

Abstract
Vitamin C (VitC) or ascorbic acid (AscA), a cofactor for collagen synthesis and a primary antioxidant, is rapidly consumed post‐wounding. Parenteral VitC administration suppresses pro‐inflammatory responses while promoting anti‐inflammatory and pro‐resolution effects in human/murine sepsis. We hypothesised that VitC could promote wound healing by altering the inflammatory, proliferative and remodelling phases of wound healing. Mice unable to synthesise VitC (Gulo−/−) were used in this study. VitC was provided in the water (sufficient), withheld from another group (deficient) and supplemented by daily intra‐peritoneal infusion (200 mg/kg, deficient + AscA) in a third group. Full thickness excisional wounds (6 mm) were created and tissue collected on days 7 and 14 for histology, quantitative polymerase chain reaction (qPCR) and Western blotting. Human neonatal dermal fibroblasts (HnDFs) were used to assess effects of In conclusion, VitC favorably on proliferation. Histological analysis showed improved wound matrix deposition and organisation in sufficient and deficient +AscA mice. Wounds from VitC sufficient and deficient + AscA mice had reduced expression of pro‐inflammatory mediators and higher expression of wound healing mediators. Supplementation of HnDF with AscA induced the expression of self‐renewal genes and promoted fibroblast proliferation. VitC favourably impacts the spatiotemporal expression of transcripts associated with early resolution of inflammation and tissue remodelling.
Keywords: Cell cycle progression, Fibroblast proliferation, Inflammation, Vitamin C, Wound healing
Introduction
Wound healing is a normal physiological process aimed at restoring the anatomical structure and function of injured skin 1. Many factors have been identified that affect wound healing, such as age, wound location, wound size, nutritional status, immune system status and underlying co‐morbidities such as diabetes and obesity 2, 3. Delayed wound healing can result in a number of complications such as increased length of hospital stay, amputations and even death 3. There is a 2% prevalence of chronic, non‐healing wounds in the general population that is associated with an annual estimated cost of more than $50 billion and this expenditure is expected to rise in the coming years 4. Hence there is an unmet need to better identify the mechanisms that delay normal wound healing.
One of the desired goals of wound repair is to adequately restore the physical barrier (wound closure) without a loss of function within a reasonable time frame. Re‐establishing the integrity of injured skin necessitates a delicate balance between four sequential yet overlapping stages: haemostasis, inflammation, proliferation and remodelling (maturation) 5. This process involves a highly ordered series of cellular events: platelet activation and fibrin clot formation at the wound site (haemostasis); polymorphonuclear neutrophil (PMN) infiltration to contain invading micro‐organisms and clear damaged matrix and tissue debris, followed by macrophage infiltration to engulf and clear apoptotic sated PMNs (inflammation); and fibroblast migration and proliferation (proliferation) to lay down the new matrix collagen that progressively matures, cross‐links and organises (remodelling/maturation) 5. These cellular events are orchestrated by chemokines and cytokines such as platelet derived growth factor (PDGF), transforming growth factor‐beta (TGF‐β), vascular endothelial growth factor (VEGF), connective tissue growth factor (CTGF), interleukin‐6 (IL‐6), interleukin‐1β (IL‐1β) and tumour necrosis factor‐alpha (TNF‐α) 6, 7 whose expressions ultimately determine the course and fate of wound healing.
Wounds create an environment of higher catabolic state 8. Following injury, the rate at which micronutrients are metabolised increases significantly often leading to critical deficiencies 8. Indeed, levels of vitamin C (VitC), a small, organic, water‐soluble micronutrient with strong antioxidant properties 9, 10, fall rapidly during inflammation. Moreover, scorbutic individuals experience delayed healing and decreased rates of collagen synthesis and maturation 11. Along with its strong antioxidant properties, VitC is an essential co‐factor for multiple enzymatic reactions and has recently been shown to suppress pro‐inflammatory processes by pleiotropic mechanisms while promoting anti‐inflammatory and pro‐resolution effects in macrophages 12, 13, 14, 15. VitC is also intimately involved in collagen metabolism and regulation and therefore many studies have focused on its particular role in wound healing 16, 17, 18, 19. Humans lack functional L‐gulono‐γ‐lactone oxidase (Gulo), the final enzyme for VitC biosynthesis, and hence are dependent upon an external supply (in diet) of VitC 9, 10. In contrast, wild‐type mice express functional Gulo and maintain high levels of VitC in their tissues. In order to better understand the role of VitC in wound healing we used humanised knock‐out mice lacking Gulo (Gulo−/−) in our studies. In these studies, we went beyond the known effects of VitC on collagen synthesis to explore the role of VitC on the spatiotemporal changes in the inflammatory, proliferative and maturation stages of wound healing.
Methods
Animals
All animal studies were performed in accordance with the Virginia Commonwealth University Animal Care and Use Committee's approved protocols. Gulo−/− mice were bred in‐house from an established homozygous colony maintained on a C57BL/6J background as previously described 14. Mice were fed ad libitum with regular chow and had free access to water supplemented with AscA (330 mg/l) renewed twice a week to yield sufficient mice. Supplements were given in de‐ionised water with 20 µl of 0·5 M EDTA/l to increase the stability of the AscA in solution. At week 10 of age, some mice were rendered VitC deficient by reducing VitC supplementation for one week (33 mg/l), followed by complete removal of VitC supplementation for an additional week. This reduced supplementation was shown to result in very low plasma VitC concentration, yet insufficient to result in scurvy 20, 21. In this study, mice were divided into three groups: VitC sufficient, VitC deficient and VitC deficient + AscA mice. The third group was given daily parenteral AscA (200 mg/kg intraperitoneal (i.p.) injection) and AscA‐supplemented drinking water (330 mg/l) for up to 14 days following wounding.
Surgical procedure
Animals were maintained under isoflurane anaesthesia. In addition, they were also injected subcutaneously with the analgesic buprenorphine (4 µg/ml) for pain management. Wounds were created using the methods described by Galiano et al. 22. Briefly, the mouse hair on the dorsum of the mice thoracic curvature was shaved with an electric clipper followed by an additional treatment with a depilatory cream (Nair), for 3 minutes, to remove any remaining fur. The surgical area was then neutralised with betadine followed by another disinfection step with alcohol swabs. Two full thickness excisional wounds were created on the back of VitC sufficient/deficient Gulo−/− mice using a sterile 6‐mm biopsy punch. Wounds were covered with non‐adherent dressing (Telfa™, Covidien) Minneapolis, MN and the animals were housed individually with ad libitum water and food access as discussed earlier. At day 7 and day 14 post‐wounding, mice were anaesthetised, blood was collected via cardiac puncture (anti‐coagulated with sodium citrate 1:10) and kept on ice to be processed for VitC analysis. Mice were then sacrificed and the wound tissue was collected using forceps and scissors. Per mouse, the tissue from one wound was fixed in formalin for 48 hours and processed for histology. The tissue from the second wound was excised, bisected and stored in liquid nitrogen followed by long‐term storage at −80°C for subsequent RNA and protein extraction.
