mice, which display delayed wound closure compared to C3H/HeN mice. Wounds were evaluated histopathologically 4, 7 or 10 days post‐infection. Photoplanimetry evaluated necrotic areas. P. aeruginosa biofilm suppressed vascular endothelial growth factor levels centrally in BALB/c wounds but increased peripheral levels 4–7 days post‐infection. Central zones of the burn wound displayed lower levels of central vascular endothelial growth factor as observed 4 and 7 days post‐infection in BALB/c mice compared to their C3H/HeN counterparts. Biofilm suppressed early, centrally located S100A8/A9 in BALB/c and centrally and peripherally later on in C3H/HeN wounds as compared to uninfected mice. Peripheral polymorphonuclear‐dominated inflammation and larger necrosis were observed in BALB/c wounds. In conclusion, P. aeruginosa biofilm modulates wounds by suppressing central, but inducing peripheral, vascular endothelial growth factor levels and reducing host response in wounds of BALB/c mice. This suppression is detrimental to the resolution of biofilm‐infected necrosis.
Keywords: Chronic wounds, Pseudomonas aeruginosa biofilm infection, S100A8/A9, Vascular endothelial growth factor, Wound necrosis
Introduction
Pseudomonas aeruginosa biofilm residing in refractory local necrotic tissue is detrimental to wound closure. The failure of chronic wounds to heal can be, at least partly, attributed to the combination of structural damage and establishment of chronic biofilm infection, which alters host responses, thereby further adding to local structural tissue damage 1. The wound is arrested in the inflammatory state of healing, and the remaining biofilm obstructs progression into the proliferative phase of wound healing. Delayed healing is thus caused by local wound infection comprising more than 105 bacteria per gram of tissue 2, 3, causing inadequate and prolonged innate immune activity 4. These factors may cause reduced bactericidal capacity of local immune cells in a perturbed microenvironment 5.
Human standard wound care includes debridement of biofilm and necrosis, either by surgical intervention or by maggot therapy 6. The removal of necrotic debris, including biofilm structures, aims to convert the chronic state of the wound. Viable, appropriately oxygenated tissue that is free of an inflammatory load will begin contraction, granulation tissue formation and collagen deposition, eventually resulting in scar formation 5. In the clinical setting, the abovementioned strategies are not always sufficient, and the remaining necrotic tissue acts as a nidus for the continuous inflammation. Schreml et al. showed a high spatial variability of human chronic venous ulcer hypoxia 7.
There is an urgent need to discover new strategies and approaches in order to optimise treatment regimens for chronic wounds. Insufficient treatment of chronic wounds may prolong the disease course and increase the socioeconomic burden. Conflicting data on the composition of chronic wound environment exist. An explanation for the gap between in vitro findings, achievements by use of animal models and clinical applicability could be the heterogeneity of clinical wounds, comorbidity among patients or the lack of suitable healing of control wounds in clinical trials. Growth factors are degraded by proteolytic environments in the chronic wounds 8, although the source of this degradation is a matter of debate. Thus, one widely accepted approach to the restoration of wound healing is substitution of these growth factors topically to recalcitrant wounds. However, there is no clear clinical success in the use of these growth factors. It is speculated that the remaining necrotic debris containing bacterial biofilm may be the cause for the lack of convincing results.
We suggest an increased focus on the interaction between the host and biofilm. P. aeruginosa is a resilient, gram‐negative rod and biofilm‐producing microorganism with high prevalence in chronic wounds 9, 10. The infection is connected to a high rate of complications, and the detrimental effect on wound healing is seen in both clinical 11 and numerous animal models 12, 13, 14, 15. In our wound model, a third‐degree full‐thickness burn wound, is induced before subcutaneous injection of P. aeruginosa biofilm, mimicking clinical wounds 16. We have previously shown that P. aeruginosa biofilm aggravates local inflammatory response in this chronic wound model, especially in the early phases of infection in the susceptible, poor‐healing BALB/c strain of mice 16. For comparison, we use the C3H/HeN mouse strain because this strain heals relatively faster than BALB/c mice 17, 18. The C3H/HeN mouse strain has been viewed as relatively resistant to P. aeruginosa biofilm infection in a lung model of chronic P. aeruginosa lung infection 19.
We hypothesised that there are qualitative differences within the different compartments of healing wounds. The peripheral hyperaemic zone appears reddish and well vascularised in contrast to the sharply demarcated central zone of coagulation (necrosis) 20. Observing mice subjected to P. aeruginosa biofilm infected wounds for a 14‐day period post‐wound infliction (10 days of infection), we found that the size of the central necrosis appeared to differ between two mouse strains as they progressed into the healing phase, indicating a role for a distinct host response in the course of central wound healing. Full‐thickness wounds heal through the initial formation of fibroblast‐derived tissue due to the loss of dermis; this process is followed by keratinocyte‐guided reepithelisation and neovascularisation. Key growth factors in wound repair and healing are vascular endothelial growth factor (VEGF) regulating endothelial cell proliferation and vascular permeability. Platelet‐derived growth factor (PDGF‐BB) has granulocyte chemotaxis properties and induces the production of matrix metalloproteinase and angiogenesis. Basic‐fibroblast growth factor (b‐FGF) is important for fibroblast and keratinocyte proliferation, wound contraction and matrix deposition.
We previously described a correlation between VEGF and levels of bacterial lipopolysaccharide in human wounds 21. Furthermore, we described a suppressive effect of biofilms on innate host defence protein S100A8/A9 in human 22 and whole murine wounds 18. To establish a foundation for topical intervention studies, there is a need for a thorough characterisation of biofilm‐infected wounds in the two abovementioned distinct compartments. Detection of favourable proteins in healing wound tissue compared to central, non‐healing necrotic tissue is needed. Thus, we assessed these key proteins in the two wound compartments in order to characterise their role in the transformation from the inflammatory phase to the reepithelisation phase. Histopathological characterisation of the distribution of inflammatory cell infiltration of polymorphonuclear leukocytes (PMNs) and mononuclear cells (MNs) in peripheral and central punch biopsies was described regarding both strains. PMNs represent the acute‐type inflammation, whereas PMN/MN ratio is the next step towards tissue regeneration 16. MN recruitment from peripheral blood to wound tissue in both strains of mice was included as these immunological cells bridge the inflammation and proliferation phases in wound repair. MN can differentiate into macrophages, which are the predominant source of growth factors to resolve inflammation and initiate skin repair 23 and stimulate fibroblasts to produce an extracellular matrix. Finally, we evaluated the size of necrosis of all wounds by digital photoplanimetry.
Materials and methods
Aim
The aim of this study was to describe the impact of P. aeruginosa biofilm on local growth factors and S100A8/A9 in the early inflammatory phase of infection and the proliferative phase in two different compartments of the same wound. We evaluated peripheral and central growth factor and host defence S100A8/A9 levels within a central (c) necrotic, non‐healing zone and a healing, peripheral (p) zone from the same wound of two immunologically different inbred strains of mice, namely BALB/c and C3H/HeN, which we previously described as relatively susceptible and resistant to P. aeruginosa infection, respectively 24, 25.