Human neonatal dermal fibroblast (HnDF) culture
Primary HnDF cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained in high glucose, Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% foetal bovine serum, penicillin (100 µg/ml) and streptomycin (100 µg/ml) under a 5% CO2 atmosphere at 37°C. All experiments were performed using cells at passages 3–5.
VitC analysis
Blood samples obtained from mice on days 7 and 14 post‐wounding were kept on ice, centrifuged and the resultant plasma was deproteinised as previously described 15. Briefly, 100 µl of plasma was deproteinised with 200 µl of cold 20% trichloroacetic acid (TCA), and treated with 200 µl of cold 0·2% dithiothreitol (DTT) to prevent oxidation. The mixture was vortexed intermittently for 2 minutes and centrifuged (10 000g, 4°C, 10 minutes). Supernatants were stored at −80°C for batch analysis using a fluorescence end‐point assay 23.
Histological staining and wound assessment
Formalin‐fixed paraffin‐embedded wound sections (3–4 µm) were cleared in xylene washes, rehydrated in a series of decreasing alcohol concentrations and brought to water. Prepared specimens were stained with Masson's Trichrome according to the manufacturer's protocol (Richard Allan Scientific, Catalogue # KTRA87019, Kalamazoo, MI).
RNA isolation and real‐time quantitative PCR (QPCR) analysis
Total RNA isolation and real‐time qPCR were performed as described previously 15. Assays were run in triplicate with no template controls and no reverse transcriptase controls. The mRNA expression from a deficient mouse or a ‘media’ well was set to ‘1·0’ and mRNA expression of all other samples was compared relative to this sample and represented as a fold change. To normalise for differences in the amount of total RNA added to each cDNA reaction and possible variation in the reverse transcriptase efficiency among the different cDNA reactions, the housekeeping gene 18S rRNA was used. Automated gene expression analysis was performed using the Comparative Quantitation module of MxPro QPCR Software Santa Clara, CA (Agilent). Both the forward and reverse primers for each target are listed in Table 1.
Table 1.
Murine and human primers used for QPCR
| Name | Sequence 5′ to 3′ |
|---|---|
| Murine IL‐1β forward | CTGAACTCAACTGTGAAATGCC |
| Murine IL‐1β reverse | CAGGTCAAAGGTTTGGAAGC |
| Murine TNF‐α forward | GATGAGAAGTTCCCAAATGGC |
| Murine TNF‐α reverse | TTGGTGGTTTGCTACGACG |
| Murine KC forward | CAATGAGCTGCGCTGTCAGTGCCTGCAG |
| Murine KC reverse | CTGAACCAAGGGAGCTTCAGGGTC |
| Murine MPO forward | CTGGATCATGACATCACCTTGACTCC |
| Murine MPO reverse | GATCTGGTTGCGAATGGTGATGTTGTTCC |
| Murine HO‐1 forward | GGTACACATCCAAGCCGAGAATGCTGAG |
| Murine HO‐1 reverse | CGGTGCAGCTCCTCAGGGAAGTAGAG |
| Murine VEGF forward | GAGACCCTGGTGGACATC |
| Murine VEGF reverse | CTTTCTTTGGTCTGCATTCAC |
| Murine CTGF forward | CCCAACTATGATGCGAGCC |
| Murine CTGF reverse | ACAGGCTTGGCGATTTTAGG |
| Murine TGF‐β forward | TGACGTCACTGGAGTTGTACGG |
| Murine TGF‐β reverse | CCACGTGGAGTTTGTTATCTTTGC |
| Murine Gal1 forward | CAGCAACCTGAATCTCAAACC |
| Murine Gal1 reverse | AGTGTAGGCACAGGTTGTTGC |
| Human IL‐6 forward | GGATTCAATGAGGAGACTTGCC |
| Human IL‐6 reverse | TCTGCAGGAACTGGATCAGG |
| Human Nanog forward | AATGTCTTCTGCTGAGATGCC |
| Human Nanog reverse | GCTGTCCTGAATAAGCAGATCC |
| Human OCT4 forward | CTTGCTGCAGAAGTGGG |
| Human OCT4 reverse | CACTCGGTTCTCGATACTGG |
| Human p21 forward | CTGTCTTGTACCCTTGTGCC |
| Human p21 reverse | CCTCTTGGAGAAGATCAGCC |
| Human p27 forward | TGGACCCAAAGACTGATCC |
| Human p27 reverse | CATTTTCTTCTGTTCTGTTGGC |
CTGF, connective tissue growth factor; HO1, hemoxygenase‐1; TGF‐β, transforming growth factor‐β; TNF‐α, tumour necrosis factor‐α; VEGF, vascular endothelial growth factor.
Western blot analysis
Wound tissue homogenates and HnDF whole cell and nuclear extracts were isolated and used for Western blot analysis as described previously 15. Nuclear extracts were isolated using the NE‐PER kit (Pierce Biotechnology, Rockford, IL). Antibodies to hemoxygenase‐1 (HO‐1) (ADI‐SPA‐896‐D, Enzo life sciences, Farmingdale, NY), OCT‐3/4 (sc‐5279, Santa Cruz Biotechnology, Santa Cruz, CA), lamin B (sc‐6216, Santa Cruz Biotechnology), and actin (sc‐1616, Santa Cruz Biotechnology) were used in this study. Optical densities of antibody‐specific bands were determined using Quantity One acquisition and analysis software (BioRad, Hercules, CA).
VitC uptake by HnDF
HnDFs were grown under normal culture conditions to confluence in 12‐well plates. Cells were then exposed to media alone or AscA (0·5 and 1 mM) for 3 hours. Media was removed and cells were washed twice with PBS. To each well, 50 µl of tissue culture grade water was added. Cells were lysed by repeated freeze–thaw cycles (×3). Lysates were treated with TCA and DTT and intracellular VitC content was determined as described above.
HnDF proliferation assay
HnDFs were seeded into 96‐well plates at an initial density of 3000 cells per well. After overnight incubation, fresh media containing 0, 0·5 or 1 mM AscA was added and incubated for 24 hours. Media was aspirated and wells washed twice with PBS. Proliferation was assessed using the CyQUANT® cell proliferation assay kit (Invitrogen, Carlsbad, CA) according to manufacturer instructions.
Healed skin tensile testing
For these set of experiments, mice were sacrificed at day 14 post‐wounding and 20 × 4 mm skin specimens were harvested along the mouse central axis. Specimens were wrapped in PBS‐moistened gauze and stored at 4°C overnight prior to testing. Dimensions of each specimen were taken three times using a digital calliper and the average was used to calculate the cross‐sectional area. Two samples were harvested per animal with the healed wound area located mid‐substance. Mechanical testing was performed at room temperature using MTS tensile testing machine and Testworks 4.06A software (MTS Systems, Eden Prairie, MN). The starting conditions were a preload of 0·01 Newtons (N), and tensile grip moving rate of 10 mm/minutes. Only samples that failed mid‐substance were included in the analysis, otherwise samples were excluded (due to failure at the clamp or samples that slipped from the clamp).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA). Data are expressed as mean ± SE. Results were compared using one‐way ANOVA. Post‐hoc Tukey test was used to carry out multi‐comparisons between groups. The level of significance for all statistical tests was P < 0·05.