Study design
A full‐thickness burn wound (442 mm2) was inflicted on a total of 54 mice using hot air as previously described 26.
Four days after burn infliction, P. aeruginosa biofilm infection was established in BALB/c (n = 18) mice and C3H/HeN (n = 18) wounds. Non‐infected sham controls from each strain served as controls (n = 18).
All mice were harvested for duplicates of peripheral and central punch biopsies of the wound zones at 4, 7 and 10 days post‐infection (DPI), corresponding to days 8, 11 and 14 after wound infliction. One set of peripheral and central punch biopsies was retrieved for the quantification of growth factor and S100A8/A9 and the other for histopathology. We evaluated the impact of infection on wound healing and host response, the course of infection, the difference between central and peripheral wound zones and the difference between the strains of mice.
Systemic mobilisation of mononuclear leukocytes was included. Anticoagulated whole blood was collected from the heart at the time of sacrifice and was kept on ice until flow cytometry analysis was performed (within 1 hour from collection). A potential clinical impact was assessed by photoplanimetric evaluation of necrosis size. Fifty‐four additional mice (27 per strain of mice, 18 uninfected and 9 sham controls) were photographed from a fixed distance at 4, 7 and 10 DPI.
Animals
Specified pathogen‐free C3H/HeN (n = 27) and BALB/c (n = 27) female mice, 10–12 weeks old, were used. Both strains were obtained from Taconic Europe A/S, Lille Skensved, Denmark. Animals were acclimatised for at least 1 week in the animal facilities before experimentation. Mice were allowed free access to chow and water and were cared for by trained personnel.
Burn wound infliction
Mice were anaesthetised subcutaneously with 0·3 ml Hyp/Mid (2·5 mg/ml Hypnorm and 1·25 mg/ml Midazolam) before induction of histologically confirmed, full‐thickness burn wounds covering 6% (442 mm2) of total body area using hot air as described previously 26.
Challenge solution
The biofilm challenge solution was prepared as described previously 16. Briefly, one colony of wild‐type P. aeruginosa strain, PA01, was transferred and grown for 18 hours at 37 °C in an LB medium (Statens Serum Institute, Copenhagen, Denmark). The overnight culture was centrifuged at 4°C at 4416 g and the pellet re‐suspended in 5 ml of serum‐bouillon (KMA Herlev Hospital, Herlev, Denmark). Protanal LF 10/60 (FMC BioPolymer N‐3002 Drammen, Norway) was dissolved in 0·9% NaCl to an alginate concentration of 1% and sterile‐filtered. The bacterial culture was diluted in a ratio of 1:20 in seaweed alginate solution; 5 ml of alginate beads were made.
Infection procedure
Mice allocated to the infection group received 100 μl of the bead challenge solution (106 CFU) injected subcutaneously beneath the burn wound 4 days after infliction of thermal lesion in order to bypass the post‐burn‐induced immunosuppression as described previously 26.
Mice were sacrificed using an intraperitoneal injection of a pentobarbiturate/lidocaine overdose 4, 7 or 10 DPI.
Collection of wound biopsies
Each wound had a central, necrotic area and a peripheral, contracting zone corresponding to the wound margin. For cytokine measurement, central and peripheral wound biopsies (n = 54) were collected aseptically using 4 ‐mm sterile biopsy punch needles (Miltex, DK) and were carefully placed in 2‐ml sterile PBS per wound. Four biopsies were retrieved from each mouse, two biopsies for cytokine analysis (central and peripheral) and two for histopathology (central and peripheral).
Samples were then stored on ice until homogenisation for 20 seconds by 14 000 rpm using a Heidolph Silent Crusher M (Heidolph Instruments, Schwabach, Germany) followed by centrifugation for 10 minutes at 1590 g. Supernatants were sterile‐filtered and stored at −80°C until analysis.
Growth factors
Levels of growth factors (VEGF, PDGF‐BB and FGF) in sterile‐filtered (0·22 μm) wound homogenates were analysed by Luminex Immunoassay (Luminex Corp., Austin, TX, USA).
Flow cytometry was performed as described by Brochmannn et al.
Detection of distribution of mononuclear leukocytes (Mononuclear Blood Count, MBC) in anticoagulated whole blood was performed using a FACSCanto™ flow cytometer (BD Biosciences, San Jose, CA, USA) with a 488‐nm argon laser and a 530/30‐nm band pass emission filter for recording of HPF fluorescence in FL‐1 27. PI fluorescence was collected through a 585/42‐nm band pass emission filter and was recorded in FL‐3. To maximise the resolution, samples were analysed at a low flow rate corresponding to 10 μl/minute. At least 10 000 events were recorded for each sample. Cytometer Setup and Tracking Beads (BD Biosciences) were used for instrument calibration, and flow data were processed and analysed by Diva (BD Biosciences).
Histopathology
Wound tissue was aseptically retrieved at each evaluation point, obtaining 4‐mm punch biopsies (n = 54) by punch needles (Miltex, Mediq, Denmark) from each central, necrotic area and each healing, peripheral area from both mouse strains in order to evaluate leucocyte subset. Samples were stored in formaldehyde until paraffin embedment, sectioning and haematoxylin and eosin (HE) staining. HE‐stained slides were scored by index as described in an earlier study 16. A pathologist blinded to group assignments estimated the dominant type of inflammation for each biopsy. Slides were scored as acute inflammatory (dominating polymorphonuclear neutrophils, PMN), mixed ratio (MN/PMN) or no inflammation seen (NI) 16. Slides were scanned (Zeiss Axio scan Z1; objective magnification 10× and 40×, NA 0·45. Software: Zeiss Zen Slidescan 2012). Brightness and contrast was adjusted in order to present the inflammatory response of the representative slides in Figure 4.
Figure 4.
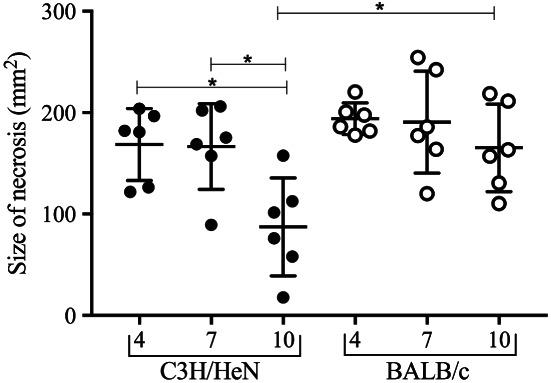
Photoplanimetric evaluation of necrosis size (mm2) in C3H/HeN (black dots) and BALB/c mice (open dots). C3H/HeN necrosis progressively diminishes in size from 4 to 10 days post‐infection (DPI) (P < 0·008) and 7 to 10 DPI (P < 0·013). In contrast, BALB/c necrosis remained equal in size through the study. At the termination of the experiment, 10 DPI, BALB/c necrosis was significantly larger than C3H/HeN necrosis (P < 0·015).