Results
Plasma VitC levels affect the progression of wound healing
To examine the effect of VitC on wound progression, three groups of Gulo−/− mice were generated and subjected to the wounding procedure described in the Methods section. As seen in Figure 1A, at day 7 post‐wounding, VitC sufficient mice had significantly higher circulating plasma VitC when compared to the deficient group. The VitC levels in the deficient +AscA group were also significantly higher than the deficient group by day 7. By day 14, circulating plasma VitC levels in the sufficient and deficient + AscA were comparable and significantly higher than the deficient group (Figure 1B). Masson's trichrome stained wound sections on day 7 demonstrated dense granulation tissue and higher collagen deposition (blue colour) from sufficient and deficient + AscA mice when compared to the wound sections from deficient mice (Figure 2A–C). In the VitC groups, the basal lamina and keratin layer were well developed and present as distinct layers. In contrast, day 7 sections from deficient mice demonstrated loose granulation tissue with reduced collagen deposition. Additionally, in deficient mice, the dermis was more cellular (and reticular) and the lamina was not yet fully distinct (Figure 2B). Most of these differences continued to be evident on day 14 post‐wounding (Figure 2D–F).
Figure 1.
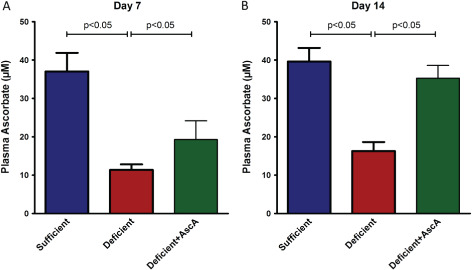
Plasma vitamin C (VitC) levels from VitC sufficient, deficient and deficient + AscA Gulo−/− mice at day 7 (A) and day 14 (B) post wounding. Daily supplementation of deficient mice with AscA post‐wounding resulted in significantly higher plasma VitC levels in the deficient + AscA mice compared to deficient mice by day 7 post wounding. These levels further rose and were comparable to plasma levels of sufficient mice at day 14 (n = 3–6 mice per group).
Figure 2.
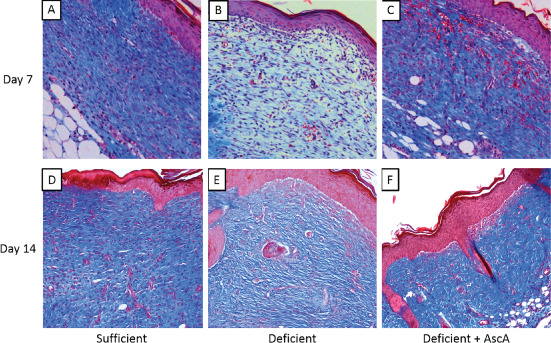
Representative Masson's trichrome stained sections from vitamin C (VitC) sufficient, deficient, and deficient + AscA Gulo−/− mice at day 7 (A–C) and day 14 (D–F) post‐wounding. VitC sufficient (A) and deficient + AscA (C) sections demonstrated more dense granulation tissue with significant collagen deposition (blue colour) and distinct lamina. In contrast, sections from the deficient mice were more cellular with loose granulation tissue and reduced collagen deposition (B). Also the lamina remained indistinct. Similar observations were evident from day‐14 sections (D–F) with the VitC deficient mice group still showing considerably reduced collagen deposition (n = 3–6 mice per group).
VitC attenuates mediators of inflammation in wound healing
Next we examined the mRNA expression of the pro‐inflammatory genes (IL‐1 β, KC, TNF‐α and MPO) in healing tissue on days 7 and 14 post‐wounding. VitC sufficiency or treatment of deficient mice with AscA significantly attenuated transcript levels of these pro‐inflammatory genes when compared to wound tissue from VitC deficient mice at day 7 (Figure 3). The transcript levels of the pro‐inflammatory signalling cytokines (IL‐1β and KC) remained elevated on day 14 in the deficient mice compared to the sufficient and deficient + AscA mice (Figure 4). MPO transcripts however were not detectable in all three groups on day 14 (data not shown). TNF‐α transcript expression on day 14 showed the same trend as IL‐1β and KC but these differences in expression did not achieve statistical significance. In light of the elevated and persistent expression of pro‐inflammatory mediator transcripts in VitC‐deficient animals, we examined whether markers of inflammation resolution might be altered. Galectin‐1 (Gal‐1) expression is associated with generation of pro‐resolving lipid mediators and successful resolution of inflammation 24. Therefore, we examined Gal‐1 expression in day‐7 wounds. Gal‐1 transcript levels were significantly higher in VitC sufficient mice compared with the deficient mice (Figure 5). Although Gal‐1 gene expression trended higher in the deficient + AscA group, it did not reach statistical significance.
Figure 3.
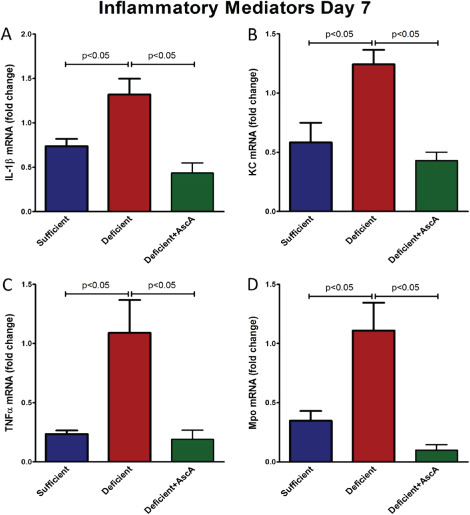
Real time quantitative polymerase chain reaction (qPCR) for IL‐1β (A), KC (B), TNF‐α (C) and MPO (D) mRNA from day‐7 wounds of vitamin C (VitC) sufficient deficient, and deficient + AscA Gulo−/− mice. Wound tissue from VitC sufficient and deficient + AscA mice demonstrated an attenuated pro‐inflammatory gene expression profile compared to wound tissue from deficient mice (n = 3–6 for each group).
Figure 4.