Macroscopic evaluation of necrosis
All mice (n = 54) were photographed at a fixed distance by a camera (CANON, EOS 650D, Kyushu, Japan) at the time of sacrifice. Necrotic areas were analysed using photoplanimetry on digital images taken at 4, 7 and 10 DPI (Image J®, vers. 1·47) by an investigator blinded to group assignment.
Colour imaging
Paraffin‐embedded samples were deparaffinised by submersion in xylene (2 × 5 minutes), 99·9% EtOH (2 × 3 minutes) and 96% EtOH (2 × 3 minutes) and rinsed in MilliQ water (3 × 3 minutes). A drop of a Peptide Nucleic Acid Fluorescence In Situ Hybridization (PNA FISH) probe specific for bacterial rRNA with a TEMRA‐5 fluorophore attached (AdvanDx, Woburn, MA) was applied to the tissue section and then covered with a coverslip. Samples were incubated for 90 minutes at 55°C on a heat plate (AdvanDx). Slides were washed in pre‐warmed washing buffer at 55°C (AdvanDx, USA) for 30 minutes, according to manufacturer's instructions. Afterwards, these slides were air‐dried in the dark. Specimen were counter‐stained with 0·3 mM DAPI (Sigma, USA) in PBS by soaking and were incubated 15 minutes in the dark at room temperature. Residue DAPI was removed by gentle rinsing with PBS. A drop of Pro‐long Gold mounting media (ThermoFisher, USA) was applied to project staining, and high‐quality coverslip were put on top and the edges sealed with clear nail polish. Slides were scanned using a Zeiss Imager.Z2, LSM 880 confocal laser scanning microscope (Zeiss, Germany) and the accompanying software Zeiss Zen 2·1 (Zeiss). Images were taken with 63×/1·4 oil objective using laser excitation at 405 nm, 488 nm and 561 nm for DAPI (Blue), tissue auto‐fluorescence (green) and TAMRA‐5, respectively. Raw images were processed in Imaris (Bitplane, Zürich, Switzerland) to create Tagged Image Formate (TIF) files of complied z‐stack images.
Statistical analysis
Statistical calculations were performed using the statistical programme GraphPad Prism (version 7·02; GraphPad Software, Inc., San Diego, CA, USA). The chi‐square test was used when comparing qualitative variables, and the ANOVA/unpaired t‐test was used when comparing quantitative variables. P ≤ 0·05 was considered statistically significant.
Ethics
The study was approved by the Animal Ethics Committee of Denmark (2012–15–2934‐000676).
Results
VEGF levels
We assessed the impact of infection, course of infection, differences between central and peripheral levels in each mouse strain and differences between the strains of mice.
BALB/c mice
Wound tissue levels of VEGF in all infected animals are displayed in Figure 1. Peripheral levels of VEGF in infected wounds were increased three to fourfold compared to uninfected controls, whereas VEGF levels were similar in central zones of infected and uninfected mice (control data displayed in Figure S1).
Figure 1.
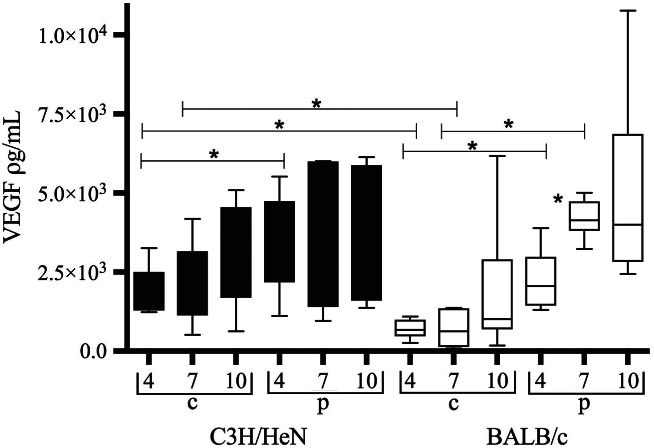
Vascular endothelial growth factor (VEGF) levels in central (c) and peripheral (p) wound biopsies from infected C3H/HeN mice (black bars) and infected BALB/c mice (white bars). Data are shown as box‐and‐whisker diagrams. VEGF levels are lower centrally than peripherally in BALB/c wounds at days 4 and 7 after infection (P < 0·004; P < 0·0001). In C3H/HeN mice, levels are lower centrally only at day 4 after infection (P < 0·05). In BALB/c mice, levels increase peripherally from 4 to 7 days after infection (P < 0·0018). Comparing the two strains of mice, central VEGF levels are reduced in BALB/c mice day 4 and 7 after infection as compared to C3H/HeN (P < 0·003; P < 0·03). Peripheral levels are equal.
The central VEGF levels were significantly lower than the peripheral levels in infected BALB/c mice 4 and 7 DPI (P < 0·004; P < 0·0001), but only at day 4 in C3H/HeN mice (P < 0·05).
Comparing the two strains of infected mice, central levels of VEGF were lower in BALB/c mice than in C3H/HeN 4 and 7 DPI (P < 0·003; P < 0·03). Peripheral levels were comparable between the strains of infected mice.
Due to missing data regarding one sample, only five values for central BALB/c at 4DPI were obtained for cytokine and S100A8/A9 analysis.
C3H/HeN mice
Biofilm infection induced central as well as peripheral VEGF levels (Figure 1).
Neither central nor peripheral VEGF levels in infected wounds changed appreciably throughout the study.
PDGF and FGF levels
The level of PDGF‐BB was increased in the central zone in both sham controls and infected wounds of BALB/c mice (Figure S2). In BALB/c mice, central wound PDGF‐BB levels were higher compared to peripheral wound levels at 7 DPI (P < 0·003). PDGF‐BB decreased significantly from 4 to 10 DPI (P < 0·008) in the central zone of wound.
PDGF‐BB levels in peripheral wound zone of BALB/c mice and both the central and peripheral wound zone in C3H/HeN mice were all below detection limits and were omitted from further analysis. FGF levels were below detection limits for both compartments of C3H/HeN mice and were omitted from further analysis. A tendency towards increased FGF levels was seen in the peripheral, compared to the central, wound zone in BALB/c (Figure S3).
Host response S100A8/A9
S100A8/A9 levels were reduced by P. aeruginosa biofilm infection centrally 4 DPI in BALB/c mice (Table 1) and 7 and 10 DPI central and peripherally in C3H/HeN mice, respectively, compared to uninfected sham controls. S100A8/A9 was reduced peripherally in C3H/HeN from 4 to 7 DPI (P < 0·024). No differences were observed between central and peripheral levels in either of the strains at any point. Levels remained stable centrally in both strains, although they were the highest in BALB/c compared to C3H/HeN mice at 4 DPI [mean (±SD) 3568 ρg/ml ± 1144 versus 2105 ρg/ml ± 461·1 ρg/ml, P < 0·02]. The same pattern was observed in the corresponding controls (6307 ρg/ml ± 434·3 versus 2497 ± 923·2, respectively).
Table 1.
S100A8/A9 in wound biopsies (ρg/ml ± SD). Impact of Pseudomonas aeruginosa biofilm infection on S100A8/A9 levels (ρg/ml ± SD) in central and peripheral wound biopsies. BALB/c mice have significantly more S100A8/A9 centrally in infected wounds (P < 0·018). C3H/HeN mice have reduced peripheral levels of S100A8/A9 from 4 to 7 days post‐infection (DPI) (P < 0·024)
| BALB/c | C3H/HeN | ||||
|---|---|---|---|---|---|
| DPI | Localisation | Infected (n = 6) | Non‐infected (n = 3) | Infected (n = 6) | Non‐infected (n = 3) |
| 4 | Central | 3568 (±1144)* | 6307 (±434·3) | 2105 (±461·1)* | 2497 (±923·2) |
| 7 | Central | 24 547 (±50 924) | 5600 (±2913) | 2390 (±1014) | 20 403 (±26 473) |
| 10 | Central | 6918 (±9693) | 4253 (±2536) | 1905 ± (701·7) | 2453 (±503·4) |
| 4 | Peripheral | 7370 (±10 417) | 4072 (±310·1) | 3152 (±1477)† | 2097 (±853·5) |
| 7 | Peripheral | 23 265 (±51 636) | 8057 (±10 103) | 1465 (±471·5)† | 3073 (±947·1) |
| 10 | Peripheral | 2947 (±1757) | 8847 (±9654) | 2387 (±900·6) | 3440 (±1488) |
Significant difference between strains.
Significant decrease in S100A8/A9 levels from 4 to 7 DPI in C3H/HeN mice.
Histopathology
Results are displayed in Table 2. At 10 DPI, significantly more acute type inflammation was observed peripherally than centrally in infected BALB/c wounds (χ 2 = 6, P < 0·05).
Table 2.
Histopathology. Histopathological evaluation of wound biopsies. Type of inflammation was scored as acute (PMNs dominating), chronic (MNs dominating), mixed type (MN/PMNs) or no inflammation seen (NI)
| 4 DPI | 7 DPI | 10 DPI | ||||
|---|---|---|---|---|---|---|
| Central | Peripheral | Central | Peripheral | Central | Peripheral | |
| C3H/HeN | ||||||
| PMN | 0 | 2 | 0 | 2 | 2 | 3 |
| PMN/MN | 5 | 4 | 5 | 3 | 2 | 3 |
| NI | 1 | 0 | 1 | 1 | 2 | 0 |
| n = 6 | n = 6 | n = 6 | n = 6 | n = 6 | n = 6 | |
| BALB/c | ||||||
| PMN | 2 | 1 | 2 | 2 | 1 | 5* |
| PMN/MN | 3 | 3 | 3 | 3 | 2 | 1 |
| NI | 1 | 2 | 1 | 1 | 3 | 0 |
| n = 6 | n = 6 | n = 6 | n = 6 | n = 6 | n = 6 | |
DPI, days post‐infection; NI, no inflammation; MN, mononuclear leukocytes; PMN, polymorphonuclear leukocytes.
P < 0·05.
Figure 2 is a representative slide from an infected BALB/c mouse, peripherally and centrally, at 10 DPI.
Figure 2.
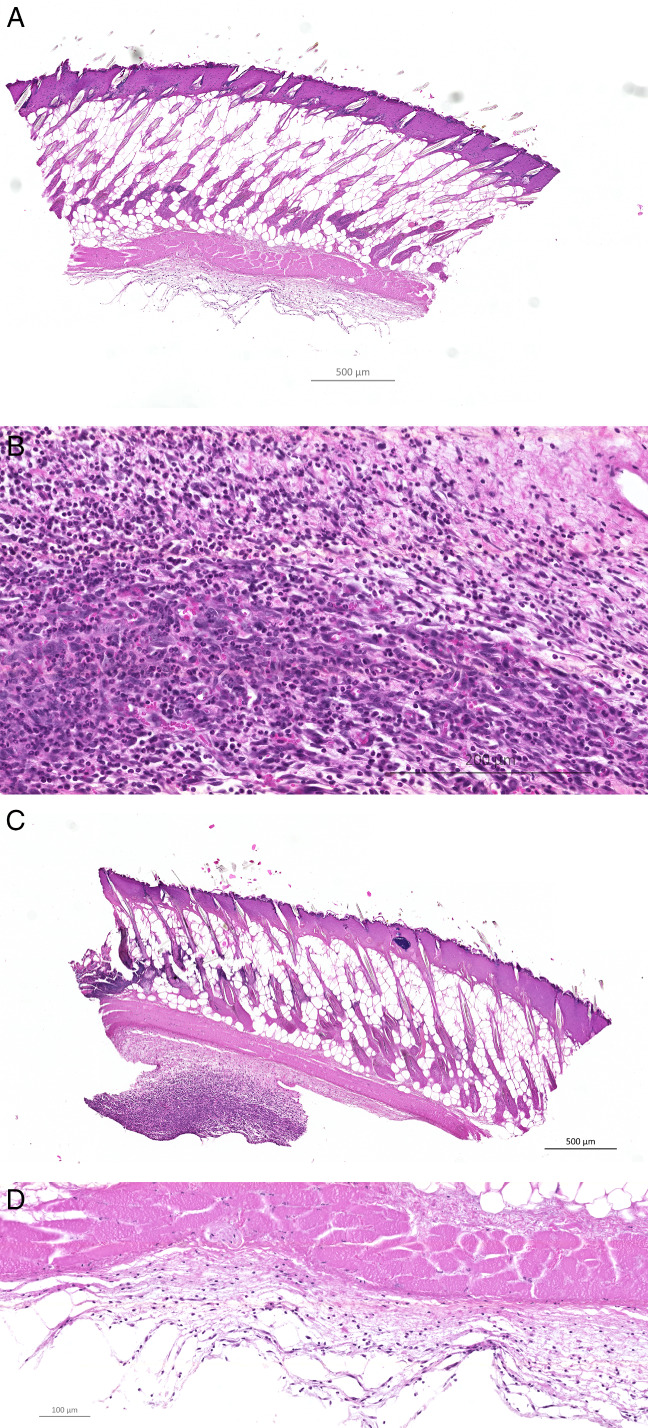
A representative display of a haematoxylin and eosin (HE)‐stained peripheral (A, B) and central (C, D) wound biopsy taken 10 DPI from the same infected BALB/c mouse (10× and magnification 40×, respectively). Significantly more of the peripheral biopsies taken from infected BALB/c mice had PMN‐dominated inflammation peripherally than central biopsies at 10DPI (P < 0·05).