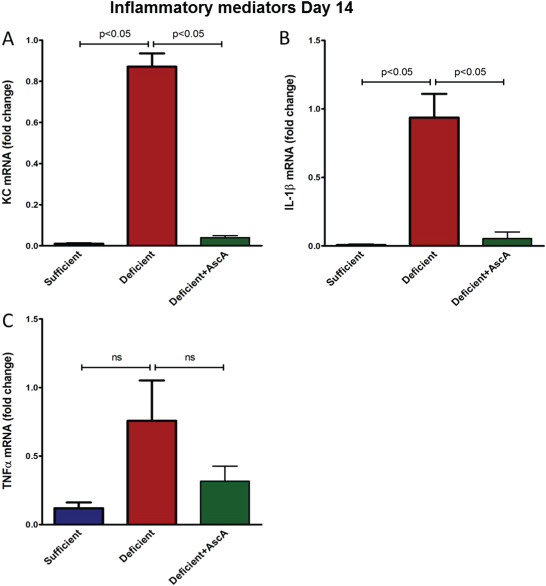
Real time quantitative polymerase chain reaction (qPCR) for IL‐1β (A), KC (B) and TNF‐α (C) mRNA from day‐14 wounds of vitamin C (VitC) sufficient deficient, and deficient + AscA Gulo−/− mice. Wounds from VitC deficient still exhibited a heightened pro‐inflammatory gene expression response compared to VitC sufficient/supplemented mice (n = 3–6 for each group, ns, not significant).
Figure 5.
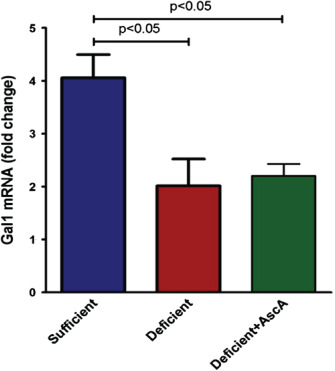
Real time quantitative polymerase chain reaction (qPCR) for Gal‐1 from day 7 wounds. Vitamin C (VitC) sufficiency is associated with significantly higher Gal‐1 expression. VitC sufficient, but not deficient + AscA, showed significantly higher Gal‐1 expression compared to VitC deficient wounds at day 7 (n = 3–6 for each group).
VitC induces genes that promote wound healing
We also examined the expression of transcripts levels of several other known modulators of wound healing including hemoxygenase‐1 (HO‐1), vascular endothelial growth factor (VEGF), connective tissue growth factor (CTGF) and transforming growth factor‐beta (TGF‐β). On day 7 post‐wounding, both VitC sufficient and deficient + AscA Gulo−/− mice showed significantly higher mRNA expression of HO‐1, VEGF and CTGF (Figure 6A–C) in comparison to the deficient mice. TGF‐β expression was significantly elevated in sufficient mice compared to deficient mice. TGF‐β expression following AscA infusion trended higher in the deficient + AscA group but did not reach statistical significance (Figure 6D). Induction at the protein level was confirmed by Western blot analysis for total HO‐1 (Figure 6E).
Figure 6.
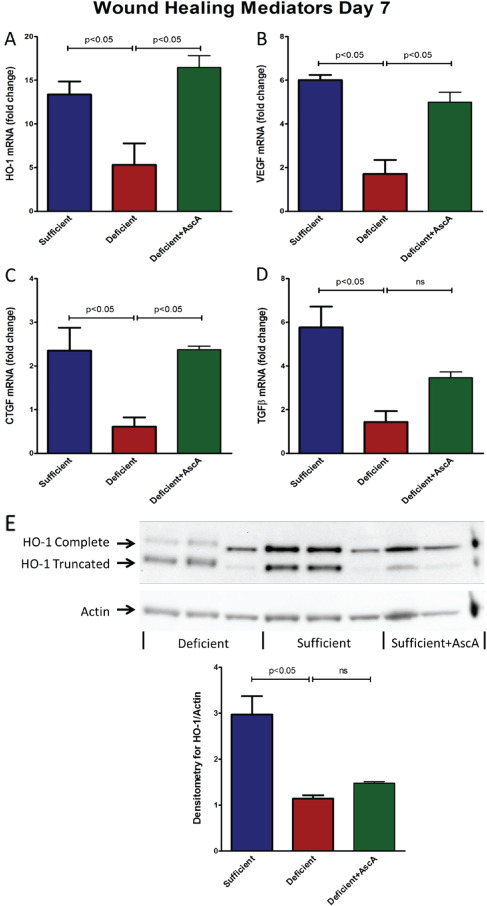
(A–D) Spatiotemporal profiling of HO‐1 and growth factors mRNA expression in day 7 wounds. Real time quantitative polymerase chain reaction (qPCR) for HO‐1 (A), VEGF (B), CTGF (C) and TGF‐β (D) mRNA from day‐7 wounds from vitamin C (VitC) sufficient, deficient, and deficient + AscA Gulo−/− mice (n = 3–6 mice/group, ns, not significant). (E): HO‐1 protein expression on day 7 post wounding. Representative Western blot for expression of HO‐1 and actin from day 7 wounds of VitC sufficient, deficient, and deficient + AscA Gulo−/− mice. The bar chart is a quantitative representation of the Western blot results using densitometry (n = 3–6 for each group, ns, not significant).
VitC and tensile strength of healed wound
Using the excisional wound model, samples collected at day 14 post‐wounding were used to carry out skin tension studies as described in the Methods section. Stress at the healing section was measured using the mid‐specimen cross‐sectional area (in kilopascals). The healed skin samples from the sufficient mice demonstrated a significantly higher stiffness compared to the other groups (Figure 7). However, there were no statistically significant differences between the deficient and deficient + AscA groups. In addition, we also measured the peak load, a measure of the tensile strength that a material can withstand before breaking. However, no significant differences were observed between the different mice groups (data not shown).
Figure 7.
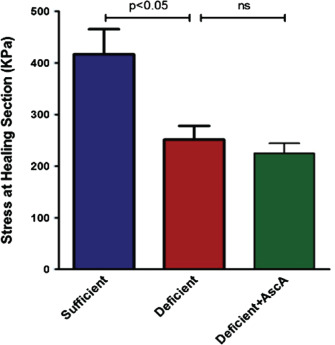
Stress at healing section of wounds collected on day 14 post‐wounding. Vitamin C (VitC) sufficient mice demonstrated a significantly higher stiffness (calculated at mid specimen) compared with the other groups. There was no statistically significant difference in the wound stiffness between the deficient and deficient + AscA groups (n = 6–12; ns, non‐significant).
VitC promotes HnDF proliferation
Confluent HnDF cells were incubated for 3 hours with two concentrations of AscA (0·5 and 1 mM). In the absence of exogenous AscA, intracellular VitC concentrations in HnDFs were extremely low. There was a dose‐ and time‐dependent increase in intracellular VitC concentrations following AscA loading (Figure 8A) suggesting that these cells have the ability to transport VitC intracellularly. Additionally, incubating sub‐confluent HnDF cells with the same AscA concentrations (0·5 and 1 mM) for 24 hours resulted in significant dose‐dependent increases in HnDF proliferation (24% and 40%, respectively) as seen in Figure 8B.
Figure 8.
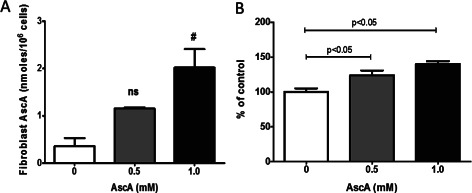
Vitamin C (VitC) (A) uptake and proliferation (B) by HnDF. (A) HnDF VitC concentrations were determined as described in Methods following exposure to AscA for 3 hours (0·5 and 1·0 mM) (n = 3 for each group, ns, not significant). (B) HnDF loading with AscA at 0·5 and 1·0 mM for 24 hours resulted in a dose‐dependent increase in cell proliferation (n = 3 for each group).