No significant difference in the histological characterisation of inflammation was found between the two infected strains of mice or in the course of infection for each strain. Inflammatory cells were located in the hypodermis for all infected biopsies (data not shown).
Count of blood mononuclear leukocytes
Only the C3H/HeN strain of mice displayed a significant reduction in mononuclear leucocytes in whole blood from 4 to 7 DPI (P ≤ 0·05) and from 4 to 10 DPI (P ≤ 0·03), depicted in Figure 3. Similar results were seen in uninfected mice (data not shown).
Figure 3.
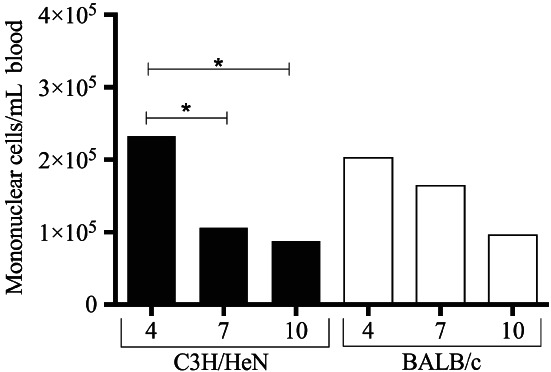
Scatter plot of peripheral blood monocyte count (MBC) in C3H/HeN (black bars) and BALB/c (white bars) mice challenged by Pseudomonas. aeruginosa biofilm‐infected burn wounds. Only C3H/HeN mice displayed a reduction in mononuclear blood count from 4 to 7 days post‐infection (DPI) (P ≤ 0·05) and 4 to 10 DPI (P ≤ 0·03). Mononuclear blood count remained stable in the BALB/c strain of mice.
Macroscopic evaluation of wound necrosis
C3H/HeN necrosis progressively diminished from day 4 to 10 DPI (P < 0·008) and day 7 to 10 DPI (P < 0·013). In contrast, BALB/c necrosis remained equal in size throughout the study. At 10 DPI, BALB/c necrosis was significantly larger than C3H/HeN necrosis (P < 0·015) (Figure 4).
The ratio necrosis/present wound size increased from 4 to 7 DPI in C3H/HeN mice but only from 4 to 10 DPI in BALB/c mice (P ≤ 0·05) (Figure S4).
Colour images
Representative slides display bacterial biofilms (red) in close proximity to inflammatory cells (blue) in the hypodermis (green) of a peripheral wound biopsy from an infected C3H/HeN mouse at 7 DPI (Figure 5A, B). Figure 5C visualises bacterial biofilms (red) and adjacent inflammatory cells (blue). Figure 5D is the bacterial biofilms without staining for inflammatory cells.
Figure 5.
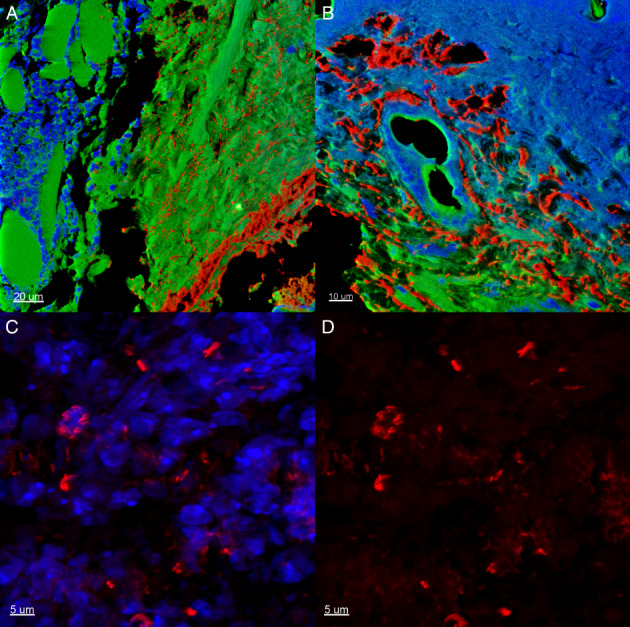
A representative section of a peripheral biopsy from a Pseudomonas aeruginosa biofilm‐infected (red colour) C3H/HeN mouse at day 7 days post‐infection (DPI). Green colour, local wound tissue; blue colour, inflammatory infiltrate. (A) Scale bar = 20 μm. (B) Scale bar = 10 μm. (C) The appearance of biofilms and inflammatory cells in clusters. Scale bar = 5 μm. (D) The appearance of biofilm in clusters. Scale bar = 5 μm.
Discussion
Normal wound healing is effectuated by growth factor‐stimulated fibroblasts and peripheral‐migrating keratinocytes near the wound edges, but little is known of the impact of P. aeruginosa biofilm on the local wound bed and the host response to this chronic infection. In this animal study, we report important new knowledge on the dynamics of the healing of P. aeruginosa biofilm‐infected wounds. The dynamics between the central and peripheral zones of the wound responses especially have the potential to provide interesting factors for healing. This can be clearly studied in representative animal models. In the present study, P. aeruginosa biofilm caused considerable induction of VEGF levels generally in the wounds. However, we observed a notable suppressive effect of biofilm infection on VEGF and S100A8/A9 centrally in the wounds of the poor‐healing BALB/c mouse strain.
In a recent clinical study, our group found a correlation between levels of lipopolysaccharide and VEGF in chronic wounds fluids 28. Tissue hypoxia strongly induces VEGF levels 29. P. aeruginosa is known to induce VEGF levels. Xue et al. 30 have shown a VEGF‐mediated, extensive, neovascularisation resulting in reduced visual acuity or even blindness at day 7 DPI in a BALB/c model of P. aeruginosa keratitis. In airway epithelium, P. aeruginosa induces VEGF synthesis in vivo, causing bronchiectasis 31. Recently, Birkenhauer et al. 32 described how P. aeruginosa movement was directed towards a larger gradient of VEGF in vitro, suggesting that, while beneficial, excess VEGF levels may aid colonisation.
Striking differences in the peripheral and central levels of VEGF in poor‐healing infected BALB/c mice were discovered in the present study. Significantly more VEGF was measured peripherally than centrally in the necrosis at 4 and 7 DPI, indicating increased oxygen depletion, which is predominantly caused by activated PMNs in P. aeruginosa lung biofilm infection 33. Besides macrophages, keratinocytes residing in the wound margin are a critical source of VEGF, and this could also be an explanation for this observation. In a comparable burn wound model of BALB/c mice, VEGF gene expression increased from day 3 to 7 or 14 days post‐wound infliction 34, although this did not take bacteriology into consideration. There are a substantial number of studies focusing on VEGF and wound healing. Drinkwater et al. found reduced angiogenesis but increased levels of VEGF gene transcript and proteins levels in human venous ulcers, suggesting an ineffectual drive in these wounds 35.
Current knowledge of VEGF dynamics and contribution to wound repair is incomplete, but it appears likely that excess hypoxia caused by PMNs could be detrimental to wound repair. James et al. found decreased oxygen levels in scabs of diabetic, wounded mice, probably caused by the metabolic activity of P. aeruginosa biofilms and consumption by PMNs 36. A more comprehensive understanding of VEGF activities during the wound‐healing process could be obtained by further research of comparable local wound microenvironments.
Comparing the two inbred strains of mice in order to evaluate host response to the infection, we observed qualitative differences between peripheral wound biopsies from infected poor‐healing BALB/c and infected good‐healing C3H/HeN mice. Central VEGF was the highest in the important early phases of healing in the C3H/HeN mice. An increase in peripheral levels of VEGF was observed in the BALB/c from 4 to 7 DPI but not the C3H/HeN strain of mice, which remained at high levels. Histologically, we observed a surplus of PMNs in the periphery of infected BALB/c wounds compared to central wounds 10 DPI. This supports the hypothesis of the continued infiltration of PMNs observed in chronic biofilm‐infected wounds 1. This finding also supports the theory that BALB/c mice endure an aggravated inflammatory response to local P. aeruginosa biofilm wound infection 16, with the new notion that there is a qualitative difference as well regarding to localisation of inflammation in the wounds. This might also explain the sustained necrosis of the BALB/c strain: gross infection will imply a continuous influx of PMNs 37, thereby keeping the wound in an inflammatory state and obstructing progression towards wound healing. Taken together, the present study provides evidence that the BALB/c strain of mice is a more suitable model for P. aeruginosa biofilm wound infection and for antibiotic intervention studies.
Several clinical studies have described the lack of matrix metalloproteinase‐dependent degradation of growth factors as an important reason for the chronicity of certain recalcitrant wounds 38, 39, 40. However, exogenous application of preferential growth factors such as PDGF‐BB has shown conflicting results in animal diabetic studies 41, 42. However, in a bipedicle ischaemic rat skin flap wound model, the topical application of PDGF‐BB to full‐thickness wounds increased wound‐healing rates 43. PDGF‐BB is the first and only approved exogenously topical drug for human diabetic wounds 44, 45. PDGF‐BB has a crucial role in initial wounding as well as throughout the healing process by stimulating fibroblasts to proliferation 16. Surprisingly, BALB/c levels of PDGF‐BB were in fact increased centrally compared to peripherally in the wounds, regardless of infection, in this study. A significant reduction in central levels was observed from 4 to 10 DPI in BALB/c mice. This could be a reflection of the coagulative necrosis induced by the burn wound infliction. However, all levels were below detection limits in the C3H/HeN strain of mice. PDGF‐BB may be an important mediator of burn wound resolution in BALB/c mice, but further research is required and is beyond the scope of this paper.
We previously reported reduced levels of S100A8/A9 in non‐healing human and in infected murine chronic wounds 18, 21, 22. The present study supports the suppressing effect of P. aeruginosa biofilm on host level of S100A8/A9, especially located towards the centre of the wounds at the earliest point of the evaluation stages (4 DPI). As the course of infection progresses, a great variety is observed in the levels of S100A/A9, and we hypothesise that this protein plays a more significant role early on in the infection. BALB/c mice had higher central levels of S100A8/A9 regardless of infection. Thorey et al. have described the induction of S100A8 and A9 proteins in BALB/c keratinocytes as a result of the infliction of incisional wounds 46.
PMNs as well as monocytes are attracted to the wound site in the acute inflammatory phase. PMNs migrate to the infection site to engulf and kill pathogens 47 followed by a pro‐inflammatory cytokine response, which subsequently activates wound keratinocyte and fibroblasts 48. Monocytes infiltrating the wound site secrete a variety of growth factors 37, among which are PDGF and VEGF, which are essential for granulation tissue formation 47. Activated macrophages are therefore important in the transition from the inflammatory phase to the generation of a provisional wound matrix. A stable count of monocytes (MBC) was noticed throughout the study in infected BALB/c mice. In contrast, in C3H/HeN mice, monocytes were reduced from 4 to 7 DPI and 4 to 10 DPI, possibly reflecting a faster genetic ability to mobilise mononuclear cells from the blood into the wound tissue in this strain as this was also observed in the uninfected animals. A similar pattern of protracted neutrophil infiltration from peripheral blood was previously observed in this model and in our airway model of chronic P. aeruginosa lung infection 18, 19.
BALB/c mice retained larger necrosis throughout the study, and reduction of necrotic areas were delayed in this strain of mice. This could be a clinical consequence of reduced induction of VEGF and innate host response centrally, as well as a lack of mononuclear extravasation.
Importantly, the present study shows a perturbing effect of P. aeruginosa biofilm on host response in wounds and provides a possible explanation for the importance of complete removal of necrotic tissue before intervention or split‐skin transplantation of chronic wounds. However, further experimental and clinical experiments are needed to substantiate this hypothesis.
In conclusion, our findings reveal how infected wounds display strikingly different growth factor levels in different compartments of the same wound during healing. Wound infection increased levels of VEGF universally in the well‐healing C3H/HeN mouse strain but primarily increased in the peripheral zone in susceptible BALB/c wounds. This may benefit P. aeruginosa in the further establishment of biofilm locally. In contrast, S100A8/A9 was partly suppressed by infection but did not display differences in production between the central and the peripheral zones. In general, the differences between the zones and the mouse strains vanished in the late stage of our observation period.
The impact of P. aeruginosa biofilm on wound host response‐determined recruitment of inflammatory cells is an area of great importance. Together with our previous results, this points towards the relevance of local application of VEGF and S100 A8/A9 as adjunctive treatment of non‐healing wounds, although the anatomical localisation of application may be important.
Supporting information
Appendix S1. Supporting information figures.
Acknowledgements
The authors thank Kasper Nørgaard Kragh at the Costerton Biofilm Center of Copenhagen University for excellent aid in the preparation of colour images. We are also grateful for the technical assistance provided by staff from the Core Facility for Integrated Microscopy, Department of Biomedical Sciences, University of Copenhagen, with regards to Zeiss Axio Scan.Z1. The authors state no conflicts of interest. No funding was received for this study.