VitC induces the expression of self‐renewal, cell‐cycle progression and fibroblast motility genes in HnDF
To further investigate the increased proliferative capacity brought by AscA, HnDF cells were incubated with AscA (0·5 and 1 mM) for 3 hours and mRNA expression of the self‐renewal genes Nanog and octamer‐binding factor 4 (OCT4) were determined by QPCR. Both Nanog and OCT4 gene expression increased in a dose‐dependent fashion following HnDF incubation with AscA (Figure 9A and B). The increased expression was not statistically significant with low level AscA (0·5 mM) but was significant with the higher AscA concentration (P < 0·05). The mRNA expression levels of the cyclin‐dependent kinase inhibitors and regulators of cell cycle, p21 and p27 were also determined (Figure 9C and D). Transcript levels of p27 were significantly decreased in a dose‐dependent fashion by AscA exposure. However p21 expression levels were unchanged. Western blot analysis confirmed the increased nuclear expression of OCT4 seen with QPCR at 24 hours post incubation with AscA (Figure 9E).
Figure 9.
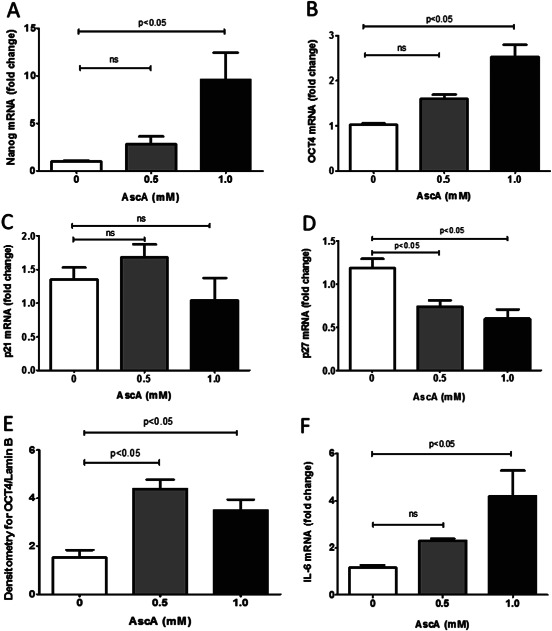
Real‐time quantitative polymerase chain reaction (qPCR) for Nanog (A), Oct4 (B), p21 (C), p27 (D) and interleukin‐6 (IL‐6) (F) from HnDF cells. (A–D and F) HnDF cells were exposed to AscA (0·5 and 1·0 mM) for 3 hours. RNA was harvested and real time QPCR for Nanog, OCT4, p21, p27 and IL‐6 was performed as described in Methods (n = 3 for each group; ns, not significant). (E) Densitometry for nuclear expression of OCT4 and lamin B from HnDF cells loaded with AscA 0·5 and 1 mM for 24 hours (n = 3–6 for each group).
The pleiotropic cytokine IL‐6 has been shown to favourably promote cell motility and matrix remodelling during the inflammatory phase of wound healing 25, 26. In addition, IL‐6 is a resolution promoting cytokine by virtue of its inhibitory effects on pro‐inflammatory signalling via activation of IL‐10 and IL‐13, and by its ability to enhance polarisation of macrophages at wound healing sites towards an anti‐inflammatory/alternatively activated phenotype 27. Therefore we examined IL‐6 mRNA expression following exposure of HnDF cells to AscA. As seen in Figure 9F, exposure of HnDF to AscA produced a dose‐dependent increase in IL‐6 mRNA expression levels; 2·5‐fold and fourfold increase with 0·5 and 1 mM AscA, respectively.
Discussion
Wounds and more generally injuries are associated with rapid micronutrient deficiencies 8. Levels of one critical micronutrient, VitC, have been shown to drop significantly (60–70%) at the wound site and not recover completely even after 14 days post‐wounding 28, 29. These observations may reflect VitC depletion by the plethora of free oxidant radicals generated in the wound microenvironment, and in part by the increased consumption of VitC in different biological processes (e.g. collagen synthesis) that are activated during the repair process. While higher oral intake is achievable, attaining significant plasma levels are limited by gastric intolerance and also by limited absorption and renal excretion thresholds 30. We and others have shown that parenteral VitC is advantageous in bypassing these limitations to produce and sustain adequate plasma levels 30, 31.
In our present study, we were able to show improved wound healing characteristics with VitC sufficiency and also with i.p. VitC repletion of Gulo(−/−) mice. Day‐7 wounds from VitC deficient mice showed a spatiotemporal gene expression characterised by persistent inflammation combined with delayed resolution and proliferation (Figures 3, 4, 5, 6). The pro‐inflammatory state persisted throughout day 14 post‐wounding in the deficient mice (Figure 4) but not in the sufficient or deficient + AscA groups (Figure 4). In contrast, VitC sufficient and supplemented Gulo(−/−) mice showed attenuated expression of pro‐inflammatory cytokines, increased expression of favourable biomarkers of wound healing, and better expression of the pro‐resolution markers of wound healing (Figures 3, 4, 5, 6). However, tangible effects on the skin tensile strength properties at day 14 post‐wounding were evident only in the VitC sufficient mice. These improvements in wound healing in the VitC sufficient and supplemented mice were corroborated by in vitro cultures of HnDF which showed that VitC supplementation led to robust uptake and fibroblast proliferation (Figure 8), induction of self‐renewal genes (Figure 9) and up‐regulation of a pro‐resolution cytokine that also plays a role in fibroblast mobility (Figure 9F).
Histological examination of day 7 and day 14 Masson's Trichrome stained sections (Figure 2) showed that wounds from VitC sufficient and supplemented mice had better matrix organisation with clear distinction of all skin layers and significant extracellular matrix deposition. In contrast, wounds from VitC deficient mice were characterised by a loose dermis that was highly cellular and accompanied by limited collagen deposition. At a molecular level, wounds from deficient mice actively expressed the pro‐inflammatory cytokines IL‐1β and TNF‐α. Additionally, wounds from deficient mice demonstrated significantly higher MPO expression compared with wounds from VitC sufficient and AscA infused mice. MPO is an enzyme almost exclusively produced by PMNs (5% of total neutrophil protein) and to some extent in monocytes and is often used as a marker of PMN infiltration 32. High MPO levels have been found to be associated with poor healing wounds 33. A similar pattern was observed with the gene expression of the PMN chemoattractant factor, KC. Murine KC is analogous to human CXCL‐1 (a functional homologue of IL‐8). We interpret the presence of these pro‐inflammatory markers as ongoing/sustained pro‐inflammatory cellular infiltration into the wound site and an extended pro‐inflammatory phase in wounds from deficient mice. In contrast, the attenuated expression of these pro‐inflammatory mediators in VitC sufficient and AscA infused mice is indicative of termination of pro‐inflammatory cell infiltration to the wound site and perhaps the end of the pro‐inflammatory phase. The expression pattern on day 14 further supports this assumption (Figure 4).