References
- 1. Bjarnsholt T, Kirketerp‐Møller K, Jensen PO, Madsen KG, Phipps R, Krogfelt K, Høiby N, Givskov M. Why chronic wounds will not heal: a novel hypothesis. Wound Repair Regen 2008;16(1):2–10. [DOI] [PubMed] [Google Scholar]
- 2. Robson MC, Mannari RJ, Smith PD, Payne WG. Maintenance of wound bacterial balance. Am J Surg 1999;178(5):399–402. [DOI] [PubMed] [Google Scholar]
- 3. Edwards R, Harding KG. Bacteria and wound healing. Curr Opin Infect Dis 2004;17(2):91–6. [DOI] [PubMed] [Google Scholar]
- 4. Pukstad BS, Ryan L, Flo TH, Stenvik J, Moseley R, Harding K, Thomas DW, Espevik T. Non‐healing is associated with persistent stimulation of the innate immune response in chronic venous leg ulcers. J Dermatol Sci 2010;59(2):115–22. [DOI] [PubMed] [Google Scholar]
- 5. Nunan R, Harding KG, Martin P. Clinical challenges of chronic wounds: searching for an optimal animal model to recapitulate their complexity. Dis Model Mech 2014;7(11):1205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gottrup F, Jorgensen B. Maggot debridement: an alternative method for debridement. Eplasty 2011;11:e33. [PMC free article] [PubMed] [Google Scholar]
- 7. Schreml S, Meier RJ, Kirschbaum M, Kong SC, Gehmert S, Felthaus O, Kuchler S, Sharpe JR, Woltje K, Weiss KT, Albert M, Seidl U, Schroder J, Morsczeck C, Prantl L, Duschl C, Pedersen SF, Gosau M, Berneburg M, Wolfbeis OS, Landthaler M, Babilas P. Luminescent dual sensors reveal extracellular pH‐gradients and hypoxia on chronic wounds that disrupt epidermal repair. Theranostics 2014;4(7):721–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wysocki AB. Wound fluids and the pathogenesis of chronic wounds. J Wound Ostomy Continence Nurs 1996;23(6):283–90. [DOI] [PubMed] [Google Scholar]
- 9. Gjødsbøl K, Christensen JJ, Karlsmark T, Jørgensen B, Klein BM, Krogfelt KA. Multiple bacterial species reside in chronic wounds: a longitudinal study. Int Wound J 2006;3(3):225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kirketerp‐Møller K, Jensen PO, Fazli M, Madsen KG, Pedersen J, Moser C, Tolker‐Nielsen T, Høiby N, Givskov M, Bjarnsholt T. Distribution, organization, and ecology of bacteria in chronic wounds. J Clin Microbiol 2008;46(8):2717–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Høgsberg T, Bjarnsholt T, Thomsen JS, Kirketerp‐Møller K. Success rate of split‐thickness skin grafting of chronic venous leg ulcers depends on the presence of Pseudomonas aeruginosa: a retrospective study. PLoS One 2011;6(5):e20492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhao G, Hochwalt PC, Usui ML, Underwood RA, Singh PK, James GA, Stewart PS, Fleckman P, Olerud JE. Delayed wound healing in diabetic (db/db) mice with Pseudomonas aeruginosa biofilm challenge: a model for the study of chronic wounds. Wound Repair Regen 2010;18(5):467–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao G, Usui ML, Underwood RA, Singh PK, James GA, Stewart PS, Fleckman P, Olerud JE. Time course study of delayed wound healing in a biofilm‐challenged diabetic mouse model. Wound Repair Regen 2012;20(3):342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watters C, Deleon K, Trivedi U, Griswold JA, Lyte M, Hampel KJ, Wargo MJ, Rumbaugh KP. Pseudomonas aeruginosa biofilms perturb wound resolution and antibiotic tolerance in diabetic mice. Med Microbiol Immunol 2012;202:131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seth AK, Geringer MR, Gurjala AN, Hong SJ, Galiano RD, Leung KP, Mustoe TA. Treatment of Pseudomonas aeruginosa biofilm‐infected wounds with clinical wound care strategies: a quantitative study using an in vivo rabbit ear model. Plast Reconstr Surg 2012;129(2):262e–74e. [DOI] [PubMed] [Google Scholar]
- 16. Trøstrup H, Thomsen K, Christophersen LJ, Hougen HP, Bjarnsholt T, Jensen PO, Kirkby N, Calum H, Høiby N, Moser C. Pseudomonas aeruginosa biofilm aggravates skin inflammatory response in BALB/c mice in a novel chronic wound model. Wound Repair Regen 2013;21(2):292–9. [DOI] [PubMed] [Google Scholar]
- 17. Li X, Gu W, Masinde G, Hamilton‐Ulland M, Xu S, Mohan S, Baylink DJ. Genetic control of the rate of wound healing in mice. Heredity (Edinb) 2001;86(Pt 6):668–74. [DOI] [PubMed] [Google Scholar]
- 18. Trøstrup Pedersen HLC, Christophersen L, Thomsen K, Jensen PØ, Hougen HP, Høiby N, Moser C. Chronic Pseudomonas aeruginosa biofilm infection impairs murine S100A8/A9 and neutrophil effector cytokines ‐ implications for delayed wound closure? Pathogens Disease 2017;75(7). [DOI] [PubMed] [Google Scholar]
- 19. Jensen PO, Moser C, Kobayashi O, Hougen HP, Kharazmi A, Hoiby N. Faster activation of polymorphonuclear neutrophils in resistant mice during early innate response to Pseudomonas aeruginosa lung infection. Clin Exp Immunol 2004;137(3):478–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jackson DM. The diagnosis of the depth of burning. Br J Surg 1953;40(164):588–96. [DOI] [PubMed] [Google Scholar]
- 21. Trøstrup H, Holstein P, Christophersen L, Jorgensen B, Karlsmark T, Hoiby N, Moser C, Agren MS. S100A8/A9 is an important host defence mediator in neuropathic foot ulcers in patients with type 2 diabetes mellitus. Arch Dermatol Res 2016;308:347–55. [DOI] [PubMed] [Google Scholar]
- 22. Trøstrup H, Lundquist R, Christensen LH, Jørgensen LN, Karlsmark T, Haab BB, Ågren MS. S100A8/A9 deficiency in nonhealing venous leg ulcers uncovered by multiplexed antibody microarray profiling. Br J Dermatol 2011;165(2):292–301. [DOI] [PubMed] [Google Scholar]
- 23. Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol 2007;127(3):514–25. [DOI] [PubMed] [Google Scholar]
- 24. Moser C, Johansen HK, Song Z, Hougen HP, Rygaard J, Hoiby N. Chronic Pseudomonas Aeruginosa lung infection is more severe in Th2 responding BALB/c mice compared to Th1 responding C3H/HeN mice. APMIS 1997;105(11):838–42. [PubMed] [Google Scholar]