The proliferation stage of wound healing is typically characterised by increased expression of the pro‐reparative growth factor cytokines such as TGF‐β, CTGF and VEGF 6. TGF‐β plays an important role at all stages of wound healing. In the initial stages, it serves as a chemotactic factor for pro‐inflammatory cells as well as fibroblasts. In later stages, TGF‐β provides a strong mitogenic signal for fibroblasts. Braiman‐Wiksman et al. have shown that high levels of TGF‐β are most beneficial after epidermal closure is complete 34. In this work, TGF‐β expression levels were significantly higher in day‐7 wounds from sufficient mice. Wounds from mice receiving daily AscA infusion showed TGF‐β levels that trended higher compared with deficient mice but did not reach statistical significance. Another growth factor in the wound milieu is CTGF whose transcription is regulated mainly by TGF‐β. It is believed that it is a downstream mediator of some of the actions of TGF‐β 35. However, TGF‐β independent regulation of CTGF has also been reported 36. Recently, Alfaro et al. (2013) reported that CTGF levels at wound sites increase during the proliferation phase 37. CTGF functions in promoting granulation tissue formation (fibroblast cell division and migration and fibroblast matrix deposition, e.g. collagen type I and fibronectin) and wound remodelling 38. In our study, VitC sufficiency and AscA supplementation were associated with significantly higher CTGF expression compared with the deficient mice at day‐7 post‐wounding (P < 0·05). Combined with histological evidence, these results indicate an active cellular and molecular proliferation in the VitC sufficient and AscA supplemented mice versus an attenuated/lagging proliferation response in deficient mice. Several studies have shown that VEGF significantly promotes angiogenesis and neovascularisation in healing wounds 5. Multiple factors such as pH, reduced oxygen tension, increased lactate as well as pro‐inflammatory cytokines have been reported to induce VEGF production at the wound site 5, 7. In our study, we observed higher VEGF expression in the VitC sufficient and AscA infused mice indicating increased angiogenesis and neovascularisation in the wounds of sufficient and AscA supplemented mice. A role for HO‐1 in wound healing has only been recently described 39. Grochot‐Przeczek et al. showed that HO‐1 inhibition adversely affects wound healing in diabetic mice while HO‐1 over‐expression promoted healing 39. Earlier, the same research group showed that HO‐1 upregulated VEGF production and therefore could indirectly promote neovascularisation. In our study, HO‐1 expression levels were significantly higher in VitC sufficient and AscA supplemented mice. In summary, the expression pattern of these growth promoting factors can be interpreted as a robust biological response to wounds in VitC sufficient mice when compared to the deficient mice. In addition, daily i.p. VitC supplementation appears to restore the growth‐promoting response in wounds of VitC deficient mice.
Physiological wound healing involves a regulated fibroblast differentiation into myofibroblasts that play an active role in reconstruction of damaged tissue following injury 40. A critical mediator of this process is Gal‐1, a lectin produced by various tissues. Recently, Lin et al. (2015) showed that Gal‐1 induced myofibroblast activation and proliferation. Moreover Gal‐1(−/−) knockout mice experienced reduced myofibroblast migration, which was corrected with topical Gal‐1 administration in excisional wound models 41. Elsewhere, Perzelova et al. showed that exposure of dermal fibroblasts to Gal‐1 stimulated extracellular matrix production that subsequently supported endothelial cell growth 42. In our studies, only VitC sufficient mice had significantly higher Gal‐1 expression levels as compared with other groups. We have previously shown that both VitC sufficiency and VitC supplementation were able to induce early Gal‐1 expression in peritoneal macrophages, which correlated with the markers of resolution 15. This suggests that VitC may be required to sustain Gal‐1 expression in healing wounds and that the level of supplementation provided to the deficient mice is probably insufficient to adequately restore Gal‐1 expression in healing wounds.
Much to our surprise, only VitC sufficient mice demonstrated significantly higher healed‐skin stiffness compared with the other groups despite the observed increased collagen deposition evident by histological analysis in the sufficient and deficient + AscA mice. Although by day 14, we had achieved plasma VitC levels in the deficient + AscA group that were comparable to the VitC sufficient mice, it is possible that the VitC levels in the healing wound may not have reached the same level as the sufficient mice. While humans have evolved over millennia to up‐regulate the VitC transporter SVCT2 at times of deficiency 43 it is unknown whether SVCT2 is up‐regulated to the same extent in the healing wounds of these knockout mice. Indeed it has been shown that the knockdown of SVCT2 in bone marrow derived stromal cells decreased wound healing, and that supplementing with VitC failed to rescue these cells 44. Therefore, we interpret these results as insufficient uptake of VitC in the healing wound and that perhaps, an additional topical application of VitC at the wound site or a higher dose of i.p. VitC could prove beneficial.
Using HnDF cells, we showed that AscA exposure resulted in a dose‐dependent and significant increase in intracellular AscA uptake and fibroblast proliferation (Figure 8). This was associated with a dose‐dependent inhibition of p27 expression (Figure 9). The inhibitors p21 and p27 suppress cell‐cycle progression by inhibiting cyclin‐dependent kinases as well as by down‐regulating expression of other genes involved with cell‐cycle progression 45. Conversely, AscA treatment was associated with a dose‐dependent increase in the expression of pluripotent and self‐renewal genes, Nanog and OCT4 46. These findings suggest a crucial role for VitC in regulating fibroblast proliferation. Although this is a novel finding in fibroblasts, there is precedence for these actions of VitC in stem cells, and in particular the adipose‐derived stem cells wherein VitC has been used to increase the yield and regenerative potential of adipose‐derived stem cells 47.
IL‐6 is a cytokine with pleotropic functions that is produced by several cell types in the wound microenvironment including fibroblasts 25, 48. It plays an important role in development of Th17 cells and is known to contribute to the inflammatory component of autoimmune diseases 49. However, recent studies suggest a pro‐resolution and anti‐inflammatory role for IL‐6 in wound‐healing environments 27. Kuhn et al. showed that inhibition of IL‐6 resulted in impaired wound healing due to decreased epithelial proliferation 50. IL‐6 knockout mice exhibit delayed wound healing (delayed re‐epithelialisation, granulation tissue formation and sub‐optimal inflammatory response) compared with wild‐type mice; this could be reverted with recombinant IL‐6 51, 52. Further, Gallucci et al. reported IL‐6 signalling to promote dermal fibroblast differentiation into myofibroblasts, increase their motility and also promote matrix remodelling 25, 53. We found that exposure of HnDF cells to AscA promoted a dose‐dependent increase in IL‐6 mRNA levels (Figure 9F). These findings suggest a novel mechanism by which VitC could promote fibroblast motility and matrix remodelling. Presumably, insufficient fibroblast VitC levels could impair IL‐6 expression and delay the resolution of wound healing.