- 25. Moser C, Hougen HP, Song Z, Rygaard J, Kharazmi A, Hoiby N. Early immune response in susceptible and resistant mice strains with chronic Pseudomonas aeruginosa lung infection determines the type of T‐helper cell response. APMIS 1999;107(12):1093–100. [DOI] [PubMed] [Google Scholar]
- 26. Calum H, Moser C, Jensen PO, Christophersen L, Maling DS, van Gennip M, Bjarnsholt T, Hougen HP, Givskov M, Jacobsen GK, Hoiby N. Thermal injury induces impaired function in polymorphonuclear neutrophil granulocytes and reduced control of burn wound infection. Clin Exp Immunol 2009;156(1):102–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brochmann RP, Toft A, Ciofu O, Briales A, Kolpen M, Hempel C, Bjarnsholt T, Hoiby N, Jensen PO. Bactericidal effect of colistin on planktonic Pseudomonas aeruginosa is independent of hydroxyl radical formation. Int J Antimicrob Agents 2014;43(2):140–7. [DOI] [PubMed] [Google Scholar]
- 28. Trøstrup HHP, Christopersen L, Jørgensen B, Karlsmark T, Høiby N, Moser C, Ågren M. S100A8/A9 is an important host defence mediator in neuropathic foot ulcers in patients with type 2 diabetes mellitus. Arch Dermatol Res 2016;308(5):347–55. [DOI] [PubMed] [Google Scholar]
- 29. Detmar M, Brown LF, Berse B, Jackman RW, Elicker BM, Dvorak HF, Claffey KP. Hypoxia regulates the expression of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) and its receptors in human skin. J Invest Dermatol 1997;108(3):263–8. [DOI] [PubMed] [Google Scholar]
- 30. Xue ML, Thakur A, Willcox M. Macrophage inflammatory protein‐2 and vascular endothelial growth factor regulate corneal neovascularization induced by infection with Pseudomonas aeruginosa in mice. Immunol Cell Biol 2002;80(4):323–7. [DOI] [PubMed] [Google Scholar]
- 31. Martin C, Thevenot G, Danel S, Chapron J, Tazi A, Macey J, Dusser DJ, Fajac I, Burgel PR. Pseudomonas aeruginosa induces vascular endothelial growth factor synthesis in airway epithelium in vitro and in vivo. Eur Respir J 2011;38(4):939–46. [DOI] [PubMed] [Google Scholar]
- 32. Birkenhauer E, Neethirajan S. A double‐edged sword: the role of VEGF in wound repair and chemoattraction of opportunist pathogens. Int J Mol Sci 2015;16(4):7159–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kolpen M, Hansen CR, Bjarnsholt T, Moser C, Christensen LD, van Gennip M, Ciofu O, Mandsberg L, Kharazmi A, Doring G, Givskov M, Høiby N, Jensen PO. Polymorphonuclear leucocytes consume oxygen in sputum from chronic Pseudomonas aeruginosa pneumonia in cystic fibrosis. Thorax 2010;65(1):57–62. [DOI] [PubMed] [Google Scholar]
- 34. Kubo H, Hayashi T, Ago K, Ago M, Kanekura T, Ogata M. Temporal expression of wound healing‐related genes in skin burn injury. Leg Med (Tokyo) 2014;16(1):8–13. [DOI] [PubMed] [Google Scholar]
- 35. Drinkwater SL, Burnand KG, Ding R, Smith A. Increased but ineffectual angiogenic drive in nonhealing venous leg ulcers. J Vasc Surg 2003;38(5):1106–12. [DOI] [PubMed] [Google Scholar]
- 36. James GA, Zhao AG, Usui M, Underwood RA, Nguyen H, Beyenal H, deLancey Pulcini E, Agostinho Hunt A, Bernstein HC, Fleckman P, Olerud J, Williamson KS, Franklin MJ, Stewart PS. Microsensor and transcriptomic signatures of oxygen depletion in biofilms associated with chronic wounds. Wound Repair Regen 2016;24:373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Martin P. Wound healing‐‐aiming for perfect skin regeneration. Science 1997;276(5309):75–81. [DOI] [PubMed] [Google Scholar]
- 38. Trengove NJ, Stacey MC, MacAuley S, Bennett N, Gibson J, Burslem F, Murphy G, Schultz G. Analysis of the acute and chronic wound environments: the role of proteases and their inhibitors. Wound Repair Regen 1999;7(6):442–52. [DOI] [PubMed] [Google Scholar]
- 39. Yager DR, Nwomeh BC. The proteolytic environment of chronic wounds. Wound Repair Regen 1999;7(6):433–41. [DOI] [PubMed] [Google Scholar]
- 40. Eming SA, Koch M, Krieger A, Brachvogel B, Kreft S, Bruckner‐Tuderman L, Krieg T, Shannon JD, Fox JW. Differential proteomic analysis distinguishes tissue repair biomarker signatures in wound exudates obtained from normal healing and chronic wounds. J Proteome Res 2010;9(9):4758–66. [DOI] [PubMed] [Google Scholar]
- 41. Park SA, Raghunathan VK, Shah NM, Teixeira L, Motta MJ, Covert J, Dubielzig R, Schurr M, Isseroff RR, Abbott NL, McAnulty J, Murphy CJ. PDGF‐BB does not accelerate healing in diabetic mice with splinted skin wounds. PLoS One 2014;9(8):e104447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li H, Fu X, Zhang L, Huang Q, Wu Z, Sun T. Research of PDGF‐BB gel on the wound healing of diabetic rats and its pharmacodynamics. J Surg Res 2008;145(1):41–8. [DOI] [PubMed] [Google Scholar]
- 43. Gowda S, Weinstein DA, Blalock TD, Gandhi K, Mast BA, Chin G, Schultz GS. Topical application of recombinant platelet‐derived growth factor increases the rate of healing and the level of proteins that regulate this response. Int Wound J 2013;12:564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Margolis DJ, Crombleholme T, Herlyn M. Clinical protocol: phase I trial to evaluate the safety of H5.020CMV.PDGF‐B for the treatment of a diabetic insensate foot ulcer. Wound Repair Regen 2000;8(6):480–93. [DOI] [PubMed] [Google Scholar]
- 45. Greenhalgh DG. Models of wound healing. J Burn Care Rehabil 2005;26(4):293–305. [DOI] [PubMed] [Google Scholar]
- 46. Thorey IS, Roth J, Regenbogen J, Halle JP, Bittner M, Vogl T, Kaesler S, Bugnon P, Reitmaier B, Durka S, Graf A, Wockner M, Rieger N, Konstantinow A, Wolf E, Goppelt A, Werner S. The Ca2+−binding proteins S100A8 and S100A9 are encoded by novel injury‐regulated genes. J Biol Chem 2001;276(38):35818–25. [DOI] [PubMed] [Google Scholar]
- 47. Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med 1999;341(10):738–46. [DOI] [PubMed] [Google Scholar]
- 48. Hubner G, Brauchle M, Smola H, Madlener M, Fassler R, Werner S. Differential regulation of pro‐inflammatory cytokines during wound healing in normal and glucocorticoid‐treated mice. Cytokine 1996;8(7):548–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting information figures.