Thus, on the basis of these in vitro studies, we speculate that in the absence of adequate VitC supplementation, the proliferation and maturation phases of wound healing would be delayed or would remain incomplete, thereby resulting in non‐healing wounds.
Conclusion
Wound repair is a complex process that requires the co‐ordination of various local cellular and biochemical events. To date, the majority of studies have used oral supplementation to augment plasma/tissue levels of VitC. However, recent studies by Padayatty and coworkers and others have shown that oral supplementation does not provide the requisite VitC levels for restoration of plasma VitC levels 21, 30. We have recently shown that parenteral routes of VitC administration are safe and effective in critical care situations in both mice and humans 31. Moreover, these doses of VitC, when administered parenterally, were sufficient to restore circulating plasma VitC levels. The current study presents new evidence that beyond its well characterised role in collagen metabolism, VitC plays a crucial role in orchestrating multiple wound‐healing processes. VitC repletion by parenteral infusion has the potential to be a safe and inexpensive therapy for enhancing tissue repair and shortening healing time.
Acknowledgements
This research was supported by grants from the Aubery Sage Mac Farlane Endowment for Acute Lung Injury Research and the VCU Johnson Center for Research.
References
- 1. Lazarus GS, Cooper DM, Knighton DR, Margolis DJ, Percoraro RE, Rodeheaver G, Robson MC. Definitions and guidelines for assessment of wounds and evaluation of healing. Wound Repair Regen 1994;2:165–70. [DOI] [PubMed] [Google Scholar]
- 2. Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, Gottrup F, Gurtner GC, Longaker MT. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen 2009;17:763–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rasik AM, Shukla A. Antioxidant status in delayed healing type of wounds. Int J Exp Pathol 2000;81:257–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fife CE, Carter MJ, Walker D, Thomson B. Wound care outcomes and associated cost among patients treated in US outpatient wound centers: data from the US wound registry. Wounds 2012;24:10–7. [PubMed] [Google Scholar]
- 5. Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci 2004;9:283–9. [DOI] [PubMed] [Google Scholar]
- 6. Shah JMY, Omar E, Pai DR, Sood S. Cellular events and biomarkers of wound healing. Indian J Plast Surg 2012;45:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev 2003;83:835–70. [DOI] [PubMed] [Google Scholar]
- 8. Demling RH. Nutrition, anabolism, and the wound healing process: an overview. Eplasty 2009;9:e9. [PMC free article] [PubMed] [Google Scholar]
- 9. Naidu KA. Vitamin C in human health and disease is still a mystery? An overview. Nutr J 2003;2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Tullio MC. The mystery of vitamin C. Nature Educ 2010;3:48. [Google Scholar]
- 11. Moores J. Vitamin C: a wound healing perspective. Br J Community Nurs 2013;18(12)(Suppl S6):S8–11. [DOI] [PubMed] [Google Scholar]
- 12. Fisher BJ, Seropian IM, Kraskauskas D, Thakkar JN, Voelkel NF, Fowler AA 3rd, Natarajan R. Ascorbic acid attenuates lipopolysaccharide‐induced acute lung injury. Crit Care Med 2011;39:1454–60. [DOI] [PubMed] [Google Scholar]
- 13. Mohammed BM, Fisher BJ, Kraskauskas D, Farkas D, Brophy DF, Fowler AA 3rd, Natarajan R. Vitamin C: a novel regulator of neutrophil extracellular trap formation. Nutrients 2013;5:3131–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fisher BJ, Kraskauskas D, Martin EJ, Farkas D, Puri P, Massey HD, Idowu MO, Brophy DF, Voelkel NF, Fowler AA 3rd, Natarajan R. Attenuation of sepsis‐induced organ injury in mice by vitamin C. JPEN J Parenter Enteral Nutr 2014;38:825–39. [DOI] [PubMed] [Google Scholar]
- 15. Mohammed BM, Fisher BJ, Huynh QK, Wijesinghe DS, Chalfant CE, Brophy DF, Fowler AA 3rd, Natarajan R. Resolution of sterile inflammation: role for vitamin C. Mediators Inflamm 2014;2014:173403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chojkier M, Houglum K, Solis‐Herruzo J, Brenner DA. Stimulation of collagen gene expression by ascorbic acid in cultured human fibroblasts. A role for lipid peroxidation? J Biol Chem 1989;264:16957–62. [PubMed] [Google Scholar]
- 17. Peterkofsky B. The effect of ascorbic acid on collagen polypeptide synthesis and proline hydroxylation during the growth of cultured fibroblasts. Arch Biochem Biophys 1972;152:318–28. [DOI] [PubMed] [Google Scholar]
- 18. Barnes MJ. Function of ascorbic acid in collagen metabolism. Ann N Y Acad Sci 1975;258:264–77. [DOI] [PubMed] [Google Scholar]
- 19. Murad S, Grove D, Lindberg KA, Reynolds G, Sivarajah A, Pinnell SR. Regulation of collagen synthesis by ascorbic acid. Proc Natl Acad Sci U S A 1981;78:2879–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim H, Bae S, Yu Y, Kim Y, Kim HR, Hwang YI, Kang JS, Lee WJ. The analysis of vitamin C concentration in organs of Gulo(−/−) mice upon vitamin C withdrawal. Immune Netw 2012;12:18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vissers MC, Bozonet SM, Pearson JF, Braithwaite LJ. Dietary ascorbate intake affects steady state tissue concentrations in vitamin C‐deficient mice: tissue deficiency after suboptimal intake and superior bioavailability from a food source (kiwifruit). Am J Clin Nutr 2011;93:292–301. [DOI] [PubMed] [Google Scholar]
- 22. Galiano RD, Michaels J 5th, Dobryansky M, Levine JP, Gurtner GC. Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen 2004;12:485–92. [DOI] [PubMed] [Google Scholar]
- 23. Babaev VR, Whitesell RR, Li L, Linton MF, Fazio S, May JM. Selective macrophage ascorbate deficiency suppresses early atherosclerosis. Free Radic Biol Med 2011;50:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rabinovich GA, Sotomayor CE, Riera CM, Bianco I, Correa SG. Evidence of a role for galectin‐1 in acute inflammation. Eur J Immunol 2000;30:1331–9. [DOI] [PubMed] [Google Scholar]
- 25. Luckett LR, Gallucci RM. Interleukin‐6 (IL‐6) modulates migration and matrix metalloproteinase function in dermal fibroblasts from IL‐6KO mice. Br J Dermatol 2007;156:1163–71. [DOI] [PubMed] [Google Scholar]
- 26. Duarte TL, Cooke MS, Jones GD. Gene expression profiling reveals new protective roles for vitamin C in human skin cells. Free Radic Biol Med 2009;46:78–87. [DOI] [PubMed] [Google Scholar]
- 27. Fernando MR, Reyes JL, Iannuzzi J, Leung G, McKay DM. The pro‐inflammatory cytokine, interleukin‐6, enhances the polarization of alternatively activated macrophages. PLoS One 2014;9:e94188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim M, Otsuka M, Yu R, Kurata T, Arakawa N. The distribution of ascorbic acid and dehydroascorbic acid during tissue regeneration in wounded dorsal skin of guinea pigs. Int J Vitam Nutr Res 1994;64:56–9. [PubMed] [Google Scholar]
- 29. Shukla A, Rasik AM, Patnaik GK. Depletion of reduced glutathione, ascorbic acid, vitamin E and antioxidant defence enzymes in a healing cutaneous wound. Free Radic Res 1997;26:93–101. [DOI] [PubMed] [Google Scholar]
- 30. Levine M, Padayatty SJ, Espey MG. Vitamin C: a concentration‐function approach yields pharmacology and therapeutic discoveries. Adv Nutr 2011;2:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fowler AA 3rd, Syed AA, Knowlson S, Sculthorpe R, Farthing D, DeWilde C, Farthing CA, Larus TL, Martin EJ, Brophy DF, Gupta S, MRICU Nursing , Fisher BJ, Natarajan R. Phase I safety trial of intravenous ascorbic acid in patients with severe sepsis. J Transl Med 2014;12:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yager DR, Kulina RA, Gilman LA. Wound fluids: a window into the wound environment? Int J Low Extrem Wounds 2007;6:262–72. [DOI] [PubMed] [Google Scholar]
- 33. Moor AN, Vachon DJ, Gould LJ. Proteolytic activity in wound fluids and tissues derived from chronic venous leg ulcers. Wound Repair Regen 2009;17:832–9. [DOI] [PubMed] [Google Scholar]
- 34. Braiman‐Wiksman L, Solomonik I, Spira R, Tennenbaum T. Novel insights into wound healing sequence of events. Toxicol Pathol 2007;35:767–79. [DOI] [PubMed] [Google Scholar]
- 35. Leask A, Abraham DJ. The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem Cell Biol 2003;81:355–63. [DOI] [PubMed] [Google Scholar]
- 36. Moussad EEA, Brigstock DR. Connective tissue growth factor: what's in a name? Mol Genet Metab 2000;71:276–92. [DOI] [PubMed] [Google Scholar]
- 37. Alfaro MP, Deskins DL, Wallus M, DasGupta J, Davidson JM, Nanney LB, Guney MA, Gannon M, Young PP. A physiological role for connective tissue growth factor in early wound healing. Lab Invest 2013;93:81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic‐Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen 2008;16:585–601. [DOI] [PubMed] [Google Scholar]
- 39. Grochot‐Przeczek A, Lach R, Mis J, Skrzypek K, Gozdecka M, Sroczynska P, Dubiel M, Rutkowski A, Kozakowska M, Zagorska A, Walczynski J, Was H, Kotlinowski J, Drukala J, Kurowski K, Kieda C, Herault Y, Dulak J, Jozkowicz A. Heme oxygenase‐1 accelerates cutaneous wound healing in mice. PLoS One 2009;4:e5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol 2007;127:526–37. [DOI] [PubMed] [Google Scholar]
- 41. Lin Y, Chen JS, Wu MH, Hsieh IS, Liang CH, Hsu CL, Hong TM, Chen YL. Galectin‐1 accelerates wound healing by regulating the neuropilin‐1/Smad3/NOX4 pathway and ROS production in myofibroblasts. J Invest Dermatol 2015;135:258–68. [DOI] [PubMed] [Google Scholar]
- 42. Perzelova V, Varinska L, Dvorankova B, Szabo P, Spurny P, Valach J, Mojzis J, Andre S, Gabius HJ, Smetana K Jr, Gal P. Extracellular matrix of galectin‐1‐exposed dermal and tumor‐associated fibroblasts favors growth of human umbilical vein endothelial cells in vitro: a short report. Anticancer Res 2014;34:3991–6. [PubMed] [Google Scholar]
- 43. Steiling H, Longet K, Moodycliffe A, Mansourian R, Bertschy E, Smola H, Mauch C, Williamson G. Sodium‐dependent vitamin C transporter isoforms in skin: distribution, kinetics, and effect of UVB‐induced oxidative stress. Free Radic Biol Med 2007;43:752–62. [DOI] [PubMed] [Google Scholar]
- 44. Sangani R, Pandya CD, Bhattacharyya MH, Periyasamy‐Thandavan S, Chutkan N, Markand S, Hill WD, Hamrick M, Isales C, Fulzele S. Knockdown of SVCT2 impairs in‐vitro cell attachment, migration and wound healing in bone marrow stromal cells. Stem Cell Res 2014;12:354–63. [DOI] [PubMed] [Google Scholar]
- 45. Coqueret O. New roles for p21 and p27 cell‐cycle inhibitors: a function for each cell compartment? Trends Cell Biol 2003;13:65–70. [DOI] [PubMed] [Google Scholar]
- 46. Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, Wong KY, Sung KW, Lee CW, Zhao XD, Chiu KP, Lipovich L, Kuznetsov VA, Robson P, Stanton LW, Wei CL, Ruan Y, Lim B, Ng HH. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 2006;38:431–40. [DOI] [PubMed] [Google Scholar]
- 47. Kim JH, Kim WK, Sung YK, Kwack MH, Song SY, Choi JS, Park SG, Yi T, Lee HJ, Kim DD, Seo HM, Song SU, Sung JH. The molecular mechanism underlying the proliferating and preconditioning effect of vitamin C on adipose‐derived stem cells. Stem Cells Dev 2014;23:1364–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goodman L, Stein GH. Basal and induced amounts of interleukin‐6 mRNA decline progressively with age in human fibroblasts. J Biol Chem 1994;269:19250–5. [PubMed] [Google Scholar]
- 49. Dayer JM, Choy E. Therapeutic targets in rheumatoid arthritis: the interleukin‐6 receptor. Rheumatology (Oxford) 2010;49:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kuhn KA, Manieri NA, Liu TC, Stappenbeck TS. IL‐6 stimulates intestinal epithelial proliferation and repair after injury. PLoS One 2014;9:e114195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gallucci RM, Simeonova PP, Matheson JM, Kommineni C, Guriel JL, Sugawara T, Luster MI. Impaired cutaneous wound healing in interleukin‐6‐deficient and immunosuppressed mice. FASEB J 2000;14:2525–31. [DOI] [PubMed] [Google Scholar]
- 52. Lin ZQ, Kondo T, Ishida Y, Takayasu T, Mukaida N. Essential involvement of IL‐6 in the skin wound‐healing process as evidenced by delayed wound healing in IL‐6‐deficient mice. J Leukoc Biol 2003;73:713–21. [DOI] [PubMed] [Google Scholar]
- 53. Gallucci RM, Lee EG, Tomasek JJ. IL‐6 modulates alpha‐smooth muscle actin expression in dermal fibroblasts from IL‐6‐deficient mice. J Invest Dermatol 2006;126:561–5688. [DOI] [PubMed] [Google Scholar]